
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Neurol., 29 August 2023
Sec. Neuromuscular Disorders and Peripheral Neuropathies
Volume 14 - 2023 | https://doi.org/10.3389/fneur.2023.1239725
 Amokelani C. Mahungu1,2
Amokelani C. Mahungu1,2 Elizabeth Steyn1
Elizabeth Steyn1 Niki Floudiotis1
Niki Floudiotis1 Lindsay A. Wilson3
Lindsay A. Wilson3 Jana Vandrovcova3Mary M. Reilly4
Jana Vandrovcova3Mary M. Reilly4 Christopher J. Record4Michael Benatar5
Christopher J. Record4Michael Benatar5 Gang Wu6
Gang Wu6 Sharika Raga2,7
Sharika Raga2,7 Jo M. Wilmshurst2,7Kireshnee Naidu1Michael Hanna3,8
Jo M. Wilmshurst2,7Kireshnee Naidu1Michael Hanna3,8 Melissa Nel1,2*†
Melissa Nel1,2*† Jeannine M. Heckmann1,2*†
Jeannine M. Heckmann1,2*†Introduction: Limited diagnostics are available for inherited neuromuscular diseases (NMD) in South Africa and (excluding muscle disease) are mainly aimed at the most frequent genes underlying genetic neuropathy (GN) and spastic ataxias in Europeans. In this study, we used next-generation sequencing to screen 61 probands with GN, hereditary spastic paraplegia (HSP), and spastic ataxias for a genetic diagnosis.
Methods: After identifying four GN probands with PMP22 duplication and one spastic ataxia proband with SCA1, the remaining probands underwent whole exome (n = 26) or genome sequencing (n = 30). The curation of coding/splice region variants using gene panels was guided by allele frequencies from internal African-ancestry control genomes (n = 537) and the Clinical Genome Resource's Sequence Variant Interpretation guidelines.
Results: Of 32 GN probands, 50% had African-genetic ancestry, and 44% were solved: PMP22 (n = 4); MFN2 (n = 3); one each of MORC2, ATP1A1, ADPRHL2, GJB1, GAN, MPZ, and ATM. Of 29 HSP probands (six with predominant ataxia), 66% had African-genetic ancestry, and 48% were solved: SPG11 (n = 3); KIF1A (n = 2); and one each of SPAST, ATL1, SPG7, PCYT2, PSEN1, ATXN1, ALDH18A1, CYP7B1, and RFT1. Structural variants in SPAST, SPG11, SPG7, MFN2, MPZ, KIF5A, and GJB1 were excluded by computational prediction and manual visualisation.
Discussion: In this preliminary cohort screening panel of disease genes using WES/WGS data, we solved ~50% of cases, which is similar to diagnostic yields reported for global cohorts. However, the mutational profile among South Africans with GN and HSP differs substantially from that in the Global North.
Little is known about the underlying genetic causes of neuromuscular disorders (NMD) among African subpopulations, especially among those from sub-Saharan Africa (1, 2). This may be due to several factors, the most important of which is limited resources. In a recent review of genetic evidence related to the causes of genetic neuropathies (GN), including hereditary motor sensory neuropathies [hereafter referred to as Charcot-Marie-Tooth disease (CMT)], hereditary spastic paraplegias (HSP), and spinal muscular atrophy (SMA) in Africans, we found reports of disease variants from only nine HSP and three CMT probands from six sub-Saharan African countries (Ivory Coast, Kenya, Mali, Nigeria, Somalia, and South Africa) (2). Six of these nine reports used next-generation sequencing to screen disease genes, while three performed single-gene testing.
In South Africa, limited genetic testing is available for non-muscle NMD and includes only PMP22 multiplex ligation-dependent probe amplification (MLPA) for GN and spinocerebellar ataxias 1, 2, 3, 6, 7, 12, 17 and Frataxin expansion screening for spastic ataxias. To address this unmet need, we have leveraged resources from two international consortia to drive the first genetic analysis of a South African cohort with inherited NMD using next-generation sequencing and virtual gene panel analysis. Here, we describe the genetic findings in patients presenting with genetic neuropathies, hereditary spastic paraplegias, and related disorders such as spastic ataxias.
Patients were eligible if they had a long history of a phenotype resembling a neuromuscular genetic disorder, either with a primary component of neuropathy or spastic paraplegia, or as part of a complex disorder with ataxia, and with or without a family history (3). The study cohort consisted of 61 probands, which were categorised using their main presenting clinical features as genetic neuropathies (GN) or hereditary spastic paraplegia (HSP), the latter group including those with associated ataxia and those with ataxia as the predominant clinical feature (designated spastic ataxia).
Almost all study participants were phenotyped and recruited opportunistically at Groote Schuur Hospital, University of Cape Town, South Africa (SA), between 2017 and 2022, whereas two HSP probands underwent prior phenotyping, and archived DNA was used. One HSP proband was assessed at the Red Cross Children's Hospital (SR, JMW).
The study was approved by the University of Cape Town Human Research Ethics Committee, and all patients gave informed consent to participate, including the sharing of case details and images.
Thirty probands were recruited for the International Centre for Genomic Medicine in Neuromuscular Diseases (ICGNMD) consortium and underwent whole exome sequencing (WES), and five HSP probands underwent whole genome sequencing (WGS) as part of the Clinical Research in ALS and Related Disorders for Therapeutic Development (CReATe) Consortium Phenotype Genotype Biomarker study (NCT02327845). An additional 25 probands underwent WGS at service providers in Cape Town, including the resequencing of five probands who had ICGNMD WES (Supplementary Table 1). One proband had an NMD gene panel test performed by Invitae.
