
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Neurol., 21 November 2023
Sec. Headache and Neurogenic Pain
Volume 14 - 2023 | https://doi.org/10.3389/fneur.2023.1282846
 Lisa McConnachie1,2
Lisa McConnachie1,2 Peter J. Goadsby3,4Robert E. Vann2Sutapa Ray2
Peter J. Goadsby3,4Robert E. Vann2Sutapa Ray2 Stephen B. Shrewsbury2
Stephen B. Shrewsbury2 Sheena K. Aurora2*
Sheena K. Aurora2*Introduction: Dihydroergotamine mesylate (DHE) is an established effective acute therapy for migraine and is often characterized by its broad receptor pharmacology. Knowledge of DHE pharmacology largely comes from studies employing older methodologies.
Objective: To assess DHE receptor activity using high-throughput methods to screen for functional ß-arrestin activity at G protein–coupled receptors (GPCRs).
Methods: Functional receptor activities of DHE and sumatriptan succinate (both 10 μM) were screened against 168 GPCRs using the gpcrMAX assay. Agonist and antagonist effects were considered significant if receptor activity was >30% or inhibited by >50%, respectively. Radiolabeled ligand binding assays were performed for DHE (0.01–300 nM for 5-HT3 and 4E; 0.3–10,000 nM for 5-HT1B, α-adrenergic2B [i.e., α2B-adrenoceptor], D2, and D5) to assess specific binding to select receptors.
Results: DHE (10 μM) exhibited agonist activity at α-adrenergic2B, CXC chemokine receptor 7 (CXCR7), dopamine (D)2/5, and 5-hydroxytryptamine (5-HT)1A/1B/2A/2C/5A receptors and antagonist activity at α-adrenergic1B/2A/2C (i.e., α1B/2A/2C-adrenoceptors), calcitonin receptor–receptor activity modifying protein 2 (CTR-RAMP2) or amylin 2 (AMY2), D1/3/4/5, and 5-HT1F receptors. Sumatriptan succinate (10 μM) exhibited agonist activity at the 5-HT1B/1E/1F/5A receptors. DHE demonstrated a half-maximal inhibitory concentration (IC50) of 149 nM at the 5-HT1F receptor and a half-maximal effective concentration (EC50) of 6 μM at the CXCR7 receptor. DHE did not bind to the 5-HT3 receptor at concentrations up to 300 nM and bound poorly to 5-HT4E and D5 receptors (IC50 of 230 and 370 nM, respectively). DHE bound strongly to the D2, 5-HT1B, and α-adrenergic2B receptors (IC50 of 0.47, 0.58, and 2.8 nM, respectively).
Conclusion: By using a high-throughput β-arrestin recruitment assay, this study confirmed the broad receptor profile of DHE and provided an update on DHE receptor pharmacology as it relates to migraine.
Migraine pathophysiology is complex, involving multiple regions of the brain, neurotransmitters, neuropeptides, ion channels, and numerous receptor pathways (1–3). An increased understanding of this pathophysiology has led to the development of novel therapeutic targets for the treatment of migraine. The development of narrowly targeted therapies for the acute treatment of migraine began in the 1980s with the advent of triptans, which are 5-hydroxytryptamine-1B/1D (5-HT1B/1D) receptor agonists, and continued with the recent approval of gepants, which are calcitonin gene-related peptide (CGRP) receptor antagonists, and lasmiditan, which is a 5-HT1F receptor agonist (4–6). By selectively targeting mediators and mechanisms shown to be involved in migraine pathophysiology, pharmacologic agents can potentially alleviate migraine symptoms, including pain, while minimizing unwanted tolerability concerns in patients (7). However, the potential benefit in the interplay between different pathways may then be left unaddressed. A comprehensive description of receptor binding, specifically which key receptors in migraine pathophysiology are activated and how various pathways may influence each other, is critical in understanding the presence or absence of clinical efficacy of migraine therapies.
Dihydroergotamine mesylate (DHE) is a familiar molecule among headache specialists and has been a mainstay for difficult-to-treat migraine, offering patients single-dose efficacy in a rapid and consistent manner (8–10). Over the course of many decades, several review articles on DHE pharmacology have been published, each suggesting that the efficacy of DHE may be attributed to its broad receptor coverage, which includes serotonergic, adrenergic, and dopaminergic receptor activity (11, 12); however, much of our understanding of DHE receptor pharmacology from these review articles are results from studies using older methodologies, performed decades ago. The most recent study was performed by Cook and colleagues, who sought to determine whether differences in the binding and functional activity of intravenous (IV) DHE and an orally inhaled DHE product, MAP0004, could explain the improved adverse event profile observed with MAP0004 (9, 12). A high DHE concentration (5 μM) was used to screen against 65 receptors, ion channels, and enzymes. Using DHE concentrations corresponding to the maximum plasma concentration (Cmax) of 1 mg of IV DHE (53 ng/mL [~0.091 μM]), 4 inhalations of MAP0004 (systemic equivalent to 0.88 mg; 4.3 ng/mL [~0.007 μM]), and 2 inhalations of MAP0004 (systemic equivalent to 0.44 mg; 1.3 ng/mL [~0.002 μM]), a customized radioligand receptor binding screening profile was then performed to determine binding activity, and functional receptor activity was determined with in vitro techniques using several signaling pathways. The receptor binding profile for IV DHE was more extensive compared to both MAP0004 doses (Table 1). With regard to functional receptor binding for IV DHE and MAP0004, functional agonist activity of DHE was demonstrated at the 5-HT1A/1B/1D receptors. Functional antagonist activity at the dopamine (D)2 receptor was reported for IV DHE and 4 MAP0004 inhalations and at the 5-HT2A receptor for IV DHE. Functional antagonist activity was also determined at the α-adrenergic1A/2A/2B receptors (i.e., α1A/2A/2B-adrenoceptors) for IV DHE, with reduced or absent antagonism for both MAP0004 doses. These results demonstrated that the interaction profile with regard to specific receptors was concentration dependent (12).
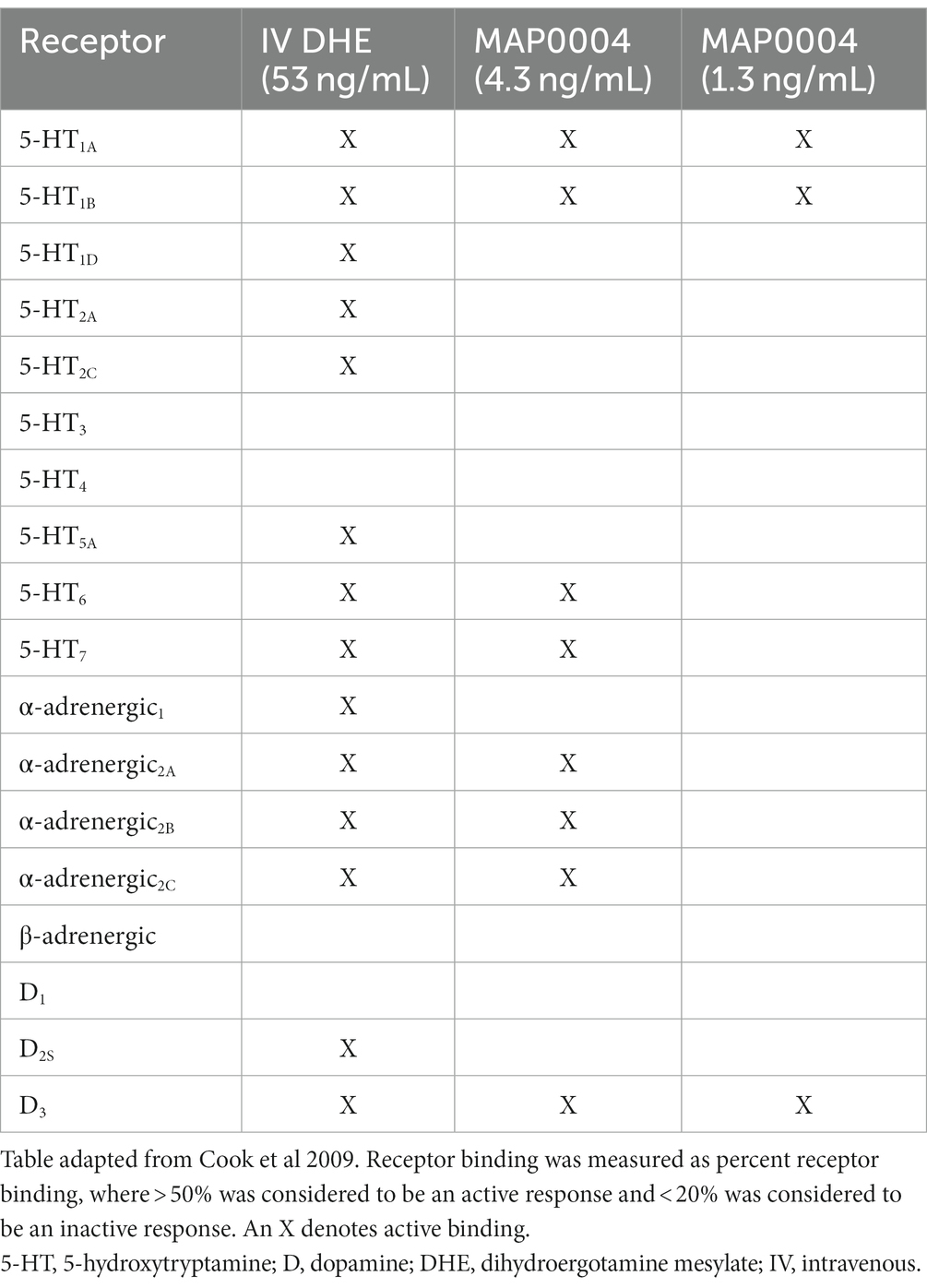
Table 1. Previously published receptor radioligand binding of IV DHE compared with MAP0004 (12).
