 Kristina M. Croucher
Kristina M. Croucher Sheila M. Fleming
Sheila M. Fleming
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Neurol., 05 January 2024
Sec. Neurogenetics
Volume 14 - 2023 | https://doi.org/10.3389/fneur.2023.1252400
This article is part of the Research TopicReviews in Cellular Neurophysiology 2022: Neurophysiological Mechanisms in the Aging BrainView all 5 articles
ATP13A2 is a lysosomal protein involved in polyamine transport with loss of function mutations associated with multiple neurodegenerative conditions. These include early onset Parkinson’s disease, Kufor-Rakeb Syndrome, neuronal ceroid lipofuscinosis, hereditary spastic paraplegia, and amyotrophic lateral sclerosis. While ATP13A2 mutations may result in clinical heterogeneity, the basal ganglia appear to be impacted in the majority of cases. The basal ganglia is particularly vulnerable to environmental exposures such as heavy metals, pesticides, and industrial agents which are also established risk factors for many neurodegenerative conditions. Not surprisingly then, impaired function of ATP13A2 has been linked to heavy metal toxicity including manganese, iron, and zinc. This review discusses the role of ATP13A2 in basal ganglia function and dysfunction, potential common pathological mechanisms in ATP13A2-related disorders, and how gene x environment interactions may contribute to basal ganglia dysfunction.
ATP13A2 is an ATPase primarily located in early and late endosomes and lysosomes. Biallelic mutations in the gene ATP13A2 cause Kufor-Rakeb Syndrome (KRS; OMIM#606693), also known as Parkinson’s disease-9 (PARK9), a juvenile form of Parkinson’s disease (PD) (1). KRS patients typically develop Parkinsonian motor symptoms and show some degree of levodopa-responsiveness (1). Following KRS, ATP13A2 was determined to be mutated in forms of neuronal ceroid lipofuscinosis (NCL), hereditary spastic paraplegia (HSP), and most recently amyotrophic lateral sclerosis (ALS) (2–7). Genetic analysis also shows that ATP13A2 variants in LRRK2 (PARK8) G2019S carriers, the most common cause of hereditary PD, are common and may modify disease onset and severity (8). In idiopathic PD and Dementia with Lewy bodies, post mortem analysis shows ATP13A2 protein levels are significantly decreased suggesting altered ATP13A2 function may be more pervasive in phenotypic PD than previously thought (9). Loss of function of ATP13A2 has also been linked to an increased sensitivity to heavy metal toxicity including manganese, iron, and zinc (10–23). Given the diverse outcomes that can result from dysfunctional ATP13A2, it is important to determine commonalties between these disorders in terms of symptom expression, peripheral and central pathology, and mechanisms of neurodegeneration in order to identify novel therapeutic strategies and targets. Currently, there is limited human pathology data on ATP13A2-related disorders but analysis of the different clinical profiles point to the basal ganglia as the central network disrupted in the majority of cases (24–32). The basal ganglia and its network (Figure 1) are particularly vulnerable to neurodegeneration and are associated with genetic and environmental factors that drive disorders such as PD, dystonia, and Huntington’s disease, among others (33). In addition, the basal ganglia are important in heavy metal transport with multiple structures negatively impacted by excessive intake, including manganese and iron. Thus, understanding how ATP13A2 contributes to basal ganglia function will be essential for the identification and development of therapeutics for ATP13A2-related disorders.
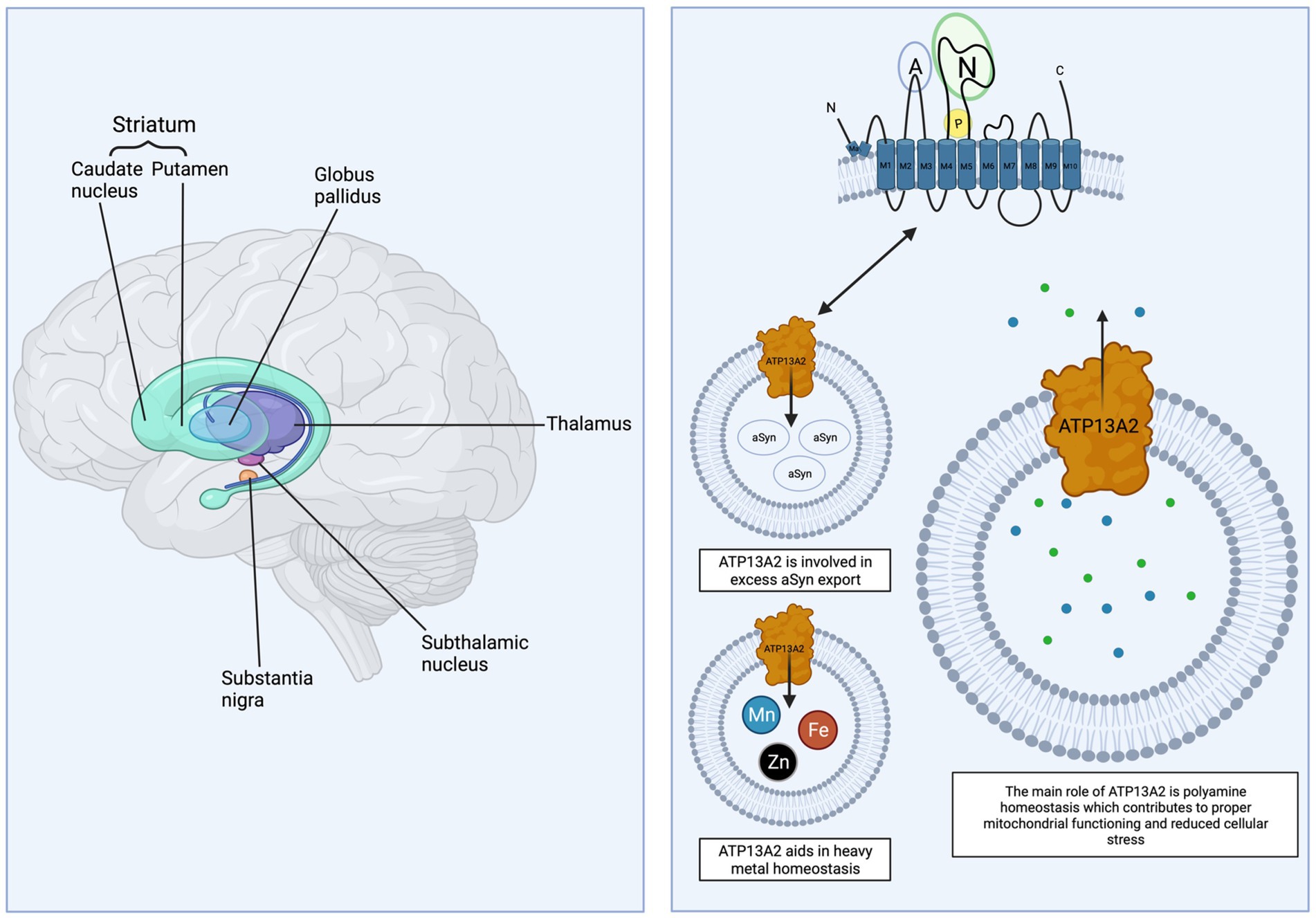
Figure 1. ATP13A2 and the basal ganglia. The basal ganglia and related nuclei (striatum, globus pallidus, subthalamic nucleus, thalamus, and substantia nigra) are vulnerable to genetic and environmental factors. Wildtype ATP13A2 is a protein with 10 transmembrane domains localized to the lysosomal membrane and is involved in polyamine transport and homeostasis, alpha-synuclein export, and intracellular heavy metal regulation. Small green and blue dots represent the polyamines spermidine and spermine. Created with BioRender.com.
Mutations in ATP13A2 are linked to the juvenile parkinsonism KRS. KRS is an autosomal recessive form of PD with similar but distinct neurological symptoms and neurodegeneration (1, 34). It was first identified in five members of a consanguineous family from Kufor-Rakeb, Jordan, with the youngest age of onset at 11 years old (30). Symptoms of KRS originally included rigidity, bradykinesia, supranuclear gaze palsy, and dementia (30). In general, KRS symptom onset occurs in young patients and the condition progresses rapidly (35). MRI brain imaging shows generalized brain atrophy beginning in the globus pallidus and pyramidal tract (30). Many KRS patients respond to levodopa, suggesting nigrostriatal dysfunction similar to what is observed in sporadic PD (30, 35). Patient follow-up performed 10 years later showed similar symptoms, but now with the addition of myoclonus and increased pyramidal signs. At the time of these studies, the link between KRS and ATP13A2 had not been made (32). Later, KRS was also identified in a Chilean population and symptoms were described in a longitudinal study (1, 24). Five family members were diagnosed with KRS between the ages of 10 and 13 with early symptoms of rigidity, frequent falls, slowed movement and speech, abnormal gait, cognitive impairment, insomnia, and upward gaze palsy. The progression of these symptoms was slower than that seen in the Jordanian family (1). Years after diagnoses, bradykinesia, resting tremor, spasticity, and myoclonus, developed. Brain imaging revealed generalized atrophy and hypointensity within the basal ganglia (24).
The genomes of the Jordanian and Chilean families were later screened to identify the genetic locus of the mutations (1). In the Jordanian family, patients had a homozygous duplication of 22 base pairs in exon 16 resulting in a frameshift and a premature stop codon (c.1632_1653dup22/p.Leu552fsTer788). In the Chilean family two compound heterozygous mutations were identified, a deletion of cytosine at the nucleotide position 3,057 in exon 26 causing a frameshift mutation (c.3057delC/p.1019GfsX1021) and a transition from guanine to adenine at the +5 position of the donor splice site in exon 13 (c.1306 + 5G > A/p.G399_L435del) (1). These mutations resulted in a loss of function of ATP13A2 which was then classified as a familial form of PD, PARK9. Since these studies, additional ATP13A2 mutations in various populations have been identified including a homozygous c.1510G > C/p.Gly504Arg mutation and the heterozygous mutations c.35C > T/p.Thr12Met or c.1597G > A/p.Gly533Arg (Table 1) (27, 39, 41). Similar to the earlier cases, patients developed basal ganglia related symptoms such as bradykinesia, rigidity, and levodopa responsiveness (27, 39, 41). Diffuse atrophy of the brain, supranuclear gaze palsy, and postural instability were observed in homozygous mutations (27). While in heterozygous mutations (ex. c.2236G > A/p.Ala746Thr), symptoms varied, but included basal ganglia-related bradykinesia, rigidity, and tremor (36–38, 40). In general, the homozygous mutations appear more severe compared to the heterozygous mutations, but symptoms can still appear in the heterozygous state with later age of onset. Further research regarding the heterozygous c.35C > T/p.Thr12Met, c.1597G > A/p.Gly533Arg, and c.2236G > A/p.Ala746Thr mutations is needed to better understand their pathogenicity, as KRS is an autosomal recessive disorder (Table 1) (1, 24, 27, 30, 35–37, 40).
