 Hannah R. Monday
Hannah R. Monday Han Chin Wang
Han Chin Wang Daniel E. Feldman
Daniel E. Feldman- Department of Molecular and Cell Biology, Helen Wills Neuroscience Institute, University of California, Berkeley, Berkeley, CA, United States
Individuals with autism spectrum disorder (ASD) exhibit a diverse range of behavioral features and genetic backgrounds, but whether different genetic forms of autism involve convergent pathophysiology of brain function is unknown. Here, we analyze evidence for convergent deficits in neural circuit function across multiple transgenic mouse models of ASD. We focus on sensory areas of neocortex, where circuit differences may underlie atypical sensory processing, a central feature of autism. Many distinct circuit-level theories for ASD have been proposed, including increased excitation–inhibition (E–I) ratio and hyperexcitability, hypofunction of parvalbumin (PV) interneuron circuits, impaired homeostatic plasticity, degraded sensory coding, and others. We review these theories and assess the degree of convergence across ASD mouse models for each. Behaviorally, our analysis reveals that innate sensory detection behavior is heightened and sensory discrimination behavior is impaired across many ASD models. Neurophysiologically, PV hypofunction and increased E–I ratio are prevalent but only rarely generate hyperexcitability and excess spiking. Instead, sensory tuning and other aspects of neural coding are commonly degraded and may explain impaired discrimination behavior. Two distinct phenotypic clusters with opposing neural circuit signatures are evident across mouse models. Such clustering could suggest physiological subtypes of autism, which may facilitate the development of tailored therapeutic approaches.
Introduction
Autism spectrum disorder (ASD) is a neurodevelopmental disorder characterized by impaired social communication, restricted or repetitive behaviors or interests, and atypical sensory processing. ASD is extremely diverse, with widespread phenotypic differences among individuals and hundreds of risk genes (1). These ASD genes do not share a single common molecular or cellular function, though they tend to be involved in chromatin remodeling, mRNA translation, and synapse function (2). A major goal in autism research has been to test whether distinct gene mutations drive convergent pathophysiology at the level of molecular pathways, synapse function, neural circuits, or neural coding, which may underlie the shared core features of the condition. Whether such convergence exists, and at what level, remains unknown. In principle, distinct gene mutations could converge on a single shared neurobiological impairment (indicating a common basis for ASD), a small set of distinct impairments (indicating different functional clusters of ASD), or no common impairment (indicating that ASD is actually a large constellation of pathophysiologically distinct conditions).
Here, we review theories of autism at the neural circuit and neural coding levels and evaluate evidence for convergence across different genetic forms of autism. We focus on the primary sensory areas of the cerebral cortex because these have been intensively studied and because atypical sensory processing is a common feature of people with ASD and has been a diagnostic criterion since DSM-5 (3). Cellular, synapse, and circuit dysfunction have been extensively studied in the primary somatosensory (S1), visual (V1), and auditory (A1) cortex in many transgenic mouse models of ASD. This review focuses on these three cortical areas because the large number of studies in different ASD models provides a strong test case for pathophysiological convergence. Circuit dysfunction in these areas may underlie atypical sensory processing in autism. Multisensory, social, and motor phenotypes in ASD likely reflect dysfunction in other cortical areas (4, 5), but may be caused by similar cellular and circuit deficits as in the sensory cortex because of shared circuit architecture and developmental principles across the neocortex.
Why focus on the sensory cortex? Sensory processing impairments in ASD
Up to 90% of people with ASD show atypical sensory processing, which is often classified into hyper-responsiveness, hypo-responsiveness, sensory avoidance, and sensory-seeking behaviors (3, 6). These behavioral symptoms suggest dysfunction in sensory brain regions, which is often observed in brain-based measurements (4).
Behaviorally, multiple sensory domains are often affected (6). Hypo-responsiveness and hyper-responsiveness can occur in the same individuals (7). Hypo-responsiveness is as pronounced as hyper-responsiveness across clinical studies (8) and in first-person accounts (9). Sensory seeking is thought to reflect underlying hyposensitivity to sensory stimuli, while sensory avoidance may be driven by hypersensitivity. Individual syndromic forms of autism also exhibit a mixture of these sensory phenotypes across different sensory modalities, depending on the type of stimulus and its social relevance (10, 11). Cluster analyses of sensory profiles of individuals with autism tend to reveal two main behavioral subgroups: one that is impaired relative to typically developing individuals across all four sensory domains (poor registration, sensitivity, sensory seeking, and sensory avoiding), and one that shows no impairment across any of the four (12–14). Recent work suggests smaller clusters of individuals with deficits in particular sensory modalities exist (15).
More specific psychophysical characterization of processing impairments is rare and usually performed in high-functioning individuals. Some of the most common findings from psychophysical studies in touch, vision, and hearing are lower tactile thresholds for detection of vibration and painful stimuli (7, 16), auditory hyper-responsiveness (17–20), difficulty in distinguishing speech in noise (21, 22), impaired temporal processing of sounds (23), elevated visual detail detection (24), weak binocular rivalry, altered visual motion processing (25), and possibly face perception deficits (26). These sensory impairments are reviewed in detail elsewhere (4, 5, 18, 27). Psychophysical differences across more severe, syndromic forms of autism are generally less known. In the sensory cortex, functional magnetic resonance imaging (fMRI) and electroencephalography (EEG) studies have identified varied phenotypes in people with ASD, including hypo- and hyperactivation by sensory stimuli. fMRI findings include hyperactivation by visual and auditory stimuli, which may relate to behavioral hypersensitivity (28), abnormal somatosensory maps, and decreased habituation in the somatosensory cortex to mildly aversive tactile stimuli (29). This suggests that the sensory cortex is a site of processing abnormalities in autism, a view that is supported by a recent study that found that, of all brain areas, primary sensory cortices show the most profound gene expression changes in people with ASD (30).
“Sensory-first” theories of autism posit that impairments in early sensory processing (including in the primary sensory cortex) may lead to widespread effects that contribute to impairments outside of the sensory domain. For example, touch and tactile perception are essential during infancy for the development of secure attachments with caregivers and continue to support social functioning throughout life (31). Impairments in somatosensation leading to tactile aversion could thereby contribute to social dysfunction and anxiety in individuals with ASD, as suggested by recent studies in mice (32). Similarly, impairments in early visual processing or in the processing of face information could drive reduced eye contact, which is an early indicator of autism, persists through adulthood, and may be relevant for social impairments in autism (4). Moreover, the mechanism of circuit disruption in sensory areas may occur commonly throughout the cortex and underlie both sensory and non-sensory features of ASD.
Genetic mouse models of ASD provide a basis for studying convergence
Nearly all widely used transgenic mouse models of ASD model severe monogenic, syndromic forms of autism, caused by loss of function in a single gene. These include fragile X syndrome (FXS, caused by loss of function in the FMR1 gene), Rett Syndrome (loss of function in the MECP2 gene), Tuberous Sclerosis Complex (loss of function in either the TSC1 or TSC2 gene), Phelan-McDermid syndrome (PMS, loss of function in the SHANK1, 2, or 3 genes), and Angelman syndrome (loss of function in the UBE3A gene). These mice exhibit abnormal social interactions and repetitive behavior, as well as sensory processing impairments reminiscent of human autism. In humans, many of these syndromes are also associated with intellectual disability, cognitive impairment, and/or seizures, and these mouse models often exhibit corresponding behavioral phenotypes. Other transgenic rare syndromic ASD models in which the sensory cortex has been substantially studied include Cntnap2–/–, Syngap1+/–, Scn1a+/–, Scn2a+/–, and Arid1b+/– mice.
These ASD models exhibit a range of sensory behavioral abnormalities, which may reflect atypical sensory processing in autism (31). In the tactile domain, many ASD models (including Mecp2 null, Shank3B–/–, Fmr1–/–, Gabrb3+/–, and Syngap1+/– mice) show impaired recognition of novel textured objects, interpreted as a deficit in texture discrimination (32–35). Syngap1+/– mice also show impaired tactile sensory detection on an operant task (34). In contrast, Shank3B–/– mice show enhanced detection of weak tactile stimuli (36), and Fmr1 mice show impaired adaptation that causes innate tactile defensiveness (37). Innate sensitivity to touch and painful stimuli is enhanced in Fmr1–/–, Shank3B–/–, Mecp2 null, Gabrb3+/–, and Cntnap2–/– mice (32, 35, 38), while pain sensitivity is decreased in Shank2–/– mice (39). In the visual domain, orientation discrimination is impaired in Fmr1–/– mice (40); visual contrast detection is impaired in Cntnap2–/– mice (41); and visual form discrimination learning is slowed in Ube3a m–/p+ mice (42). In contrast, a variety of innate visual detection behaviors are enhanced, including the optokinetic reflex in Mecp2 duplication mice (43), and visually evoked fidget responses in Fmr1–/– mice (44). Visual acuity is also enhanced in Mecp2 duplication mice (45). In the auditory domain, Fmr1–/– and Cntnap2–/– mice show increased acoustic startle (19, 46) and audiogenic seizures (47), and Fmr1–/– mice show normal tone detection performance on an operant task, but reduced reaction times that suggest increased perceived loudness (48).
This diversity of sensory deficits parallels the broad range of sensory phenotypes in people with ASD, but is there any convergence in sensory deficits across different mouse models? While behavioral task design varies across studies, a strong trend is apparent in the evidence. Across mouse models, innate sensory detection-related behaviors are reliably elevated (e.g., paw withdrawal to a touch stimulus, acoustic startle, and visually evoked fidget behavior), operantly trained detection behaviors show a mix of enhancement and impairment (e.g., operant visual or whisker touch detection), and innate or operant sensory discrimination behaviors are reduced or impaired (e.g., textured novel object recognition and operantly trained visual discrimination). This suggests that subcortical sensory processing (subserving innate detection) may be hypersensitive across many models, whereas cortical processing (mediating more complex discrimination and some operantly learned detection behaviors) may be degraded.
Using these mouse models, many studies have measured molecular, cellular, and circuit correlates of ASD in the sensory cortex. These provide the main data for evaluating possible pathophysiological convergence in brain function across genetic forms of ASD. Mouse models also provide a platform for testing potential therapeutic approaches to normalize cellular and circuit function and behavior. However, it is important to recognize that these mice all model severe syndromic forms of autism that are only a small fraction of autism cases in humans and do not include milder, higher-functioning forms of autism. Lessons from existing mouse models are therefore unlikely to be relevant across the entire autism spectrum.
Theories of sensory circuit dysfunction in ASD
This section will describe and evaluate common mechanistic theories of cortical circuit dysfunction in ASD (schematized in Figure 1). This overview will position us to evaluate, for each theory, the evidence for convergence across various genetic mouse models of autism.
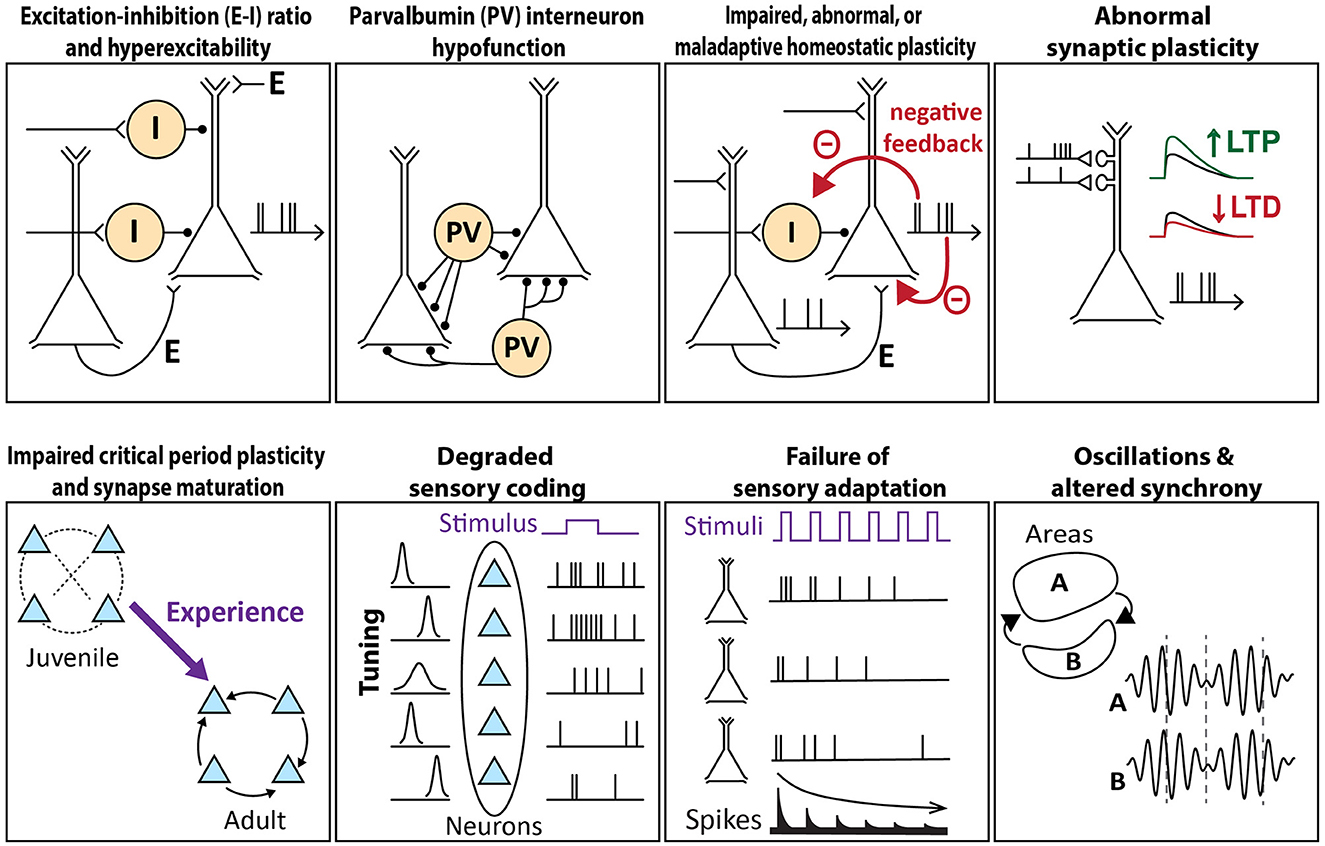
Figure 1. Schema of circuit mechanisms underlying sensory processing that are theorized to go awry in autism. Each image illustrates the normal circuit function, a breakdown of which may contribute to sensory features of autism.
Excitation–inhibition (E–I) ratio and hyperexcitability
One of the first cellular-circuit theories of ASD was the E–I ratio hypothesis. This hypothesis proposes that in ASD, cortical circuits exhibit reduced synaptic inhibition or increased synaptic excitation onto pyramidal (PYR) cells, and that the resulting increase in E–I ratio drives circuit hyperexcitability and excess PYR spiking (49, 50). In support of this idea, a reduction of GABAergic markers is commonly observed in the brains of people with autism, which could potentially drive hyperexcitability, and may explain why autism is associated with epilepsy in 30% of individuals (51). In the sensory cortex, circuit hyperexcitability could predict sensory hypersensitivity in autism (52). But atypical sensory processing in autism is not just hypersensitivity, and indeed, hyposensitivity and sensory seeking are common, so circuit hyperexcitability is unlikely to provide a full explanation. Moreover, the diversity of inhibitory cell types and circuits in the cortex complicates the idea of a simple “E–I ratio,” and it is now understood that transient imbalances and delays between E and I are a critical aspect of normal information processing in the cortex. While inhibition is often reduced in the ASD mouse cortex, this only in some cases leads to consequential changes in PYR spiking (53). Therefore, the E–I ratio and hyperexcitability hypothesis does not appear to account for sensory cortex dysfunction in the majority of autism mouse models.