There have been advances in receptor assay methodology since the study by Cook and colleagues (12). Traditional receptor binding studies, which often require secondary functional activity assays to establish agonism or antagonism, are useful tools to determine the activity of a drug at a specific receptor (12, 13). A more updated approach for assessing receptor activity includes high-throughput methods assessing reporter protein activity, such as ß-arrestin, to screen rapidly for ligand activity, as opposed to binding, at various G protein–coupled receptors (GPCRs) (14). ß-arrestin is a ubiquitously expressed protein that plays an important role in cell signaling, and recruitment of ß-arrestin following ligand binding to GPCRs is well characterized (14–18). Several high-throughput screening approaches that rely on the detection of ß-arrestin recruitment (14, 17) to evaluate unknown ligand binding to GPCRs have been developed (eg, the PRESTO-Tango platform (19) or the PathHunter® Assay (20)) and can be performed relatively rapidly and efficiently. One advantage of these assays is that the activity proximal to, rather than downstream of, specific G protein activation is measured, which may minimize false positives resulting from off-target effects due to downstream signaling cascades (21). Results from these assays provide a comprehensive assessment of binding and activity and are useful for understanding the potential of both on- and off-target effects (14). A potential disadvantage of these approaches is that they are typically performed using one concentration of the test ligand, and follow-up evaluations are required should positive results be obtained in the screening assay. The objective of the present study was to build on previous work to further update our understanding of DHE receptor activity and provide a relevant clinical context for the mechanism of action of DHE as an acute therapy for migraine.
Functional receptor activity of DHE and sumatriptan succinate was screened against 168 GPCRs with the gpcrMAX assay panel, which utilizes the PathHunter β-arrestin enzyme fragment complementation technology (Eurofins DiscoverX; Fremont, CA; Table 2). The gpcrMAX panel evaluates ß-arrestin recruitment and was carried out in agonist and antagonist modes using 10 μM each of DHE and sumatriptan succinate. The human plasma Cmax of DHE depends on the dose, formulation, and route of administration. A phase 1 study assessed the pharmacokinetics of 1.45 mg of INP104 (DHE delivered by Precision Olfactory Delivery; Impel Pharmaceuticals, Seattle, WA), 1.0 mg of IV DHE, and 2.0 mg of DHE nasal spray (Migranal®, Bausch Health Companies, Inc. or its affiliates, Bridgewater, NJ). Human plasma Cmax was ~2 nM (1.3 ng/mL), ~24 nM (14.2 ng/mL), and ~ 0.5 nM (0.3 ng/mL) for INP104, IV DHE, and DHE nasal spray, respectively (24, 25). Similarly, the Cmax was ~173–182 nM (51–53.8 ng/mL), ~54 nM (16 ng/mL), and ~ 240–245 nM (71–72.4 ng/mL) for 100 mg of oral sumatriptan, 20 mg of sumatriptan nasal spray, and 6 mg of subcutaneous sumatriptan, respectively (26–28).
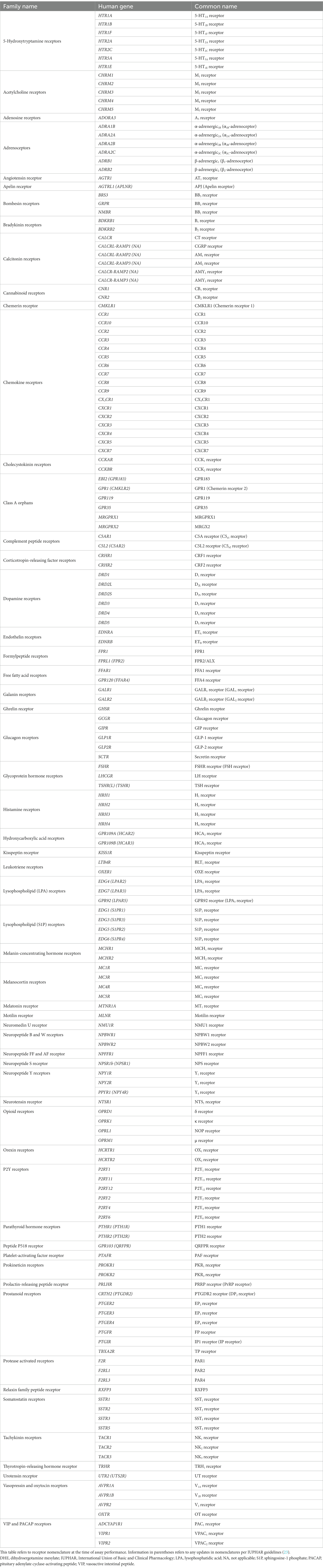
Table 2. Receptors included in the screening for functional receptor activity of DHE (10 μM) (22).
PathHunter cell lines were expanded from freezer stocks according to standard procedures and cells were seeded in a total volume of 20 μL into white-walled, 384-well microplates in duplicate and incubated at 37°C prior to testing. For agonist activity, cells expressing the various receptors were incubated with 10 μM DHE mesylate or 10 μM sumatriptan succinate. Intermediate dilution (1% vehicle concentration) of sample stocks was performed to generate a 5× sample in assay buffer, of which 5 μL was added to the cells and incubated for 90 or 180 min at 37°C or room temperature, depending on the specific receptor, as established by manufacturer optimization protocols (see Supplementary Table 1). For antagonist activity, cells were preincubated with an antagonist for 30 min, followed by an agonist challenge at 80% of the maximal effective concentration (EC80). Intermediate dilution of sample stocks was performed to generate 5× sample in assay buffer, of which 5 μL was added to cells and incubated at 37°C or room temperature for 30 min. This was followed by an addition of 5 μL of 6× EC80 agonist in assay buffer to the cells, which were incubated at 37°C or room temperature for 90 or 180 min. Assay signal for agonist and antagonist modes was generated through a single addition of 12.5 or 15 μL (50% v/v) of PathHunter Detection reagent cocktail, followed by a 1-h incubation at room temperature. Further experimental details can be found in the Supplementary File. Microplates were read with a PerkinElmer EnVision (Shelton, CT) instrument for chemiluminescent signal detection. The gpcrMAX panel does not include a cell line expressing human 5-HT1D.
Because the gpcrMAX panel assessed ß-arrestin recruitment with a single concentration of DHE, a range of concentrations was used to determine DHE binding to 4 select GPCRs (5-HT1B, α-adrenergic2B, D2, and D5). The 5-HT3 and 5-HT4E receptors were also evaluated because they had not been evaluated in the gpcrMAX assay. All assays were performed in duplicate (ie, 2 replicates per assay, per standard manufacturer protocol).
Binding of DHE to the human 5-HT3 receptor was evaluated via a radioligand binding assay in transfected human recombinant HEK-293 cells and performed by Eurofins Panlabs Discovery Services (New Taipei City, Taiwan). Cell membrane homogenates (30 μg protein) were incubated for 60 min at 25°C with 0.69 nM [3H]GR-65630 in the absence or presence of DHE in a buffer containing 50 mM Tris–HCl (pH 7.4), 5 mM MgCl2, and 1 mM ethylenediaminetetraacetic acid (29). The experiment was conducted in a 96-well plate format with 200 μL total volume and 8 concentrations of DHE, ranging from 0.01 to 300 nM. This concentration range was selected to cover the human plasma Cmax of DHE administered by multiple routes. Nonspecific binding was determined in the presence of 10 μM MDL 72222. Following incubation, the samples were filtered rapidly under vacuum through glass fiber filters (GF/B, Packard; Kennesaw, GA) presoaked with 0.3% polyethyleneimine and rinsed several times with ice-cold 50 mM Tris–HCl using a 96-sample cell harvester (UniFilter, Packard). The filters were dried and then counted for radioactivity in a scintillation counter (TopCount, Packard) using a scintillation cocktail (MicroScint 0, Packard). Results are expressed as percent inhibition of the control radioligand–specific binding. The standard reference compound was MDL 72222, which was tested in each experiment at several concentrations to obtain a competition curve from which its half-maximal inhibitory concentration (IC50) was calculated.
Binding of DHE to the human 5-HT4E receptor was similarly evaluated via a radioligand binding assay in transfected human recombinant Chinese hamster ovary (CHO) cells and performed by Eurofins Cerep SA (Le Bois L’Evêque, France). Cell membrane homogenates (140 μg protein) were incubated for 60 min at 37°C with 0.3 nM [3H]GR 113808 in the absence or presence of DHE in a buffer containing 50 mM HEPES/Tris (pH 7.4) and 1 μM pargyline (30). The experiment was conducted in a 96-well plate format with 200 μL total volume, and 8 concentrations of DHE (0.01–300 nM) were evaluated. Nonspecific binding was determined in the presence of 100 μM serotonin. Following incubation, the same protocol as described above for 5-HT3 binding was employed. The standard reference compound for 5-HT4E binding is serotonin.
Binding of DHE to 5-HT1B, α-adrenergic2B, D2, and D5 receptors was evaluated via a radioligand binding assay in human recombinant Chem-1 cells, CHO cells, HEK-293 cells, and GH4 cells, respectively, and performed by Eurofins Cerep SA. The incubation time was 60 min at room temperature (or 37°C for 5-HT1B) with [3H]RX 821002, [3H]7-OH-DPAT, [3H]SCH 23390, [3H]GR125743 for α-adrenergic2B, D2, D5, and 5-HT1B, respectively (31–34). Concentrations of DHE ranged from 0.3 to 10,000 nM. Nonspecific binding was determined in the presence of (−)epinephrine (100 μM), butaclamol (10 μM), SCH 23390 (10 μM), and serotonin (10 μM) for α-adrenergic2B, D2, D5, and 5-HT1B, respectively.
DHE and sumatriptan succinate activity were analyzed using CBIS data analysis suite (ChemInnovation; San Diego, CA). Measurement of agonist and antagonist activity in the assay was calculated as percent activity of relative luminescence units (from positive control). Significance of agonist/antagonist activity was determined based upon prespecified criteria provided by Eurofins DiscoverX. In brief, receptor activity >30% was considered a significant agonist effect. Receptor inhibition >50% was considered a significant inhibitory effect. Please refer to the Supplementary File for more detail.