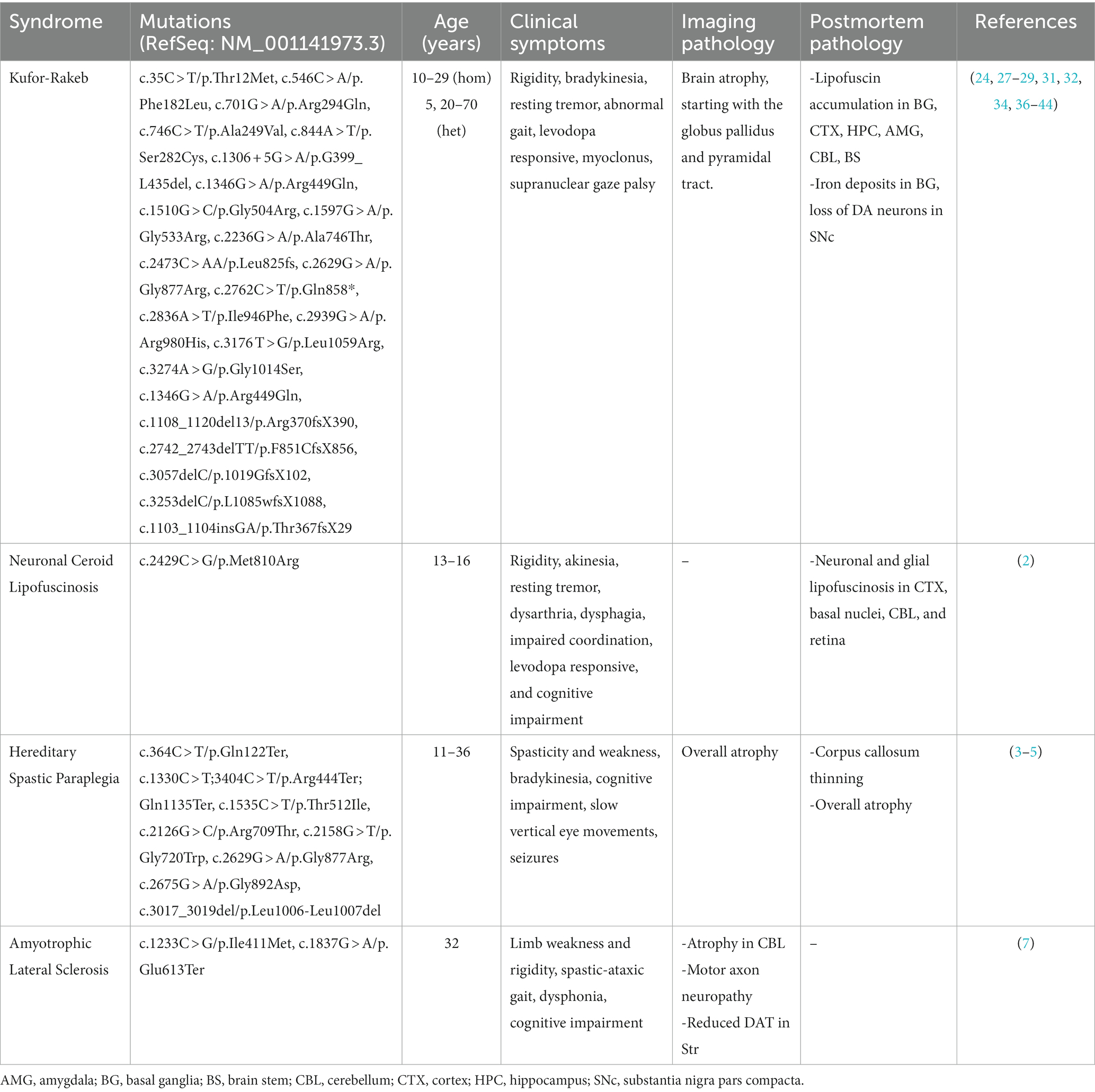
Table 1. Clinical syndromes associated with mutations in ATP13A2.
Recently, the first and only postmortem KRS study was completed on a patient with a homozygous ATP13A2 missense mutation (34). In this case, symptoms appeared at approximately 12 years of age and included rigidity and akinesia, upward gaze palsy, and spasticity. Later in life, the patient suffered from severe levodopa-induced dyskinesias, hallucinations, and irritability. Postmortem analysis revealed loss of pigmented neurons in the substantia nigra, lipofuscin accumulation in many brain regions including basal ganglia, iron accumulation in basal ganglia, and temporal lobe atrophy. This offers the first confirmation of basal ganglia pathology and substantia nigra degeneration in KRS (Table 1) (34).
Mutations in ATP13A2 are also linked to NCL, a lysosomal storage disorder. NCLs are a group of degenerative diseases characterized by accumulation of autofluorescent lysosomal storage material within lysosomes (2, 6, 45, 46). NCL symptoms can include basal ganglia dysfunction, seizures, visual impairments, cerebellar ataxia, and dementia (2, 6, 45, 46). A homozygous mutation in ATP13A2 (c.2429C > G/p.Met810Arg) was identified in a Belgian family with NCL (2). Symptoms included akinesia and rigidity in addition to gait impairments, myoclonus, and alterations in mood. Similar to KRS, levodopa responsiveness was noted along with the development of levodopa-induced dyskinesias (2). Postmortem analysis revealed widespread lipofuscinosis throughout the brain in neurons and glia (Table 1) (2).
HSP is a neurodegenerative condition characterized by progressive limb spasticity (3–5). Similar to both KRS and NCL, the clinical presentation can be quite heterogenous where, in addition to limb spasticity, seizures and cognitive impairment can also develop (3–5). The first family identified with ATP13A2-associated HSP showed a variety of symptoms in addition to adult-onset of limb spasticity, with some developing bradykinesia and rigidity, cognitive deficits, and supranuclear gaze palsy (4). Brain imaging revealed cerebellar and cortical atrophy and in one case decreased dopamine transporter density in the putamen (4). Since then, several families have been identified with ATP13A2-related HSP (3–5). Again, symptoms vary but can include bradykinesia, resting tremor, neuropsychiatric dysfunction, cognitive impairments, dysarthria, dysphagia, and oculomotor impairments in addition to limb spasticity and cerebellar symptoms (Table 1) (3–5).
Most recently, mutations in ATP13A2 have been linked to ALS (7, 47). ALS is characterized by progressive degeneration of motor neurons leading to motor weakness, impaired breathing, and ultimately death (48). Two mutations in ATP13A2, c.1837G > A/p.Glu613Ter and c.1233C > G/p.Ile411Met, were identified in two family members, resulting in ATP13A2 loss of function (7). These cases presented with limb spasticity, dysphonia, ataxic gait, and cognitive impairment. Initially, they were diagnosed with HSP but as the condition progressed, ALS-related symptoms developed. While brain imaging showed cerebellar atrophy, dopamine transporter analysis revealed a bilateral reduction in uptake in the putamen (Table 1) (7). Thus, despite the heterogenous nature of clinical symptoms and pathology in ATP13A2-associated diseases, the basal ganglia are affected in the majority of the cases.
P-type ATPases are a large family of proteins involved in the transport of cations and other substrates across cell membranes through the utilization of energy from ATP hydrolysis (49). Of these, P5-type ATPases are only expressed in eukaryotes and are the least characterized of the P-type ATPases. Of the P5-types, ATP13A2 is most abundant in the brain (49). Although there are limited studies on ATP13A2 expression in the human brain, high expression in neurons in the ventral midbrain including the substantia nigra and in the basal ganglia (globus pallidus and putamen), cortex, and hippocampus has been shown (1). However, more work is needed to identify expression levels in different brain regions across species. In vitro studies show that ATP13A2 localizes to intracellular vesicular compartments including lysosomes and early and late endosomes implicating it in protein handling and degradation (1, 12, 21, 50, 51).
ATP13A2 is a 1,180 amino acid ATPase with 10 transmembrane domains (1, 35). Molecular analysis shows ATP13A2 is a P5B-type ATPase with the N- and C- termini residing in the cytosol (Figure 1). The ATP13A2 N-terminus hydrophobic Ma region does not span the membrane and remains cytosolic (52, 53). The N- terminus and the Ma domain are important for targeting of ATP13A2 to lysosomes as they are hydrophobic. This hydrophobicity encourages interactions with lipids, specifically phosphatidic acid (PA) and phosphatidylinositol(3,5)bisphosphate [Pi(3,5)P2], which are present at high concentrations in endosomal and lysosomal membranes (52). These two lipids bind to three distinct regions in the N-terminus, which partially includes the Ma domain, to regulate ATP13A2 activity by stimulating autophosphorylation. Although PA and PI(3,5)P2 are necessary for ATP13A2 activation, they are not the transported substrates (52). Biochemical studies show that ATP13A2 activity depends on these signaling lipids and it is important to note that both are involved in vesicular trafficking, membrane fission and fusion, and autophagy, mechanisms known to be involved in multiple neurodegenerative disorders (54–58). The conformational states of ATP13A2 have also been recently identified and will facilitate the development of targeted mechanistic therapeutics (59–62).