Parvalbumin (PV) interneuron hypofunction
The parvalbumin (PV) hypothesis of ASD posits that cortical dysfunction originates from the hypofunction of PV-positive interneuron circuits (54). PV interneurons represent ~40% of cortical interneurons and provide powerful perisomatic inhibition to PYR cells (55). PV cells have critical information processing functions in the sensory cortex, including gain modulation, sharpening sensory tuning, generating gamma rhythms, enforcing precise spike timing, and modulating noise correlations. In post-mortem brains of people with ASD, PV is the most downregulated mRNA transcript, and the number of PV-positive neurons is diminished (56, 57). In studies of ASD mouse models, reduced PV cell number, reduced PV protein expression, or impaired PV circuit function are commonly featured (58, 59). PV cell and circuit hypofunction could detrimentally elevate the PYR firing rate, and even if the PYR firing rate remains normal, it could muddy neural codes by broadening tuning, impairing gain regulation, disturbing gamma synchronization, or degrading spike timing. PV neurons also regulate critical period plasticity, and thus PV hypofunction could alter or impair activity-dependent circuit development, producing abnormal or persistently immature circuits (60). Ample evidence exists for PV hypofunction in ASD, which could drive many of the sensory phenotypes. PV function may be altered directly by some ASD gene mutations. Alternatively, because PV circuits are highly plastic as part of the brain's endogenous homeostatic mechanisms, PV hypofunction could arise as a compensatory mechanism that is recruited in response to abnormal cortical activity (61).
Impaired, abnormal, or maladaptive homeostatic plasticity
Homeostatic plasticity is a negative-feedback process that dynamically adjusts excitation, inhibition, and/or intrinsic excitability to stabilize network activity. Impaired or abnormal homeostatic plasticity has been proposed to drive atypical firing rates or patterns, disrupting the flow of information in cortical circuits and driving sensory processing impairment in ASD (62). Multiple homeostatic mechanisms exist in cortical circuits, and function together to actively maintain the PYR cell firing rate near a setpoint value that may represent the optimal firing rate for encoding information (63). These mechanisms include synaptic scaling, which multiplicatively adjusts excitatory or inhibitory synaptic weights on single PYR cells; homeostatic plasticity of PYR cell intrinsic excitability; and PV circuit plasticity, in which the gain of PV interneuron circuits is adjusted in response to mean activity in local networks. These processes work at different time scales, but all have the net effect of preventing the mean firing rates of local PYR cells from deviating from their setpoint.
If homeostatic plasticity is impaired or abnormal in ASD, networks would lose the ability to maintain stable PYR firing rates. In support of this theory, ASD risk genes span a range of cellular processes, including activity-dependent transcription, translation, energy metabolism, synaptic function, and intrinsic excitability, that are expected to contribute to homeostatic plasticity (64). This suggests that ASD-linked mutations may abolish or dysregulate key forms of neuronal homeostasis, so that PYR activity is destabilized or abnormal (62). Alternatively, mutations may alter homeostatic plasticity to make it maladaptive, in the sense that it could succeed in stabilizing firing rates, but at the cost of degrading some other critical aspect of neural coding (53). Several genes associated with syndromic autism have been linked to the failure of homeostatic mechanisms, suggesting that impaired or abnormal homeostasis could be a convergent explanation for multiple forms of ASD in humans (65). In this view, the diverse array of synaptic and intrinsic physiological abnormalities observed across different ASD mouse models may reflect the failure of homeostasis to reset normal values for these parameters.
Abnormal synaptic plasticity
Impairments in long-term synaptic plasticity, including long-term potentiation (LTP) and long-term depression (LTD) at excitatory synapses onto PYR cells, are prevalent across mouse models of autism. These impairments may not only impact learning and memory but also drive sensory dysfunction by impairing the use-dependent refinement of circuits in the sensory cortex. Impaired LTD could lead to excessive synaptic strength and sensory hypersensitivity, while impaired LTP may prevent the formation or strengthening of connections necessary for normal sensory processing or integration (66). Many ASD-associated gene mutations are predicted to influence synaptic plasticity directly or indirectly, for example by dysregulating protein synthesis and degradation (2, 67).
A specific synaptic plasticity-related theory is the metabotropic glutamate receptor (mGluR) hypothesis for fragile X syndrome (68). In Fmr1 null mice, mGluR-mediated LTD is exaggerated due to the absence of the fragile X protein (FXP, encoded by the Fmr1 gene), leading to increased protein synthesis levels (69). Excessive LTD is associated with an increased prevalence of long, thin dendritic spines with weaker synapses, which may reflect immature circuits that generate weak or imprecise sensory codes. Other mouse models of ASD also have immature spine phenotypes and altered mGluR signaling and/or mGluR-LTD (70–72), and it appears that dysregulation of protein synthesis and mGluR-LTD in either direction may lead to ASD-related behaviors (73). Activity-dependent protein synthesis subserves both LTP and LTD and is dysregulated in numerous ASD models (74, 75). Decoupling protein synthesis from network activity will affect not only mGluR-mediated forms of plasticity but also have numerous consequences for sensory circuit development and function. Recent reviews discuss these synaptic plasticity and protein synthesis-related hypotheses in depth (76–78).
Impaired critical period plasticity and synapse maturation
Sensory cortical areas undergo robust activity-dependent plasticity during critical periods in development, when environmental input drives synapse maturation and refines and stabilizes circuits. In the impaired critical period hypothesis, ASD pathophysiology disrupts critical period plasticity, perturbing normal circuit maturation to produce long-lasting changes in circuit organization and behavior (79). Such critical period disruption could be due to impairments in cellular plasticity mechanisms, abnormal sensory experience, or abnormal circuit activity patterns. Impaired or dysregulated synaptic plasticity rules are well described in several ASD mouse models (see “Abnormal synaptic plasticity”). In addition, PV hypofunction is likely to disrupt critical period plasticity because PV interneurons are critical for regulating the timing of critical periods. Because PV neurons tend to sharpen the sensory tuning of PYR cells, PV hypofunction could also impair the sensory-guided development of precise sensory circuits, undermining the development of typical sensory processing (80).
Children often begin showing behavioral signs of ASD during these critical windows of development, although structural and physiological changes in neural circuitry could begin as early as infancy (81). Multiple mouse models of ASD show disrupted critical periods (82–84), altered critical period timing (85), and critical period-related synaptic impairments that include impaired cellular plasticity, delayed maturation of inhibitory circuitry, and abnormal retention of immature dendritic spines and silent synapses (86–90). Whether these abnormalities reflect a specific deficit in critical period plasticity or a more general impairment of plasticity throughout life, remains unclear.
Degraded sensory coding
The local circuit and synaptic alterations described above are all likely to lead to impairments in the neural coding of sensory information. Degraded coding could take many forms, including broadened sensory tuning of individual neurons, blurred sensory maps, reduced signal-to-noise ratio of sensory responses relative to spontaneous activity, increased trial-to-trial sensory response variability, and altered firing correlations that reduce information carried by population codes. We define degraded coding as changes in sensory coding that reduce the information available for sensory detection or discrimination. Such changes would dim, blur, or distort perception and could underlie both atypical sensory processing and downstream behavioral phenotypes such as sensory seeking, sensory aversion, or insistence on sameness. The degraded coding hypothesis supposes that the specific genetic, cellular, or circuit origin of the deficits is less relevant than the coding deficit itself, so that convergence across genetic forms of ASD occurs on the neural coding level.
Many mouse models of ASD exhibit degraded sensory coding in the form of reduced signal-to-noise for sensory responses (34, 53, 91, 92), increased trial-to-trial response variability (93–95), abnormal sensory maps (83, 96–98), degraded sensory tuning (40, 93–95, 99), or abnormal firing correlations (100–105). In people with ASD, studies have reported impaired sensory discrimination for touch and vision (40, 106, 107), impaired detection of speech in noise (21, 108), altered sensory-evoked event-related potentials (ERPs) (18), increased trial-to-trial variability of sensory-evoked ERPs (109), and altered topography of cortical sensory maps (110, 111). These suggest degraded sensory coding, though more quantitative psychophysical measurements are needed.
Degraded sensory coding is distinct from the classical E–I ratio theory, which proposes hyperexcitability and excess spiking in ASD, leading to sensory hypersensitivity (49) and an overly intense sensory world (52). Sensory hyper-reactivity and aversion in people with ASD may reflect either excessive psychophysical intensity or a heightened affective reaction but a normal perception of intensity (20, 47, 112). Across ASD mouse models, even those with increased E–I ratio in the sensory cortex, normal or below-normal neural sensory-evoked spiking is common (34, 40, 53, 91, 99, 113) while excess spiking is relatively rare (36, 94, 114). Thus, degraded coding may be more prevalent than hyperexcitability in the sensory cortex, at least in mice.
Failure of sensory adaptation
Adaptation to repeated or continuous stimuli is a common feature of sensory processing and serves to increase coding efficiency and enhance the representation of novel stimuli by reducing spiking to repeated, predictable stimuli. This theory proposes that in ASD, cellular and circuit abnormalities cause a failure of adaptation so that repeated stimuli evoke abnormally strong cortical spiking. This may lead to sensory processing impairments, sensory hypersensitivity, and behavioral avoidance of ongoing stimuli. Failure of adaptation will preferentially affect coding in sensory contexts with dense ongoing stimuli, which are common in natural environments. Consistent with this theory, individuals with ASD can show reduced adaptation to audio-visual stimulation (115) and tactile stimuli (29, 107, 116).
In ASD mice, evidence for this theory comes from Fmr1–/– mice, which show impaired spike adaptation to repeated whisker stimuli in L2/3 of the S1 cortex and behavioral avoidance of repetitive whisker stimulation, termed ‘tactile defensiveness' (37). Fmr1–/– mice also have impaired habituation in A1 to repeated sounds (117). In Fmr1–/– mice, pharmacological or pharmacogenetic enhancement of PV cell spiking increases response adaptation by PYR cells to repeated whisker stimuli and reduces tactile defensiveness (61). This suggests that impaired adaptation in Fmr1–/– mice arises at least in part from PV hypofunction in the sensory cortex, although other mechanisms could also contribute. Whether other ASD models have impaired sensory adaptation is unknown, though Ube3A m–/p+ mice have changes in inhibitory synaptic transmission that potentially suggest changes in response adaptation (118).
Oscillations and altered synchrony
Cortical sensory areas exhibit rhythmic oscillations at a wide range of frequencies (119). Both the amplitude and phase of oscillations, particularly in the gamma (>30 Hz) and alpha (8–12 Hz) bands, have been correlated with sensory perception in humans (120–125). In standard models, gamma rhythms (thought to be generated by PV interneuron networks) create a temporal scaffold for local sensory processing and information relay that is necessary for sensory perception, while alpha rhythms are part of an attentional suppression mechanism. EEG and magnetoencephalography (MEG) studies have reported atypical oscillations in both the gamma and alpha bands in humans with ASD (126). In multiple studies, visual-evoked gamma rhythm was weakened in individuals with ASD (127–134), suggesting a processing impairment. Yet other studies found no difference or increased gamma power (135–138), so there is no complete consensus (139). Some studies have shown weaker alpha power or aberrant alpha power modulation in ASD children, which may indicate impaired suppression of responses to irrelevant stimuli (129, 140). In addition to these local effects, synchronization between the left and right hemisphere V1 is reported to be disrupted across a wide range of frequencies (141–143), which may correlate with impaired perceptual integration (143).
Oscillations have been studied less in mouse models of ASD. Divergent effects on gamma power in the auditory cortex have been reported, from reduced (138, 144, 145), to increased (146), to not different from wild type (147). Some studies have reported that gamma phase locking is impaired (138, 144, 145). Thus, gamma oscillations may be abnormal in sensory cortex in humans with ASD, and interhemispheric coherence and gamma phase locking may be reduced, but whether these effects are reliably observed in ASD mouse models remains unclear. More work needs to be done to discern common oscillation and synchrony phenotypes in mice and to identify the underlying circuit mechanisms.
Convergence across mouse models of autism
Here, we examine the evidence for each of these circuit-level theories across ASD mouse models to evaluate the extent to which distinct genetic forms of ASD converge on any of these circuit-level deficits. Of the theories summarized above, sufficient empirical studies exist across mouse models to begin to evaluate convergence for four theories. These are the E–I ratio and hyperexcitability hypothesis, the PV hypofunction hypothesis, the impaired homeostasis hypothesis, and the degraded sensory coding hypothesis.
E–I ratio and hyperexcitability
Elevated E–I ratio is theorized to drive hyperexcitability and excess spiking in the sensory cortex in ASD mouse models, leading to behavioral hypersensitivity. At the local circuit level, synaptic E–I ratio is demonstrably increased across many ASD models, including in S1 for Fmr1–/–, Cntnap2–/–, Tsc2+/–, and 16p11.2 deletion (53), driven by a large reduction in synaptic inhibition coupled to a weaker drop in excitation onto L2/3 pyramidal cells. Remarkably similar synaptic findings are reported in V1 for Mecp2 null and Ube3A m–/p+ mouse lines (93, 118). However, rather than cause excess spiking, the increase in E–I ratio in the four mouse models from Antoine et al. (53) was shown to quantitatively predict normal net synaptic depolarization and to be associated with normal firing rate in L2/3 pyramidal cells measured in vivo. This shows that while an increased E–I ratio is common, it does not necessarily drive hyperactive cortical circuits in ASD mice.
There is convincing evidence for neural hyperactivity in primary sensory cortices in just three mouse lines: Scn1a+/–, Shank3B–/–, and some cortical areas in Fmr1–/–. Scn1a is a major sodium channel in forebrain GABAergic interneurons, and Scn1a+/– mice show reduced excitability of PV and somatostatin interneurons that reduces recurrent inhibition and leads to strong hyperexcitability and epilepsy, including in S1 (148–152). Shank3B–/– mice have increased spontaneous and whisker-evoked calcium responses in L2/3 pyramidal neurons in S1 and reduced activity in interneurons, and this excess pyramidal activity is associated with behavioral hyper-reactivity to weak whisker stimulation in a whisker detection task (36). Spontaneous spiking in L5 of S1 is also strongly elevated in Shank3B–/– (153) and hyperexcitability is present in V1 (154). However, one study did not observe neural hyperactivity in S1 using c-fos (33). Fmr1–/– mice show excess sensory-evoked spiking in some sensory areas, including in A1 and forepaw S1 (94, 114). These mice show audiogenic seizures, but these are due to circuit hyperexcitability in the inferior colliculus, not the auditory cortex (155). In slice physiology experiments in Fmr1–/– mice, thalamocortical circuits evoke sustained up-states in S1, indicating local circuit hyperexcitability (156). Together, these results indicate hyperactivity in A1 and S1 in Fmr1–/– mice. However, hyperexcitability is not present in Fmr1–/y in other sensory areas, for example in whisker S1, where spiking to preferred whisker stimuli is normal or slightly depressed with broadened single-neuron tuning, leading to blurred somatotopic maps (53, 97, 157). Excess spikes are also not apparent in V1 (40). Thus, Fmr1–/y mice appear to show hyperexcitability only in some cortical areas.
Cntnap2–/– mouse models present somewhat weaker evidence for hyperexcitability, which is inconsistent across studies. Cntnap2–/– mice show increased c-fos expression in S1 following whisker stimulation (158), but no change in spontaneous or whisker-evoked spiking in S1 measured with extracellular recordings (53). In V1, neurons are hyporesponsive to visual stimuli, and mice show behavioral hyposensitivity and impaired discrimination (41). In A1, Cntnap2–/– mice show reduced spontaneous activity and slightly increased sound-evoked spiking responses (159), which are associated behaviorally with increased startle and impaired auditory filtering (160). Thus, Cntnap2–/– rodents show a range of spike rate phenotypes but not consistent evidence for excess spiking.