Study participants with either demyelinating or intermediate conduction velocities (CVs) on electrophysiological testing underwent the MLPA (Salsa MLPA kit P405 CMT1, MRC-Holland, Amsterdam) for PMP22 copy number analysis by the SA National Health Laboratory Services (NHLS) prior to WES. Those with spastic ataxia had undergone repeat expansion testing for spinocerebellar ataxia (SCA) types 1, 2, 3, 6, 7, 12, 17 and Frataxin (NHLS). No further testing for large deletions or duplications was performed through MLPA analysis.
Whole genome sequencing data from various projects were aggregated to generate an internal African-ancestry database for allele frequency filtering (n = 537): 100 individuals from the AWI-Gen study (4) and 347 individuals from the H3Africa Genotyping Chip project (5), 39 individuals from the Simons Genome Diversity project (6), 24 individuals from the South African Human Genome program (7), and 27 individuals from other studies (1, 8). DNA was sequenced by various service providers using both PCR and PCR-free sequencing kits, where 100–150 bp read-length sequencing libraries were obtained and sequenced to a coverage of 30× (Supplementary Table 1).
DNA from whole blood was extracted for next-generation sequencing (NGS) using previously described methods (9). WES or WGS on NMD probands was performed using various library kits and sequencing platforms (Supplementary Table 1).
The NGS data were analysed at UCT using the Ilifu Cloud Computing Facility. WES and WGS reads were aligned to the NCBI GRCh38 reference genome with ALT contigs using alt-aware alignment, followed by joint variant calling according to the Genome Analysis Toolkit (GATK) best-practice guidelines for exomes and genomes (documented in https://github.com/grbot/varcall), which was performed separately for WES and WGS datasets.
Both the case and control joint called VCF files were uploaded to the NHGRI AnVIL (10) into various workspaces, which were then loaded into seqr, a rare disease analysis platform developed by the BROAD Institute (11). As part of the seqr's loading pipeline, multiallelic sites were split and then annotated with VEP version 95. High and moderate impact variants (missense, nonsense, essential and extended splice site, frameshift, and in frame) were assessed for both de novo/dominant and recessive inheritance patterns using gnomAD minor allele frequency (MAF) thresholds of 0.001 and 0.01, respectively. Further curation was restricted to variants occurring in genes represented in the following PanelApp Australia gene panels (12): hereditary neuropathy CMT isolated and complex (version 1.63), optic atrophy (version 1.17), hereditary spastic paraplegia superpanel (version 2.64), and ataxia superpanel (version 3.6). Panel selection was guided by clinical phenotype, although probands were deemed unsolved only when the genes from all four panels screened negative for candidate variants.
In the absence of ClinGen CMT and HSP variant curation expert panel guidelines, candidate variants were curated in the ClinGen Variant Curation Interface (VCI) (13) according to the American College of Medical Genetics (ACMG) guidelines (14) with the modifications recommended by the ClinGen Sequence Variant Interpretation working group (15) (detailed in Supplementary Table 2). In addition to the gnomAD database, the rarity of variants was confirmed by assessing their frequency in an internal dataset of African-ancestry control genomes [n = 537, 151 (28%) from South Africa; see Methods 2.2]. We used the Mastermind search engine (16) to perform comprehensive literature searches for candidate variants and contacted various genetic testing laboratories for additional information regarding their ClinVar variant submissions (e.g., phenotype information and internal laboratory variant frequency information), which proved helpful in classifying variants. The NGS read support for each variant was verified (read depth, genotype quality, and allelic balance), and selected variants were validated by Sanger sequencing. The interpretations for each classified variant (pathogenic, likely pathogenic, or uncertain significance) have been submitted to ClinVar (https://www.ncbi.nlm.nih.gov/clinvar/).
In unsolved cases (those without short disease-causing variants such as single nucleotide variants and small insertions/deletions in the various gene panels), the following genes were screened for structural variants using a combination of computational prediction by ClinSV (17) and manual visualisation using the Integrative Genomics Viewer (IGV) (18) and Samplot (19): SPAST, SPG11, SPG7, MFN2, MPZ, KIF5A, and GJB1. To control for batch effects related to the library preparation and sequencing instruments, exomes and genomes from the same sequencing batch were compared to identify split reads, discordant pairs, and coverage anomalies indicative of structural variation.
We utilised GraphPad Prism v9.5.1 to perform statistical analysis on the probands' ages of onset. We assessed the continuous variable (ages of onset) using the nonparametric Mann–Whitney U-test for skewed data by comparing the ages of onset between solved and unsolved cases as median values with their interquartile ranges (IQR).
Probands self-categorised themselves according to South Africa census population categories as South African Coloured (SAC, n = 25), Black African (n = 10), and European-genetic ancestry or White (n = 25). Furthermore, our cohort included one proband with Indian-genetic ancestry. The majority (57%) of our study cohort was of African-genetic ancestry (n = 35, combined B and SAC; Table 1). This information is relevant for genomic analysis as the African ancestry probands in our cohort have no ancestry-matched population data represented in the gnomAD database, which impacts the effectiveness of variant frequency filtering to identify rare variants in these individuals. The SAC ancestry group is an admixed population primarily residing in the Western Cape Province of South Africa whose genetic ancestry is mainly from Khoisan (hunter-gatherers) and Black African populations (60%) with fewer contributions from South-East Asians (20%) and Europeans (20%).