DHE (10 μM) exhibited agonist activity at α-adrenergic2B, CXC chemokine receptor 7 (CXCR7), D2/5, and 5-HT1A/1B/2A/2C/5A receptors (Table 3). DHE (10 μM) exhibited antagonist activity at α-adrenergic1B/2A/2C (i.e., α1B/2A/2C-adrenoceptors), calcitonin receptor–receptor activity modifying protein 2 (CTR-RAMP2) or amylin 2 (AMY2), D1/3/4/5, and 5-HT1F receptors (Table 4). Sumatriptan succinate (10 μM) exhibited agonist activity at 5-HT1B/1E/1F/5A receptors and no antagonist activity at any receptor tested (Table 5). In the initial screening, DHE exhibited fairly strong antagonist activity at the 5-HT1F receptor and agonist activity at the CXCR7 receptor. Because of this, a more-thorough evaluation of ß-arrestin recruitment was performed to determine the activity of DHE at these receptors. The IC50 for DHE was 149 nM at the 5-HT1F receptor, and the EC50 was 6 μM at the CXCR7 receptor.
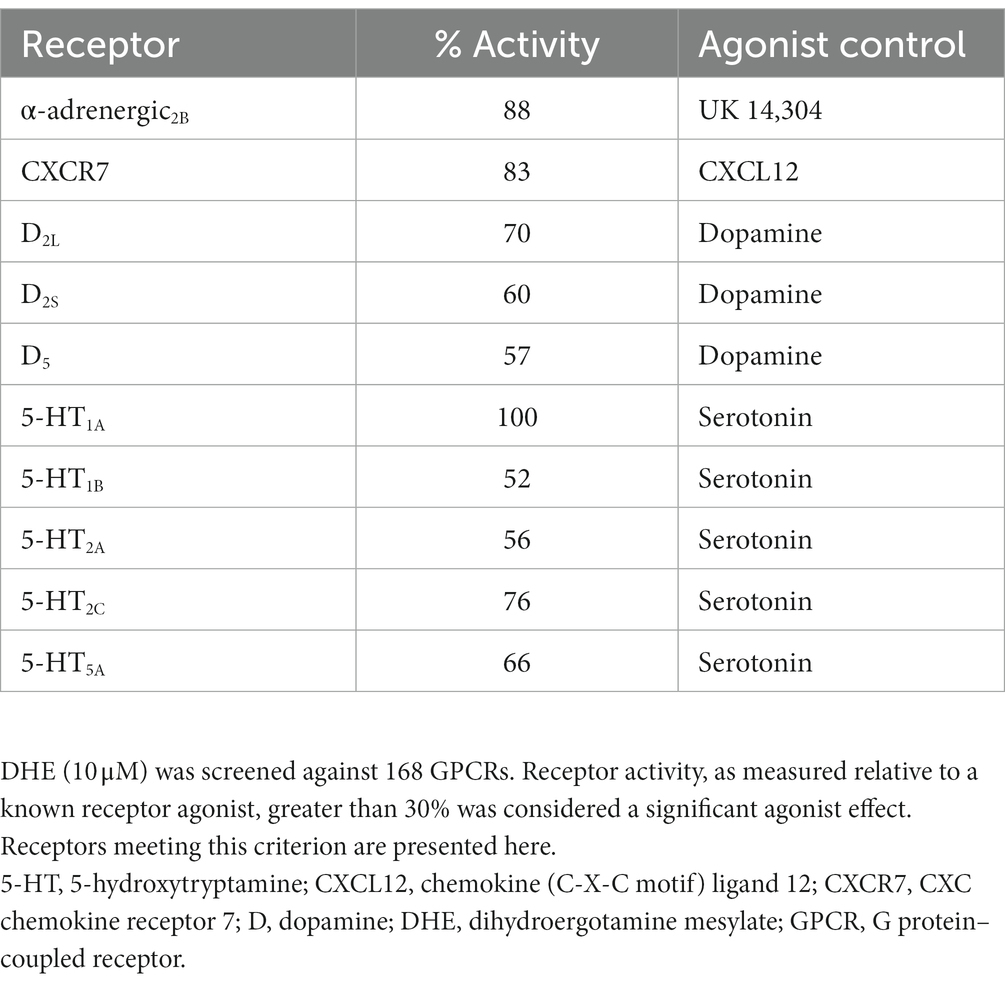
Table 3. gpcrMAX agonist mode results for DHE.
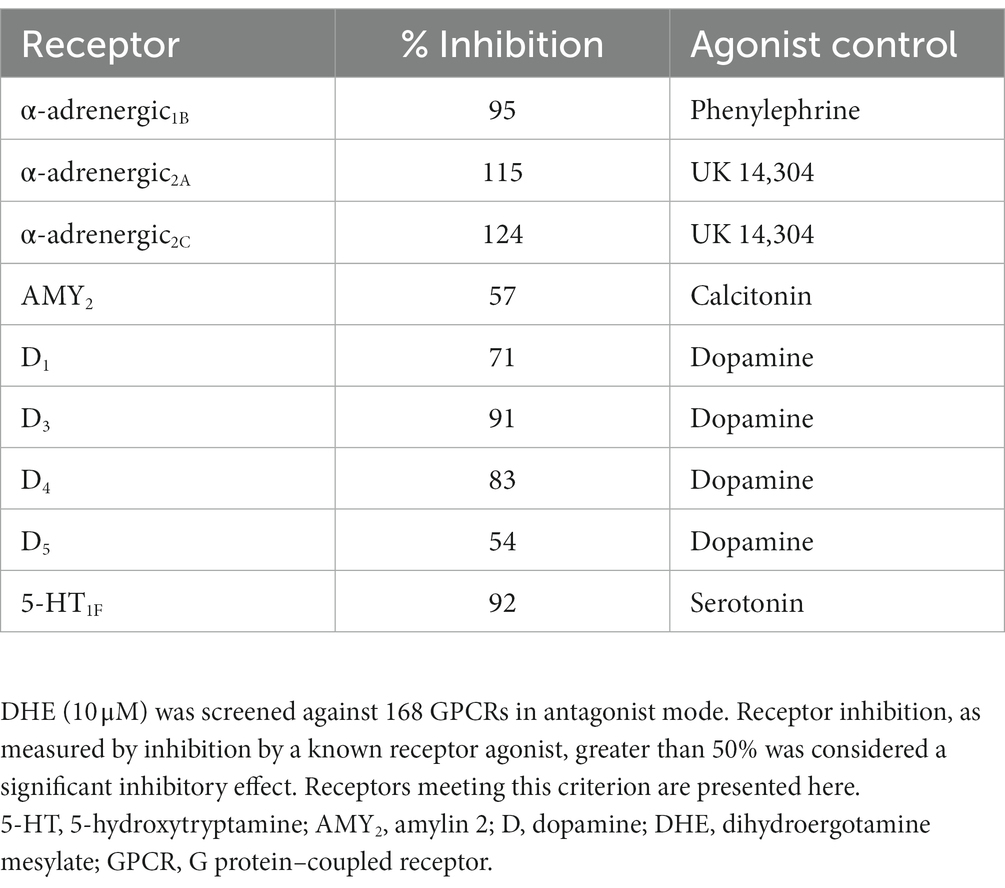
Table 4. gpcrMAX antagonist mode results for DHE.
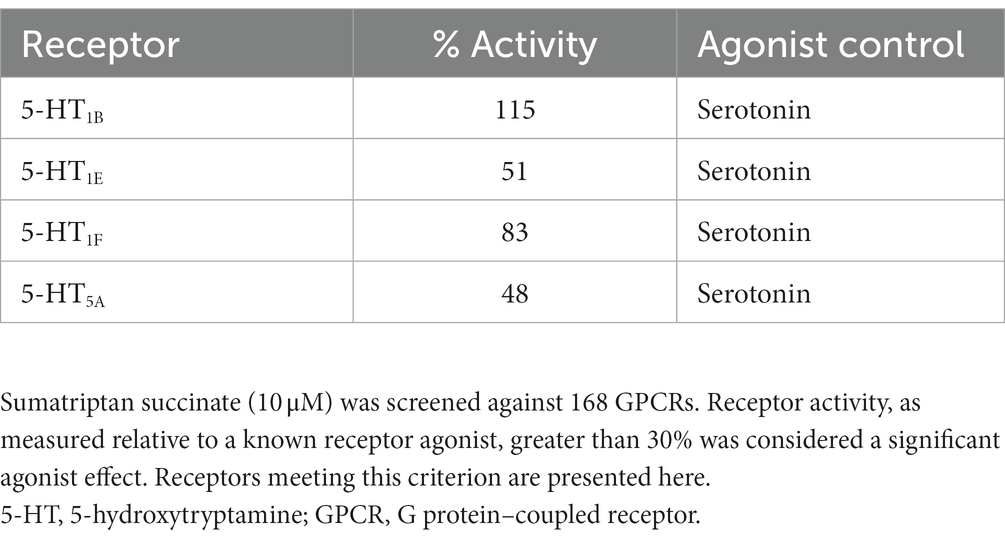
Table 5. gpcrMAX agonist mode results for sumatriptan succinate.
DHE did not exhibit binding to the 5-HT3 receptor at concentrations up to 300 nM and bound poorly to 5-HT4E and D5 receptors, with IC50 values of 230 and 370 nM, respectively (Table 6). DHE exhibited stronger binding to the D2, 5-HT1B, and α-adrenergic2B receptors, with IC50 values of 0.47, 0.58, and 2.8 nM, respectively (Figure 1; Table 6).