Other mouse lines, including Mecp2 and Syngap1, show substantial evidence of decreased cortical excitability. In both Mecp2 null and Mecp2 duplication mice, multiple reports show a decreased E–I ratio, involving both reduced synaptic excitation (91) and increased inhibition associated with increased PV cell number and/or PV expression in V1, S1, and A1 (85, 161, 162). These changes are associated with a strong reduction in sensory-evoked neural responses in V1 (43, 45, 163). A different study in Mecp2 null mice found an increased E–I ratio in V1 as a result of a preferential reduction of inhibition over excitation, but still observed abnormally weak visual-evoked spiking (93). In contrast, A1 of Mecp2 knockout mice shows hyperexcitable auditory responses (164). The reason for discrepancies between various Mecp2 studies is not clear but could be related to age differences (163, 165). Hypoactivity is also evident in Syngap1+/– mice, which displayed reduced whisker-evoked activity in L2/3 pyramidal cells in S1, due to reduced whisker-evoked synaptic input and reduced intrinsic excitability (34). This neural hypoactivity was correlated with poor performance in tactile object detection and discrimination.
Thus, Shank3B–/–, Scn1a+/–, and Fmr1–/– in some brain regions form a phenotypic cluster that exhibits increased E–I ratio and excess spiking and often correlates with hypersensitivity in sensory detection tasks (Figure 2). In other sensory areas, Fmr1–/– has normal or reduced spiking, rather than increased spiking. Cntnap2–/– shows mixed results, and in at least one study, Cntnap2–/– is similar to Fmr1–/y, 16p11.2 del, and Tsc2+/– mice in showing elevated synaptic E–I ratio but no increased spiking (53), with Ube3a m–/p+ showing similar results. These latter genotypes may have abnormal cortical circuit function due to reduced inhibition, but they do not exhibit overt hyperexcitability. In contrast, Syngap1+/– and most Mecp2 null studies indicate a second phenotypic cluster, which generally shows a reduced E–I ratio and reduced spiking in the sensory cortex. Both clusters exhibit impairments in sensory processing (described below), but only the first cluster shows excess spiking.
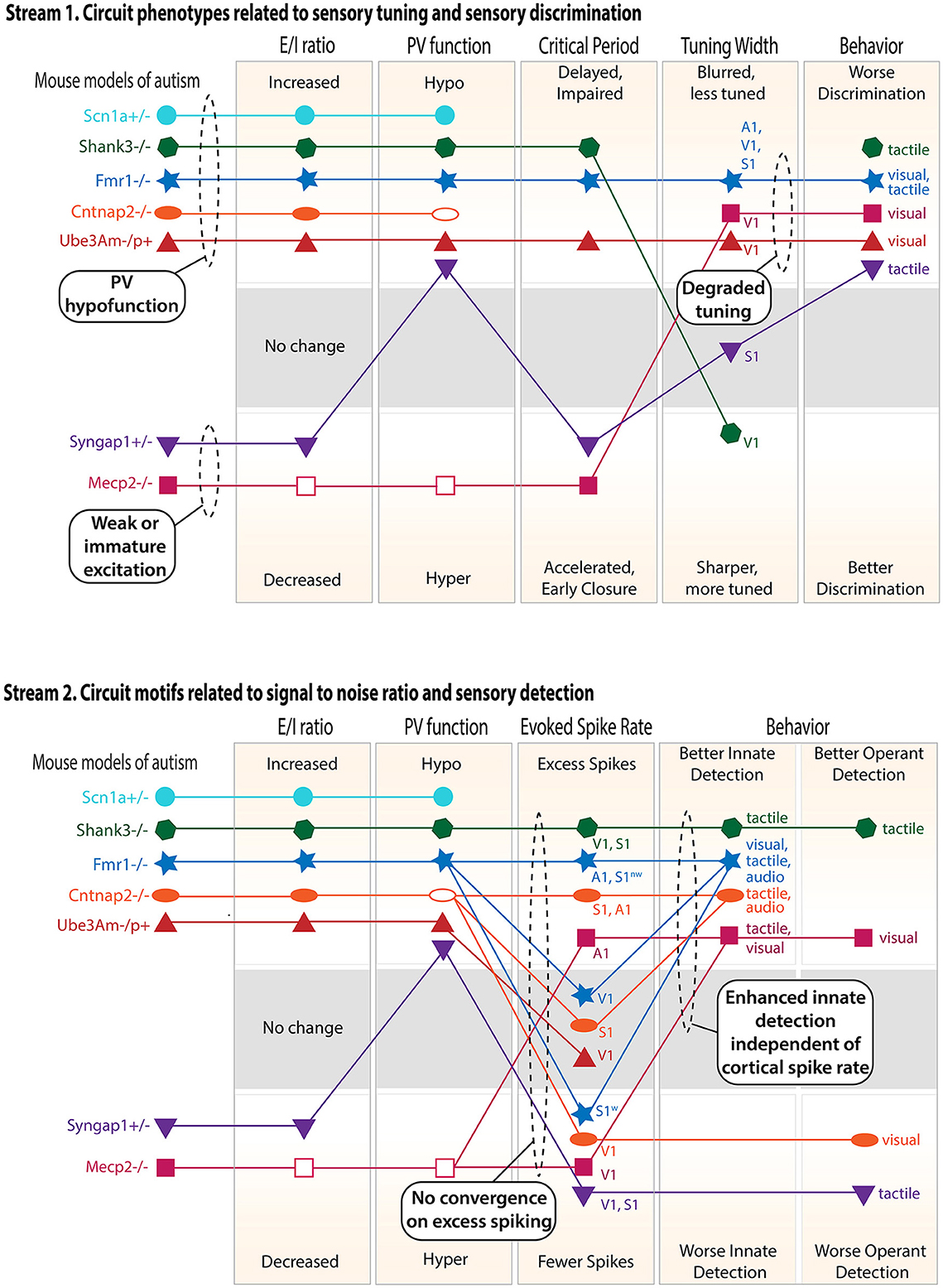
Figure 2. Cluster analysis of the circuit, neural coding, and behavioral phenotypes across different genetic mouse models of ASD. Only findings from V1, S1, and A1 are considered, and only mouse models with a substantial number of published studies are included. Top panel: circuit phenotypes related to sensory tuning and sensory discrimination behavior. Discrimination behavior results include both innate discrimination behavior and operantly trained discrimination behavior (see text). Within each rectangle, the axis is organized so that the naively predicted consequences of a high E–I ratio are above and the consequences of a low E–I ratio are below. When multiple studies in the same cortical area gave conflicting results, an unfilled symbol was used to indicate the major reported effect. When multiple studies in different sensory regions or modalities gave different results, multiple points were plotted and labeled accordingly. Lines show relationships within each mouse model. S1w denotes whisker S1, and S1nw denotes non-whisker S1. Bottom panel: circuit phenotypes related to signal-to-noise ratio and sensory detection behaviors. Dashed ovals highlight convergence across ASD models or logical conclusions that can be drawn across models.
Therefore, the expectation that an increased E–I ratio drives excess spiking and that this leads to sensory hypersensitivity holds only rarely, and just for genes in the first cluster (Figure 2). These represent ASD genes that are essential for interneuron function but have a smaller role in PYR cells (e.g., Scn1a, which encodes NaV1.1, the main voltage-gated sodium channel in cortical interneurons). Loss of function of these genes is likely to weaken inhibition and increase the E–I ratio very substantially without a sufficient compensating drop in excitation, thus driving excess PYR spiking. Other ASD genes are associated with a modestly elevated E–I ratio, which appears to degrade neural coding without an elevation of net spiking in the sensory cortex, or a decreased E–I ratio, which correlates with reduced spiking (as in the case of the Syngap1+/– and Mecp2 null clusters) (Figure 2). Overall, increased E–I ratio does not systematically predict elevated spike rates. This may be because in many cases, a modest increase in E–I ratio associated with normal spike rates may actually reflect an endogenous homeostatic adjustment of E–I ratio that is recruited to preserve the mean firing rate or other aspects of sensory processing (53). Consistent with these results from mice, an analysis of syndromic ASD individuals shows that cortical hyperactivity vs. hypoactivity does not correlate well with sensory hypersensitivity vs. hyposensitivity, arguing further against the monolithic E–I ratio hypothesis (166).
Hypofunctional parvalbumin inhibitory circuits
Parvalbumin (PV) circuit dysfunction is strongly linked to autism, both in people with ASD and across genetically distinct ASD mouse models (54). PV is a calcium-binding protein whose expression in PV interneurons is believed to be activity-dependent (167). PV cell number, PV expression level, and PV circuit function have all been shown to be perturbed in ASD (58). Given the many impacts of PV interneurons on circuit function and neural coding, including sharpening sensory tuning, regulating E–I ratio and sensory response gain, contributing to sensory adaptation, and generating gamma oscillations, PV circuit dysfunction could drive multiple impairments in sensory processing and perception (80).
Reduced PV cell number, assayed by anti-PV immunostaining, is observed in sensory cortex in many ASD models, including Fmr1–/– (61, 168, 169), Shank3B–/– (170), Neuroligin-3–/– (171), Arid1b+/– (172), and some studies of Cntnap2–/– (173, 174) but not others (175, 176). In S1, PV loss in Shank3B–/– and Cntnap2–/– mice is strongest in the hemisphere corresponding to the mouse's dominant paw (177). Loss of PV cells could be due to their failure to develop properly or through selective apoptosis. Both of these factors are known to contribute to GABAergic cell loss in Arid1b+/– mice (172). In S1 of Fmr1–/– mice, PV cells die off via apoptosis during early postnatal development due to insufficient PV cell activity (61). In other ASD models, it remains unclear whether PV cell number is truly reduced or whether PV cell hypoactivity causes PV protein expression to fall below detectable levels (170). The use of alternative markers for PV cells, such as labels targeting the perineuronal net, can resolve this issue.
Those PV cells that remain are hypoactive in many mouse models. In S1 slices, feedforward L4-L2/3 inhibition, which is known to be PV-mediated, is reduced in Fmr1–/–, Cntnap2–/–, 16p11.2 del, and Tsc2+/– mice, and whisker stimuli evoke 50% fewer spikes than normal in L2/3 fast-spiking (presumed PV) units in Fmr1–/–, Cntnap2–/–, and 16p11.2 del mice (53). Whisker-evoked spiking in PV cells is also greatly reduced in Syngap1+/– mice (34) and Shank3B–/– mice (36). In V1, reduced visual-evoked activity is observed in PV interneurons in Fmr1–/– mice (40) and in one study in Mecp2 null mice (93). Reduced sensory responses in PV cells can have diverse causes, including reduced PV intrinsic excitability in Tsc2+/– mice (178) and Scn1a+/– mice (148), and delayed development of intrinsic excitability and excitatory synaptic input to PV cells in Fmr1–/– mice (89, 156).
The prediction in the cases above is that PV hypofunction would promote excess spiking, but as discussed in the “E–I ratio and hyperexcitability” section, this is not always true. Instead, PV hypofunction can lead to either network hyperexcitability or other types of downstream effects, including dysregulation of critical period plasticity, broadening of sensory tuning in PYR cells, or failure of sensory adaptation, as discussed below. Two clear examples of this divergence are Shank3B–/– and Fmr1–/– mice, which both exhibit PV hypofunction and increased E–I ratio. In Shank3B-/–, this leads to excess spiking in S1 and enhanced tactile detection (36, 40), but in Fmr1–/–, this leads to variable findings of excess, normal, or slightly reduced spiking in S1 (53, 97, 114, 157), and no excess spiking in V1 but broadened orientation tuning of PYR cells that impairs behavioral discrimination (36, 40).
Unlike the mice above that show PV hypofunction, Mecp2 null mice generally exhibit PV hyperfunction, which contributes to an overall increase in inhibition in the sensory cortex, particularly in the juvenile period. These mice show elevated PV expression and excitatory synaptic input to PV cells in S1 and excess PV synapses and PV inhibitory transmission in V1 (85, 162, 163). Some evidence suggests early PV hyperfunction may give way to PV hypofunction in adulthood (93, 163).
Because PV cells regulate critical periods in the sensory cortex, a reduction in PV number or a delay in PV circuit maturation could drive consequential changes in the developmental refinement of neural circuitry. This has been examined most closely in Fmr1 null mice, where PV interneurons in S1 show delayed development of intrinsic excitability and synaptic connections that are rescued by treatment with a TrkB agonist (89, 168). Delayed PV maturation also occurs in A1, where it involves delayed perineuronal net formation, the rescue of which is sufficient to improve network hyperactivity (169). In S1, Fmr1 null mice exhibit a delayed developmental transition from depolarizing to hyperpolarizing GABA, and correcting this imbalance restores the precision of the somatosensory map (179). Critical period plasticity is also impaired in Ube3A m–/p+ mice (84). In contrast, Mecp2+/– mice show an accelerated critical period in the visual cortex, which is attributed to elevated GABAergic activity (85, 161).
Given broad GABAergic changes in ASD brains, multiple studies have focused on manipulating all or broad subsets of interneuron types (MGE-derived vs. CGE-derived) to determine their role in ASD phenotypes. Deletion of Shank3B from forebrain interneurons is sufficient to drive whisker hypersensitivity, as is acutely reducing inhibition using chemogenetics in wild-type mice (36). Scn1a is primarily expressed in interneurons, and its selective deletion from Dlx1/2+ interneurons recapitulates the full knockout ASD phenotype (148). In maternal Ube3A m–/p+ mice, re-expression of Ube3A selectively in GABAergic cells corrects increased spiking and improves orientation tuning deficits (99). It is unknown whether these effects are due to effects on PV cells alone or other interneuron types.
Specific manipulation of the PV interneuron population has been extensively studied in Fmr1–/– mice across sensory areas. Activation of PV cells using chemogenetics restored visual responsiveness and orientation tuning and improved performance on an orientation discrimination task (40). Similarly, in S1 of Fmr1–/– mice, pharmacological or chemogenetic activation of PV cells restored normal sensory adaptation in pyramidal cells and rescued tactile defensiveness behavior (61). Interestingly, Fmr1 re-expression selectively in excitatory neurons during P14–P21 was sufficient to rescue the reduction in PV cell density and activation in A1 (180), suggesting Fmr1 mutation in excitatory neurons drives changes in inhibitory circuits. Haploinsufficiency of the PV gene itself causes autism-like behaviors and neural activity phenotypes, and rescuing PV levels is sufficient to rescue social behavior (181, 182). Thus, selective loss or restoration of ASD genes in PV cells or GABAergic interneurons can directly drive or rescue ASD circuits and behavioral phenotypes.
Whether Mecp2 acts exclusively in PV neurons to drive ASD-related phenotypes is unclear because of conflicting results in the literature. In some studies, Mecp2 loss from GABAergic neurons recapitulates Rett syndrome features and reduces levels of GABA synthesis enzymes (93, 183). PV-specific deletion of Mecp2 resulted in a decreased evoked spike rate in PV cells upon visual stimulation, indicating PV hypofunction and impaired V1 critical period plasticity (88). These studies are consistent with the idea that PV hypofunction (either caused cell-autonomously by Mecp2 loss in PV cells or secondary to loss in excitatory cells) can contribute to Rett phenotypes. But in another study, deletion of Mecp2 from forebrain excitatory but not inhibitory neurons led to seizures and a cell-autonomous reduction in GABAergic transmission, suggesting Mecp2 loss has its primary effect in excitatory neurons (184). Mecp2 is also expressed in other interneuron types, where its deletion may have complementary effects (185).