The polyamines spermidine and spermine are highly regulated in cells and bind to nucleic acids to aid in optimal cell function including gene transcription and translation, cell cycle progression, oxidative stress response, and metabolism (61, 63, 64). Within the human brain, polyamine concentration decreases with age in multiple regions including basal ganglia structures (putamen, globus pallidus, and subthalamic nucleus) and cerebellum (65). Alterations in the polyamine pathway are also linked to PD (66). Studies by Pinto et al. (67) and De La Hera et al. (68) were the first to suggest ATP13A2 may be involved in polyamine transport. It is now confirmed that ATP13A2 transports the polyamines spermidine and spermine and functions as a H+/K+-ATPase to regulate polyamine levels (Figure 1) (64, 69–71). Specifically, ATP13A2 transports polyamines from the lysosome to the cytosol to maintain polyamine homeostasis (69, 71). Loss of ATP13A2 function subsequently leads to toxic polyamine accumulation within the lysosome (64). Polyamine accumulation may then impact other key cellular functions including protein degradation and mitochondrial function.
Although there are a limited number of studies on the expression profile of ATP13A2 across species, it is found to be abundant within basal ganglia structures and in regions that provide important innervation to the basal ganglia including substantia nigra and cortex.
Several heavy metals preferentially accumulate within basal ganglia structures and are linked to multiple neurodegenerative conditions (72). Heavy metal transporters such as divalent metal transporter 1 (DMT1) are abundant in basal ganglia structures and facilitate metal homeostasis (73). Excessive exposure to heavy metals and/or genetic mutations to metal transporters can impair heavy metal handling and transport leading to motor and cognitive impairments in humans (74–77). ATP13A2 function appears to be important in maintaining heavy metal balance (Figure 1) as loss of function mutations are linked to increased susceptibility to manganese, iron, and zinc toxicity.
Manganese (Mn) is an essential metal involved in multiple cellular functions including but not limited to energy metabolism, antioxidant response, the immune response, and development (78–80). Mn is ubiquitous in the environment and thus, Mn deficiency is rare. In contrast, excessive exposure to Mn, especially in certain occupations such as mining and welding, is a significant health risk and can cause manganism, an age-related neurodegenerative condition. Manganism is characterized by PD-like motor symptoms and cognitive impairment but is distinct from classical PD as the motor deficits are typically not responsive to levodopa and additional impairments such as dystonia and “cock-walk” gait develop. It has been shown that Mn preferentially accumulates in the basal ganglia affecting primarily the globus pallidus (74).
Mn is transported by a variety of metal transporters, including but not limited to DMT1, dopamine transporter (DAT), L-type calcium channels, transferrin, and transferrin receptor (81–83). Mn enters the brain primarily through DMT1 and transferrin/transferrin receptors [transferrin-dependent pathway; (81, 84)]. DMT1 expression in nonhuman primate brain shows high levels in the caudate nucleus, putamen, internal and external globus pallidus, and moderate expression in the substantia nigra pars compacta, thalamus and subthalamic nucleus (85). DAT is shown to transport Mn during excess exposure and is highly expressed in the striatum (86). The compounded effect of DMT1 and DAT transport of Mn during excess exposure contributes to the preferential accumulation within basal ganglia structures.
Intracellular Mn toxicity is associated with multiple mechanisms also involved in neurodegenerative diseases such as mitochondrial dysfunction, ER stress, impaired protein degradation, oxidative stress, and apoptosis (75, 87, 88). Since manganism does not develop in everyone exposed to high Mn levels, it suggests that genetic susceptibility may be an important contributing factor. Indeed, loss of function mutations in the Mn efflux transporter Slc30a10 cause an inherited form of Mn-induced Parkinsonism without excessive exposure (77). ATP13A2 may be another genetic susceptibility factor in Mn toxicity. Polymorphisms in ATP13A2 were shown to influence Mn toxicity in an elderly population (76). Mn toxicity and ATP132 have been extensively examined in different cell systems, yeast, and in vivo (Table 2). In cultured human neuroblastoma cells (NLF cell line), overexpression of ATP13A2 results in cellular protection against high concentrations of Mn compared to mutated forms of ATP13A2 (c.546C > A/p.Phe182Leu, c.1510G > C/p.Gly504Arg and c.1537G > A/p.Asp513Asn) (12). While in cultured rat primary cortical neurons, wildtype and c.1537G > A/p.Asp513Asn ATP13A2 expression protect against Mn toxicity, c.1510G > C/p.Gly504Arg and c.546C > A/p.Phe182Leu mutants do not (13). In yeast, excess Mn is sequestered to the vacuole and mutated Ypk9 (yeast homolog of ATP13A2) showed a higher sensitivity to Mn toxicity than cells that expressed wildtype Ypk9 (15, 89). Similarly, ATP13A2 overexpression in C. elegans dopaminergic neurons protects against Mn toxicity, further indicating an important link between ATP13A2 and Mn homeostasis in the basal ganglia and substantia nigra (23). In Atp13a2 knockout mice, low dose Mn exposure resulted in alterations in sensorimotor function, increased accumulation of Mn in the brain, and increased insoluble alpha-synuclein in the ventral midbrain (14). Taken together, these studies suggest an important role for ATP13A2 in Mn homeostasis (Table 2).
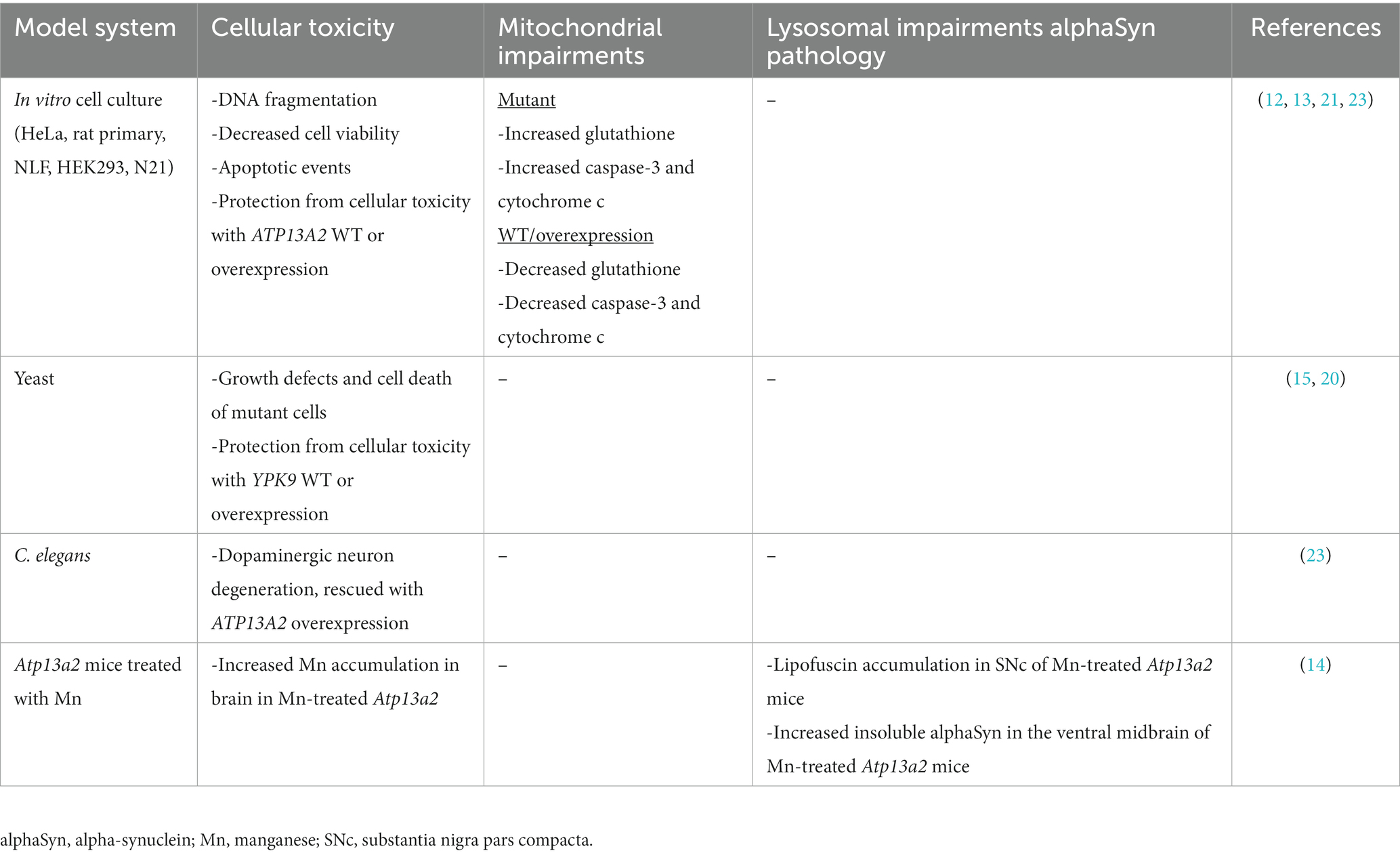
Table 2. Manganese toxicity in ATP13A2 models.
Iron (Fe) is an essential metal important in vital cellular functions such as oxygen transport, electron transport, and neurotransmitter synthesis (90). Iron accumulation in the brain increases with age and is found primarily in basal ganglia regions such as the globus pallidus, putamen, and substantia nigra (91, 92). Iron is transported into the brain using a similar mechanism to Mn transferrin-dependent transport. Transferrin receptors are moderately expressed in the putamen, caudate nucleus, globus pallidus, and substantia nigra in humans (85, 93). In rodents transferrin receptors are also expressed in striatum, thalamus, and cerebellum (94). Once inside the cell, Fe is then released into the cytoplasm with the help of DMT1 (81, 82, 94). In the basal ganglia, Fe is important in DNA synthesis, mitochondrial respiration, oxygen transportation, and neurotransmitter synthesis, especially dopamine.
Dysregulation of iron is associated with several neurological conditions including PD and Neurodegeneration with Brain Iron Accumulation (NBIA). NBIA involves disorders in which iron accumulates within the basal ganglia and presents with motor and cognitive symptoms including but not limited to abnormal gait, dystonia, parkinsonism, spasticity, seizures, and impaired cognitive function (95, 96). NBIA is typically diagnosed based on clinical symptoms and MRI imaging (T2*-weighted). In addition to PD, mutations in ATP13A2 are linked to NBIA, suggesting ATP13A2-linked disorders may be considered a form of NBIA (97). For example, in a patient with a homozygous ATP13A2 mutation (c.1103_1104insGA/p.Thr367ArgfsX29) and clinical symptoms resembling NBIA, T2*-weighted MRI analysis showed hypointensities indicative of iron accumulation in the basal ganglia (31). Iron accumulation in the basal ganglia was also reported in the Chilean family with KRS (24). Furthermore, the first postmortem analysis of KRS showed iron accumulation in the basal ganglia however, the deposits were sparse and no axonal spheroids typical of some NBIA were observed (34). Although limited, in vitro work indicates ATP13A2 can protect against iron toxicity supporting a potential role for ATP13A2 in iron homeostasis within the basal ganglia (Table 3) (19).