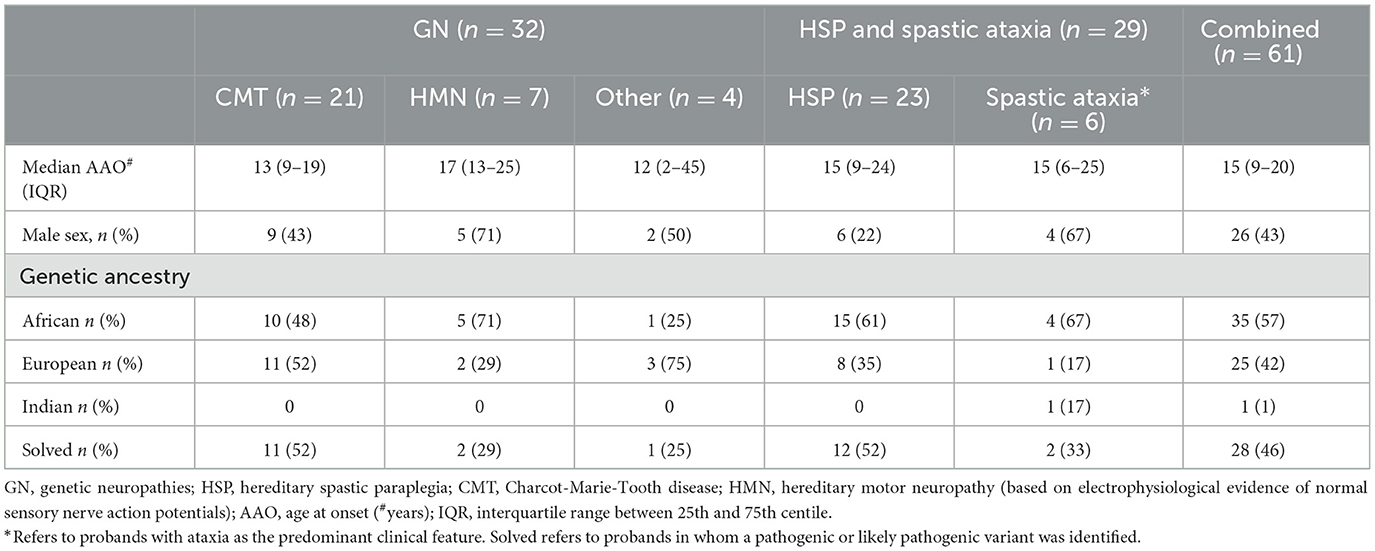
Table 1. Characteristics of South African patients with genetic neuropathies (GN), hereditary spastic paraplegia (HSP), and spastic ataxia.
The cohort comprised 61 individuals, of whom 32 were broadly categorised as GN and 29 as HSP, although in six of the 29 probands, ataxia was the predominant clinical feature. Among those with GN, most had CMT (21/32, 66%) with electrophysiological evidence of both sensory and motor impairment, and 7/32 (22%) had hereditary motor neuropathy (HMN) with normal sensory nerve action potentials (SNAPs) on electrophysiological testing (Table 1). Only four individuals had a demyelinating CMT1 phenotype with motor conduction velocities <38 m/s, and one individual had severe hypertrophic nerves and was previously recorded as having a “demyelinating picture”; she was too disabled at recruitment for electrophysiological testing. One individual had ataxia, neuropathy, and cognitive impairment without telangiectasia.
HSP categorisation referred to a predominant spastic phenotype involving the legs more than arms (n = 8), which may be complicated by other neurological signs (e.g., ataxia, neuropathy; n = 15), whereas six probands were classified as having spastic ataxia if the clinical picture was predominantly ataxia with additional spasticity (Table 1).
Apart from detecting four PMP22 gene duplications in GN (CMT1) and 1 ATXN1 expansion mutation among HSP probands, the remaining probands underwent WES and/or WGS for gene panel analysis. With the rigorous and objective application of ACMG evidence codes (described in methods 2.6 and Supplementary Table 2), we identified 10 pathogenic, eight likely pathogenic, and 11 variants of uncertain significance (VUS) in the overall cohort (Tables 2, 3), which were assessed for quality by inspection of IGV read pile-ups or validated using Sanger sequencing (Supplementary Figure 1). Probands were deemed to be solved where a pathogenic or likely pathogenic variant was identified, while a subset of patients harbouring a VUS was deemed to be probably solved (n = 6, Figure 1). In these cases, there was insufficient pathogenic evidence for a confident pathogenic classification by ACMG criteria, no conflicting benign evidence (apart from computational predictors), and the proband's phenotype was consistent with the genetic findings. Notably, five of these six “probably solved” cases harboured a novel variant not previously implicated in disease and absent from 537 African-ancestry control genomes. There was no difference in the proportion of solved cases between the GN and HSP groups (44 vs. 48%, p = 0.7; Figure 1).