In summary, PV circuit hypofunction is a convincing feature of many ASD models, including Fmr1–/–, Shank3B–/–, Cntnap2–/–, Scn1a+/–, and Ube3A m–/p+ (Figure 2). This can be due to the loss of PV cells, decreased PV synaptic connectivity, reduced PV intrinsic excitability, or reduced PV expression itself. In some cases, PV hypofunction is driven by mutations in genes acting in PV cells specifically, while in other cases it may be a secondary, adaptive response to alterations in excitatory networks, e.g., via PV circuit homeostasis. PV hypofunction does not manifest in the same circuit- or sensory deficits in all ASD mouse models, and instead drives a variable set of circuit consequences ranging from excess spiking in PYR cells (Shank3B, Scn1a, some Fmr1 studies) to broader sensory tuning in PYR cells without excess spiking (other Fmr1 studies) to delayed or impaired critical periods (Fmr1–/–, Ube3A m–/p+). Mecp2 studies are mixed, but some suggest that Mecp2 null mice exhibit enhanced, not reduced, PV circuit function.
In spite of strong evidence for GABAergic dysfunction in humans with ASD (186) and successful rescue of ASD-related behaviors in mice using GABA modulators or PV circuit modulation (detailed above), clinical trials using GABAergic modulators to correct inhibition have not shown a substantial effect in treating sensory issues in children with ASD (59, 187). A possible explanation is that circuit dysfunction specifically reflects the hypofunction of PV cells and not other interneurons, suggesting that therapies should focus on PV-specific modulation. However, the relative contribution of early developmental PV impairment (which impairs critical period circuit refinement) vs. ongoing adult PV circuit impairment for ASD phenotypes remains unknown. Thus, new approaches using chemogenetics, photoactivatable proteins, small molecules, and gene therapies to selectively target PV cells in precise time windows may prove useful in treating ASDs and other neuropsychiatric disorders (188, 189).
Altered homeostasis
Impaired homeostatic plasticity has been theorized to underlie ASD, but studies evaluating this hypothesis are still somewhat scarce in the sensory cortex. Many ASD genes are involved in the activity-dependent regulation of network excitability (64), so the breakdown of homeostatic plasticity in ASD is a plausible mechanism of sensory circuit dysfunction.
Synaptic scaling is the most well-characterized homeostatic plasticity mechanism and has been extensively studied in ASD. Synaptic scaling is impaired in many mouse models, including Fmr1–/–, Mecp2–/–, Cntnap2–/–, Syngap1+/–, and Shank3B–/– (190–194). The classical form of synaptic scaling is up-scaling of excitatory synapses, induced in cultured neurons in response to silencing the network with TTX or glutamatergic blockers. In up-scaling, the synaptic strength of excitatory synapses is increased due to the insertion of α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid receptor (AMPAR). Up-scaling is absent in neuronal cultures from V1 of Shank3B–/– mice (194), and is reduced in cultures from Cntnap2–/– and Mecp2 knockdown neurons (190, 191). Synaptic scaling may fail for Shank3B–/– and Cntnap2–/– due to the role of these genes as postsynaptic scaffolding molecules and for Mecp2 because of its role as a transcriptional regulator. Interestingly, Fmr1–/– mice have impaired excitatory and GABAergic synaptic scaling in cultures from the hippocampus (192, 193), but synaptic scaling is normal in cultures from the cortex (195), which is surprising given that the fragile X protein is a key regulator of AMPAR receptor transcription and translation (196).
Homeostatic plasticity of intrinsic excitability is also induced by network silencing in neuronal cultures and is impaired in two ASD mouse models, Shank3B–/– (194) and Fmr1–/– (195). This may reflect direct and indirect interactions between Shank3 and FXP with ion channels that are regulated during homeostatic intrinsic plasticity. Whether other ASD mouse models show similar deficits has not been tested.
In vivo, homeostatic synaptic scaling, homeostatic plasticity of intrinsic excitability in pyramidal cells, and homeostatic plasticity in PV circuits work together to actively stabilize the mean firing rate of cortical PYR cells, which can be observed experimentally in response to sensory manipulations. For example, sustained monocular visual deprivation causes an initial rapid reduction in firing rate in PYR cells in V1, due to Hebbian plasticity mechanisms that suppress cortical responses to the closed eye, but after several days, firing rates begin to climb to restore pre-deprivation firing rates (197). This restoration, termed “firing rate homeostasis,” is due to homeostatic synaptic scaling, homeostatic intrinsic plasticity, and downregulation of PV circuit activity (198–200). Similar mechanisms occur in S1 to maintain stable firing rates for several days after whisker deprivation (201–203). In Shank3B–/– mice, firing rate homeostasis is abolished or substantially delayed (194). Visual deprivation also fails to induce synaptic scaling in V1 in vivo in Mecp2–/– and Cntnap2–/– mice (190, 191). Whether PV circuit homeostasis is impaired in ASD remains unknown, but it could be an important aspect of cortical pathophysiology that destabilizes pyramidal firing rates and sensory coding. Thus, more studies are needed, but existing evidence points to a major impairment in homeostatic plasticity in V1 across several ASD models.
Degraded sensory coding
Population imaging of neural activity and high-density single-unit recording in ASD mouse models provide strong evidence for neural coding disruptions in the sensory cortex. As discussed above, relatively few ASD mouse models show excess sensory-evoked spiking. Instead, mean sensory-evoked spike rate is often similar to wild type, as in whisker S1 of Cntnap2–/–, 16p11.2 del, and Tsc2+/– mice (53), V1 of Fmr1–/– mice and Ube3a m–/p+ mice (40, 118), or is weaker than wild type, as in S1 of Syngap1+/– mice and Fmr1–/– mice (34, 53), and V1 of Mecp2 null mice (93). But multiple aspects of neural coding are abnormal in these cases, including elevated spontaneous firing, increased trial-to-trial variability, broadened sensory tuning, blurred sensory maps, and abnormal adaptation. These coding phenotypes likely reflect cortical circuit dysfunction and may contribute to altered sensory behavior in ASD.
Spontaneous firing represents baseline noise in the absence of sensory stimuli and affects the signal-to-noise ratio for stimulus encoding. Some ASD models exhibit altered spontaneous activity, with Shank3B–/– mice exhibiting increased and Syngap1+/– mice exhibiting decreased spontaneous activity in S1 (34, 36). In contrast, spontaneous firing is largely normal in S1 and V1 in Fmr1 and En2 null mice (37, 40, 53, 83, 97, 114). Trial-to-trial reliability of sensory-evoked responses also affects coding accuracy and thus impacts behavioral detection and discrimination of stimuli. Trial-to-trial variability in sensory responses is increased in A1 and V1 of Fmr1, Cacna2d3, and Mecp2 null mice (93–95), which is predicted to worsen sensory performance. This is consistent with excess trial-to-trial variability measured in visual, auditory, and somatosensory-evoked ERPs in people with ASD (109).
Degraded sensory tuning of single neurons (i.e., broader tuning) will also degrade sensory perception, by blurring differences in population activity across different stimuli. In Fmr1–/– mice, frequency tuning in A1 (94), whisker tuning in L2/3 of S1 (97), and orientation tuning in L2/3 of V1 (40) are all broader compared to the control. For Mecp2, two studies in Mecp2–/– and Mecp2 overexpression mice found broader tuning in V1 (43, 93), but other studies found normal tuning width (45, 85). Broader orientation tuning was also found in V1 of Ube3a m–/p+ mice (99), and broader whisker tuning in S1 of 15q duplication mice (92). Thus, broadened sensory tuning is a common motif across S1, A1, and V1 (Figure 2). But this phenotype is not universal, with PTEN and Cacna2d3–/– mice showing normal sensory tuning (95, 204), and Shank3+/– mice showing overly narrow orientation tuning in V1 (205). Broader tuning is expected to correlate with impaired behavioral discrimination of sensory stimuli, and such impairments have been observed for tactile stimuli (33, 34, 206), and for visual orientation (40) across several ASD mouse models.
Topographic maps are another important feature of primary sensory cortices and are also impacted in multiple ASD mouse models. The somatotopic whisker map is blurred in Fmr1–/– mice (53, 97, 157, 179), and the tonotopic map in the auditory cortex is blurred in Mecp2 overexpression mice (98), Fmr1–/– mice (94) and in the VPA rat model (96). The binocular area of V1 is expanded in En2 null mice (83). These examples of blurring or altered map topography are consistent with alterations in map topography in humans with ASD (110, 111). Thus, ASD mouse models commonly show broadened single-neuron sensory tuning and blurred or altered sensory maps. These phenotypes are likely to result in impairments in sensory discrimination.
Other aspects of neural coding are also disrupted in multiple ASD mouse models, including abnormal sensory adaptation in Fmr1–/– mice (37), which can be rescued by PV cell activation (61), and abnormal firing synchrony between neurons in local networks in Fmr1 and Cntnap2 null mice (100–105). Decoding of sensory information by downstream regions may be impaired by abnormal activity correlations across areas, which have been observed in Shank3 null macaques and in humans with ASD (207, 208), and abnormal gamma rhythms, as observed in Fmr1 and Cntnap2 null mice and in people with fragile X syndrome (104, 144, 145, 180, 209).
Thus, degraded neural coding is evident on multiple levels across many mouse models, even when sensory-evoked firing rates are near normal. These coding deficits are predicted to reduce the amount of sensory information available for detection and discrimination at the level of the primary sensory cortex, which may create a dim, blurred, or confusing sensory world. Across ASD mice, there is no single uniform type of coding degradation, but degraded sensory tuning and blurred maps are the most common. These phenotypes are predicted either from an acute lack of PV inhibition that normally sharpens sensory tuning or from a developmental failure of experience-dependent strengthening, refinement, and consolidation of synapses during critical periods. In the latter case, sensory enrichment or training that engages natural plasticity mechanisms could potentially restore coding precision, perhaps in concert with treatments that enhance the capacity for synaptic plasticity.
What are we missing beyond the primary sensory cortex?
In this review, we have only considered circuit and coding abnormalities in the primary sensory cortex, but other brain areas are also likely to contribute to ASD sensory phenotypes. For example, multiple ASD mouse models show hyperexcitability in peripheral somatosensory receptor neurons that contribute to behavioral touch hypersensitivity and may drive downstream circuit changes in the somatosensory cortex, including homeostatic changes to compensate for the increased upstream sensory drive (35). These peripheral sensory changes could also drive secondary social behavioral impairments due to distorted or aversive touch-related social cues during critical periods for social behavior (27). Other subcortical sensory circuits could contribute to elevated sensory responsiveness in ASD, potentially including the brainstem, amygdala, or cerebellum (which builds ongoing predictive models of sensory input).
We also did not review theories of abnormal functional connectivity between brain areas, which have been suggested by fMRI and EEG studies in people with ASD. Such abnormalities could alter the propagation and integration of sensory information in the brain, but they include a wide variety of phenotypes across brain areas and ages (210) and are currently difficult to map onto underlying circuit mechanisms. One interesting form of this theory is that sensory impairments in autism arise from atypical hierarchical processing, in which there is an over-reliance on bottom-up sensory processing and weakened top-down modulation, leading to impaired ability to contextualize sensory input (31, 211). While this idea has some interesting support (212), the prevalence, circuit mechanisms, and functional consequences remain unclear.
Synthesis: convergence and clustering across ASD mouse models
The circuit, coding, and behavioral phenotypes for the best-studied ASD mouse models presented above are plotted in Figure 2 as a means of visualizing convergence and clustering across mouse models. This analysis also allows predictive relationships between theories to be visualized. Results are separated into two logical streams in order to better visualize associations. Each stream focuses on a different potential effect of inhibition on circuit function, neural coding, and sensory behavior.
Stream 1—circuit properties related to sensory tuning and discrimination
PV circuits impact the sensory tuning of pyramidal cells. This occurs both during development, where PV inhibition gates the critical periods that refine excitatory circuits to create appropriate sensory tuning and sensory maps, and in the adult brain, where PV inhibition is recruited by sensory stimuli and acts to acutely sharpen PYR cell sensory tuning. Thus, abnormal PV circuit function in ASD, either in development or adulthood, may be expected to alter the sensory tuning of pyramidal cells, which in turn is likely to impact sensory discrimination behavior. To test for these relationships, the first stream plots the E–I ratio, PV function, critical periods, sensory tuning, and sensory discrimination behavior, including both innate (e.g., texture novel object recognition) and operantly trained discrimination behavior.
Key lessons from this analysis:
1. “PV hypofunction” and “weak or immature excitation” clusters. By examining E–I ratio, PV circuit function, and critical period plasticity, two clusters of mouse models emerge. the first cluster (Shank3B–/–, Fmr1–/–, Ube3Am–/p+, and Cntnap2–/–) shows elevated E–I ratio, weakened PV circuits, and delayed or impaired critical periods, which is a predicted consequence of PV hypofunction. This may be considered a “PV hypofunction” cluster, and Scn1a+/– is likely to also be in this cluster. The second cluster (Mecp2–/– and Syngap1+/–) shows the opposite effects: decreased E–I ratio and accelerated early closure of critical periods, sometimes with hyperfunction of PV cells (Mecp2–/–), but not always (Syngap1+/–). This may be considered a “weak or immature excitation” cluster.
2. All models show impaired sensory discrimination behavior. This behavioral phenotype can arise through different circuit mechanisms because it is associated with both PV hypofunction and weak excitation clusters.
3. Degraded sensory tuning is common and predicts impaired discrimination behavior. Multiple ASD models show degraded sensory tuning in the sensory cortex (and also blurred maps, not explicitly plotted in the figure). All of these models show impaired sensory discrimination. These are likely to be causally linked because degraded tuning reduces the information available in neural population codes to support sensory discrimination, and circuit manipulation in the primary sensory cortex that restores neural tuning can rescue behavioral discrimination phenotypes (40). Mouse models from both the PV hypofunction cluster and the weak excitation cluster can show degraded sensory coding, suggesting this is a common failure mode of cortical development or computation.
Stream 2—circuit properties related to signal-to-noise ratio and sensory detection
PV circuits, and inhibition more generally, also impact spontaneous firing and the gain and signal-to-noise ratio of sensory-evoked responses. These aspects of neural coding are essential for stimulus detection. Thus, abnormal PV circuit function or abnormal inhibition in ASD may be expected to alter sensory detection behavior (for example, to generate sensory hypersensitivity, as in the original E–I ratio hypothesis). To test for these relationships, the second stream plots the E–I ratio, PV function, sensory-evoked spike rate, and sensory detection behavior, including both innate sensory detection behavior (e.g., paw withdrawal to a calibrated touch stimulus or acoustic startle) and operantly trained detection behavior.
Key lessons from this analysis:
1. Elevated E–I ratio and PV hypofunction do not predict excess PYR spiking. Instead, some models show excess spiking (although not in all studies or all brain areas), and others show normal or decreased spiking. The strongest or most consistent excess spiking phenotypes are in Shank3+/– and in some but not all sensory cortical areas in Fmr1–/y. Scn1a+/– has highly elevated spontaneous activity and is also likely to exhibit excess spiking, but this has not been tested. Other models show clear evidence for normal or decreased spiking. Among the mouse models with reduced E–I ratio (i.e., those in the “weak or immature excitation” cluster (Syngap1+/– and Mecp2–/–), there is a trend toward lower PYR spiking, but this is also not absolute. Thus, while some phenotypic clustering exists at the level of E-I ratio and PV function, there is no clear convergence at the level of PYR firing rates in the sensory cortex. Those models that show excess spiking may be those in which gene mutation decreases PV and other interneuron functions most dramatically, perhaps via direct regulation of interneuron intrinsic excitability (e.g., Shank3B+/– and Scn1a+/–), or in which gene mutation also impairs synaptic and cellular homeostasis, so that firing rate cannot be normalized by homeostatic mechanisms (e.g., Fmr1–/y and Shank3B–/–).