Table 3. Iron and zinc toxicity in ATP13A2 models.
Zinc is another essential metal involved in numerous cellular processes including synthesis of DNA and proteins (17, 22). While zinc deficiency is well studied, less is understood about the mechanisms of excess and accumulated zinc (98). Zn is most notably transported by zinc-regulated zinc transporter 1, ZIP8/ZIP14, and DMT1 (81, 82). Zinc accumulation has been shown in the basal ganglia and substantia nigra in sporadic PD patients and is linked to loss of function mutations in ATP13A2 (16, 17, 22, 95, 98–100). Analysis in PARK9 patient-associated cultures showed increased sensitivity to zinc, lysosomal dysfunction, mitochondrial alterations, and increased alpha-synuclein (Table 3). In addition, overexpression of ATP13A2 reduced these pathological features in vitro (16, 22, 98, 100). While in vitro human-derived ATP13A2 models have been investigated, there are no imaging or postmortem studies to date to demonstrate alterations in zinc homeostasis in patients.
Taken together, clinical, in vivo, and in vitro studies suggest long-term impairment in ATP13A2 function may impair the basal ganglia’s ability to maintain metal homeostasis.
Mutations in ATP13A2 are associated with diverse disorders of overlapping symptoms and with heavy metals that share common transport mechanisms. Thus, it should expected that ATP13A2 mutations affect key pathological systems, such as mitochondrial function and lysosome-mediated protein degradation, involved in most neurodegenerative disorders.
At some stage in every neurodegenerative disease there is mitochondrial dysfunction. Determining where in the brain mitochondrial dysfunction occurs, when it happens, and how it begins are critical questions for every neurodegenerative condition. Mutated ATP13A2 is linked to multiple mitochondrial defects (Tables 3, 4). Studies in PARK9 fibroblasts and ATP13A2 knockdown in cortical neurons collectively reveal impaired autophagic flux and the following mitochondrial defects: reduced mitochondrial membrane potential, reduced ATP synthesis, increased respiration rate, increased fragmentation, and reactive oxygen species (ROS) (107, 108). While overexpression of ATP13A2 confers resistance against the mitochondrial complex 1 inhibitors rotenone and MPP+ (12). In addition, the ATP13A2 associated lipids PI(3,5)P2 and PA when pharmacologically inhibited, result in mitochondrial stress and toxicity in mutant cells exposed to rotenone (109). These data suggest loss of function mutations in ATP13A2 are associated with mitochondrial defects that could lead to increased susceptibility to environmental insults such as heavy metal and pesticide exposures and ultimately to neurodegenerative disease.
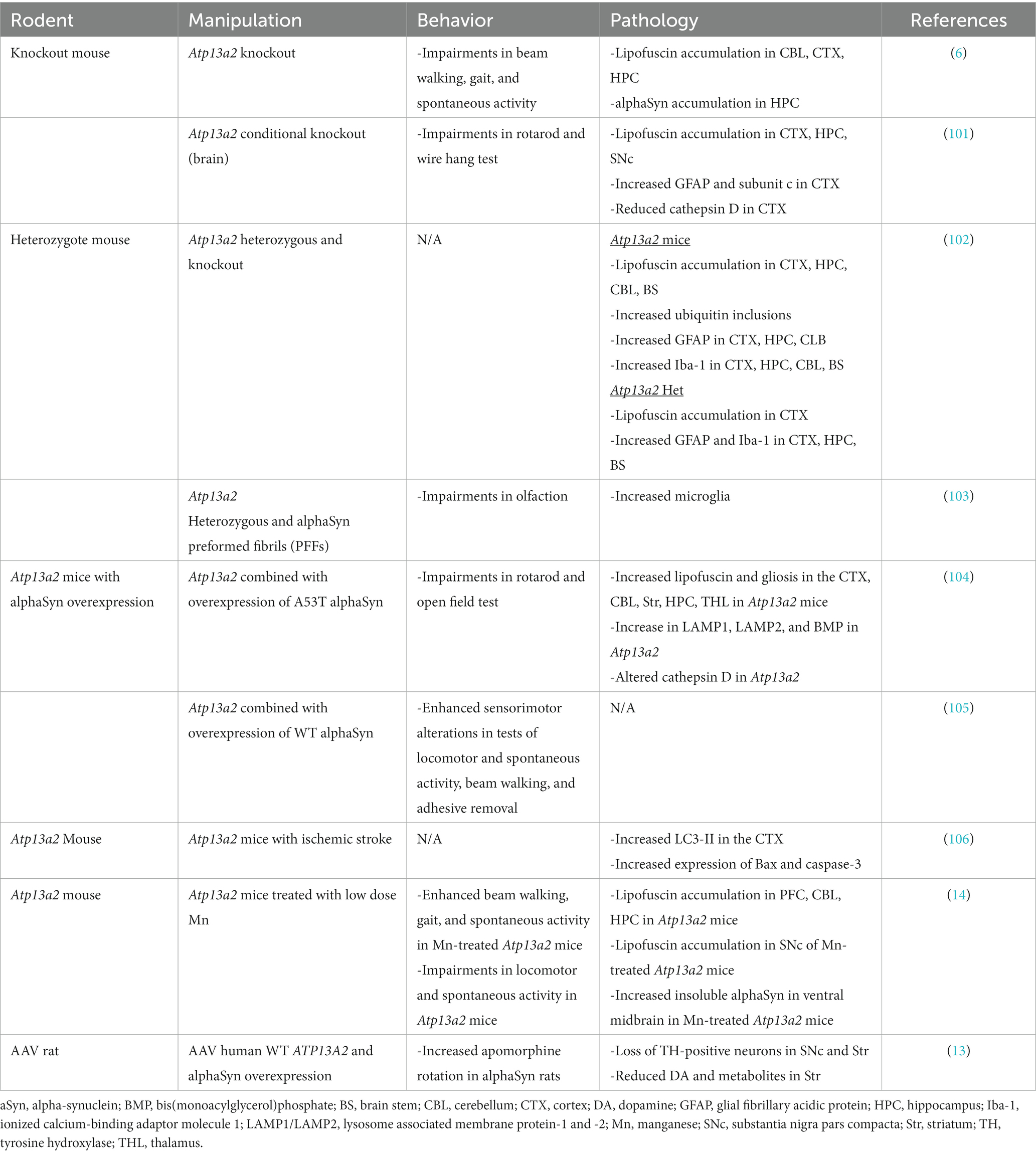
Table 4. Consequences of impaired ATP13A2 function in vivo in rodents.
Excess exposure to the heavy metals implicated in ATP13A2 function all negatively impact mitochondrial function (Tables 2, 3). Alaimo et al. (110) showed that Mn can cause dysregulation of fusion and fission, processes important in mitochondrial dynamics. An imbalance of these systems can result in ROS accumulation and cell death. Excess iron is also associated with mitochondrial dysfunction and increased ROS and is strongly linked to neurodegeneration (111). Zinc is shown to inhibit mitochondrial function causing increased ROS, energy impairments, and cytotoxicity (16, 17).
ATP13A2 transports polyamines out of the lysosome into the cytoplasm to maintain polyamine levels in cells (19, 112). Accumulation of polyamines is toxic as lysosomes can rupture when polyamine concentration is too high resulting in detrimental effects on the cell (64). ATP13A2 mutations impair export of polyamines, resulting in lysosomal polyamine accumulation, reduced cytosolic polyamine levels and mitochondrial ROS cytotoxicity. Thus, ATP13A2 seems to be important in mediating polyamine levels which then further supports optimal mitochondrial function (64, 112).
Similar to mitochondrial dysfunction, impaired protein degradation systems such as the autophagy lysosomal pathway underly multiple neurodegenerative diseases (113, 114). Autophagy lysosomal defects are prominent in PD, NCL, and Gaucher’s disease, among others. Early investigation into the effect of ATP13A2 mutations on lysosomal function showed wildtype ATP13A2 localizes to the lysosome but that mutated ATP13A2 can localize to the endoplasmic reticulum (ER) causing ER stress and decreased lysosomal function (1, 12, 21, 50, 51, 115). Studies in ATP13A2 patient-derived fibroblasts and in knockdown of ATP13A2 in dopaminergic cell lines show multiple lysosomal anomalies including reduced degradation of lysosomal substrates, alterations in acidification, decreased clearance of autophagosomes, and impaired proteolytic processing of lysosomal enzymes (113). In mice, loss of Atp13a2 function results in enhanced lipofuscinosis, accumulation of the substrates p62, cathepsin D, and ubiquitin (Table 4) (6, 104). ATP13A2 is also important for exosome secretion, where loss of function is associated with decreased exosomes, and overexpression promotes exosomal generation, release, and functioning (115). Collectively, impaired ATP13A2 function is linked to lysosome dysfunction, impaired exosome secretion, and autophagic flux (Table 4) (1, 6, 12, 21, 50, 51, 104, 113, 115).