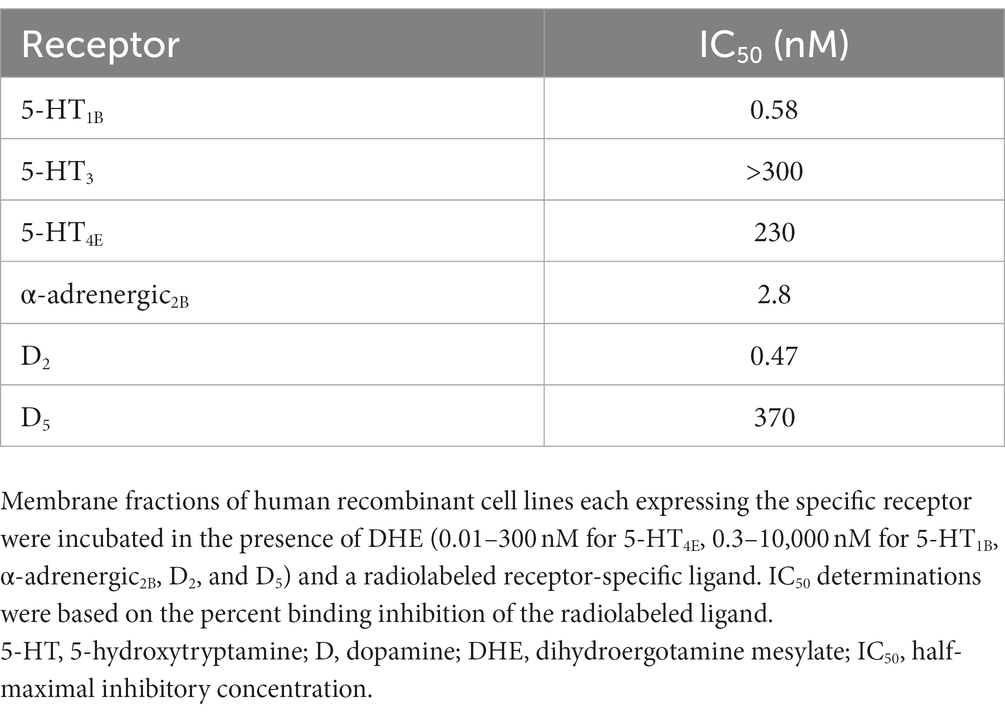
Table 6. Radiolabeled ligand binding assay results for DHE.
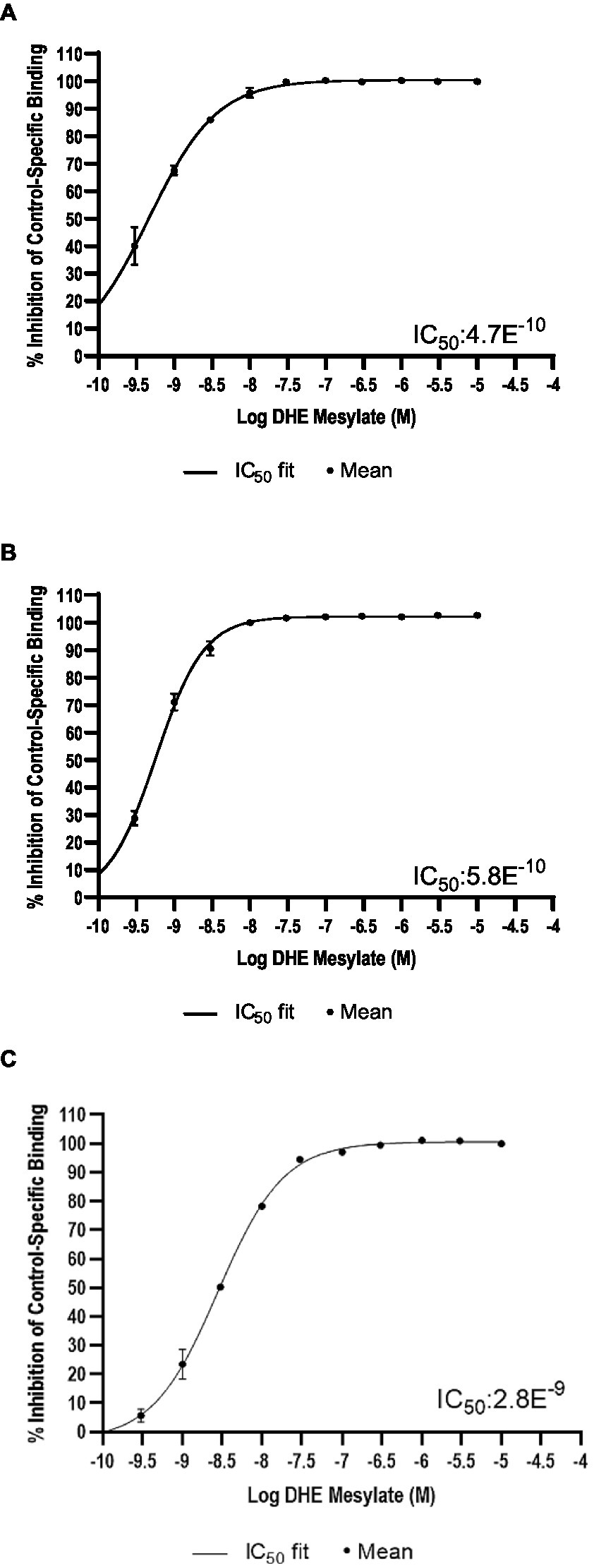
Figure 1. Radioligand binding results. Percent inhibition of radioligand binding to (A) the D2* receptor, (B) the 5-HT1B receptor, and (C) the α-adrenergic2B receptor in the presence of DHE. *Analysis was for D2S. 5-HT, 5-hydroxytryptamine; D, dopamine; DHE, dihydroergotamine mesylate; IC50, half-maximal inhibitory concentration.
DHE has a long history as an efficacious acute therapy for migraine, with the explanation for its efficacy being its broad receptor pharmacology (8, 12). An update in our understanding of how DHE may acutely treat migraine is appropriate, particularly as there have been advancements in receptor binding methodology and new DHE products are being added to the research and development pipeline. Here we report updated data on DHE receptor pharmacology using the gpcrMAX assay panel, a high-throughput screening assay of GPCR ligands employing ß-arrestin recruitment. DHE (10 μM) was screened for functional activity at 168 GPCRs, demonstrating agonist activity across multiple receptor classes, including α-adrenergic2B, CXCR7, D2L/2S/5, and 5-HT1A/1B/2A/2C/5A receptors (Table 7). This contrasted with sumatriptan succinate (10 μM), which demonstrated agonist activity at 4 receptors within a single class, 5-HT1B/1E/1F/5A. DHE also demonstrated antagonist activity at 9 receptors, including α-adrenergic1B/2A/2C, AMY2, D1/3/4/5, and 5-HT1F receptors, across several receptor classes. The concentration used for this assay was high compared to DHE plasma concentrations; however, it was an assay requirement to ensure no potential receptor interaction was missed. Based on results from the screening assay, further assessment of DHE binding at therapeutically relevant concentrations was performed at select GPCRs to add clinical context.
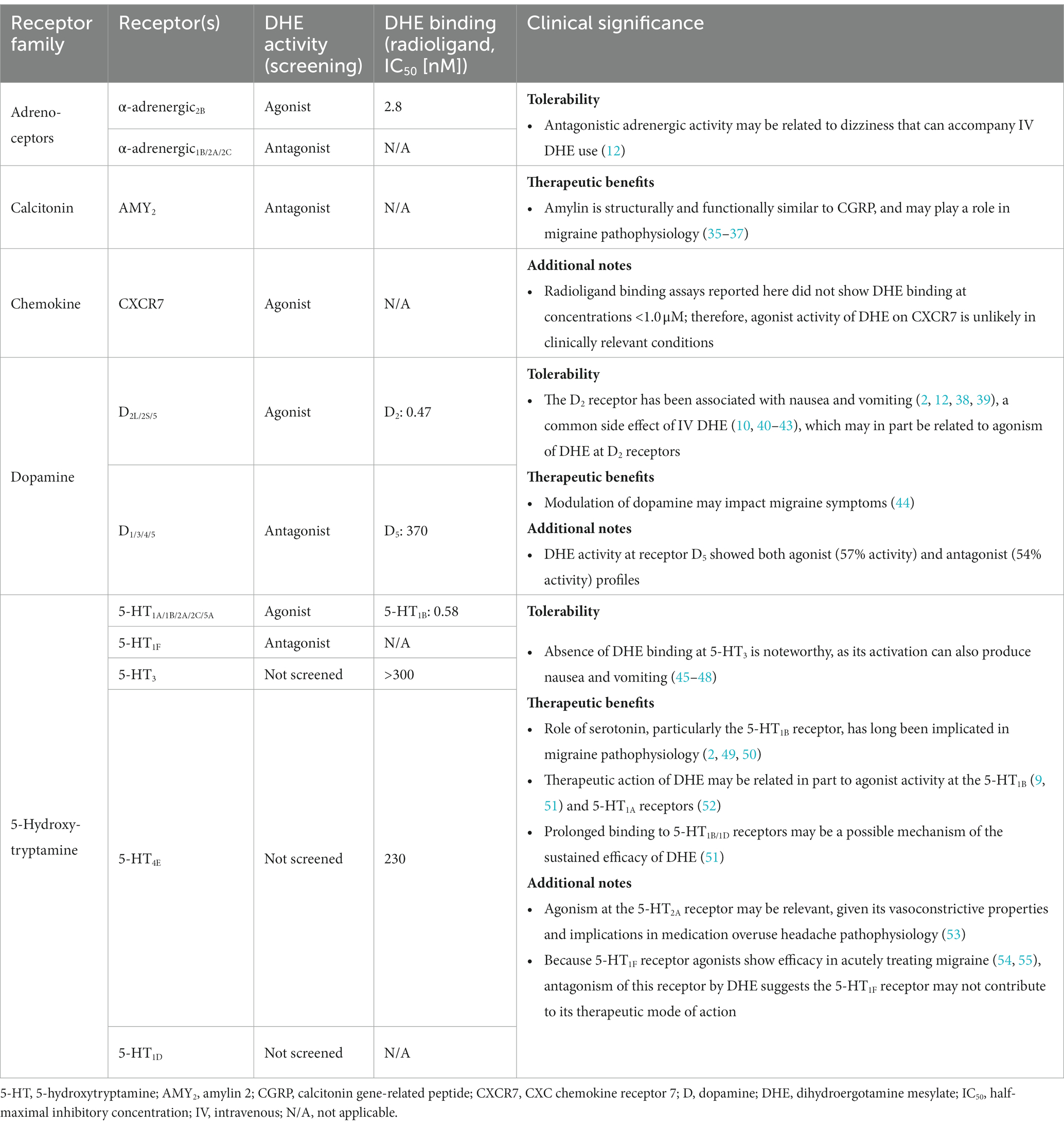
Table 7. Summary of DHE activity at assayed receptors and clinical significance.