2. Innate sensory detection behavior is uniformly elevated, whether the sensory-evoked firing rate is increased or not. Innate detection behavior is enhanced across all ASD models in this review (i.e., lower detection thresholds or heightened behavioral responses). This occurs independent of whether the sensory-evoked firing rate is elevated in the sensory cortex or not. We suggest that this is because innate detection behaviors are often not cortically dependent, so this enhancement may be driven by subcortical circuit alterations in ASD (35), or by the breakdown of cortical modulation of subcortical circuits. Operant sensory detection behavior is more variable, sometimes being enhanced (e.g., Shank3B–/–) and sometimes being reduced (e.g., Syngap1+/– and Cntnap2–/–). While operant sensory detection behavior correlates well with PYR spike rates in the sensory cortex, innate sensory detection behavior does not. Overall, only a few mouse models show consistent elevation (Shank3B–/–) or reduction (Syngap1+/–) at all three levels of E–I ratio, PYR spiking, and sensory behavioral detection.
Strikingly, the literature reveals a consistent pattern of degraded sensory discrimination behavior but enhanced innate detection behavior across all ASD mouse models that have been tested. Why might this pattern exist? We hypothesize that because sensory discrimination behavior is typically cortically mediated, impaired discrimination reflects degraded sensory coding that is common in the sensory cortex across many ASD models. In contrast, innate detection behavior is often not cortically dependent, and Stream 2 shows that innate detection phenotypes do not correlate with spiking phenotypes in the sensory cortex. Thus, enhanced sensitivity for innate detection behaviors may not reflect excess cortical spiking but hypersensitivity of subcortical sensory pathways, which is known to occur in several ASD models (35). These severe syndromic forms of ASD may therefore reflect degraded cortical function, with the enhancement of subcortically driven behaviors due to loss of cortical modulation. Alternatively, discrimination behavior could be impaired simply because discrimination is more processing-intensive than detection and would be more affected by any minimal circuit dysfunction.
This meta-analysis reveals common circuit and sensory features across ASD mouse models and the presence of phenotypic clusters based on neurophysiology that can be labeled as “PV hypofunction” vs. “weak/immature excitation.” Ideally, future research will attempt to fill in the gaps across these mouse models and identify other patterns of convergence. Notably, recent studies are attempting to address this problem in individuals with autism by integrating large genetic datasets, functional connectivity studies, and sensory behavioral profiles to determine whether the expression of ASD-related genes can predict clusters of neuropathophysiology and behavior (15, 213, 214). Such approaches leverage the massive amounts of data available and newly developed machine learning tools but rely heavily on being able to compare and normalize data across studies (215). Thus, a key aspect of future research in human and mouse models is the creation and usage of standardized quantitative sensory behavior assays that integrate and compare naturalistic stimuli with social and non-social valence. Understanding convergence and divergence is decidedly useful from a therapeutic standpoint, as it may allow us to predict which individuals will benefit from which therapies. Moreover, by identifying shared domains of impairment in syndromic models the autism field can establish biomarkers of different forms of circuit impairment, which could be applied to stratify candidates with idiopathic autism into particular clusters of ASD pathophysiology and inform treatments options.
Author contributions
HM: Writing—original draft. HW: Writing—original draft. DF: Funding acquisition, Writing—review and editing.
Funding
This work was supported by the NIH F32NS126310 to HM and NIH R01NS105333 and Simons Foundation Autism Research Initiative (SFARI) Investigator Award 703906 to DF.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher's note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Ramaswami G, Geschwind DH. “Chapter 21 - Genetics of autism spectrum disorder” In:Geschwind DH, Paulson HL, Klein C, , editors. Handbook of Clinical Neurology. Neurogenetics, Part I. Elsevier (2018). p. 321–329.
2. Huguet G, Ey E, Bourgeron T. The genetic landscapes of autism spectrum disorders. Annu Rev Genomics Hum Genet. (2013) 14:191–213. doi: 10.1146/annurev-genom-091212-153431
3. Levy SE, Mandell DS, Schultz RT. Autism. Lancet. (2009) 374:1627–38. doi: 10.1016/S0140-6736(09)61376-3
4. Marco EJ, Hinkley LBN, Hill SS, Nagarajan SS. Sensory processing in autism: a review of neurophysiologic findings. Pediatr Res. (2011) 69:48–54. doi: 10.1203/PDR.0b013e3182130c54
5. Robertson CE, Baron-Cohen S. Sensory perception in autism. Nat Rev Neurosci. (2017) 18:671–84. doi: 10.1038/nrn.2017.112
6. Leekam SR, Nieto C, Libby SJ, Wing L, Gould J. Describing the sensory abnormalities of children and adults with autism. J Autism Dev Disord. (2007) 37:894–910. doi: 10.1007/s10803-006-0218-7
7. Cascio C, McGlone F, Folger S, Tannan V, Baranek G, Pelphrey KA, et al. Tactile perception in adults with autism: a multidimensional psychophysical study. J Autism Dev Disord. (2008) 38:127–37. doi: 10.1007/s10803-007-0370-8
8. Ben-Sasson A, Hen L, Fluss R, Cermak SA, Engel-Yeger B, Gal E, et al. meta-analysis of sensory modulation symptoms in individuals with autism spectrum disorders. J Autism Dev Disord. (2009) 39:1–11. doi: 10.1007/s10803-008-0593-3
9. Jones RSP, Quigney C, Huws JC. First-hand accounts of sensory perceptual experiences in autism: a qualitative analysis. J Intellect Dev Disabil. (2003) 28:112–21. doi: 10.1080/1366825031000147058
10. Hudac CM, Friedman NR, Ward VR, Estreicher RE, Dorsey GC, Bernier RA. Characterizing sensory phenotypes of subgroups with a known genetic etiology pertaining to diagnoses of autism spectrum disorder and intellectual disability. J Autism Dev Disord. (2023)1–16. doi: 10.1007/s10803-023-05897-9
11. Lyons-Warren AM, McCormack MC, Holder JL. Sensory processing phenotypes in phelan-McDermid syndrome and SYNGAP1-related intellectual disability. Brain Sci. (2022) 12:137. doi: 10.3390/brainsci12020137
12. Brockevelt BL, Nissen R, Schweinle WE, Kurtz E, Larson KJ. A comparison of the sensory profile scores of children with autism and an age- and gender-matched sample. S D Med J S D State Med Assoc. (2013) 66:459–465.
13. Simpson K, Adams D, Alston-Knox C, Heussler HS, Keen D. Exploring the sensory profiles of children on the autism spectrum using the short sensory profile-2 (SSP-2). J Autism Dev Disord. (2019) 49:2069–79. doi: 10.1007/s10803-019-03889-2
14. Uljarević M, Lane A, Kelly A, Leekam S. Sensory subtypes and anxiety in older children and adolescents with autism spectrum disorder. Autism Res Off J Int Soc Autism Res. (2016) 9:1073–8. doi: 10.1002/aur.1602
15. Lyons-Warren AM, Wangler MF, Wan Y-W. Cluster analysis of short sensory profile data reveals sensory-based subgroups in autism spectrum disorder. Int J Mol Sci. (2022) 23:13030. doi: 10.3390/ijms232113030
16. Blakemore S-J, Tavassoli T, Calò S, Thomas RM, Catmur C, Frith U, et al. Tactile sensitivity in Asperger syndrome. Brain Cogn. (2006) 61:5–13. doi: 10.1016/j.bandc.2005.12.013
17. Danesh AA, Howery S, Aazh H, Kaf W, Eshraghi AA. Hyperacusis in autism spectrum disorders. Audiol Res. (2021) 11:547–56. doi: 10.3390/audiolres11040049
18. Rotschafer SE. Auditory discrimination in autism spectrum disorder. Front Neurosci. (2021) 15:651209. doi: 10.3389/fnins.2021.651209
19. Rotschafer SE, Razak KA. Auditory processing in fragile x syndrome. Front Cell Neurosci. (2014) 8:19. doi: 10.3389/fncel.2014.00019
20. Williams ZJ, He JL, Cascio CJ, Woynaroski TG. A review of decreased sound tolerance in autism: definitions, phenomenology, and potential mechanisms. Neurosci Biobehav Rev. (2021) 121:1–17. doi: 10.1016/j.neubiorev.2020.11.030
21. Alcántara JI, Weisblatt EJL, Moore BCJ, Bolton PF. Speech-in-noise perception in high-functioning individuals with autism or Asperger's syndrome. J Child Psychol Psychiatry. (2004) 45:1107–14. doi: 10.1111/j.1469-7610.2004.t01-1-00303.x
22. Groen WB, van Orsouw L, Huurne N, Swinkels S, van der Gaag R-J, Buitelaar JK, Zwiers MP. Intact spectral but abnormal temporal processing of auditory stimuli in autism. J Autism Dev Disord. (2009) 39:742–50. doi: 10.1007/s10803-008-0682-3
23. Foss-Feig JH, Schauder KB, Key AP, Wallace MT, Stone WL. Audition-specific temporal processing deficits associated with language function in children with autism spectrum disorder. Autism Res Off J Int Soc Autism Res. (2017) 10:1845–56. doi: 10.1002/aur.1820
24. Plaisted K, O'Riordan M, Baron-Cohen S. Enhanced visual search for a conjunctive target in autism: a research note. J Child Psychol Psychiatry. (1998) 39:777–83.
25. Orekhova EV, Manyukhina VO, Galuta IA, Prokofyev AO, Goiaeva DE, Obukhova TS, et al. Gamma oscillations point to the role of primary visual cortex in atypical motion processing in autism. PLoS ONE. (2023) 18:e0281531. doi: 10.1371/journal.pone.0281531
26. Simmons DR, Robertson AE, McKay LS, Toal E, McAleer P, Pollick FE. Vision in autism spectrum disorders. Vision Res. (2009) 49:2705–39. doi: 10.1016/j.visres.2009.08.005
27. Orefice LL. Peripheral somatosensory neuron dysfunction: emerging roles in autism spectrum disorders. Neuroscience. (2020) 445:120–9. doi: 10.1016/j.neuroscience.2020.01.039
28. Green SA, Rudie JD, Colich NL, Wood JJ, Shirinyan D, Hernandez L, et al. Over reactive brain responses to sensory stimuli in youth with autism spectrum disorders. J Am Acad Child Adolesc Psychiatry. (2013) 52:1158–72. doi: 10.1016/j.jaac.2013.08.004
29. Green SA, Hernandez L, Tottenham N, Krasileva K, Bookheimer SY, Dapretto M. Neurobiology of sensory over responsivity in youth with autism spectrum disorders. JAMA Psychiatry. (2015) 72:778–86. doi: 10.1001/jamapsychiatry.2015.0737
30. Gandal MJ, Haney JR, Wamsley B, Yap CX, Parhami S, Emani PS, et al. Broad transcriptomic dysregulation occurs across the cerebral cortex in ASD. Nature. (2022) 611:532–9. doi: 10.1038/s41586-022-05377-7
31. Balasco L, Provenzano G, Bozzi Y. Sensory abnormalities in autism spectrum disorders: a focus on the tactile domain from genetic mouse models to the clinic. Front Psychiatry. (2020) 10:1016. doi: 10.3389/fpsyt.2019.01016
32. Orefice LL, Mosko JR, Morency DT, Wells MF, Tasnim A, Mozeika SM. Targeting peripheral somatosensory neurons to improve tactile-related phenotypes in ASD Models. Cell. (2019) 178:867–86. doi: 10.1016/j.cell.2019.07.024
33. Balasco L, Pagani M, Pangrazzi L, Chelini G, Ciancone Chama AG, Shlosman E. Abnormal whisker-dependent behaviors and altered cortico-hippocampal connectivity in shank3b-/- Mice. Cereb Cortex N Y N. (2022) 32:3042–56. doi: 10.1093/cercor/bhab399
34. Michaelson SD, Ozkan ED, Aceti M, Maity S, Llamosas N, Weldon M, et al. SYNGAP1 heterozygosity disrupts sensory processing by reducing touch-related activity within somatosensory cortex circuits. Nat Neurosci. (2018) 21:1–13. doi: 10.1038/s41593-018-0268-0
35. Orefice LL, Zimmerman AL, Chirila AM, Sleboda SJ, Head JP, Ginty DD. Peripheral mechanosensory neuron dysfunction underlies tactile and behavioral deficits in mouse models of ASDs. Cell. (2016) 166:299–313. doi: 10.1016/j.cell.2016.05.033
36. Chen Q, Deister CA, Gao X, Guo B, Lynn-Jones T, Chen N, et al. Dysfunction of cortical GABAergic neurons leads to sensory hyper-reactivity in a Shank3 mouse model of ASD. Nat Neurosci. (2020) 23:520–32. doi: 10.1038/s41593-020-0598-6
37. He CX, Cantu DA, Mantri SS, Zeiger WA, Goel A, Portera-Cailliau C. Tactile defensiveness and impaired adaptation of neuronal activity in the Fmr1 knock-out mouse model of autism. J Neurosci. (2017) 37:6475–87. doi: 10.1523/JNEUROSCI.0651-17.2017
38. Dawes JM, Weir GA, Middleton SJ, Patel R, Chisholm KI, Pettingill P, et al. Immune or genetic-mediated disruption of CASPR2 causes pain hypersensitivity due to enhanced primary afferent excitability. Neuron. (2018) 97:806-822.e10. doi: 10.1016/j.neuron.2018.01.033
39. Ko H-G, Oh S-B, Zhuo M, Kaang B-K. Reduced acute nociception and chronic pain in Shank2-/- mice. Mol Pain. (2016) 12:56. doi: 10.1177/1744806916647056
40. Goel A, Cantu DA, Guilfoyle J, Chaudhari GR, Newadkar A, Todisco B, et al. Impaired perceptual learning in a mouse model of Fragile X syndrome is mediated by parvalbumin neuron dysfunction and is reversible. Nat Neurosci. (2018) 21:1404–11. doi: 10.1038/s41593-018-0231-0
41. Del Rosario J, Speed A, Arrowood H, Motz C, Pardue M, Haider B. Diminished cortical excitation and elevated inhibition during perceptual impairments in a mouse model of autism. Cereb Cortex N Y N. (2021) 31:3462–74. doi: 10.1093/cercor/bhab025
42. Leach PT, Crawley JN. Touchscreen learning deficits in Ube3a, Ts65Dn and Mecp2 mouse models of neurodevelopmental disorders with intellectual disabilities. Genes Brain Behav. (2018) 17:e12452. doi: 10.1111/gbb.12452