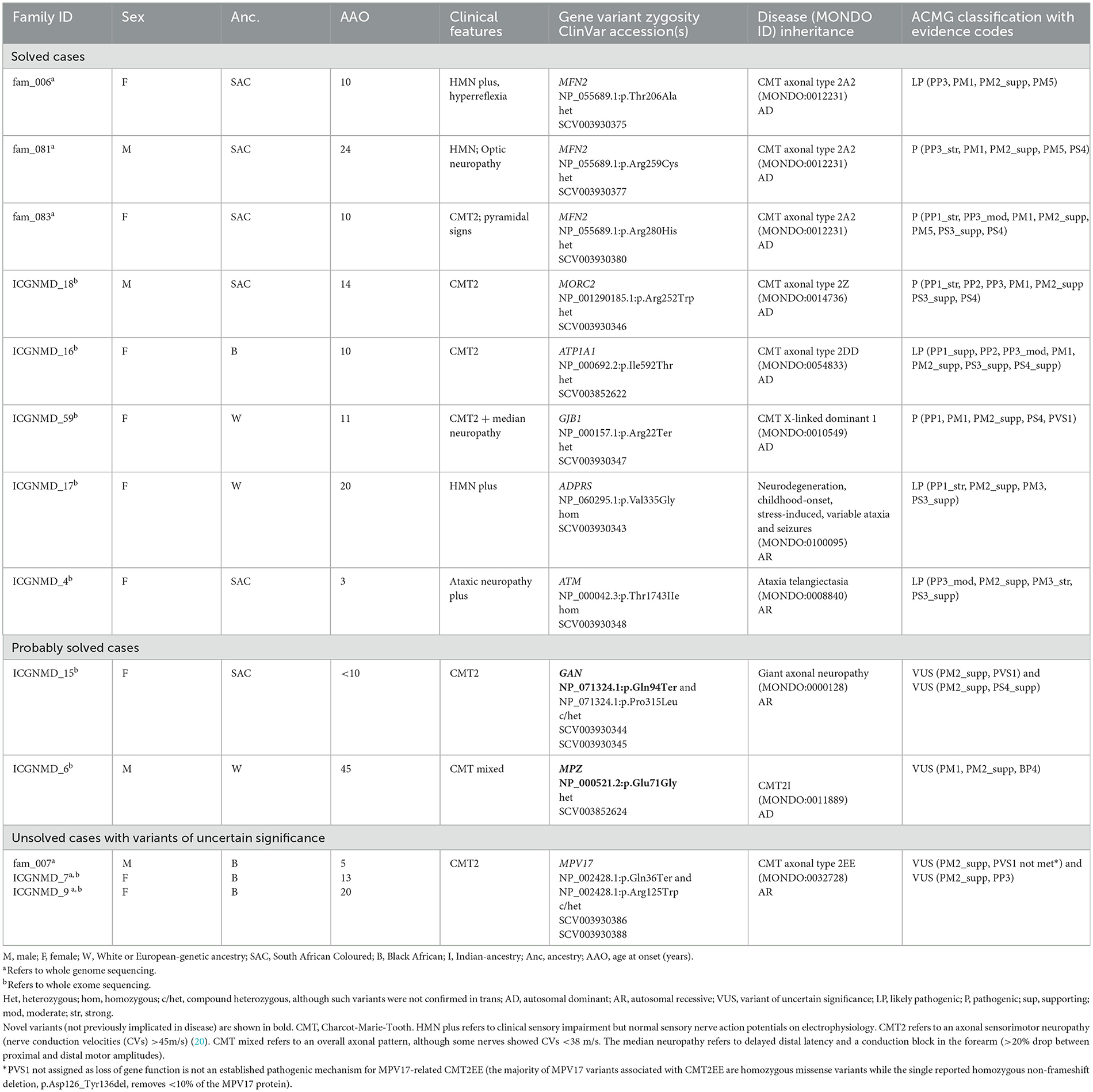
Table 2. Variants identified by whole exome and whole genome sequencing in probands with genetic neuropathies.
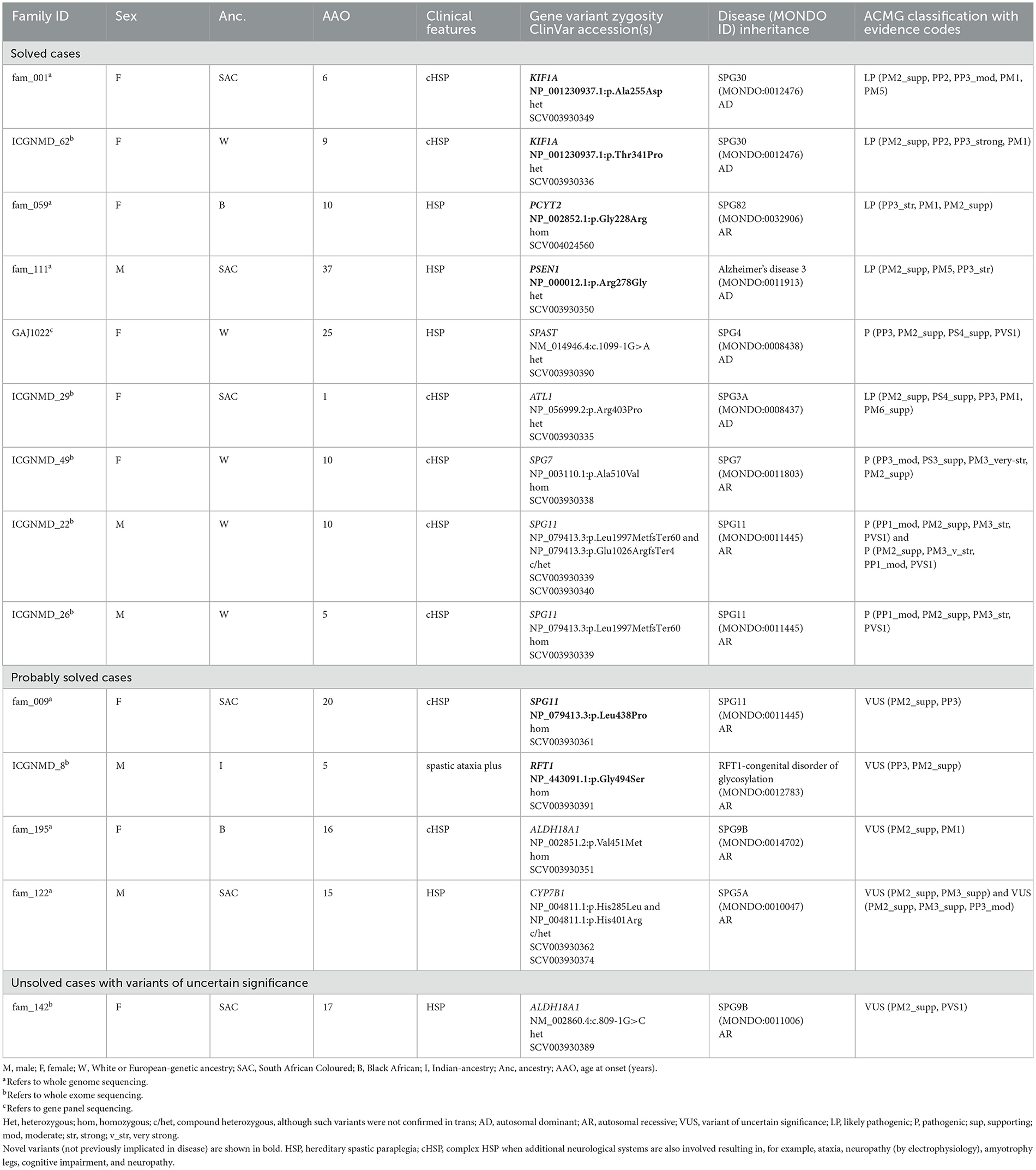
Table 3. Variants identified by whole exome and whole genome sequencing in probands with hereditary spastic paraplegia and/or spastic ataxia.