The binding (IC50) and agonist activity at the 5-HT1B receptor were expected, and align with previously published data (11, 12, 56–59). Evidence supporting a role for serotonin in migraine pathophysiology is extensive, and the 5-HT1B receptor has long been implicated, notably in the setting of triptans, by exerting its therapeutic effect on migraine symptoms via mediation of vasoconstriction in cranial and cerebral arteries and inhibition of relevant neural pathways (2, 49, 50). Sustained efficacy of DHE up to 48 h has been reported (9), and a study by Kori and colleagues (51) determined that prolonged binding to 5-HT1B/1D receptors may be a possible mechanism for the sustained efficacy of DHE when used to treat migraine acutely. The dissociation half-lives of DHE on human 5-HT1B (DHE: 1.38 h; sumatriptan: 0.17 h) and 5-HT1D (DHE: 1.28 h; sumatriptan: 0.09 h) were approximately 10 times longer than those of sumatriptan, and DHE bound to these receptors 8–14 h longer than did sumatriptan. Importantly, our data confirm strong binding of DHE at the 5-HT1B receptor with clinically relevant doses, suggesting that the therapeutic action of DHE may be due, at least in part, to agonist activity at the 5-HT1B receptor. In agreement with previous work (12), our data show that DHE displayed agonist activity at the 5-HT1A receptor. Newman-Tancredi and colleagues (60) assessed binding and agonist efficacy of DHE using recombinant human 5-HT1A receptors expressed in CHO cells and determined that DHE bound strongly and displayed high-efficacy agonism (ie, Emax ≥ 90% relative to 5-HT) at 5-HT1A receptors at nanomolar concentrations. Work by Hanoun and colleagues (52) characterized the action of DHE at the 5-HT1A receptor in the rat brain, determining that DHE and its metabolite have an inhibitory influence on neuronal excitability and may potentially reduce anxiety via partial agonism at the 5-HT1A receptor.
The D2 receptor has been implicated in the pathophysiology of nausea and vomiting, which are frequent symptoms that accompany migraine (2, 12, 38, 39). Nausea is also a common side effect of IV DHE use and most likely associated with the high Cmax observed with its use (12, 40–43); therefore, it was not surprising that this study demonstrated DHE agonism and strong binding at the D2 receptor. Pretreatment with an antiemetic is a well-established option for preventing the nausea and vomiting associated with IV DHE use (41, 43). Some DHE products have reported a low rate of nausea, which may be due to their lower peak concentrations, (25, 61) suggesting D2 agonism by DHE may not necessarily result in increased nausea. Absence of DHE binding at the 5-HT3 receptor up to 300 nM is another noteworthy finding of this study, as the activation of 5-HT3 receptors can also produce nausea and vomiting (45–48). Interestingly, Cook and colleagues demonstrated antagonist activity at the D2 receptor with concentrations equivalent to the Cmax of IV DHE and inhaled DHE (12). These discrepancies with the current study may be the result of differences in methodology (ß-arrestin recruitment vs. GPCR Ca2+ influx screening), antagonist or agonist cutoff requirements, or concentrations of DHE used (12). Our findings of agonism at the D2 receptor align with what has been reported in the literature (57). Further, according to a recent cross-sectional study, modulating the dopaminergic system should be considered for migraine treatment, as 32.6% of individuals with migraine experienced dopaminergic symptoms (eg, yawning, somnolence, nausea) during an attack. Attacks in these individuals were of longer duration and were more disabling than attacks in individuals without dopaminergic symptoms (44). Lastly, our assays detected both an agonistic (57% receptor activity) and antagonistic (54% receptor activity) profile for D5. Whether this finding suggests that DHE modulates the D5 receptor in a complex manner or is a result of the high concentration of DHE utilized (10 μM) and/or the defined cutoffs for determining significant agonist/antagonist activity (>30% or 50% receptor activity relative to the known receptor agonist or antagonist, respectively), would need to be assessed further in the future.
Interestingly, our study showed that DHE has demonstrated antagonist activity at the AMY2 receptor, 1 of 3 receptors for amylin, a peptide that is structurally and functionally similar to CGRP (35, 36). Recently, a randomized clinical trial showed that a synthetic amylin analogue, pramlintide, can induce migraine-like attacks in patients with migraine (62). Moreover, a recent prospective study reported higher interictal plasma amylin levels in patients with chronic migraine compared to healthy controls (37). The canonical focus on CGRP in migraine expanded to include amylin when it was discovered that 2 s-generation gepants antagonized both the CGRP receptor and amylin 1 (AMY1) receptor, the latter of which has been shown to be stimulated by CGRP and amylin with equal potency in vitro (63–65). These studies highlight an underappreciated role for and clinical relevance of amylin in migraine pathophysiology. In our study, DHE antagonized the AMY2 receptor, a high-affinity receptor for amylin (66), which may be therapeutically and clinically relevant to patients with migraine with high levels of interictal amylin signaling. Whether DHE has antagonism at the AMY1 receptor is a limitation of the current study, as it was not investigated due to lack of availability in the gpcrMAX assay. In future studies, it would be interesting to further delineate the role and interaction of DHE, amylin receptors, and migraine pathophysiology.
Data presented here revealed agonist activity of 10 μM DHE at the α-adrenergic2B receptor and strong binding of therapeutically relevant doses of DHE, which was an unexpected finding. According to the literature, DHE binds to α- and β-adrenergic receptors (11, 56, 57); however, Cook and colleagues contrastingly reported functional antagonism at the α-adrenergic2B receptor in addition to α-adrenergic1A and α-adrenergic2A receptors at a DHE concentration correlating to Cmax for IV DHE and low or absent adrenergic antagonism for the MAP0004 doses, citing antagonism at the α-adrenergic receptors as a possible mechanism for the dizziness that accompanies DHE use (12). Our data demonstrated DHE antagonism at α-adrenergic1B,2A,2C receptors at the 10 μM concentration, aligning with the Cook study. Interestingly, there are reports of an association between vasopressor effects and activation of vascular α-adrenergic1 and α-adrenergic2 receptors. Early work by Roquebert and Grenié (67, 68) reported that DHE elicited vasopressor effects in pithed rats, which were mediated by partial agonist activity at the α-adrenergic2 receptor but not the α-adrenergic1 receptor; however, this study did not consider the strong binding DHE exhibits at the 5-HT2A receptor, a finding not known at the time. Rivera-Mancilla and colleagues (68) assessed the vasopressor responses to DHE following α-adrenergic1 and α-adrenergic2 receptor antagonist administration in pithed rats pretreated with ritanserin, an antagonist with very strong binding at 5-HT2A receptors and very weak binding at α-adrenergic1 and α-adrenergic2 receptors, to eliminate the possibility of vasoconstriction mediated by the 5-HT2A receptor. Results showed that vasopressor responses were present following administration of DHE, which were inhibited by both α-adrenergic1 and α-adrenergic2 receptor antagonists, theorizing the involvement of α-adrenergic1A,1B,1D and α-adrenergic2A,2B,2C receptors. However, the binding of DHE to the α-adrenergic2B receptor was lower than to the α-adrenergic2A and α-adrenergic2C receptors (68). González-Hernández and colleagues (69) also utilized a pithed rat model to demonstrate that DHE blocks vasodepressor sensory CGRPergic outflow via the activation of the α-adrenergic2 receptor and 5-HT1B/1D receptors. These findings were further corroborated by Villalón and colleagues (70), who reported vasoconstrictive properties of DHE mediated primarily—although, importantly, not exclusively—by 5-HT1B and α-adrenergic2A/2C receptors in a canine model. Kalkman and colleagues (71) compared the vasoconstrictive effects of DHE and ergotamine in rat aorta, demonstrating that ergotamine contracted rat aorta and behaved as a partial 5-HT2A receptor agonist, whereas DHE was an insurmountable 5-HT2A receptor antagonist. Cook and colleagues also reported that DHE was an antagonist at the 5-HT2A receptor with 5 μM of DHE and at a DHE concentration correlating to Cmax for IV DHE (~0.091 μM), with limited antagonism or an absence of functional activity with 4 MAP0004 inhalations (~0.007 μM) and 2 MAP0004 inhalations (~0.002 μM), respectively, suggesting it was unlikely that the 5-HT2A receptor mediated coronary contraction (12). In contrast, we report agonist activity of 10 μM DHE at the 5-HT2A receptor, which was a surprising finding. DHE product labels warn of potential cardiovascular (CV) and peripheral ischemic events, possibly attributed to agonist activity at the 5-HT1B receptor, which can cause vasoconstriction of coronary arteries (72–74). However, it has been shown that the vasoconstrictive effects induced by DHE are more pronounced in the meningeal arteries than in the coronary arteries, suggesting patients without CV disease may not have this limitation or contraindication (75), and some DHE products have not reported increases in blood pressure in clinical studies (76). In addition to its vasoconstrictive properties, the 5-HT2A receptor has been implicated in medication overuse headache (MOH) pathophysiology (53), although DHE is not known to be associated with high rates of MOH in the clinic (77). Agonism at the 5-HT2B receptor has been implicated in drug-induced valvular heart disease (78). While Cook and colleagues showed no 5-HT2B agonism for both MAP0004 doses, but agonism for IV DHE (12), this receptor was not screened in our study.