43. Ash RT, Palagina G, Fernandez-Leon JA, Park J, Seilheimer R, Lee S, et al. Increased reliability of visually-evoked activity in area v1 of the mecp2-duplication mouse model of autism. J Neurosci Off J Soc Neurosci. (2022) 42:6469–82. doi: 10.1523/JNEUROSCI.0654-22.2022
44. Yang C, Tian Y, Su F, Wang Y, Liu M, Wang H, et al. Restoration of FMRP expression in adult V1 neurons rescues visual deficits in a mouse model of fragile X syndrome. Protein Cell. (2022) 13:203–19. doi: 10.1007/s13238-021-00878-z
45. Zhang D, Yu B, Liu J, Jiang W, Xie T, Zhang R, et al. Altered visual cortical processing in a mouse model of MECP2 duplication syndrome. Sci Rep. (2017) 7:6468. doi: 10.1038/s41598-017-06916-3
46. Nielsen DM, Derber WJ, McClellan DA, Crnic LS. Alterations in the auditory startle response in Fmr1 targeted mutant mouse models of fragile X syndrome. Brain Res. (2002) 927:8–17. doi: 10.1016/s0006-8993(01)03309-1
47. Rais M, Binder DK, Razak KA, Ethell IM. Sensory processing phenotypes in fragile X syndrome. ASN Neuro. (2018) 10:1759091418801092. doi: 10.1177/1759091418801092
48. Auerbach BD, Manohar S, Radziwon K, Salvi R. Auditory hypersensitivity and processing deficits in a rat model of fragile X syndrome. Neurobiol Dis. (2021) 161:105541. doi: 10.1016/j.nbd.2021.105541
49. Rubenstein JLR, Merzenich MM. Model of autism: increased ratio of excitation/inhibition in key neural systems. Genes Brain Behav. (2003) 2:255–67. doi: 10.1034/j.1601-183x.2003.00037.x
50. Sohal VS, Rubenstein JLR. Excitation-inhibition balance as a framework for investigating mechanisms in neuropsychiatric disorders. Mol Psychiatry. (2019) 24:1248–57. doi: 10.1038/s41380-019-0426-0
51. Bolton PF, Carcani-Rathwell I, Hutton J, Goode S, Howlin P, Rutter M. Epilepsy in autism: features and correlates. Br J Psychiatry J Ment Sci. (2011) 198:289–94. doi: 10.1192/bjp.bp.109.076877
52. Markram K, Markram H. The intense world theory – a unifying theory of the neurobiology of autism. Front Hum Neurosci. (2010) 4:224. doi: 10.3389/fnhum.2010.00224
53. Antoine MW, Langberg T, Schnepel P, Feldman DE. Increased excitation-inhibition ratio stabilizes synapse and circuit excitability in four autism mouse models. Neuron. (2019) 101:648–61. doi: 10.1016/j.neuron.2018.12.026
54. Filice F, Janickova L, Henzi T, Bilella A, Schwaller B. The parvalbumin hypothesis of autism spectrum disorder. Front Cell Neurosci. (2020) 14:577525. doi: 10.3389/fncel.2020.577525
55. Rudy B, Fishell G, Lee S, Hjerling-Leffler J. Three groups of interneurons account for nearly 100% of neocortical GABAergic neurons. Dev Neurobiol. (2011) 71:45–61. doi: 10.1002/dneu.20853
56. Ariza J, Rogers H, Hashemi E, Noctor SC, Martínez-Cerdeño V. The number of chandelier and basket cells are differentially decreased in prefrontal cortex in autism. Cereb Cortex N Y N. (2018) 28:411–20. doi: 10.1093/cercor/bhw349
57. Hashemi E, Ariza J, Rogers H, Noctor SC, Martínez-Cerdeño V. The number of parvalbumin-expressing interneurons is decreased in the prefrontal cortex in autism. Cereb Cortex N Y N. (2017) 27:1931–43. doi: 10.1093/cercor/bhw021
58. Contractor A, Ethell IM, Portera-Cailliau C. Cortical interneurons in autism. Nat Neurosci. (2021) 24:1648–59. doi: 10.1038/s41593-021-00967-6
59. Nomura T. Interneuron dysfunction and inhibitory deficits in autism and fragile X syndrome. Cells. (2021) 10:2610. doi: 10.3390/cells10102610
60. Reh RK, Dias BG, Nelson CA, Kaufer D, Werker JF, Kolb B, et al. Critical period regulation across multiple timescales. Proc Natl Acad Sci U S A. (2020) 117:23242–51. doi: 10.1073/pnas.1820836117
61. Kourdougli N, Suresh A, Liu B, Juarez P, Lin A, Chung DT. Improvement of sensory deficits in fragile X mice by increasing cortical interneuron activity after the critical period. Neuron. (2023) 25:S0896–6273. doi: 10.1016/j.neuron.2023.06.009
62. Nelson SB, Valakh V. Excitatory/inhibitory balance and circuit homeostasis in autism spectrum disorders. Neuron. (2015) 87:684–98. doi: 10.1016/j.neuron.2015.07.033
63. Turrigiano G. Homeostatic synaptic plasticity: local and global mechanisms for stabilizing neuronal function. Cold Spring Harb Perspect Biol. (2012) 4:a005736. doi: 10.1101/cshperspect.a005736
64. Mullins C, Fishell G, Tsien RW. Unifying views of autism spectrum disorders: a consideration of auto regulatory feedback loops. Neuron. (2016) 89:1131–56. doi: 10.1016/j.neuron.2016.02.017
65. Genç Ö, An J-Y, Fetter RD, Kulik Y, Zunino G, Sanders SJ. Homeostatic plasticity fails at the intersection of autism-gene mutations and a novel class of common genetic modifiers. eLife. (2020) 9:775. doi: 10.7554/eLife.55775
66. Monday HR, Younts TJ, Castillo PE. Long-term plasticity of neurotransmitter release: emerging mechanisms and contributions to brain function and disease. Annu Rev Neurosci. (2018) 41:299–322. doi: 10.1146/annurev-neuro-080317-062155
67. Bourgeron T. From the genetic architecture to synaptic plasticity in autism spectrum disorder. Nat Rev Neurosci. (2015) 16:551–63. doi: 10.1038/nrn3992
68. Bear MF, Huber KM, Warren ST. The mGluR theory of fragile X mental retardation. Trends Neurosci. (2004) 27:370–7. doi: 10.1016/j.tins.2004.04.009
69. Huber KM, Gallagher SM, Warren ST, Bear MF. Altered synaptic plasticity in a mouse model of fragile X mental retardation. Proc Natl Acad Sci U S A. (2002) 99:7746–50. doi: 10.1073/pnas.122205699
70. Bateup HS, Johnson CA, Denefrio CL, Saulnier JL, Kornacker K, Sabatini BL. Excitatory/inhibitory synaptic imbalance leads to hippocampal hyperexcitability in mouse models of tuberous sclerosis. Neuron. (2013) 78:510–22. doi: 10.1016/j.neuron.2013.03.017
71. Dang R, Qi J, Liu A, Ren Q, Lv D, Han L, et al. Regulation of hippocampal long term depression by Neuroligin 1. Neuropharmacology. (2018) 143:205–16. doi: 10.1016/j.neuropharm.2018.09.035
72. Lee K, Vyas Y, Garner CC, Montgomery JM. Autism-associated Shank3 mutations alter mGluR expression and mGluR-dependent but not NMDA receptor-dependent long-term depression. Synap N Y N. (2019) 73:e22097. doi: 10.1002/syn.22097
73. Auerbach BD, Osterweil EK, Bear MF. Mutations causing syndromic autism define an axis of synaptic pathophysiology. Nature. (2011) 480:63–8. doi: 10.1038/nature10658
74. Klein ME, Monday H, Jordan BA. Proteostasis and RNA binding proteins in synaptic plasticity and in the pathogenesis of neuropsychiatric disorders. Neural Plast. (2016) 2016:3857934. doi: 10.1155/2016/3857934
75. Santini E, Klann E. Reciprocal signaling between translational control pathways and synaptic proteins in autism spectrum disorders. Sci Signal. (2014) 7:re10. doi: 10.1126/scisignal.2005832
76. Bagni C, Zukin RS, A. Synaptic perspective of fragile X syndrome and autism spectrum disorders. Neuron. (2019) 101:1070–88. doi: 10.1016/j.neuron.2019.02.041
77. Forrest MP, Parnell E, Penzes P. Dendritic structural plasticity and neuropsychiatric disease. Nat Rev Neurosci. (2018) 19:215–34. doi: 10.1038/nrn.2018.16
78. Zieger HL, Choquet D. Nanoscale synapse organization and dysfunction in neurodevelopmental disorders. Neurobiol Dis. (2021) 158:105453. doi: 10.1016/j.nbd.2021.105453
79. LeBlanc JJ, Fagiolini M. Autism: a “critical period” disorder? Neural Plast. (2011) 2011:921680. doi: 10.1155/2011/921680
80. Rupert DD, Shea SD. Parvalbumin-Positive Interneurons Regulate Cortical Sensory Plasticity in Adulthood and Development Through Shared Mechanisms. Front Neural Circuits. (2022) 16:886629. doi: 10.3389/fncir.2022.886629
81. Varcin KJ, Jeste SS. The emergence of autism spectrum disorder (ASD): insights gained from studies of brain and behaviour in high-risk infants. Curr Opin Psychiatry. (2017) 30:85–91. doi: 10.1097/YCO.0000000000000312
82. Aceti M, Creson TK, Vaissiere T, Rojas C, Huang W-C, Wang Y-X, et al. Syngap1 haploin sufficiency damages a postnatal critical period of pyramidal cell structural maturation linked to cortical circuit assembly. Biol Psychiatry. (2015) 77:805–15. doi: 10.1016/j.biopsych.2014.08.001
83. Allegra M, Genovesi S, Maggia M, Cenni MC, Zunino G, Sgadò P, et al. Altered GABAergic markers, increased binocularity and reduced plasticity in the visual cortex of Engrailed-2 knockout mice. Front Cell Neurosci. (2014) 8:163. doi: 10.3389/fncel.2014.00163
84. Yashiro K, Riday TT, Condon KH, Roberts AC, Bernardo DR, Prakash R, et al. Ube3a is required for experience-dependent maturation of the neocortex. Nat Neurosci. (2009) 12:777–83. doi: 10.1038/nn.2327
85. Krishnan K, Wang B-S, Lu J, Wang L, Maffei A, Cang J, et al. MeCP2 regulates the timing of critical period plasticity that shapes functional connectivity in primary visual cortex. Proc Natl Acad Sci U S A. (2015) 112:E4782–4791. doi: 10.1073/pnas.1506499112
86. Bureau I, Shepherd GMG, Svoboda K. Circuit and plasticity defects in the developing somatosensory cortex of Fmr1 knock-out mice. J Neurosci. (2008) 28:5178–88. doi: 10.1523/JNEUROSCI.1076-08.2008
87. Harlow EG, Till SM, Russell TA, Wijetunge LS, Kind P, Contractor A. Critical period plasticity is disrupted in the barrel cortex of FMR1 knockout mice. Neuron. (2010) 65:385–98. doi: 10.1016/j.neuron.2010.01.024
88. He L, Liu N, Cheng T, Chen X, Li Y, Shu Y, et al. Conditional deletion of Mecp2 in parvalbumin-expressing GABAergic cells results in the absence of critical period plasticity. Nat Commun. (2014) 5:5036. doi: 10.1038/ncomms6036
89. Nomura T, Musial TF, Marshall JJ, Zhu Y, Remmers CL, Xu J, et al. Delayed maturation of fast-spiking interneurons is rectified by activation of the TrkB receptor in the mouse model of fragile X syndrome. J Neurosci. (2017) 37:11298–310. doi: 10.1523/JNEUROSCI.2893-16.2017
90. Song YJ, Xing B, Barbour AJ, Zhou C, Jensen FE. Dysregulation of GABAA receptor-mediated neurotransmission during the auditory cortex critical period in the fragile X syndrome mouse model. Cereb Cortex. (2022) 32:197–215. doi: 10.1093/cercor/bhab203
91. Dani VS, Chang Q, Maffei A, Turrigiano GG, Jaenisch R, Nelson SB. Reduced cortical activity due to a shift in the balance between excitation and inhibition in a mouse model of Rett Syndrome. Proc Natl Acad Sci. (2005) 102:12560–5. doi: 10.1073/pnas.0506071102
92. Nakai N, Nagano M, Saitow F, Watanabe Y, Kawamura Y, Kawamoto A, et al. Serotonin rebalances cortical tuning and behavior linked to autism symptoms in 15q11-13 CNV mice. Sci Adv. (2017) 3:e1603001. doi: 10.1126/sciadv.1603001
93. Banerjee A, Rikhye RV, Breton-Provencher V, Tang X, Li C, Li K, et al. Jointly reduced inhibition and excitation underlies circuit-wide changes in cortical processing in Rett syndrome. Proc Natl Acad Sci U S A. (2016) 113:E7287–96. doi: 10.1073/pnas.1615330113
94. Rotschafer S, Razak K. Altered auditory processing in a mouse model of fragile X syndrome. Brain Res. (2013) 1506:12–24. doi: 10.1016/j.brainres.2013.02.038
95. Wadle SL, Schmitt TTX, Engel J, Kurt S, Hirtz JJ. Altered population activity and local tuning heterogeneity in auditory cortex of Cacna2d3-deficient mice. Biol Chem. (2023) 404:607–17. doi: 10.1515/hsz-2022-0269
96. Anomal RF, de Villers-Sidani E, Brandão JA, Diniz R, Costa MR, Romcy-Pereira RN. Impaired processing in the primary auditory cortex of an animal model of autism. Front Syst Neurosci. (2015) 9:158. doi: 10.3389/fnsys.2015.00158
97. Juczewski K, von Richthofen H, Bagni C, Celikel T, Fisone G, Krieger P. Somatosensory map expansion and altered processing of tactile inputs in a mouse model of fragile X syndrome. Neurobiol Dis. (2016) 96:201–15. doi: 10.1016/j.nbd.2016.09.007
98. Zhou C, Yan S, Qian S, Wang Z, Shi Z, Xiong Y, et al. Atypical response properties of the auditory cortex of awake MECP2-overexpressing mice. Front Neurosci. (2019) 13:439. doi: 10.3389/fnins.2019.00439
99. Wallace ML, van Woerden GM, Elgersma Y, Smith SL, Philpot BD. Ube3a loss increases excitability and blunts orientation tuning in the visual cortex of Angelman syndrome model mice. J Neurophysiol. (2017) 118:634–46. doi: 10.1152/jn.00618.2016
100. Cheyne JE, Zabouri N, Baddeley D, Lohmann C. Spontaneous activity patterns are altered in the developing visual cortex of the Fmr1 knockout mouse. Front Neural Circuits. (2019) 13:57. doi: 10.3389/fncir.2019.00057
101. Ethridge LE, White SP, Mosconi MW, Wang J, Pedapati EV, Erickson CA, et al. Neural synchronization deficits linked to cortical hyper-excitability and auditory hypersensitivity in fragile X syndrome. Mol Autism. (2017) 8:22. doi: 10.1186/s13229-017-0140-1
102. Gonçalves JT, Anstey JE, Golshani P, Portera-Cailliau C. Circuit level defects in the developing neocortex of Fragile X mice. Nat Neurosci. (2013) 16:903–9. doi: 10.1038/nn.3415
103. Hays SA, Huber KM, Gibson JR. Altered neocortical rhythmic activity states in Fmr1 KO Mice Are Due to enhanced mGluR5 signaling and involve changes in excitatory circuitry. J Neurosci. (2011) 31:14223–34. doi: 10.1523/JNEUROSCI.3157-11.2011
104. Lazaro MT, Taxidis J, Shuman T, Bachmutsky I, Ikrar T, Santos R. Reduced prefrontal synaptic connectivity and disturbed oscillatory population dynamics in the CNTNAP2 model of autism. Cell Rep. (2019) 27:2567–78. doi: 10.1016/j.celrep.2019.05.006