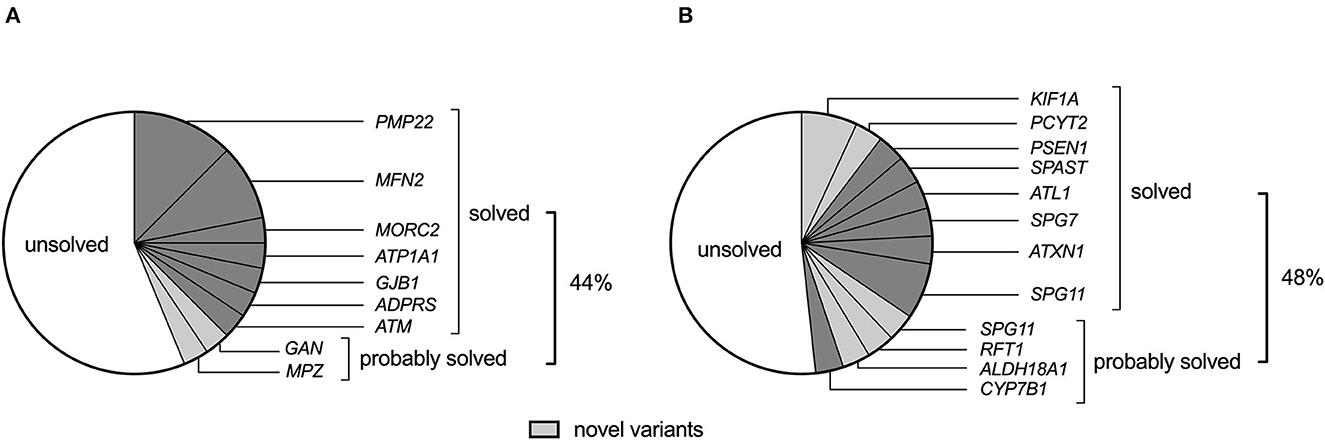
Figure 1. The genetic landscape of (A) genetic neuropathy (GN) and (B) hereditary spastic paraplegia (HSP) (including spastic ataxias) in a South African cohort of 61 probands. Novel variants (light grey) refer to variants that have not been previously associated with the disease.
The GN group comprised 31 probands, including 21 with CMT2 (axonal), seven with HMN, and four probands clinically classified as “other.” The latter group consisted of an individual from an autosomal dominant (AD) inherited optic neuropathy family, a proband with AD inherited gait difficulty since infancy, flat feet, and a small fibre neuropathy, a case with a small fibre neuropathy, and a proband with childhood-onset ataxic neuropathy (without telangiectasia). The proportion of solved GN cases did not differ by ancestry (African 8/16, 50% vs. European 6/16, 38%, p = 0.8). Furthermore, the ages at onset between solved (median 13, IQR 10–21) and unsolved (median 15, IQR 9–20) cases were similar (p = 0.7). Of the 14 solved GN cases, 11 (79%) had heterozygous pathogenic variants (compatible with AD inheritance), and 3 (21%) had homozygous/compound heterozygous variants [compatible with autosomal recessive (AR) inheritance].
The genes harbouring pathogenic mutations differed by ancestry; as expected, PMP22 duplications were a frequent cause of GN in the European-ancestry CMT cases (three of 11). These cases had typical CMT1 features, with symptoms of ankle instability as an adolescent or young adult, degrees of distal lower limb atrophy, and sensorimotor demyelinating neuropathy more prominent in the legs than arms. Three of the four probands with PMP22 duplications had European ancestry, and one had African (identified as SAC) ancestry. However, MFN2 mutations were the most common among those with African genetic ancestry (three of 10, Table 2).
We identified two pathogenic missense MFN2 variants (p.Arg280His, p.Arg259Cys) and a likely pathogenic MFN2 p.Thr206Ala variant in three probands with African ancestry and CMT2 (axonal neuropathy; Table 2). The proband carrying the MFN2 p.Arg280His variant presented at age 10 with CMT2 and spasticity in her legs. The MFN2 p.Arg259Cys variant was found in a CMT2 proband who developed optic neuropathy in adulthood.