Another surprising finding was antagonist activity of DHE at the 5-HT1F receptor. The initial screening assay demonstrated strong antagonist activity (92%) using 10 μM of DHE; however, this antagonist activity was found to be somewhat limited when further assessment using therapeutically relevant doses of DHE demonstrated an IC50 of 149 nM, indicating weak binding. Ergotamine has also shown weak binding at the 5-HT1F receptor (79). The 5-HT1F receptor agonist, lasmiditan, has shown efficacy in acutely treating migraine in several clinical studies (54, 55). The literature has also shown that DHE binds to the 5-HT1F receptor (11, 26, 56–58). Although the Cook study did not evaluate binding at the 5-HT1F receptor to compare findings to our study (12), the limited antagonist activity of DHE at the 5-HT1F receptor in the present study could suggest that DHE does not demonstrate efficacy through activity at this receptor or that the DHE-binding kinetics are biased to the β-arrestin signaling pathways (80, 81). High agonist activity at the CXCR7 receptor (83%) using 10 μM of DHE was another unexpected result during the initial screening assay. Meyrath and colleagues (82) recently reported findings that CXCR7 (currently known as ACKR3) is an atypical scavenge receptor for a wide variety of opioid peptides, reducing their availability for the classical opioid receptors. The radioligand binding assay in this study revealed that DHE did not exert activity at CXCR7 at concentrations <1.0 μM, suggesting it is unlikely that DHE is active at CXCR7 under clinically relevant conditions.
This study has some limitations. First, some receptors that are important in migraine pathophysiology, such as 5-HT1D and AMY1, were not screened because a human cell line expressing the receptors was unavailable for the assay for technical reasons or lack of availability. Second, agonist and antagonist activity of DHE and sumatriptan succinate at GCPRs was evaluated only via ß-arrestin recruitment. Because GCPRs can signal through several pathways, utilizing a single signaling pathway in this current study, ß-arrestin, may result in incomplete detection of functional activity of DHE and/or sumatriptan succinate (83, 84). Further, there is the possibility of a biased signaling that can further confound results (83).
Using a new methodology to screen against 168 GPCRs via high-throughput assay, the receptor binding work presented here provides an update to our understanding of DHE receptor pharmacology. Similar to what has been reported in the literature, DHE in this study demonstrated broad receptor pharmacology, binding at several receptors across receptor classes, including agonist activity at α-adrenergic2B, CXCR7, D2/5, and 5-HT1A/1B/2A/2C/5A receptors, and antagonist activity at α-adrenergic1B/2A/2C, AMY2, D1/3/4/5, and 5-HT1F receptors. The antimigraine efficacy of DHE may be explained by agonism and strong binding of therapeutic doses at the 5-HT1B receptor (5-HT1D was not available in the GPCR assay), as well as slow dissociation, whereas the side effect profile of DHE may be attributed to agonist activity at the D2, α-adrenergic2B, and 5-HT2A receptors. The exact interplay between activation and inhibition of multiple receptor pathways in the migraine cycle, extending beyond individual attacks and embracing organ systems beyond the central nervous system, has yet to be fully elucidated.
The datasets presented in this article are not readily available because raw data may include IP information that is not readily available for distribution without NDAs in place. Requests to access the datasets should be directed to c2F1cm9yYUBpbXBlbHBoYXJtYS5jb20=.
Ethical approval was not required for the studies on humans in accordance with the local legislation and institutional requirements because only commercially available established cell lines were used.
LM: Writing – original draft, Writing – review & editing, Conceptualization, Data curation, Formal analysis. PG: Writing – original draft, Writing – review & editing, Conceptualization. RV: Writing – original draft, Writing – review & editing, Conceptualization. SR: Writing – original draft, Writing – review & editing, Conceptualization. SS: Writing – original draft, Writing – review & editing, Conceptualization. SA: Conceptualization, Supervision, Writing – original draft, Writing – review & editing.
The author(s) declare financial support was received for the research, authorship, and/or publication of this article. This study was funded by Impel Pharmaceuticals Inc.
Writing and editorial assistance was provided by IMPRINT Science, New York, NY, USA, and supported by Impel Pharmaceuticals Inc. The authors are fully responsible for the content, editorial decisions, and opinions expressed in the current article.
LM is a full-time employee of Priovant Therapeutics and is a stockholder in Impel Pharmaceuticals. She was formerly a full-time employee of Impel Pharmaceuticals. PG reports, over the last 36 months, personal fees for consulting with Impel Pharmaceuticals, and grants and personal fees from Eli Lilly and Company, a grant from Celgene, and personal fees from AEON Biopharma, Allergan/AbbVie, Amgen, BioDelivery Sciences International, Biohaven Pharmaceuticals Inc., CoolTech LLC, Dr. Reddy’s, Epalex, GlaxoSmithKline, Lundbeck, Novartis, Praxis, Sanofi, Satsuma, and Teva Pharmaceuticals, and personal fees for advice through Gerson Lehrman Group, Guidepoint, SAI MedPartners, Vector Metric, and fees for educational materials from CME Outfitters, Omnia Education, WebMD, and publishing royalties or fees from Massachusetts Medical Society, Oxford University Press, UpToDate, and Wolters Kluwer. SR and SA are full-time employees of Impel Pharmaceuticals and stockholders in Impel Pharmaceuticals. SS was formerly a full-time employee of and an officer of Impel Pharmaceuticals. He remains a stockholder. RV was formerly a full-time employee of Impel Pharmaceuticals and remains a stockholder.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
The Supplementary material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fneur.2023.1282846/full#supplementary-material
5-HT, 5-hydroxytryptamine; AMY, amylin; CGRP, calcitonin gene-related peptide; CXCR7, CXC chemokine receptor 7; D, dopamine; DHE, dihydroergotamine mesylate; EC50, half-maximal effective concentration; EC80, 80% of maximal effective concentration; IC50, half-maximal inhibitory concentration; IV, intravenous.
1. Burstein, R, Noseda, R, and Borsook, D. Migraine: multiple processes, complex pathophysiology. J Neurosci. (2015) 35:6619–29. doi: 10.1523/JNEUROSCI.0373-15.2015
2. Goadsby, PJ, Holland, PR, Martins-Oliveira, M, Hoffmann, J, Schankin, C, and Akerman, S. Pathophysiology of migraine: a disorder of sensory processing. Physiol Rev. (2017) 97:553–622. doi: 10.1152/physrev.00034.2015
4. Humphrey, PP, Feniuk, W, Perren, MJ, Beresford, IJ, Skingle, M, and Whalley, ET. Serotonin and migraine. Ann N Y Acad Sci. (1990) 600:587–98. doi: 10.1111/j.1749-6632.1990.tb16912.x
5. Moreno-Ajona, D, Villar-Martínez, MD, and Goadsby, PJ. Targets for migraine treatment: beyond calcitonin gene-related peptide. Curr Opin Neurol. (2021) 34:363–72. doi: 10.1097/wco.0000000000000935
6. Moreno-Ajona, D, Villar-Martínez, MD, and Goadsby, PJ. New generation gepants: migraine acute and preventive medications. J Clin Med. (2022) 11:11. doi: 10.3390/jcm11061656
7. Olesen, J, Tfelt-Hansen, P, and Ashina, M. Finding new drug targets for the treatment of migraine attacks. Cephalalgia. (2009) 29:909–20. doi: 10.1111/j.1468-2982.2008.01837.x
8. Silberstein, SD, Shrewsbury, SB, and Hoekman, J. Dihydroergotamine (DHE) – then and now: a narrative review. Headache. (2020) 60:40–57. doi: 10.1111/head.13700
9. Aurora, SK, Silberstein, SD, Kori, SH, Tepper, SJ, Borland, SW, Wang, M, et al. MAP0004, orally inhaled DHE: a randomized, controlled study in the acute treatment of migraine. Headache. (2011) 51:507–17. doi: 10.1111/j.1526-4610.2011.01869.x
10. Kellerman, DJ, Forst, A, Combs, DL, Borland, S, and Kori, S. Assessment of the consistency of absorption of dihydroergotamine following oral inhalation: pooled results from four clinical studies. J Aerosol Med Pulm Drug Deliv. (2013) 26:297–306. doi: 10.1089/jamp.2012.0999
11. Tfelt-Hansen, P, Saxena, PR, Dahlöf, C, Pascual, J, Láinez, M, Henry, P, et al. Ergotamine in the acute treatment of migraine: a review and European consensus. Brain. (2000) 123:9–18. doi: 10.1093/brain/123.1.9
12. Cook, RO, Shrewsbury, SB, and Ramadan, NM. Reduced adverse event profile of orally inhaled DHE (MAP0004) vs IV DHE: potential mechanism. Headache. (2009) 49:1423–34. doi: 10.1111/j.1526-4610.2009.01510.x
13. Weissman, A, Keefer, J, Miagkov, A, Sathyamoorthy, M, Perschke, S, and Wang, FL. Cell-asbed screening assays In: JB Taylor and DJ Triggle, editors. Comprehensive medicinal chemistry II. Oxford, UK: Elsevier (2007). 617–46. doi: 10.1016/B0-08-045044-X/00102-4
14. Wang, T, Li, Z, Cvijic, ME, Krause, C, Zhang, L, and Sum, CS. Measurement of beta-arrestin recruitment for GPCR targets In: S Markossian, GS Sittampalam, A Grossman, K Brimacombe, M Arkin, and D Auld, et al., editors. Assay guidance manual. Bethesda (MD): Eli Lilly & Company and the National Center for Advancing Translational Sciences (2004). 829–40.