In conjunction with mitochondrial and lysosomal defects, loss of ATP13A2 function is shown to increase alpha-synuclein accumulation (52, 107, 108, 113, 116–118). Alpha-synuclein is a presynaptic protein involved in synaptic transmission, vesicular trafficking, and plasticity and it is the major component of Lewy bodies, the hallmark pathology in PD, Multiple System Atrophy, and Dementia with Lewy Bodies (119, 120). Studies show ATP13A2 is involved in the exosomal externalization of alpha-synuclein (Figure 1), indicating a potentially important role in PD and other synucleinopathies (16, 22). While the in vitro work establishing a relationship between loss of function of ATP13A2 and alpha-synuclein is compelling, in vivo studies paint a more inconsistent picture (Table 4) (6, 13–15, 104, 113, 116). Differential effects are observed in Atp13a2 null (13a2) mouse lines, as one study found abnormal alpha-synuclein accumulation in the brain while the other did not (6, 104). The mouse line with increased abnormal alpha-synuclein in the brain also exhibited increased triton-insoluble alpha-synuclein in the ventral midbrain in response to systemic manganese administration and enhanced sensorimotor deficits when combined with alpha-synuclein overexpression (Table 4) (14, 105). However, no acceleration of pathology was observed when a mutated form of alpha-synuclein (A53T) was overexpressed (104). In addition, viral co-overexpression of Atp13a2 and alpha-synuclein did not protect against alpha-synuclein toxicity in the substantia nigra in rats (13). There are several methodological differences between the studies to note though including the timing (Atp13a2 may need to precede alpha-synuclein overexpression) and level of overexpression of Atp13a2. In viral vector studies and in crossbreeding studies the promoter and type of alpha-synuclein being expressed (mutated or wildtype) are known to yield differential phenotypes and pathology (14). Clinically, the one postmortem case of KRS did not show Lewy body pathology (34). However, this is not unprecedented as other genetic forms of PD such as LRRK2 have cases with Lewy body pathology and without (121–125). ATP13A2 variants are common in LRRK2 carriers and may modify disease onset and progression (8). More in vivo studies are needed to elucidate the relationship between ATP13A2 and alpha-synuclein.
ATP13A2’s role in polyamine transport, lysosomal function, and mitochondrial function suggests that when its function is impaired it leaves the basal ganglia particularly vulnerable to different types of insults be it heavy metal toxicity or alpha-synuclein toxicity. Understanding how these interactions develop and lead to basal ganglia dysfunction and neurodegeneration would inform multiple basal ganglia conditions and identify much needed novel targets for therapy.
KC and SF conceptualized and wrote this work. All authors edited and approved the final version of the manuscript.
The author(s) declare financial support was received for the research, authorship, and/or publication of this article. This work was supported by the National Institutes of Health (ES031124 to SF) and Department of Defense (W81XWH-19-0772 to SF).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
1. Ramirez, A , Heimbach, A , Gründemann, J , Stiller, B , Hampshire, D , Cid, LP, et al. Hereditary parkinsonism with dementia is caused by mutations in ATP13A2, encoding a lysosomal type 5 P-type ATPase. Nat Genet. (2006) 38:1184–91. doi: 10.1038/ng1884
2. Bras, J , Verloes, A , Schneider, SA , Mole, SE , and Guerreiro, RJ . Mutation of the parkinsonism gene ATP13A2 causes neuronal ceroid-lipofuscinosis. Hum Mol Genet. (2012) 21:2646–50. doi: 10.1093/hmg/dds089
3. Estiar, MA , Leveille, E , Spiegelman, D , Dupre, N , Trempe, J-F , Rouleau, GA, et al. Clinical and genetic analysis of ATP13A2 in hereditary spastic paraplegia expands the phenotype. Mol Genet Genomic Med. (2020) 8:e1052. doi: 10.1002/mgg3.1052
4. Estrada-Cuzcano, A , Martin, S , Chamova, T , Synofzik, M , Timmann, D , Holemans, T, et al. Loss-of-function mutations in the ATP13A2/PARK9 gene cause complicated hereditary spastic paraplegia (SPG78). Brain. (2017) 140:287–305. doi: 10.1093/brain/aww307
5. Kara, E , Tucci, A , Manzoni, C , Lynch, DS , Elpidorou, M , Bettencourt, C, et al. Genetic and phenotypic characterization of complex hereditary spastic paraplegia. Brain. (2016) 139:1904–18. doi: 10.1093/brain/aww111
6. Schultheis, PJ , Fleming, SM , Clippinger, AK , Lewis, J , Tsunemi, T , Giasson, B, et al. Atp13a2-deficient mice exhibit neuronal ceroid lipofuscinosis, limited α-synuclein accumulation and age-dependent sensorimotor deficits. Hum Mol Genet. (2013) 22:2067–82. doi: 10.1093/hmg/ddt057
7. Spataro, R , Kousi, M , Farhan, SMK , Willer, JR , Ross, JP , Dion, PA, et al. Mutations in ATP13A2 (PARK9) are associated with an amyotrophic lateral sclerosis-like phenotype, implicating this locus in further phenotypic expansion. Hum Genomics. (2019) 13:19. doi: 10.1186/s40246-019-0203-9
8. Lubbe, SJ , Escott-Price, V , Gibbs, JR , Nalls, MA , Bras, J , Price, TR, et al. Additional rare variant analysis in Parkinson’s disease cases with and without known pathogenic mutations: evidence for oligogenic inheritance. Hum Mol Genet. (2016) 25:ddw348–ddw5489. doi: 10.1093/hmg/ddw348
9. Murphy, KE , Cottle, L , Gysbers, AM , Cooper, AA , and Halliday, GM . ATP13A2 (PARK9) protein levels are reduced in brain tissue of cases with Lewy bodies. Acta Neuropathol Commun. (2013) 1:11. doi: 10.1186/2051-5960-1-11
10. Anand, N , Holcom, A , Broussalian, M , Schmidt, M , Chinta, SJ , Lithgow, GJ, et al. Dysregulated iron metabolism in C. elegans catp-6/ATP13A2 mutant impairs mitochondrial function. Neurobiol Dis. (2020) 139:104786. doi: 10.1016/j.nbd.2020.104786
11. Baesler, J , Kopp, JF , Pohl, G , Aschner, M , Haase, H , Schwerdtle, T, et al. Zn homeostasis in genetic models of Parkinson’s disease in Caenorhabditis elegans. J Trace Elem Med Biol. (2019) 55:44–9. doi: 10.1016/j.jtemb.2019.05.005
12. Covy, JP , Waxman, EA , and Giasson, BI . Characterization of cellular protective effects of ATP13A2/PARK9 expression and alterations resulting from pathogenic mutants. J Neurosci Res. (2012) 90:2306–16. doi: 10.1002/jnr.23112
13. Daniel, G , Musso, A , Tsika, E , Fiser, A , Glauser, L , Pletnikova, O, et al. α-Synuclein-induced dopaminergic neurodegeneration in a rat model of Parkinson’s disease occurs independent of ATP13A2 (PARK9). Neurobiol Dis. (2015) 73:229–43. doi: 10.1016/j.nbd.2014.10.007
14. Fleming, SM , Santiago, NA , Mullin, EJ , Pamphile, S , Karkare, S , Lemkuhl, A, et al. The effect of manganese exposure in Atp13a2-deficient mice. Neurotoxicology. (2018) 64:256–66. doi: 10.1016/j.neuro.2017.06.005
15. Gitler, AD , Chesi, A , Geddie, ML , Strathearn, KE , Hamamichi, S , Hill, KJ, et al. Alpha-synuclein is part of a diverse and highly conserved interaction network that includes PARK9 and manganese toxicity. Nat Genet. (2009) 41:308–15. doi: 10.1038/ng.300
16. Kong, SMY , Chan, BKK , Park, J-S , Hill, KJ , Aitken, JB , Cottle, L, et al. Parkinson’s disease-linked human PARK9/ATP13A2 maintains zinc homeostasis and promotes α-Synuclein externalization via exosomes. Hum Mol Genet. (2014) 23:2816–33. doi: 10.1093/hmg/ddu099
17. Park, J-S , Koentjoro, B , Veivers, D , Mackay-Sim, A , and Sue, CM . Parkinson’s disease-associated human ATP13A2 (PARK9) deficiency causes zinc dyshomeostasis and mitochondrial dysfunction. Hum Mol Genet. (2014) 23:2802–15. doi: 10.1093/hmg/ddt623
18. Rajagopalan, S , Rane, A , Chinta, SJ , and Andersen, JK . Regulation of ATP13A2 via PHD2-HIF1α signaling is critical for cellular iron homeostasis: implications for Parkinson’s disease. J Neurosci. (2016) 36:1086–95. doi: 10.1523/JNEUROSCI.3117-15.2016
19. Rinaldi, DE , Corradi, GR , Cuesta, LM , Adamo, HP , and de Tezanos Pinto, F . The Parkinson-associated human P5B-ATPase ATP13A2 protects against the iron-induced cytotoxicity. Biochim Biophys Acta. (2015) 1848:1646–55. doi: 10.1016/j.bbamem.2015.04.008
20. Schmidt, K , Wolfe, DM , Stiller, B , and Pearce, DA . Cd2+, Mn2+, Ni2+ and Se2+ toxicity to Saccharomyces cerevisiae lacking YPK9p the orthologue of human ATP13A2. Biochem Biophys Res Commun. (2009) 383:198–202. doi: 10.1016/j.bbrc.2009.03.151