The proband carrying the MFN2 p.Thr206Ala variant was diagnosed with childhood-onset CMT2 with prominent motor neuropathy and generalised hyperreflexia in the upper limbs and bulbar region. In her 4th decade, she required a wheelchair for mobility. At age 53, her mother was asymptomatic without clinical features, but her sister had moderately severe axonal sensorimotor neuropathy at age 31 (Figure 2A). Although rare cases of incomplete penetrance of MFN2 pathogenic variants have been reported (21), at least three cases have been reported with symptom onset after the age of 53 years (22, 23) and with the variability of age at symptom onset within a family (24). The MFN2 p.Thr206Ala variant occurs in the conserved dynamin-like GTPase domain together with other pathogenic variants, and the likely pathogenic MFN2 p.Thr206Ile variant has been observed in multiple CMT2 probands (25).
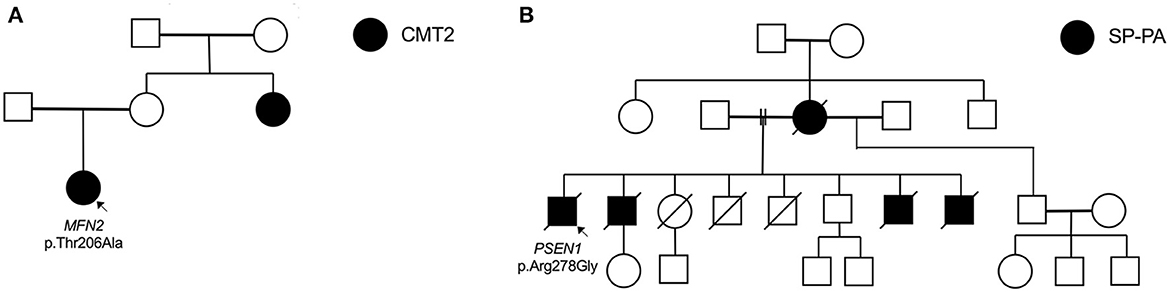
Figure 2. Selected pedigrees (genotyping only performed in probands). (A) Family of proband (fam_006) with heterozygous MFN2 mutation. (B) Family of proband (fam_111) with novel PSEN1 mutation presenting with spastic paraparesis (SP) followed by progressive aphasia (PA).
A pathogenic MORC2 p.Arg252Trp variant was found in a proband without any family history (Table 2). During adolescence, the proband had trouble running, and the phenotype has progressed to resemble that of CMT2Z with sensory neuropathy, severe distal amyotrophy with distal greater than proximal weakness, truncal weakness, and type II respiratory failure requiring non-invasive nocturnal ventilation. Similar to our case, most CMT2Z patients are wheelchair-bound between their 20s and 40s. He displayed clinical “post-exertional” fasciculations in the legs and an axonal sensorimotor neuropathic process with active denervation.
A likely pathogenic ATP1A1 p.Ile592Thr variant was detected in an African-ancestry proband with an axonal length-dependent severe sensorimotor neuropathy with prominent peroneal muscular atrophy (foot drop) manifesting in adolescence with clumsy gait (Table 2). This variant was previously described in a three-generational American family (26).
A known pathogenic variant in GJB1 p.Arg22Ter was identified in a woman with European ancestry who developed a clumsy gait at age 12 (Table 2). She was examined at 51 and again at 81. She has pes cavus, distal limb amyotrophy, Achilles' tendon contractures, moderately severe axonal length-dependent sensorimotor neuropathy (but median nerve conduction velocities were 37 m/s), and proximal hip girdle weakness. She has required foot orthoses and crutches for mobilisation since her seventh decade. The severity of her CMT is compatible with previously reported female carriers (27).
We identified a homozygous ADPRS p.Val335Gly variant in a European ancestry proband with severe distal HMN (Table 2). This rare variant was previously described in three unrelated probands (28, 29). This proband had severe axonal HMN with distal amyotrophy, proximal weakness, hyperreflexia (with extensor plantar responses at age 29), dysphagia, and increasing type II respiratory failure, from which she died in her fifth decade. Her brother manifested childhood-onset HMN and died in his third decade of complications secondary to a fall.
This patient with early childhood-onset ataxic neuropathy (without telangiectasia) had a homozygous likely pathogenic ATM p.Thr1743Ile variant (Table 2). This variant was previously described in trans with other pathogenic ATM variants in a few European ataxia-telangiectasia probands (30, 31).
We detected biallelic GAN variants (novel p.Gln94Ter and known p.Pro315Leu) in an African-ancestry proband with CMT (Table 2). Our case has an affected sibling (not examined) but non-consanguineous parents. Apart from moderately severe sensorimotor axonal neuropathy and pes cavus, which started in childhood and progressed to severe proximal weakness, which required mobilising with a wheelchair in her fifth decade, she also has diaphragmatic weakness and autonomic nervous system involvement with hyperhidrosis and constipation. GAN encodes gigaxonin and missense variants, resulting in a phenotypic continuum ranging from severe (with central nervous system neurodegeneration) to a milder phenotype, such as our case (32).
Mutations in the MPZ gene result in autosomal dominant CMT, presenting as a phenotype spectrum from early onset severe CMT1 to late-onset CMT2. We identified a two-generation European ancestry family presenting with symptoms in middle age with the novel MPZ p.Glu71Gly variant (Table 2). Although this variant has not been reported, a pathogenic null variant (MPZ p.Glu71Ter) at the same amino acid location was described in a CMT family with incomplete penetrance (33). Furthermore, a VUS (because there was insufficient evidence to support its pathogenicity) at the same amino acid position (p.Glu71Lys) was detected in a CMT1 case (34).