105. O'Donnell C, Gonçalves JT, Portera-Cailliau C, Sejnowski TJ. Beyond excitation/inhibition imbalance in multidimensional models of neural circuit changes in brain disorders. Elife. (2017) 6:e26724. doi: 10.7554/eLife.26724
106. He JL, Wodka E, Tommerdahl M, Edden RAE, Mikkelsen M, Mostofsky SH, et al. Disorder-specific alterations of tactile sensitivity in neurodevelopmental disorders. Commun Biol. (2021) 4:1–15. doi: 10.1038/s42003-020-01592-y
107. Puts NAJ, Wodka EL, Tommerdahl M, Mostofsky SH, Edden RAE. Impaired tactile processing in children with autism spectrum disorder. J Neurophysiol. (2014) 111:1803–11. doi: 10.1152/jn.00890.2013
108. Schelinski S, von Kriegstein K. Brief report: speech-in-noise recognition and the relation to vocal pitch perception in adults with autism spectrum disorder and typical development. J Autism Dev Disord. (2020) 50:356–63. doi: 10.1007/s10803-019-04244-1
109. Dinstein I, Heeger DJ, Lorenzi L, Minshew NJ, Malach R, Behrmann M. Unreliable evoked responses in autism. Neuron. (2012) 75:981–91. doi: 10.1016/j.neuron.2012.07.026
110. Coskun MA, Varghese L, Reddoch S, Castillo EM, Pearson DA, Loveland KA, et al. How somatic cortical maps differ in autistic and typical brains. Neuroreport. (2009) 20:175–9. doi: 10.1097/WNR.0b013e32831f47d1
111. Frey H-P, Molholm S, Lalor EC, Russo NN, Foxe JJ. Atypical cortical representation of peripheral visual space in children with an autism spectrum disorder. Eur J Neurosci. (2013) 38:2125–38. doi: 10.1111/ejn.12243
112. Smith H, Lane C, Al-Jawahiri R. Freeth M. Sensory processing in 16p112 deletion and 16p112 duplication Autism. Res Off J Int Soc Autism Res. (2022) 15:2081–98. doi: 10.1002/aur.2802
113. Guo B, Chen J, Chen Q, Ren K, Feng D, Mao H, et al. Anterior cingulate cortex dysfunction underlies social deficits in Shank3 mutant mice. Nat Neurosci. (2019) 22:1223–34. doi: 10.1038/s41593-019-0445-9
114. Zhang Y, Bonnan A, Bony G, Ferezou I, Pietropaolo S, Ginger M, et al. Dendritic channelopathies contribute to neocortical and sensory hyperexcitability in Fmr1(-/y) mice. Nat Neurosci. (2014) 17:1701–9. doi: 10.1038/nn.3864
115. Millin R, Kolodny T, Flevaris AV, Kale AM, Schallmo M-P, Gerdts J, et al. Reduced auditory cortical adaptation in autism spectrum disorder. Elife. (2018) 7:e36493. doi: 10.7554/eLife.36493
116. Tannan V, Holden JK, Zhang Z, Baranek GT, Tommerdahl MA. Perceptual metrics of individuals with autism provide evidence for disinhibition. Autism Res. (2008) 1:223–30. doi: 10.1002/aur.34
117. Lovelace JW, Wen TH, Reinhard S, Hsu MS, Sidhu H, Ethell IM, et al. Matrix metalloproteinase-9 deletion rescues auditory evoked potential habituation deficit in a mouse model of Fragile X Syndrome. Neurobiol Dis. (2016) 89:126–35. doi: 10.1016/j.nbd.2016.02.002
118. Wallace ML, Burette AC, Weinberg RJ, Philpot BD. Maternal loss of Ube3a produces an excitatory/inhibitory imbalance through neuron type-specific synaptic defects. Neuron. (2012) 74:793–800. doi: 10.1016/j.neuron.2012.03.036
119. Buzsáki G, Draguhn A. Neuronal oscillations in cortical networks. Science. (2004) 304:1926–9. doi: 10.1126/science.1099745
120. Ai L, Ro T. The phase of prestimulus alpha oscillations affects tactile perception. J Neurophysiol. (2014) 111:1300–7. doi: 10.1152/jn.00125.2013
121. Busch NA, Dubois J, Van Rullen R. The phase of ongoing EEG oscillations predicts visual perception. J Neurosci. (2009) 29:7869–76. doi: 10.1523/JNEUROSCI.0113-09.2009
122. Gruber WR, Klimesch W, Sauseng P, Doppelmayr M. Alpha phase synchronization predicts P1 and N1 latency and amplitude size. Cereb Cortex N Y N. (2005) 15:371–7. doi: 10.1093/cercor/bhh139
123. Iemi L, Chaumon M, Crouzet SM, Busch NA. Spontaneous neural oscillations bias perception by modulating baseline excitability. J Neurosci Off J Soc Neurosci. (2017) 37:807–19. doi: 10.1523/JNEUROSCI.1432-16.2016
124. Samaha J, Iemi L, Haegens S, Busch NA. Spontaneous brain oscillations and perceptual decision-making. Trends Cogn Sci. (2020) 24:639–53. doi: 10.1016/j.tics.2020.05.004
125. Strauß A, Henry MJ, Scharinger M, Obleser J. Alpha phase determines successful lexical decision in noise. J Neurosci Off J Soc Neurosci. (2015) 35:3256–62. doi: 10.1523/JNEUROSCI.3357-14.2015
126. Simon DM, Wallace MT. Dysfunction of sensory oscillations in autism spectrum disorder. Neurosci Biobehav Rev. (2016) 68:848–61. doi: 10.1016/j.neubiorev.2016.07.016
127. Edgar JC, Khan SY, Blaskey L, Chow VY, Rey M, Gaetz W, et al. Neuro magnetic oscillations predict evoked-response latency delays and core language deficits in autism spectrum disorders. J Autism Dev Disord. (2015) 45:395–405. doi: 10.1007/s10803-013-1904-x
128. Khan S, Michmizos K, Tommerdahl M, Ganesan S, Kitzbichler MG, Zetino M, et al. Somatosensory cortex functional connectivity abnormalities in autism show opposite trends, depending on direction and spatial scale. Brain J Neurol. (2015) 138:1394–409. doi: 10.1093/brain/awv043
129. Milne E, Scope A, Pascalis O, Buckley D, Makeig S. Independent component analysis reveals atypical electroencephalographic activity during visual perception in individuals with autism. Biol Psychiatry. (2009) 65:22–30. doi: 10.1016/j.biopsych.2008.07.017
130. Rojas DC, Maharajh K, Teale P, Rogers SJ. Reduced neural synchronization of gamma-band MEG oscillations in first-degree relatives of children with autism. BMC Psychiatry. (2008) 8:66. doi: 10.1186/1471-244X-8-66
131. Snijders TM, Milivojevic B, Kemner C. Atypical excitation-inhibition balance in autism captured by the gamma response to contextual modulation. NeuroImage Clin. (2013) 3:65–72. doi: 10.1016/j.nicl.2013.06.015
132. Stroganova TA, Orekhova EV, Prokofyev AO, Tsetlin MM, Gratchev VV, Morozov AA, et al. High-frequency oscillatory response to illusory contour in typically developing boys and boys with autism spectrum disorders. Cortex J Devoted Study Nerv Syst Behav. (2012) 48:701–17. doi: 10.1016/j.cortex.2011.02.016
133. Sun L, Grützner C, Bölte S, Wibral M, Tozman T, Schlitt S, et al. Impaired gamma-band activity during perceptual organization in adults with autism spectrum disorders: evidence for dysfunctional network activity in frontal-posterior cortices. J Neurosci Off J Soc Neurosci. (2012) 32:9563–73. doi: 10.1523/JNEUROSCI.1073-12.2012
134. Wilson TW, Rojas DC, Reite ML, Teale PD, Rogers SJ. Children and adolescents with autism exhibit reduced MEG steady-state gamma responses. Biol Psychiatry. (2007) 62:192–7. doi: 10.1016/j.biopsych.2006.07.002
135. Brown C, Gruber T, Boucher J, Rippon G, Brock J. Gamma abnormalities during perception of illusory figures in autism. Cortex J Devoted Study Nerv Syst Behav. (2005) 41:364–76. doi: 10.1016/s0010-9452(08)70273-9
136. Buard I, Rogers SJ, Hepburn S, Kronberg E, Rojas DC. Altered oscillation patterns and connectivity during picture naming in autism. Front Hum Neurosci. (2013) 7:742. doi: 10.3389/fnhum.2013.00742
137. Côté V, Knoth IS, Agbogba K, Vannasing P, Côté L, Major P, et al. Differential auditory brain response abnormalities in two intellectual disability conditions: SYNGAP1 mutations and Down syndrome. Clin Neurophysiol. (2021) 132:1802–12. doi: 10.1016/j.clinph.2021.03.054
138. Gandal MJ, Edgar JC, Ehrlichman RS, Mehta M, Roberts TPL, Siegel SJ. Validating γ oscillations and delayed auditory responses as translational biomarkers of autism. Biol Psychiatry. (2010) 68:1100–6. doi: 10.1016/j.biopsych.2010.09.031
139. David N, Schneider TR, Peiker I, Al-Jawahiri R, Engel AK, Milne E. Variability of cortical oscillation patterns: a possible endophenotype in autism spectrum disorders? Neurosci Biobehav Rev. (2016) 71:590–600. doi: 10.1016/j.neubiorev.2016.09.031
140. Murphy JW, Foxe JJ, Peters JB, Molholm S. Susceptibility to distraction in autism spectrum disorder: probing the integrity of oscillatory alpha-band suppression mechanisms. Autism Res Off J Int Soc Autism Res. (2014) 7:442–58. doi: 10.1002/aur.1374
141. Isler JR, Martien KM, Grieve PG, Stark RI, Herbert MR. Reduced functional connectivity in visual evoked potentials in children with autism spectrum disorder. Clin Neurophysiol Off J Int Fed Clin Neurophysiol. (2010) 121:2035–43. doi: 10.1016/j.clinph.2010.05.004
142. Lazarev VV, Pontes A, Mitrofanov AA, deAzevedo LC. Reduced interhemispheric connectivity in childhood autism detected by electroencephalographic photic driving coherence. J Autism Dev Disord. (2015) 45:537–47. doi: 10.1007/s10803-013-1959-8
143. Peiker I, David N, Schneider TR, Nolte G, Schöttle D, Engel AK. Perceptual integration deficits in autism spectrum disorders are associated with reduced interhemispheric gamma-band coherence. J Neurosci Off J Soc Neurosci. (2015) 35:16352–61. doi: 10.1523/JNEUROSCI.1442-15.2015
144. Jonak CR, Lovelace JW, Ethell IM, Razak KA, Binder DK. Multielectrode array analysis of EEG biomarkers in a mouse model of Fragile X Syndrome. Neurobiol Dis. (2020) 138:104794. doi: 10.1016/j.nbd.2020.104794
145. Lovelace JW, Ethell IM, Binder DK, Razak KA. Translation-relevant EEG phenotypes in a mouse model of Fragile X Syndrome. Neurobiol Dis. (2018) 115:39–48. doi: 10.1016/j.nbd.2018.03.012
146. Sinclair D, Featherstone R, Naschek M, Nam J, Du A, Wright S. GABA-B agonist baclofen normalizes auditory-evoked neural oscillations and behavioral deficits in the fmr1 knockout mouse model of fragile X syndrome. eNeuro. (2017) 4: 1–7. doi: 10.1523/ENEURO.0380-16.2017
147. Janz P, Bainier M, Marashli S, Schoenenberger P, Valencia M, Redondo RL. Neurexin1α knockout rats display oscillatory abnormalities and sensory processing deficits back-translating key endophenotypes of psychiatric disorders. Transl Psychiatry. (2022) 12:455. doi: 10.1038/s41398-022-02224-1
148. Han S, Tai C, Westenbroek RE, Yu FH, Cheah CS, Potter GB. Autistic-like behaviour in Scn1a+/– mice and rescue by enhanced GABA-mediated neurotransmission. Nature. (2012) 489:385–90. doi: 10.1038/nature11356
149. Tai C, Abe Y, Westenbroek RE, Scheuer T, Catterall WA. Impaired excitability of somatostatin- and parvalbumin-expressing cortical interneurons in a mouse model of Dravet syndrome. Proc Natl Acad Sci. (2014) 111:E3139–48. doi: 10.1073/pnas.1411131111
150. Favero M, Sotuyo NP, Lopez E, Kearney JA, Goldberg EM, A. Transient developmental window of fast-spiking interneuron dysfunction in a mouse model of dravet syndrome. J Neurosci. (2018) 38:7912–27. doi: 10.1523/JNEUROSCI.0193-18.2018
151. Tran CH, Vaiana M, Nakuci J, Somarowthu A, Goff KM, Goldstein N, et al. Interneuron desynchronization precedes seizures in a mouse model of Dravet syndrome. J Neurosci Off J Soc Neurosci. (2020) 40:2764–75. doi: 10.1523/JNEUROSCI.2370-19.2020
152. Ogiwara I, Miyamoto H, Morita N, Atapour N, Mazaki E, Inoue I, et al. Nav11 localizes to axons of parvalbumin-positive inhibitory interneurons: a circuit basis for epileptic seizures in mice carrying an Scn1a gene mutation. J Neurosci. (2007) 27:5903–14. doi: 10.1523/JNEUROSCI.5270-06.2007
153. Peixoto RT, Wang W, Croney DM, Kozorovitskiy Y, Sabatini BL. Early hyperactivity and precocious maturation of corticostriatal circuits in Shank3B–/– mice. Nat Neurosci. (2016) 19:716–24. doi: 10.1038/nn.4260
154. Pagano J, Landi S, Stefanoni A, Nardi G, Albanesi M, Bauer HF, et al. Shank3 deletion in PV neurons is associated with abnormal behaviors and neuronal functions that are rescued by increasing GABAergic signaling. Mol Autism. (2023) 14:28. doi: 10.1186/s13229-023-00557-2
155. Gonzalez D, Tomasek M, Hays S, Sridhar V, Ammanuel S, Chang C-W, et al. Audiogenic seizures in the Fmr1 knock-out mouse are induced by fmr1 deletion in subcortical, VGlut2-expressing excitatory neurons and require deletion in the inferior colliculus. J Neurosci Off J Soc Neurosci. (2019) 39:9852–63. doi: 10.1523/JNEUROSCI.0886-19.2019
156. Gibson JR, Bartley AF, Hays SA, Huber KM. Imbalance of neocortical excitation and inhibition and altered UP states reflect network hyperexcitability in the mouse model of fragile X syndrome. J Neurophysiol. (2008) 100:2615–26. doi: 10.1152/jn.90752.2008
157. Arnett MT, Herman DH, McGee AW. Deficits in tactile learning in a mouse model of fragile X syndrome. PLoS ONE. (2014) 9:e109116. doi: 10.1371/journal.pone.0109116
158. Balasco L, Pagani M, Pangrazzi L, Chelini G, Viscido F, Chama AGC, et al. Somatosensory cortex hyperconnectivity and impaired whisker-dependent responses in Cntnap2–/– mice. Neurobiol Dis. (2022) 169:105742. doi: 10.1016/j.nbd.2022.105742
159. Scott KE, Mann RS, Schormans AL, Schmid S, Allman BL. Hyperexcitable and immature-like neuronal activity in the auditory cortex of adult rats lacking the language-linked CNTNAP2 gene. Cereb Cortex N Y N. (2022) 32:4797–817. doi: 10.1093/cercor/bhab517
160. Scott KE, Schormans AL, Pacoli KY, De Oliveira C, Allman BL, Schmid S. Altered auditory processing, filtering, and reactivity in the cntnap2 knock-out rat model for neurodevelopmental disorders. J Neurosci Off J Soc Neurosci. (2018) 38:8588–604. doi: 10.1523/JNEUROSCI.0759-18.2018