21. Tan, J , Zhang, T , Jiang, L , Chi, J , Hu, D , Pan, Q, et al. Regulation of intracellular manganese homeostasis by Kufor-Rakeb syndrome-associated ATP13A2 protein. J Biol Chem. (2011) 286:29654–62. doi: 10.1074/jbc.M111.233874
22. Tsunemi, T , and Krainc, D . Zn2+ dyshomeostasis caused by loss of ATP13A2/PARK9 leads to lysosomal dysfunction and alpha-synuclein accumulation. Hum Mol Genet. (2014) 23:2791–801. doi: 10.1093/hmg/ddt572
23. Ugolino, J , Dziki, KM , Kim, A , Wu, JJ , Vogel, BE , and Monteiro, MJ . Overexpression of human Atp13a2Isoform-1 protein protects cells against manganese and starvation-induced toxicity. PLoS One. (2019) 14:e0220849. doi: 10.1371/journal.pone.0220849
24. Behrens, MI , Brüggemann, N , Chana, P , Venegas, P , Kägi, M , Parrao, T, et al. Clinical spectrum of Kufor-Rakeb syndrome in the Chilean kindred with ATP13A2 mutations. Mov Disord. (2010) 25:1929–37. doi: 10.1002/mds.22996
25. Brüggemann, N , Hagenah, J , Reetz, K , Schmidt, A , Kasten, M , Buchmann, I, et al. Recessively inherited parkinsonism: effect of ATP13A2 mutations on the clinical and neuroimaging phenotype. Arch Neurol. (2010) 67:1357–63. doi: 10.1001/archneurol.2010.281
26. Chen, C-M , Lin, C-H , Juan, H-F , Hu, F-J , Hsiao, Y-C , Chang, H-Y, et al. ATP13A2 variability in Taiwanese Parkinson’s disease. Am J Med Genet B Neuropsychiatr Genet. (2011) 156B:720–9. doi: 10.1002/ajmg.b.31214
27. Di Fonzo, A , Chien, HF , Socal, M , Giraudo, S , Tassorelli, C , Iliceto, G, et al. ATP13A2 missense mutations in juvenile parkinsonism and young onset Parkinson disease. Neurology. (2007) 68:1557–62. doi: 10.1212/01.wnl.0000260963.08711.08
28. Djarmati, A , Hagenah, J , Reetz, K , Winkler, S , Behrens, MI , Pawlack, H, et al. ATP13A2 variants in early-onset Parkinson’s disease patients and controls. Mov Disord. (2009) 24:2104–11. doi: 10.1002/mds.22728
29. Eiberg, H , Hansen, L , Korbo, L , Nielsen, I , Svenstrup, K , Bech, S, et al. Novel mutation in ATP13A2 widens the spectrum of Kufor-Rakeb syndrome (PARK9). Clin Genet. (2012) 82:256–63. doi: 10.1111/j.1399-0004.2011.01745.x
30. Najimal-Din, AS , Wriekat, A , Mubaidin, A , Dasouki, M , and Hiari, M . Pallido-pyramidal degeneration, supranuclear upgaze paresis and dementia: Kufor-Rakeb syndrome. Acta Neurol Scand. (1994) 89:347–52. doi: 10.1111/j.1600-0404.1994.tb02645.x
31. Schneider, SA , Paisan-Ruiz, C , Quinn, NP , Lees, AJ , Houlden, H , Hardy, J, et al. ATP13A2 mutations (PARK9) cause neurodegeneration with brain Iron accumulation. Mov Disord. (2010) 25:979–84. doi: 10.1002/mds.22947
32. Williams, DR , Hadeed, A , Al-Din, ASN , Wreikat, A-L , and Lees, AJ . Kufor Rakeb disease: autosomal recessive, levodopa-responsive parkinsonism with pyramidal degeneration, supranuclear gaze palsy, and dementia. Mov Disord. (2005) 20:1264–71. doi: 10.1002/mds.20511
33. Tambasco, N , Romoli, M , and Calabresi, P . Selective basal ganglia vulnerability to energy deprivation: experimental and clinical evidences. Prog Neurobiol. (2018) 169:55–75. doi: 10.1016/j.pneurobio.2018.07.003
34. Chien, HF , Rodriguez, RD , Bonifati, V , Nitrini, R , Pasqualucci, CA , Gelpi, E, et al. Neuropathologic findings in a patient with juvenile-onset levodopa-responsive parkinsonism due to ATP13A2 mutation. Neurology. (2021) 97:763–6. doi: 10.1212/WNL.0000000000012705
35. Hampshire, D , Roberts, E , Crow, Y , Bond, J , Mubaidin, A , Wriekat, A, et al. Kufor-Rakeb syndrome, pallido-pyramidal degeneration with supranuclear upgaze paresis and dementia, maps to 1p36. J Med Genet. (2001) 38:680–2. doi: 10.1136/jmg.38.10.680
36. Chan, AYY , Baum, L , Tang, NLS , Lau, CYK , Ng, PW , Hui, KF, et al. The role of the Ala746Thr variant in the ATP13A2 gene among Chinese patients with Parkinson’s disease. J Clin Neurosci. (2013) 20:761–2. doi: 10.1016/j.jocn.2012.05.052
37. Fei, Q-Z , Cao, L , Xiao, Q , Zhang, T , Zheng, L , Wang, X-J, et al. Lack of association between ATP13A2 Ala746Thr variant and Parkinson’s disease in Han population of mainland China. Neurosci Lett. (2010) 475:61–3. doi: 10.1016/j.neulet.2010.03.018
38. Funayama, M , Tomiyama, H , Wu, R-M , Ogaki, K , Yoshino, H , Mizuno, Y, et al. Rapid screening of ATP13A2 variant with high-resolution melting analysis. Mov Disord. (2010) 25:2434–7. doi: 10.1002/mds.23106
39. Li, G , Zhang, Z , Xia, H , and Yang, X . Analysis of Thr12Met and Ala1144Thr mutations of the ATP13A2 gene in Parkinson’s disease patients in Xinjiang Uygur and Han ethnic groups. Med Sci Monit. (2014) 20:2177–82. doi: 10.12659/MSM.892821
40. Lin, CH , Tan, EK , Chen, ML , Tan, LC , Lim, HQ , Chen, GS, et al. Novel ATP13A2 variant associated with Parkinson disease in Taiwan and Singapore. Neurology. (2008) 71:1727–32. doi: 10.1212/01.wnl.0000335167.72412.68
41. Wang, D , Gao, H , Li, Y , Jiang, S , and Yang, X . ATP13A2 gene variants in patients with Parkinson’s disease in Xinjiang. Biomed Res Int. (2020) 2020:6954820. doi: 10.1155/2020/6954820
42. Crosiers, D , Ceulemans, B , Meeus, B , Nuytemans, K , Pals, P , Van Broeckhoven, C, et al. Juvenile dystonia-parkinsonism and dementia caused by a novel ATP13A2 frameshift mutation. Parkinsonism Relat Disord. (2011) 17:135–8. doi: 10.1016/j.parkreldis.2010.10.011
43. Fong, CY , Rolfs, A , Schwarzbraun, T , Klein, C , and O’Callaghan, FJK . Juvenile parkinsonism associated with heterozygous frameshift ATP13A2 gene mutation. Eur J Paediatr Neurol. (2011) 15:271–5. doi: 10.1016/j.ejpn.2011.01.001
44. Yang, X , and Xu, Y . Mutations in the ATP13A2 gene and Parkinsonism: a preliminary review. Biomed Res Int. (2014) 2014:371256. doi: 10.1155/2014/371256
45. Farias, FHG , Zeng, R , Johnson, GS , Wininger, FA , Taylor, JF , Schnabel, RD, et al. A truncating mutation in ATP13A2 is responsible for adult-onset neuronal ceroid lipofuscinosis in Tibetan terriers. Neurobiol Dis. (2011) 42:468–74. doi: 10.1016/j.nbd.2011.02.009
46. Wöhlke, A , Philipp, U , Bock, P , Beineke, A , Lichtner, P , Meitinger, T, et al. A one base pair deletion in the canine ATP13A2 gene causes exon skipping and late-onset neuronal ceroid lipofuscinosis in the Tibetan terrier. PLoS Genet. (2011) 7:e1002304. doi: 10.1371/journal.pgen.1002304
47. Vacchiano, V , Bartoletti-Stella, A , Rizzo, G , Avoni, P , Parchi, P , Salvi, F, et al. Frequency of Parkinson’s disease genes and role of PARK2 in amyotrophic lateral sclerosis: an NGS study. Genes. (2022) 13:1306. doi: 10.3390/genes13081306
48. López-Pingarrón, L , Almeida, H , Soria-Aznar, M , Reyes-Gonzales, MC , Terrón, MP , and García, JJ . Role of oxidative stress on the etiology and pathophysiology of amyotrophic lateral sclerosis (ALS) and its relation with the enteric nervous system. Curr Issues Mol Biol. (2023) 45:3315–32. doi: 10.3390/cimb45040217
49. Schultheis, PJ , Hagen, TT , O’Toole, KK , Tachibana, A , Burke, CR , McGill, DL, et al. Characterization of the P5 subfamily of P-type transport ATPases in mice. Biochem Biophys Res Commun. (2004) 323:731–8. doi: 10.1016/j.bbrc.2004.08.156
50. Park, J-S , Mehta, P , Cooper, AA , Veivers, D , Heimbach, A , Stiller, B, et al. Pathogenic effects of novel mutations in the P-type ATPase ATP13A2 (PARK9) causing Kufor-Rakeb syndrome, a form of early-onset parkinsonism. Hum Mutat. (2011) 32:956–64. doi: 10.1002/humu.21527
51. Ugolino, J , Fang, S , Kubisch, C , and Monteiro, MJ . Mutant Atp13a2 proteins involved in parkinsonism are degraded by ER-associated degradation and sensitize cells to ER-stress induced cell death. Hum Mol Genet. (2011) 20:3565–77. doi: 10.1093/hmg/ddr274
52. Holemans, T , Sørensen, DM , van Veen, S , Martin, S , Hermans, D , Kemmer, GC, et al. A lipid switch unlocks Parkinson’s disease-associated ATP13A2. Proc Natl Acad Sci U S A. (2015) 112:9040–5. doi: 10.1073/pnas.1508220112
53. Sørensen, DM , Buch-Pedersen, MJ , and Palmgren, MG . Structural divergence between the two subgroups of P5 ATPases. Biochim Biophys Acta. (2010) 1797:846–55. doi: 10.1016/j.bbabio.2010.04.010
54. Blackwood, RA , Smolen, JE , Transue, A , Hessler, RJ , Harsh, DM , Brower, RC, et al. Phospholipase D activity facilitates Ca2+−induced aggregation and fusion of complex liposomes. Am J Phys. (1997) 272:C1279–85. doi: 10.1152/ajpcell.1997.272.4.C1279