Eighteen GN cases (58%) remained without a genetic diagnosis (Figure 1), which included a screening of the SPAST, SPG11, SPG7, MFN2, MPZ, KIF5A, and GJB1 genes for structural variants. Half of the unsolved cases were CMT (n = 10), of which nine were axonal neuropathies, and one had severe hypertrophic neuropathy. Five of the unsolved cases had HMN (ages at onset between 9 and 32 years and normal sensory nerve action potentials on electrophysiology together with evidence of motor axonopathy), and three had other neuropathies, including AD optic neuropathy, small fibre neuropathy, and AD infantile gait difficulty with pes planus and numbness (normal electrophysiology).
Of the nine unsolved probands with axonal neuropathies, five were Black African patients from the Western and Eastern Cape regions of South Africa who speak isiXhosa, with an average age of disease onset of 13 years (range 7–20). In all five of these CMT2 probands, we identified the heterozygous p.Gln36Ter MPV17 variant (reported to be pathogenic for Navajo neurohepatopathy in homozygosity, ClinVar SCV000282035.1), and three of these cases also had an additional missense MPV17 p.Arg125Trp variant (not confirmed in trans). Biallelic homozygous MPV17 variants have been reported to result in juvenile-onset isolated peripheral sensorimotor neuropathy (CMT2EE) without hepatocerebral involvement, as occurs in autosomal recessive (AR) mitochondrial DNA depletion syndrome 6 (Navajo neurohepatopathy). The MPV17 p.Gln36Ter variant has a minor allele frequency (MAF) >0.001 in two independent SAB control samples from Cape Town, South Africa: 11/1,500 alleles in newborns (MAF 0.007, one in 68 carrier frequency) (35) and 6/400 alleles in another study (MAF 0.015, one in 33 carrier frequency) (36) (personal communication L. Majara) and has not been found in Black individuals from other parts of South Africa (internal database). The p.Arg125Trp variant is reported in gnomAD v2.1.1 (MAF 0.0002) with the highest frequency in the African/African-American subpopulation, although our internal control database has an even higher frequency (MAF 0.005). In our internal control database, we have not identified the p.Gln36Ter and p.Arg125Trp variants in the same individual. However, only 58/537 (11%) control samples were ancestry-matched to the small geographical cluster of p.Gln36Ter carriers as determined by ancestry principal component analysis (data not shown). Further pathogenic evidence is required to assert that compound heterozygous MPV17 p.Gln36Ter/p.Arg125Trp VUS variants explain a localised cluster of CMT2 in South Africa, particularly since a high regional prevalence of CMT2 has not been reported, the clinical phenotype among our three reported cases is dissimilar (Supplementary Table 3), there is a lack of ancestry-matched control data to determine the frequency of p.Gln36Ter/p.Arg125Trp co-occurrence in controls and the strength of the evidence supporting the MPV17-CMT2EE gene-disease dyad have not yet been evaluated by a ClinGen gene curation expert panel.
Of the 29 cases with HSP or spastic ataxias, 48% were solved. The median age of onset was 15 years (IQR 9–24) for HSP and 15 years (IQR 6–25) for spastic ataxias (Table 1). The proportion of solved HSP cases did not differ by ancestry (African 9/20, 45% vs. European 5/9, 56%, p = 0.5). Furthermore, the ages at onset between resolved (median 13, IQR 8–24) and unresolved (median 16, IQR 9–28) cases were similar (p = 0.3). In an African ancestry case with spastic ataxia and without a family history, a heterozygous ATXN1 trinucleotide expansion was detected to designate SCA1. Of the 14 solved HSP cases, six (43%) had heterozygous pathogenic variants (compatible with AD inheritance), and eight (57%) had homozygous/compound heterozygous variants (compatible with AR inheritance).
Among the African-ancestry probands, variants in rare HSP genes such as PCYT2 and PSEN1 were identified, whereas the European-ancestry probands harboured variants in common HSP genes: SPG11 (n = 2/5), SPAST (n = 1/5), SPG7 (n = 1/5) and a novel variant in KIF1A (n = 1/5; Table 3).
We detected two AD-HSP30 probands in our cohort harbouring novel variants in the KIF1A mutational hotspot region in the motor domain (amino acids 1–361) (37) (Table 3). A likely pathogenic KIF1A p.Ala255Asp variant was identified in a mother and daughter with a complicated HSP phenotype with symptom onset in childhood and additional cerebellar dysfunction (gait ataxia, horizontal and downbeat nystagmus), mild intellectual impairment, optic neuropathy, and a sensorimotor axonal neuropathy with acral leg ulceration. These are common features in HSP30 (37, 38). A second likely pathogenic KIF1A p.Thr341Pro variant was detected in a proband whose clinical features fit those of HSP30.
We detected a novel homozygous PCYT2 p.Gly246Arg variant in a Black individual with complex HSP, optic neuropathy, and length-dependent sensorimotor neuropathy (Table 3). She manifested gait unsteadiness in childhood, progressive optic neuropathy since adolescence and spastic dysarthria and seizures in adulthood. She was completely blind in middle age, which underscores the prominent role of PCYT2 in ocular homeostasis via the CDP-ethanolamine pathway (39).
A novel PSEN1 p.Arg278Gly pathogenic variant was found in an AD African ancestry family (mother and four sons) presenting with progressive spastic paraparesis starting in their fourth decade and followed within months by progressive non-fluent aphasia (and later frontal dysexecutive dysfunction; Table 3, Figure 2B). The condition progressed to akinetic mutism and death over 8 to 10 years from symptom onset.