15. DeWire, SM, Ahn, S, Lefkowitz, RJ, and Shenoy, SK. Beta-arrestins and cell signaling. Annu Rev Physiol. (2007) 69:483–510. doi: 10.1146/annurev.physiol.69.022405.154749
16. Jean-Charles, PY, Kaur, S, and Shenoy, SK. GPCR signaling via beta-arrestin-dependent mechanisms. J Cardiovasc Pharmacol. (2017) 70:142–58. doi: 10.1097/FJC.0000000000000482
17. Spillmann, M, Thurner, L, Romantini, N, Zimmermann, M, Meger, B, Behe, M, et al. New insights into arrestin recruitment to GPCRs. Int J Mol Sci. (2020) 21:4949. doi: 10.3390/ijms21144949
18. Lohse, MJ, Benovic, JL, Codina, J, Caron, MG, and Lefkowitz, RJ. Beta-arrestin: a protein that regulates beta-adrenergic receptor function. Science. (1990) 248:1547–50. doi: 10.1126/science.2163110
19. Kroeze, WK, Sassano, MF, Huang, XP, Lansu, K, McCorvy, JD, Giguère, PM, et al. PRESTO-Tango as an open-source resource for interrogation of the druggable human GPCRome. Nat Struct Mol Biol. (2015) 22:362–9. doi: 10.1038/nsmb.3014
20. Eurofins . PathHunter® build your own β-Arrestin assays. (n.d.). Available at: https://www.discoverx.com/tools-resources/document-resource-library/documents/pathhunter-%CE%B2-arrestin-retroparticles (Accessed February 28, 2023).
21. Zhao, X, Jones, A, Olson, KR, Peng, K, Wehrman, T, Park, A, et al. A homogeneous enzyme fragment complementation-based beta-arrestin translocation assay for high-throughput screening of G-protein-coupled receptors. J Biomol Screen. (2008) 13:737–47. doi: 10.1177/1087057108321531
22. Eurofins . GPCR assay formats and services offered. (n.d.). Available at: https://www.discoverx.com/services/drug-discovery-development-services/gpcr-screening-profiling-services/gpcrscan-gpcr-profiling/gpcrmax (Accessed October 27, 2022).
23. Guide to Pharmacology IUPHAR GPCR nomenclature. (n.d.). Available at: https://www.guidetopharmacology.org/GRAC/GPCRListForward?class=A (Accessed October 18, 2023).
24. National Center for Biotechnology Information . PubChem compound summary for CID 10531, Dihydroergotamine. (n.d.). Available at: https://pubchem.ncbi.nlm.nih.gov/compound/Dihydroergotamine (Accessed August 16, 2022).
25. Shrewsbury, SB, Jeleva, M, Satterly, KH, Lickliter, J, and Hoekman, J. STOP 101: a phase 1, randomized, open-label, comparative bioavailability study of INP104, dihydroergotamine mesylate (DHE) administered intranasally by a I123 precision olfactory delivery (POD®) device, in healthy adult subjects. Headache. (2019) 59:394–409. doi: 10.1111/head.13476
26. Dahlöf, C, and Maassen Van Den Brink, A. Dihydroergotamine, ergotamine, methysergide and sumatriptan - basic science in relation to migraine treatment. Headache. (2012) 52:707–14. doi: 10.1111/j.1526-4610.2012.02124.x
27. Fowler, PA, Lacey, LF, Thomas, M, Keene, ON, Tanner, RJ, and Baber, NS. The clinical pharmacology, pharmacokinetics and metabolism of sumatriptan. Eur Neurol. (1991) 31:291–4. doi: 10.1159/000116756
28. National Center for Biotechnology Information . PubChem compound summary for CID 5358, Sumatriptan. (n.d.). Available at: https://pubchem.ncbi.nlm.nih.gov/compound/Sumatriptan (Accessed August 16, 2022).
29. Kilpatrick, GJ, Jones, BJ, and Tyers, MB. Binding of the 5-HT3 ligand, [3H]GR65630, to rat area postrema, vagus nerve and the brains of several species. Eur J Pharmacol. (1989) 159:157–64. doi: 10.1016/0014-2999(89)90700-0
30. Mialet, J, Berque-Bestel, I, Eftekhari, P, Gastineau, M, Giner, M, Dahmoune, Y, et al. Isolation of the serotoninergic 5-HT4(e) receptor from human heart and comparative analysis of its pharmacological profile in C6-glial and CHO cell lines. Br J Pharmacol. (2000) 129:771–81. doi: 10.1038/sj.bjp.0703101
31. Doménech, T, Beleta, J, and Palacios, JM. Characterization of human serotonin 1D and 1B receptors using [3H]-GR-125743, a novel radiolabelled serotonin 5HT1D/1B receptor antagonist. Naunyn Schmiedeberg's Arch Pharmacol. (1997) 356:328–34. doi: 10.1007/pl00005058
32. Sunahara, RK, Guan, HC, O'Dowd, BF, Seeman, P, Laurier, LG, Ng, G, et al. Cloning of the gene for a human dopamine D5 receptor with higher affinity for dopamine than D1. Nature. (1991) 350:614–9. doi: 10.1038/350614a0
33. Devedjian, JC, Esclapez, F, Denis-Pouxviel, C, and Paris, H. Further characterization of human alpha 2-adrenoceptor subtypes: [3H]RX821002 binding and definition of additional selective drugs. Eur J Pharmacol. (1994) 252:43–9. doi: 10.1016/0014-2999(94)90573-8
34. Gonzalez, AM, and Sibley, DR. [3H]7-OH-DPAT is capable of labeling dopamine D2 as well as D3 receptors. Eur J Pharmacol. (1995) 272:R1–3. doi: 10.1016/0014-2999(94)00738-s
35. Cooper, GJ . Amylin compared with calcitonin gene-related peptide: structure, biology, and relevance to metabolic disease. Endocr Rev. (1994) 15:163–201. doi: 10.1210/edrv-15-2-163
36. Rees, TA, Hendrikse, ER, Hay, DL, and Walker, CS. Beyond CGRP: the calcitonin peptide family as targets for migraine and pain. Br J Pharmacol. (2021) 179:381–99. doi: 10.1111/bph.15605
37. Irimia, P, Martínez-Valbuena, I, Mínguez-Olaondo, A, Domínguez-Vivero, C, Sánchez-Arias, JA, Martínez-Vila, E, et al. Interictal amylin levels in chronic migraine patients: a case-control study. Cephalalgia. (2021) 41:604–12. doi: 10.1177/0333102420977106
38. Munjal, S, Singh, P, Reed, ML, Fanning, K, Schwedt, TJ, Dodick, DW, et al. Most bothersome symptom in persons with migraine: results from the Migraine in America Symptoms and Treatment (MAST) study. Headache. (2020) 60:416–29. doi: 10.1111/head.13708
39. Lipton, RB, Buse, DC, Saiers, J, Fanning, KM, Serrano, D, and Reed, ML. Frequency and burden of headache-related nausea: results from the American Migraine Prevalence and Prevention (AMPP) study. Headache. (2013) 53:93–103. doi: 10.1111/j.1526-4610.2012.02292.x
40. Kellerman, D, Kori, S, and Forst, A. Nausea associated with dihydroergotamine is a function of maximum concentration and not route of administration (P03.238). Neurology. (2012) 78:P03.238. doi: 10.1212/WNL.78.1_MeetingAbstracts.P03.238
41. Silberstein, SD, and Kori, SH. Dihydroergotamine: a review of formulation approaches for the acute treatment of migraine. CNS Drugs. (2013) 27:385–94. doi: 10.1007/s40263-013-0061-2
42. Shrewsbury, SB, Cook, RO, Taylor, G, Edwards, C, and Ramadan, NM. Safety and pharmacokinetics of dihydroergotamine mesylate administered via a Novel (tempo) inhaler. Headache. (2008) 48:355–67. doi: 10.1111/j.1526-4610.2007.01006.x
43. Chou, DE, Tso, AR, and Goadsby, PJ. Aprepitant for the management of nausea with inpatient IV dihydroergotamine. Neurology. (2016) 87:1613–6. doi: 10.1212/wnl.0000000000003206
44. Barbanti, P, Aurilia, C, Egeo, G, Fofi, L, Guadagni, F, and Ferroni, P. Dopaminergic symptoms in migraine: a cross-sectional study on 1148 consecutive headache center-based patients. Cephalalgia. (2020) 40:1168–76. doi: 10.1177/0333102420929023
45. Smith, HS, Cox, LR, and Smith, EJ. 5-HT3 receptor antagonists for the treatment of nausea/vomiting. Ann Palliat Med. (2012) 1:115–20. doi: 10.3978/j.issn.2224-5820.2012.07.07
46. Tyers, MB, and Freeman, AJ. Mechanism of the anti-emetic activity of 5-HT3 receptor antagonists. Oncology. (1992) 49:263–8. doi: 10.1159/000227054
47. Kohler, DR, and Goldspiel, BR. Ondansetron: a serotonin receptor (5-HT3) antagonist for antineoplastic chemotherapy-induced nausea and vomiting. DICP. (1991) 25:367–80. doi: 10.1177/106002809102500408
48. Zhong, W, Shahbaz, O, Teskey, G, Beever, A, Kachour, N, Venketaraman, V, et al. Mechanisms of nausea and vomiting: current knowledge and recent advances in intracellular emetic signaling systems. Int J Mol Sci. (2021) 22:5797. doi: 10.3390/ijms22115797
49. Buzzi, MG, Carter, WB, Shimizu, T, Heath, H 3rd, and Moskowitz, MA. Dihydroergotamine and sumatriptan attenuate levels of CGRP in plasma in rat superior sagittal sinus during electrical stimulation of the trigeminal ganglion. Neuropharmacology. (1991) 30:1193–200. doi: 10.1016/0028-3908(91)90165-8
50. Hoskin, KL, Kaube, H, and Goadsby, PJ. Sumatriptan can inhibit trigeminal afferents by an exclusively neural mechanism. Brain. (1996) 119:1419–28. doi: 10.1093/brain/119.5.1419
51. Kori, S, Zhang, J, Kellerman, D, Armer, T, and Goadsby, PJ. Sustained pain relief with dihydroergotamine in migraine is potentially due to persistent binding to 5-HT1B and 5-HT 1D receptors. J Headache Pain. (2013) 14:75. doi: 10.1186/1129-2377-14-S1-P75
52. Hanoun, N, Saurini, F, Lanfumey, L, Hamon, M, and Bourgoin, S. Dihydroergotamine and its metabolite, 8′-hydroxy-dihydroergotamine, as 5-HT1A receptor agonists in the rat brain. Br J Pharmacol. (2003) 139:424–34. doi: 10.1038/sj.bjp.0705258
53. Supornsilpchai, W, le Grand, SM, and Srikiatkhachorn, A. Involvement of pro-nociceptive 5-HT2A receptor in the pathogenesis of medication-overuse headache. Headache. (2010) 50:185–97. doi: 10.1111/j.1526-4610.2009.01591.x
54. Kuca, B, Silberstein, SD, Wietecha, L, Berg, PH, Dozier, G, and Lipton, RB. Lasmiditan is an effective acute treatment for migraine: a phase 3 randomized study. Neurology. (2018) 91:e2222–32. doi: 10.1212/wnl.0000000000006641
55. Goadsby, PJ, Wietecha, LA, Dennehy, EB, Kuca, B, Case, MG, Aurora, SK, et al. Phase 3 randomized, placebo-controlled, double-blind study of lasmiditan for acute treatment of migraine. Brain. (2019) 142:1894–904. doi: 10.1093/brain/awz134