55. Dong, X , Shen, D , Wang, X , Dawson, T , Li, X , Zhang, Q, et al. PI(3,5)P(2) controls membrane trafficking by direct activation of mucolipin ca(2+) release channels in the endolysosome. Nat Commun. (2010) 1:38. doi: 10.1038/ncomms1037
56. Hsu, AL , Ching, TT , Sen, G , Wang, DS , Bondada, S , Authi, KS, et al. Novel function of phosphoinositide 3-kinase in T cell Ca2+ signaling. A phosphatidylinositol 3,4,5-trisphosphate-mediated Ca2+ entry mechanism. J Biol Chem. (2000) 275:16242–50. doi: 10.1074/jbc.M002077200
57. Weigert, R , Silletta, MG , Spanò, S , Turacchio, G , Cericola, C , Colanzi, A, et al. CtBP/BARS induces fission of Golgi membranes by acylating lysophosphatidic acid. Nature. (1999) 402:429–33. doi: 10.1038/46587
58. Fang, Y , Vilella-Bach, M , Bachmann, R , Flanigan, A , and Chen, J . Phosphatidic acid-mediated mitogenic activation of mTOR signaling. Science. (2001) 294:1942–5. doi: 10.1126/science.1066015
59. Mateeva, T , Klähn, M , and Rosta, E . Structural dynamics and catalytic mechanism of ATP13A2 (PARK9) from simulations. J Phys Chem B. (2021) 125:11835–47. doi: 10.1021/acs.jpcb.1c05337
60. Mu, J , Xue, C , Fu, L , Yu, Z , Nie, M , Wu, M, et al. Conformational cycle of human polyamine transporter ATP13A2. Nat Commun. (2023) 14:1978. doi: 10.1038/s41467-023-37741-0
61. Sim, SI , von Bülow, S , Hummer, G , and Park, E . Structural basis of polyamine transport by human ATP13A2 (PARK9). Mol Cell. (2021) 81:4635–4649.e8. doi: 10.1016/j.molcel.2021.08.017
62. Tillinghast, J , Drury, S , Bowser, D , Benn, A , and Lee, KPK . Structural mechanisms for gating and ion selectivity of the human polyamine transporter ATP13A2. Mol Cell. (2021) 81:4650–4662.e4. doi: 10.1016/j.molcel.2021.10.002
63. Heinick, A , Urban, K , Roth, S , Spies, D , Nunes, F , Phanstiel, O, et al. Caenorhabditis elegans P5B-type ATPase CATP-5 operates in polyamine transport and is crucial for norspermidine-mediated suppression of RNA interference. FASEB J. (2010) 24:206–17. doi: 10.1096/fj.09-135889
64. van Veen, S , Martin, S , Van den Haute, C , Benoy, V , Lyons, J , Vanhoutte, R, et al. ATP13A2 deficiency disrupts lysosomal polyamine export. Nature. (2020) 578:419–24. doi: 10.1038/s41586-020-1968-7
65. Vivó, M , de Vera, N , Cortés, R , Mengod, G , Camón, L , and Martínez, E . Polyamines in the basal ganglia of human brain. Influence of aging and degenerative movement disorders. Neurosci Lett. (2001) 304:107–11. doi: 10.1016/s0304-3940(01)01776-1
66. Lewandowski, NM , Ju, S , Verbitsky, M , Ross, B , Geddie, ML , Rockenstein, E, et al. Polyamine pathway contributes to the pathogenesis of Parkinson disease. Proc Natl Acad Sci U S A. (2010) 107:16970–5. doi: 10.1073/pnas.1011751107
67. Pinto, F. De T , Corradi, GR , Hera, DP , and Adamo, HP . (2012). CHO cells expressing the human P₅-ATPase ATP13A2 are more sensitive to the toxic effects of herbicide paraquat. Neurochem Int. 60, 243–248. doi: 10.1016/j.neuint.2012.01.002
68. De La Hera, DP , Corradi, GR , Adamo, HP , and De Tezanos Pinto, F . Parkinson’s disease-associated human P5B-ATPase ATP13A2 increases spermidine uptake. Biochem J. (2013) 450:47–53. doi: 10.1042/BJ20120739
69. Fujii, T , Nagamori, S , Wiriyasermkul, P , Zheng, S , Yago, A , Shimizu, T, et al. Parkinson’s disease-associated ATP13A2/PARK9 functions as a lysosomal H+,K+-ATPase. Nat Commun. (2023) 14:1–11. doi: 10.1038/s41467-023-37815-z
70. Houdou, M , Jacobs, N , Coene, J , Azfar, M , Vanhoutte, R , Van den Haute, C, et al. Novel green fluorescent polyamines to analyze ATP13A2 and ATP13A3 activity in the mammalian polyamine transport system. Biomol Ther. (2023) 13:337. doi: 10.3390/biom13020337
71. Li, P , Wang, K , Salustros, N , Grønberg, C , and Gourdon, P . Structure and transport mechanism of P5B-ATPases. Nat Commun. (2021) 12:3973. doi: 10.1038/s41467-021-24148-y
72. Chen, P , Miah, MR , and Aschner, M . Metals and neurodegeneration. F1000Res. (2016) 5:F1000 Faculty Rev-366. doi: 10.12688/f1000research.7431.1
73. Au, C , Benedetto, A , and Aschner, M . Manganese transport in eukaryotes: the role of DMT1. Neurotoxicology. (2008) 29:569–76. doi: 10.1016/j.neuro.2008.04.022
74. Guilarte, TR . Manganese neurotoxicity: new perspectives from behavioral, neuroimaging, and neuropathological studies in humans and non-human primates. Front Aging Neurosci. (2013) 5:23. doi: 10.3389/fnagi.2013.00023
75. Kwakye, GF , Paoliello, MMB , Mukhopadhyay, S , Bowman, AB , and Aschner, M . Manganese-induced parkinsonism and Parkinson’s disease: shared and distinguishable features. Int J Environ Res Public Health. (2015) 12:7519–40. doi: 10.3390/ijerph120707519
76. Rentschler, G , Covolo, L , Haddad, AA , Lucchini, RG , Zoni, S , and Broberg, K . ATP13A2 (PARK9) polymorphisms influence the neurotoxic effects of manganese. Neurotoxicology. (2012) 33:697–702. doi: 10.1016/j.neuro.2012.01.007
77. Tuschl, K , Clayton, PT , Gospe, SM , Gulab, S , Ibrahim, S , Singhi, P, et al. Syndrome of hepatic cirrhosis, dystonia, polycythemia, and hypermanganesemia caused by mutations in SLC30A10, a manganese transporter in man. Am J Hum Genet. (2012) 90:457–66. doi: 10.1016/j.ajhg.2012.01.018
78. Roth, J , Ponzoni, S , and Aschner, M . Manganese homeostasis and transport. Met Ions Life Sci. (2013) 12:169–201. doi: 10.1007/978-94-007-5561-1_6
79. Roth, JA , Feng, L , Dolan, KG , Lis, A , and Garrick, MD . Effect of the iron chelator desferrioxamine on manganese-induced toxicity of rat pheochromocytoma (PC12) cells. J Neurosci Res. (2002) 68:76–83. doi: 10.1002/jnr.10207
80. Roth, JA , Horbinski, C , Higgins, D , Lein, P , and Garrick, MD . Mechanisms of manganese-induced rat pheochromocytoma (PC12) cell death and cell differentiation. Neurotoxicology. (2002) 23:147–57. doi: 10.1016/s0161-813x(01)00077-8
81. Forero-Rodríguez, LJ , Josephs-Spaulding, J , Flor, S , Pinzón, A , and Kaleta, C . Parkinson’s disease and the metal-microbiome-gut-brain axis: A systems toxicology approach. Antioxidants. (2021) 11:71. doi: 10.3390/antiox11010071
82. Martinez-Finley, EJ , Chakraborty, S , Fretham, SJB , and Aschner, M . Cellular transport and homeostasis of essential and nonessential metals. Metallomics. (2012) 4:593–605. doi: 10.1039/c2mt00185c
83. Pyatha, S , Kim, H , Lee, D , and Kim, K . Association between heavy metal exposure and Parkinson’s disease: A review of the mechanisms related to oxidative stress. Antioxidants (Basel). (2022) 11:2467. doi: 10.3390/antiox11122467
84. Roth, JA , and Garrick, MD . Iron interactions and other biological reactions mediating the physiological and toxic actions of manganese. Biochem Pharmacol. (2003) 66:1–13. doi: 10.1016/s0006-2952(03)00145-x
85. Huang, E , Ong, WY , and Connor, JR . Distribution of divalent metal transporter-1 in the monkey basal ganglia. Neuroscience. (2004) 128:487–96. doi: 10.1016/j.neuroscience.2004.06.055
86. Erikson, KM , John, CE , Jones, SR , and Aschner, M . Manganese accumulation in striatum of mice exposed to toxic doses is dependent upon a functional dopamine transporter. Environ Toxicol Pharmacol. (2005) 20:390–4. doi: 10.1016/j.etap.2005.03.009
87. Milatovic, D , Yin, Z , Gupta, RC , Sidoryk, M , Albrecht, J , Aschner, JL, et al. Manganese induces oxidative impairment in cultured rat astrocytes. Toxicol Sci. (2007) 98:198–205. doi: 10.1093/toxsci/kfm095
88. Morcillo, P , Cordero, H , Ijomone, OM , Ayodele, A , Bornhorst, J , Gunther, L, et al. Defective mitochondrial dynamics underlie manganese-induced neurotoxicity. Mol Neurobiol. (2021) 58:3270–89. doi: 10.1007/s12035-021-02341-w
89. Chesi, A , Kilaru, A , Fang, X , Cooper, AA , and Gitler, AD . The role of the Parkinson’s disease gene PARK9 in essential cellular pathways and the manganese homeostasis network in yeast. PLoS One. (2012) 7:e34178. doi: 10.1371/journal.pone.0034178
90. Chen, P , Chakraborty, S , Peres, TV , Bowman, AB , and Aschner, M . Manganese-induced neurotoxicity: from C. elegans to humans. Toxicol Res. (2015) 4:191–202. doi: 10.1039/C4TX00127C
91. Hallgren, B , and Sourander, P . The effect of age on the non-haemin iron in the human brain. J Neurochem. (1958) 3:41–51. doi: 10.1111/j.1471-4159.1958.tb12607.x
92. Schenck, JF , and Zimmerman, EA . High-field magnetic resonance imaging of brain iron: birth of a biomarker? NMR Biomed. (2004) 17:433–45. doi: 10.1002/nbm.922
93. Griffiths, PD , and Crossman, AR . Autoradiography of transferrin receptors in the human brain. Neurosci Lett. (1996) 211:53–6. doi: 10.1016/0304-3940(96)12719-1