161. Krishnan K, Lau BYB, Ewall G, Huang ZJ, Shea SD. MECP2 regulates cortical plasticity underlying a learned behaviour in adult female mice. Nat Commun. (2017) 8:14077. doi: 10.1038/ncomms14077
162. Morello N, Schina R, Pilotto F, Phillips M, Melani R, Plicato O. Loss of Mecp2 causes atypical synaptic and molecular plasticity of parvalbumin-expressing interneurons reflecting rett syndrome-like sensorimotor defects. eNeuro. (2018) 5:2018. doi: 10.1523/ENEURO.0086-18.2018
163. Durand S, Patrizi A, Quast KB, Hachigian L, Pavlyuk R, Saxena A, et al. Receptor regulation prevents regression of visual cortical function in the absence of Mecp2. Neuron. (2012) 76:1078–90. doi: 10.1016/j.neuron.2012.12.004
164. Engineer CT, Rahebi KC, Borland MS, Buell EP, Centanni TM, Fink MK, et al. Degraded neural and behavioral processing of speech sounds in a rat model of Rett syndrome. Neurobiol Dis. (2015) 83:26–34. doi: 10.1016/j.nbd.2015.08.019
165. Ramocki MB, Zoghbi HY. Failure of neuronal homeostasis results in common neuropsychiatric phenotypes. Nature. (2008) 455:912–8. doi: 10.1038/nature07457
166. Neklyudova A, Smirnov K, Rebreikina A, Martynova O, Sysoeva O. Electrophysiological and behavioral evidence for hyper- and hyposensitivity in rare genetic syndromes associated with autism. Genes. (2022) 13:671. doi: 10.3390/genes13040671
167. Donato F, Rompani SB, Caroni P. Parvalbumin-expressing basket-cell network plasticity induced by experience regulates adult learning. Nature. (2013) 504:272–6. doi: 10.1038/nature12866
168. Selby L, Zhang C, Sun Q-Q. Major defects in neocortical GABAergic inhibitory circuits in mice lacking the fragile X mental retardation protein. Neurosci Lett. (2007) 412:227–32. doi: 10.1016/j.neulet.2006.11.062
169. Wen TH, Afroz S, Reinhard SM, Palacios AR, Tapia K, Binder DK, et al. Genetic Reduction of matrix metalloproteinase-9 promotes formation of perineuronal nets around parvalbumin-expressing interneurons and normalizes auditory cortex responses in developing fmr1 knock-out mice. Cereb Cortex. (2018) 28:3951–64. doi: 10.1093/cercor/bhx258
170. Filice F, Vörckel KJ, Sungur AÖ, Wöhr M, Schwaller B. Reduction in parvalbumin expression not loss of the parvalbumin-expressing GABA interneuron subpopulation in genetic parvalbumin and shank mouse models of autism. Mol Brain. (2016) 9:10. doi: 10.1186/s13041-016-0192-8
171. Gogolla N, Leblanc JJ, Quast KB, Südhof TC, Fagiolini M, Hensch TK. Common circuit defect of excitatory-inhibitory balance in mouse models of autism. J Neurodev Disord. (2009) 1:172–81. doi: 10.1007/s11689-009-9023-x
172. Jung E-M, Moffat JJ, Liu J, Dravid SM, Gurumurthy CB, Kim W-Y. Arid1b haploinsufficiency disrupts cortical interneuron development and mouse behavior. Nat Neurosci. (2017) 20:1694–707. doi: 10.1038/s41593-017-0013-0
173. Peñagarikano O, Abrahams BS, Herman EI, Winden KD, Gdalyahu A, Dong H, et al. Absence of CNTNAP2 leads to epilepsy, neuronal migration abnormalities, and core autism-related deficits. Cell. (2011) 147:235–46. doi: 10.1016/j.cell.2011.08.040
174. Vogt D, Cho KKA, Shelton SM, Paul A, Huang ZJ, Sohal VS, et al. Mouse Cntnap2 and human CNTNAP2 ASD alleles cell autonomously regulate PV+ cortical interneurons. Cereb Cortex. (2018) 28:3868–79. doi: 10.1093/cercor/bhx248
175. Lauber E, Filice F, Schwaller B. Dysregulation of parvalbumin expression in the Cntnap2-/- mouse model of autism spectrum disorder. Front Mol Neurosci. (2018) 11:262. doi: 10.3389/fnmol.2018.00262
176. Scott R, Sánchez-Aguilera A, van Elst K, Lim L, Dehorter N, Bae SE, et al. Loss of Cntnap2 causes axonal excitability deficits, developmental delay in cortical myelination, and abnormal stereotyped motor behavior. Cereb Cortex. (2019) 29:586–97. doi: 10.1093/cercor/bhx341
177. Deemyad T, Puig S, Papale AE, Qi H, LaRocca GM, Aravind D. Lateralized decrease of Parvalbumin+ cells in the somatosensory cortex of ASD models is correlated with unilateral tactile hypersensitivity. Cereb Cortex N Y N. (2022) 32:554–68. doi: 10.1093/cercor/bhab233
178. Bernard C, Exposito-Alonso D, Selten M, Sanalidou S, Hanusz-Godoy A, Aguilera A, et al. Cortical wiring by synapse type–specific control of local protein synthesis. Science. (2022) 378:eabm7466. doi: 10.1126/science.abm7466
179. He Q, Arroyo ED, Smukowski SN, Xu J, Piochon C, Savas JN, et al. Critical period inhibition of NKCC1 rectifies synapse plasticity in the somatosensory cortex and restores adult tactile response maps in fragile X mice. Mol Psychiatry. (2019) 24:1732–47. doi: 10.1038/s41380-018-0048-y
180. Lovelace JW, Rais M, Palacios AR, Shuai XS, Bishay S. Deletion of Fmr1 from forebrain excitatory neurons triggers abnormal cellular, EEG, and Behavioral Phenotypes in the auditory cortex of a mouse model of fragile X syndrome. Cereb Cortex N Y N. (2020) 30:969–88. doi: 10.1093/cercor/bhz141
181. Filice F, Lauber E, Vörckel KJ, Wöhr M, Schwaller B. 17-β estradiol increases parvalbumin levels in Pvalb heterozygous mice and attenuates behavioral phenotypes with relevance to autism core symptoms. Mol Autism. (2018) 9:15. doi: 10.1186/s13229-018-0199-3
182. Wöhr M, Orduz D, Gregory P, Moreno H, Khan U, Vörckel KJ, et al. Lack of parvalbumin in mice leads to behavioral deficits relevant to all human autism core symptoms and related neural morphofunctional abnormalities. Transl Psychiatry. (2015) 5:e525. doi: 10.1038/tp.2015.19
183. Chao H-T, Chen H, Samaco RC, Xue M, Chahrour M, Yoo J, et al. Dysfunction in GABA signalling mediates autism-like stereotypies and Rett syndrome phenotypes. Nature. (2010) 468:263–9. doi: 10.1038/nature09582
184. Zhang W, Peterson M, Beyer B, Frankel WN, Zhang Z. Loss of MeCP2 from forebrain excitatory neurons leads to cortical hyperexcitation and seizures. J Neurosci. (2014) 34:2754–63. doi: 10.1523/JNEUROSCI.4900-12.2014
185. Ito-Ishida A, Ure K, Chen H, Swann JW, Zoghbi HY. Loss of MeCP2 in Parvalbumin-and somatostatin-expressing neurons in mice leads to distinct rett syndrome-like phenotypes. Neuron. (2015) 88:651–8. doi: 10.1016/j.neuron.2015.10.029
186. Morin-Parent F, Champigny C, Lacroix A, Corbin F, Lepage J-F. Hyperexcitability and impaired intracortical inhibition in patients with fragile-X syndrome. Transl Psychiatry. (2019) 9:1–9. doi: 10.1038/s41398-019-0650-z
187. Canitano R, Palumbi R. Excitation/inhibition modulators in autism spectrum disorder: current clinical research. Front Neurosci. (2021) 15:753274. doi: 10.3389/fnins.2021.753274
188. Kavalali ET, Monteggia LM. Rapid homeostatic plasticity and neuropsychiatric therapeutics. Neuropsychopharmacol Off Publ Am Coll Neuropsychopharmacol. (2023) 48:54–60. doi: 10.1038/s41386-022-01411-4
189. Winkel F, Ryazantseva M, Voigt MB, Didio G, Lilja A, Llach Pou M, et al. Pharmacological and optical activation of TrkB in Parvalbumin interneurons regulate intrinsic states to orchestrate cortical plasticity. Mol Psychiatry. (2021) 26:7247–56. doi: 10.1038/s41380-021-01211-0
190. Blackman MP, Djukic B, Nelson SB, Turrigiano GG, A. Critical and cell-autonomous role for MeCP2 in synaptic scaling up. J Neurosci. (2012) 32:13529–36. doi: 10.1523/JNEUROSCI.3077-12.2012
191. Fernandes D, Santos SD, Coutinho E, Whitt JL, Beltrão N, Rondão T, et al. Disrupted AMPA receptor function upon genetic- or antibody-mediated loss of autism-associated CASPR2. Cereb Cortex. (2019) 29:4919–31. doi: 10.1093/cercor/bhz032
192. Sarti F, Zhang Z, Schroeder J, Chen L. Rapid suppression of inhibitory synaptic transmission by retinoic acid. J Neurosci. (2013) 33:11440–50. doi: 10.1523/JNEUROSCI.1710-13.2013
193. Soden ME, Chen L. Fragile X protein FMRP is required for homeostatic plasticity and regulation of synaptic strength by retinoic acid. J Neurosci Off J Soc Neurosci. (2010) 30:16910–21. doi: 10.1523/JNEUROSCI.3660-10.2010
194. Tatavarty V, Torrado Pacheco A, Groves Kuhnle C, Lin H, Koundinya P. Autism-associated Shank3 Is essential for homeostatic compensation in rodent V1. Neuron. (2020) 106:769–77. doi: 10.1016/j.neuron.2020.02.033
195. Bülow P, Murphy TJ, Bassell GJ, Wenner P. Homeostatic intrinsic plasticity is functionally altered in Fmr1 KO cortical neurons. Cell Rep. (2019) 26:1378–83. doi: 10.1016/j.celrep.2019.01.035
196. Hwang J-Y, Monday HR, Yan J, Gompers A, Buxbaum AR, Sawicka KJ, et al. CPEB3-dependent increase in GluA2 subunits impairs excitatory transmission onto inhibitory interneurons in a mouse model of fragile X. Cell Rep. (2022) 39:110853. doi: 10.1016/j.celrep.2022.110853
197. Hengen KB, Lambo ME, Van Hooser SD, Katz DB, Turrigiano GG. Firing rate homeostasis in visual cortex of freely behaving rodents. Neuron. (2013) 80:335–42. doi: 10.1016/j.neuron.2013.08.038
198. Hengen KB, Lambo ME, Hooser SDV, Katz DB, Turrigiano GG. Firing rate homeostasis in visual cortex of freely behaving rodents. Neuron. (2013) 80:335–42.
199. Lambo ME, Turrigiano GG. Synaptic and Intrinsic Homeostatic Mechanisms Cooperate to Increase L2/3 Pyramidal Neuron Excitability during a Late Phase of Critical Period Plasticity. J Neurosci Off J Soc Neurosci. (2013) 33:8810–9.
200. Wu YK, Hengen KB, Turrigiano GG, Gjorgjieva J. Homeostatic mechanisms regulate distinct aspects of cortical circuit dynamics. Proc Natl Acad Sci U S A. (2020) 117:24514–25. doi: 10.1073/pnas.1918368117
201. Gainey MA, Aman JW, Feldman DE. Rapid disinhibition by adjustment of PV intrinsic excitability during whisker map plasticity in mouse S1. J Neurosci. (2018) 38:4749–61. doi: 10.1523/JNEUROSCI.3628-17.2018
202. Gainey MA, Feldman DE. Multiple shared mechanisms for homeostatic plasticity in rodent somatosensory and visual cortex. Philos Trans R Soc Lond B Biol Sci. (2017) 372:20160157. doi: 10.1098/rstb.2016.0157
203. Li L, Gainey MA, Goldbeck JE, Feldman DE. Rapid homeostasis by disinhibition during whisker map plasticity. Proc Natl Acad Sci U S A. (2014) 111:1616–21. doi: 10.1073/pnas.1312455111
204. Garcia-Junco-Clemente P, Chow DK, Tring E, Lazaro MT, Trachtenberg JT, Golshani P. Overexpression of calcium-activated potassium channels underlies cortical dysfunction in a model of PTEN-associated autism. Proc Natl Acad Sci U S A. (2013) 110:18297–302. doi: 10.1073/pnas.1309207110
205. Ortiz-Cruz CA, Marquez EJ, Linares-García CI, Perera-Murcia GR, Ramiro-Cortés Y. Haploinsufficiency of Shank3 increases the orientation selectivity of V1 neurons. Sci Rep. (2022) 12:22230. doi: 10.1038/s41598-022-26402-9
206. Pyronneau A, He Q, Hwang J-Y, Porch M, Contractor A, Zukin RS. Aberrant Rac1-cofilin signaling mediates defects in dendritic spines, synaptic function, and sensory perception in fragile X syndrome. Sci Signal. (2017) 10:eaan0852. doi: 10.1126/scisignal.aan0852
207. Chang Y-S, Owen JP, Desai SS, Hill SS, Arnett AB, Harris J, et al. Autism and sensory processing disorders: shared white matter disruption in sensory pathways but divergent connectivity in social-emotional pathways. PLoS ONE. (2014) 9:e103038. doi: 10.1371/journal.pone.0103038
208. Zhou Y, Sharma J, Ke Q, Landman R, Yuan J, Chen H, et al. Atypical behaviour and connectivity in SHANK3-mutant macaques. Nature. (2019) 570:326–31. doi: 10.1038/s41586-019-1278-0
209. Wang W, Liu J, Shi S, Liu T, Ma L, Ma X, et al. Altered resting-state functional activity in patients with autism spectrum disorder: a quantitative meta-analysis. Front Neurol. (2018) 9:556. doi: 10.3389/fneur.2018.00556
210. Uddin L, Supekar K, Menon V. Reconceptualizing functional brain connectivity in autism from a developmental perspective. Front Hum Neurosci. (2013) 7: 458. doi: 10.3389/fnhum.2013.00458
211. Pellicano E, Burr D. When the world becomes “too real”: a Bayesian explanation of autistic perception. Trends Cogn Sci. (2012) 16:504–10. doi: 10.1016/j.tics.2012.08.009
212. Ursino M, Serra M, Tarasi L, Ricci G, Magosso E, Romei V. Bottom-up vs. top-down connectivity imbalance in individuals with high-autistic traits: an electroencephalographic study. Front Syst Neurosci. (2022) 16:932128. doi: 10.3389/fnsys.2022.932128
213. Buch AM, Vértes PE, Seidlitz J, Kim SH, Grosenick L, Liston C. Molecular and network-level mechanisms explaining individual differences in autism spectrum disorder. Nat Neurosci. (2023) 26:650–63. doi: 10.1038/s41593-023-01259-x
214. Lefebvre A, Tillmann J, Cliquet F, Amsellem F, Maruani A, Leblond C, et al. Tackling hypo and hyper sensory processing heterogeneity in autism: From clinical stratification to genetic pathways. Autism Res Off J Int Soc Autism Res. (2023) 16:364–78. doi: 10.1002/aur.2861
Keywords: autism, sensory, cortex, theory, excitability, neural coding, circuit, inhibition
Citation: Monday HR, Wang HC and Feldman DE (2023) Circuit-level theories for sensory dysfunction in autism: convergence across mouse models. Front. Neurol. 14:1254297. doi: 10.3389/fneur.2023.1254297
Received: 06 July 2023; Accepted: 14 August 2023;
Published: 07 September 2023.
Edited by:
Jun Egawa, Niigata University, JapanReviewed by:
Yuri Bozzi, University of Trento, ItalyCorette J. Wierenga, Radboud University, Netherlands
Copyright © 2023 Monday, Wang and Feldman. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Hannah R. Monday, aGFubmFoLm1vbmRheUBiZXJrZWxleS5lZHU=; Daniel E. Feldman, ZGZlbGRtYW5AYmVya2VsZXkuZWR1