56. Silberstein, SD . The pharmacology of ergotamine and dihydroergotamine. Headache. (1997) 37:S15–25.
57. Silberstein, SD, and McCrory, DC. Ergotamine and dihydroergotamine: history, pharmacology, and efficacy. Headache. (2003) 43:144–66. doi: 10.1046/j.1526-4610.2003.03034.x
58. Saper, JR, and Silberstein, S. Pharmacology of dihydroergotamine and evidence for efficacy and safety in migraine. Headache. (2006) 46 Suppl 4:S171–81. doi: 10.1111/j.1526-4610.2006.00601.x
59. Sullivan, HJ, Tursi, A, Moore, K, Campbell, A, Floyd, C, and Wu, C. Binding interactions of ergotamine and dihydroergotamine to 5-hydroxytryptamine receptor 1B (5-HT1b) using molecular dynamics simulations and dynamic network analysis. J Chem Inf Model. (2020) 60:1749–65. doi: 10.1021/acs.jcim.9b01082
60. Newman-Tancredi, A, Conte, C, Chaput, C, Verrièle, L, Audinot-Bouchez, V, Lochon, S, et al. Agonist activity of antimigraine drugs at recombinant human 5-HT1A receptors: potential implications for prophylactic and acute therapy. Naunyn Schmiedeberg's Arch Pharmacol. (1997) 355:682–8. doi: 10.1007/pl00005000
61. Smith, TR, Winner, P, Aurora, SK, Jeleva, M, Hocevar-Trnka, J, and Shrewsbury, SB. STOP 301: a phase 3, open-label study of safety, tolerability, and exploratory efficacy of INP104, Precision Olfactory Delivery (POD®) of dihydroergotamine mesylate, over 24/52 weeks in acute treatment of migraine attacks in adult patients. Headache. (2021) 61:1214–26. doi: 10.1111/head.14184
62. Ghanizada, H, Al-Karagholi, MA, Walker, CS, Arngrim, N, Rees, T, Petersen, J, et al. Amylin analog pramlintide induces migraine-like attacks in patients. Ann Neurol. (2021) 89:1157–71. doi: 10.1002/ana.26072
63. Pan, KS, Siow, A, Hay, DL, and Walker, CS. Antagonism of CGRP signaling by rimegepant at two receptors. Front Pharmacol. (2020) 11:1240. doi: 10.3389/fphar.2020.01240
64. Moore, E, Fraley, ME, Bell, IM, Burgey, CS, White, RB, Li, CC, et al. Characterization of ubrogepant: a potent and selective antagonist of the human calcitonin gene–related peptide receptor. J Pharmacol Exp Ther. (2020) 373:160–6. doi: 10.1124/jpet.119.261065
65. Walker, CS, Eftekhari, S, Bower, RL, Wilderman, A, Insel, PA, Edvinsson, L, et al. A second trigeminal CGRP receptor: function and expression of the AMY1 receptor. Ann Clin Transl Neurol. (2015) 2:595–608. doi: 10.1002/acn3.197
66. Gingell, JJ, Simms, J, Barwell, J, Poyner, DR, Watkins, HA, Pioszak, AA, et al. An allosteric role for receptor activity-modifying proteins in defining GPCR pharmacology. Cell Discov. (2016) 2:16012. doi: 10.1038/celldisc.2016.12
67. Roquebert, J, and Grenié, B. Alpha 2-adrenergic agonist and alpha 1-adrenergic antagonist activity of ergotamine and dihydroergotamine in rats. Arch Int Pharmacodyn Ther. (1986) 284:30–7.
68. Rivera-Mancilla, E, Aviles-Rosas, VH, Manrique-Maldonado, G, Altamirano-Espinoza, AH, Villanueva-Castillo, B, MaassenVanDenBrink, A, et al. The role of alpha1-and alpha2-adrenoceptor subtypes in the vasopressor responses induced by dihydroergotamine in ritanserin-pretreated pithed rats. J Headache Pain. (2017) 18:104. doi: 10.1186/s10194-017-0812-4
69. González-Hernández, A, Lozano-Cuenca, J, Marichal-Cancino, BA, MaassenVanDenBrink, A, and Villalón, CM. Dihydroergotamine inhibits the vasodepressor sensory CGRPergic outflow by prejunctional activation of α(2)-adrenoceptors and 5-HT(1) receptors. J Headache Pain. (2018) 19:40. doi: 10.1186/s10194-018-0869-8
70. Villalón, CM, Centurión, D, Willems, EW, Arulmani, U, Saxena, PR, and Valdivia, LF. 5-HT1B receptors and alpha 2A/2C-adrenoceptors mediate external carotid vasoconstriction to dihydroergotamine. Eur J Pharmacol. (2004) 484:287–90. doi: 10.1016/j.ejphar.2003.11.026
71. Kalkman, HO, and Schneider, F. Effects of ergotamine and dihydroergotamine on 5-hydroxytryptamine-2A receptors in the isolated rat aorta. Pharmacology. (1996) 53:351–5. doi: 10.1159/000139450
73. Villalón, CM, De Vries, P, Rabelo, G, Centurión, D, Sánchez-López, A, and Saxena, P. Canine external carotid vasoconstriction to methysergide, ergotamine and dihydroergotamine: role of 5-HT1B/1D receptors and alpha2-adrenoceptors. Br J Pharmacol. (1999) 126:585–94. doi: 10.1038/sj.bjp.0702324
75. Labruijere, S, Chan, KY, de Vries, R, van den Bogaerdt, AJ, Dirven, CM, Danser, AJ, et al. Dihydroergotamine and sumatriptan in isolated human coronary artery, middle meningeal artery and saphenous vein. Cephalalgia. (2015) 35:182–9. doi: 10.1177/0333102414544977
76. Craig, K, Jeleva, M, Hocevar-Trnka, J, Aurora, SK, Shrewsbury, SB, et al. Cardiovascular safety results of INP104 (POD-DHE) from the STOP 301 phase 3 study. Presented at: Headache Update of the Diamond Clinic; July 15–18, 2021; Lake Buena Vista, FL, USA (n.d.).
77. Saper, JR, Silberstein, S, Dodick, D, and Rapoport, A. DHE in the pharmacotherapy of migraine: potential for a larger role. Headache. (2006) 46 Suppl 4:S212–20. doi: 10.1111/j.1526-4610.2006.00605.x
78. Roth, BL . Drugs and valvular heart disease. N Engl J Med. (2007) 356:6–9. doi: 10.1056/NEJMp068265
79. Rubio-Beltrán, E, Labastida-Ramírez, A, Villalón, CM, and MaassenVanDenBrink, A. Is selective 5-HT(1F) receptor agonism an entity apart from that of the triptans in antimigraine therapy? Pharmacol Ther. (2018) 186:88–97. doi: 10.1016/j.pharmthera.2018.01.005
80. Klein Herenbrink, C, Sykes, DA, Donthamsetti, P, Canals, M, Coudrat, T, Shonberg, J, et al. The role of kinetic context in apparent biased agonism at GPCRs. Nat Commun. (2016) 7:10842. doi: 10.1038/ncomms10842
81. Sanchez, JE, Kc, GB, Franco, J, Allen, WJ, Garcia, JD, and Sirimulla, S. BiasNet: a model to predict ligand bias toward GPCR signaling. J Chem Inf Model. (2021) 61:4190–9. doi: 10.1021/acs.jcim.1c00317
82. Meyrath, M, Szpakowska, M, Zeiner, J, Massotte, L, Merz, MP, Benkel, T, et al. The atypical chemokine receptor ACKR3/CXCR7 is a broad-spectrum scavenger for opioid peptides. Nat Commun. (2020) 11:3033. doi: 10.1038/s41467-020-16664-0
83. Smith, JS, Lefkowitz, RJ, and Rajagopal, S. Biased signalling: from simple switches to allosteric microprocessors. Nat Rev Drug Discov. (2018) 17:243–60. doi: 10.1038/nrd.2017.229
84. Eurofins DiscoverX Products . Insights into GPCR drug discovery & development – exploring GPCR-ligand interactions and signaling pathways with binding and functional assays. (n.d.). Available at: https://www.discoverx.com/content/uploads/2023/07/eurofins-discoverx-21342.pdf (Accessed October 12, 2023).
Keywords: migraine, dihydroergotamine, receptor, pharmacology, binding
Citation: McConnachie L, Goadsby PJ, Vann RE, Ray S, Shrewsbury SB and Aurora SK (2023) New characterization of dihydroergotamine receptor pharmacology in the context of migraine: utilization of a β-arrestin recruitment assay. Front. Neurol. 14:1282846. doi: 10.3389/fneur.2023.1282846
Edited by:
Milena De Felice, The University of Sheffield, United KingdomReviewed by:
Carlo Baraldi, University of Modena and Reggio Emilia, ItalyCopyright © 2023 McConnachie, Goadsby, Vann, Ray, Shrewsbury and Aurora. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Sheena K. Aurora, c2F1cm9yYUBpbXBlbHBoYXJtYS5jb20=; c2hlYXVyQHlhaG9vLmNvbQ==
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.