94. Burdo, JR , Menzies, SL , Simpson, IA , Garrick, LM , Garrick, MD , Dolan, KG, et al. Distribution of divalent metal transporter 1 and metal transport protein 1 in the normal and Belgrade rat. J Neurosci Res. (2001) 66:1198–207. doi: 10.1002/jnr.1256
95. Dexter, DT , Carayon, A , Javoy-Agid, F , Agid, Y , Wells, FR , Daniel, SE, et al. Alterations in the levels of iron, ferritin and other trace metals in Parkinson’s disease and other neurodegenerative diseases affecting the basal ganglia. Brain. (1991) 114:1953–75. doi: 10.1093/brain/114.4.1953
96. Gregory, A , and Hayflick, S . Neurodegeneration with brain Iron accumulation disorders overview In: MP Adam, GM Mirzaa, RA Pagon, SE Wallace, LJ Bean, and KW Gripp, et al., editors. GeneReviews®. Seattle, Seattle (WA): University of Washington (1993)
97. Schneider, SA , Dusek, P , Hardy, J , Westenberger, A , Jankovic, J , and Bhatia, KP . Genetics and pathophysiology of neurodegeneration with brain Iron accumulation (NBIA). Curr Neuropharmacol. (2013) 11:59–79. doi: 10.2174/157015913804999469
98. Gao, H , Sun, H , Yan, N , Zhao, P , Xu, H , Zheng, W, et al. ATP13A2 declines zinc-induced accumulation of α-Synuclein in a Parkinson’s disease model. Int J Mol Sci. (2022) 23:8035. doi: 10.3390/ijms23148035
99. Dexter, DT , Wells, FR , Lee, AJ , Agid, F , Agid, Y , Jenner, P, et al. Increased nigral iron content and alterations in other metal ions occurring in brain in Parkinson’s disease. J Neurochem. (1989) 52:1830–6. doi: 10.1111/j.1471-4159.1989.tb07264.x
100. Park, J-S , Davis, RL , and Sue, CM . Mitochondrial dysfunction in Parkinson’s disease: new mechanistic insights and therapeutic perspectives. Curr Neurol Neurosci Rep. (2018) 18:21. doi: 10.1007/s11910-018-0829-3
101. Sato, S , Koike, M , Funayama, M , Ezaki, J , Fukuda, T , Ueno, T, et al. Lysosomal storage of subunit c of mitochondrial ATP Synthase in brain-specific Atp13a2-Deficient mice. Am J Pathol. (2016) 186:3074–82. doi: 10.1016/j.ajpath.2016.08.006
102. Rayaprolu, S , Seven, YB , Howard, J , Duffy, C , Altshuler, M , Moloney, C, et al. Partial loss of ATP13A2 causes selective gliosis independent of robust lipofuscinosis. Mol Cell Neurosci. (2018) 92:17–26. doi: 10.1016/j.mcn.2018.05.009
103. Johnson, ME , Bergkvist, L , Stetzik, L , Steiner, JA , Meyerdirk, L , Schulz, E, et al. Heterozygous GBA D409V and ATP13a2 mutations do not exacerbate pathological α-synuclein spread in the prodromal preformed fibrils model in young mice. Neurobiol Dis. (2021) 159:105513. doi: 10.1016/j.nbd.2021.105513
104. Kett, LR , Stiller, B , Bernath, MM , Tasset, I , Blesa, J , Jackson-Lewis, V, et al. α-Synuclein-independent histopathological and motor deficits in mice lacking the endolysosomal parkinsonism protein Atp13a2. J Neurosci. (2015) 35:5724–42. doi: 10.1523/JNEUROSCI.0632-14.2015
105. Dirr, ER , Ekhator, OR , Blackwood, R , Holden, JG , Masliah, E , Schultheis, PJ, et al. Exacerbation of sensorimotor dysfunction in mice deficient in Atp13a2 and overexpressing human wildtype alpha-synuclein. Behav Brain Res. (2018) 343:41–9. doi: 10.1016/j.bbr.2018.01.029
106. Yu, J , Cui, M , Wang, W , Hu, K , and Cai, G . ATP13A2 knockout does not affect the infarct size in mice with acute ischemic stroke. CNS Neurosci Ther. (2012) 18:1027–9. doi: 10.1111/cns.12023
107. Grünewald, A , Arns, B , Seibler, P , Rakovic, A , Münchau, A , Ramirez, A, et al. ATP13A2 mutations impair mitochondrial function in fibroblasts from patients with Kufor-Rakeb syndrome. Neurobiol Aging. (2012) 33:1843.e1–7. doi: 10.1016/j.neurobiolaging.2011.12.035
108. Gusdon, AM , Zhu, J , Van Houten, B , and Chu, CT . ATP13A2 regulates mitochondrial bioenergetics through macroautophagy. Neurobiol Dis. (2012) 45:962–72. doi: 10.1016/j.nbd.2011.12.015
109. Martin, S , van Veen, S , Holemans, T , Demirsoy, S , van den Haute, C , Baekelandt, V, et al. Protection against mitochondrial and metal toxicity depends on functional lipid binding sites in ATP13A2. Parkinsons Dis. (2016) 2016:9531917. doi: 10.1155/2016/9531917
110. Alaimo, A , Gorojod, RM , Miglietta, EA , Villarreal, A , Ramos, AJ , and Kotler, ML . Manganese induces mitochondrial dynamics impairment and apoptotic cell death: A study in human Gli36 cells. Neuroscience Letters. (2013) 554:76–81. doi: 10.1016/j.neulet.2013.08.061
111. Genoud, S , Roberts, BR , Gunn, AP , Halliday, GM , Lewis, SJG , Ball, HJ, et al. Subcellular compartmentalisation of copper, iron, manganese, and zinc in the Parkinson’s disease brain. Metallomics. (2017) 9:1447–55. doi: 10.1039/c7mt00244k
112. Vrijsen, S , Besora-Casals, L , van Veen, S , Zielich, J , Van den Haute, C , Hamouda, NN, et al. ATP13A2-mediated endo-lysosomal polyamine export counters mitochondrial oxidative stress. Proc Natl Acad Sci U S A. (2020) 117:31198–207. doi: 10.1073/pnas.1922342117
113. Dehay, B , Ramirez, A , Martinez-Vicente, M , Perier, C , Canron, M-H , Doudnikoff, E, et al. Loss of P-type ATPase ATP13A2/PARK9 function induces general lysosomal deficiency and leads to Parkinson disease neurodegeneration. Proc Natl Acad Sci U S A. (2012) 109:9611–6. doi: 10.1073/pnas.1112368109
114. Hara, T , Nakamura, K , Matsui, M , Yamamoto, A , Nakahara, Y , Suzuki-Migishima, R, et al. Suppression of basal autophagy in neural cells causes neurodegenerative disease in mice. Nature. (2006) 441:885–9. doi: 10.1038/nature04724
115. Ramonet, D , Podhajska, A , Stafa, K , Sonnay, S , Trancikova, A , Tsika, E, et al. PARK9-associated ATP13A2 localizes to intracellular acidic vesicles and regulates cation homeostasis and neuronal integrity. Hum Mol Genet. (2012) 21:1725–43. doi: 10.1093/hmg/ddr606
116. Usenovic, M , Knight, AL , Ray, A , Wong, V , Brown, KR , Caldwell, GA, et al. Identification of novel ATP13A2 interactors and their role in α-synuclein misfolding and toxicity. Hum Mol Genet. (2012) 21:3785–94. doi: 10.1093/hmg/dds206
117. Usenovic, M , and Krainc, D . Lysosomal dysfunction in neurodegeneration: the role of ATP13A2/PARK9. Autophagy. (2012) 8:987–8. doi: 10.4161/auto.20256
118. Usenovic, M , Tresse, E , Mazzulli, JR , Taylor, JP , and Krainc, D . Deficiency of ATP13A2 leads to lysosomal dysfunction, α-synuclein accumulation, and neurotoxicity. J Neurosci. (2012) 32:4240–6. doi: 10.1523/JNEUROSCI.5575-11.2012
119. Lotharius, J , and Brundin, P . Pathogenesis of Parkinson’s disease: dopamine, vesicles and alpha-synuclein. Nat Rev Neurosci. (2002) 3:932–42. doi: 10.1038/nrn983
120. Spillantini, MG , Schmidt, ML , Lee, VM , Trojanowski, JQ , Jakes, R , and Goedert, M . Alpha-synuclein in Lewy bodies. Nature. (1997) 388:839–40. doi: 10.1038/42166
121. Mamais, A , Raja, M , Manzoni, C , Dihanich, S , Lees, A , Moore, D, et al. Divergent α-synuclein solubility and aggregation properties in G2019S LRRK2 Parkinson’s disease brains with Lewy body pathology compared to idiopathic cases. Neurobiol Dis. (2013) 58:183–90. doi: 10.1016/j.nbd.2013.05.017
122. Poulopoulos, M , Cortes, E , Vonsattel, J-PG , Fahn, S , Waters, C , Cote, LJ, et al. Clinical and pathological characteristics of LRRK2 G2019S patients with PD. J Mol Neurosci. (2012) 47:139–43. doi: 10.1007/s12031-011-9696-y
123. Tezuka, T , Taniguchi, D , Sano, M , Shimada, T , Oji, Y , Tsunemi, T, et al. Pathophysiological evaluation of the LRRK2 G2385R risk variant for Parkinson’s disease. NPJ Parkinsons Dis. (2022) 8:97. doi: 10.1038/s41531-022-00367-y
124. Zhao, Y , Perera, G , Takahashi-Fujigasaki, J , Mash, DC , Vonsattel, JPG , Uchino, A, et al. Reduced LRRK2 in association with retromer dysfunction in post-mortem brain tissue from LRRK2 mutation carriers. Brain. (2018) 141:486–95. doi: 10.1093/brain/awx344
Keywords: Parkinson’s disease, Kufor-Rakeb Syndrome, neuronal ceroid lipofuscinosis, manganese, iron, zinc, mitochondria, alpha-synuclein
Citation: Croucher KM and Fleming SM (2024) ATP13A2 (PARK9) and basal ganglia function. Front. Neurol. 14:1252400. doi: 10.3389/fneur.2023.1252400
Edited by:
Lorraine Clark, Columbia University, United StatesReviewed by:
Barbara Garavaglia, IRCCS Carlo Besta Neurological Institute Foundation, ItalyCopyright © 2024 Croucher and Fleming. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Sheila M. Fleming, c2ZsZW1pbmcxQG5lb21lZC5lZHU=
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.