Although a common SPAST c.1099-1G>A variant was found in a European-ancestry proband (Table 3), which accounts for 60%−80% of AD-HSP cases in European and Asian cohorts (40, 41), we did not find SPAST variants (including structural variants) in our African-ancestry patients (n = 8 with HSP) even after visual inspection of WGS data on IGV and by using ClinSV.
We detected a likely pathogenic ATL1 p.Arg403Pro variant in the exon 12 mutational hotspot (42) in an individual with early childhood-onset HSP (Table 3). This is the second most common AD-HSP in the global north/European cohorts (43).
The common homozygous SPG7 p.Ala510Val pathogenic variant was detected in a European-ancestry individual (Table 3). Although this variant has a high population frequency on gnomAD (gnomAD v2 exomes 0.48%/gnomAD v3 genomes 0.61% in the European non-Finnish population), it is found to segregate with disease in multiple families and is described as a common founder mutation in SPG7 cases of British ancestry (44).
SPG11 is the most common subtype of autosomal recessive HSP with cognitive impairment (45). Two pathogenic SPG11 mutations, homozygous SPG11 p.Leu1997MetfsTer60, and compound heterozygous variants, p.Leu1997MetfsTer60 and p.Glu1026ArgfsTer4, were detected in two probands (46, 47) (Table 3).
A novel homozygous missense SPG11 p.Leu438Pro variant was curated as a good disease candidate in an African ancestry (SAC) proband (Table 3). Although loss of function variants have been implicated in SPG11 disease, there are reports in the literature of SPG11 probands with homozygous missense SPG11 variants (48, 49).
A homozygous missense variant p.Gly494Ser in exon 13 of the RFT1 gene was classified as a VUS for RFT1-congenital disorder of glycosylation (CDG) with autosomal recessive inheritance (Table 3). The proband is of Indian ancestry, and her parents were consanguineous. RFT1-CDG was first recognised as a severe congenital disorder of N-linked glycosylation characterised by developmental delay, failure to thrive, myoclonic encephalopathy, seizures, and sensorineural hearing loss. However, a milder variant CDG phenotype was reported, resembling our patient's phenotype (50, 51). Our patient had a childhood-onset intellectual disability, adult-onset cerebellar ataxia, spasticity, mild sensorineural hearing impairment, inverted nipples, motor > sensory length-dependent axonal polyneuropathy, and spontaneous painful ulcers developing in the distal leg > forearm (Figure 3). Her brain MR imaging at age 35 showed periventricular white matter signal hyperintensities on FLAIR imaging and cerebellar atrophy. RFT1-CDG probands are known to suffer from clotting abnormalities. Interestingly, despite a negative clotting dysfunction screen, this proband's spontaneous, painful, and ulcerating skin lesions in her extremities improved substantially on oral anticoagulant therapy.
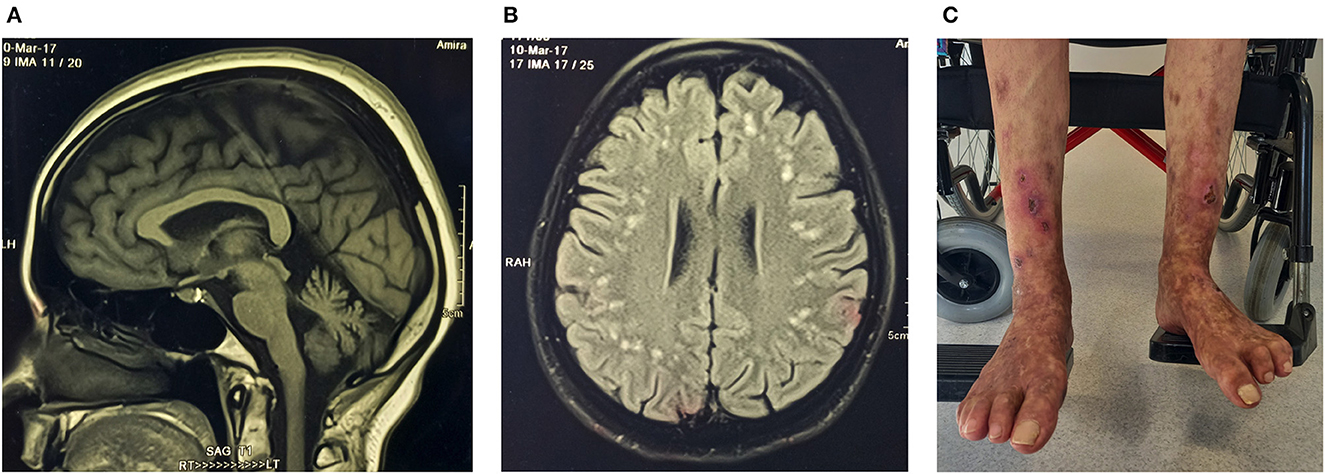
Figure 3. Proband (ICGNMD_8) with a milder phenotype of RFT1-congenital disorder of glycosylation. Brain imaging at age 35 years. (A) Sagittal T1-weighted MRI-brain scan showing cerebellar atrophy. (B) An axial brain FLAIR MRI image showing widespread discrete high signal intensity lesions in the subcortical white matter. Apart from the RFT1-disorders known to have coagulation factor abnormalities, the patient had no other cardiovascular risk factors to account for these white matter abnormalities of presumed vascular origin. (C) Spontaneous painful lesions with subsequent ulceration, which occurred in the distal extremities (before anticoagulation). Since oral anticoagulants were started for possible clotting abnormalities as a cause, the lesions have disappeared in the forearms and are substantially less in the legs (age 49).
ALDH18A1 is known to result in SPG9A (autosomal dominant) and SPG9B (autosomal recessive) HSP (52). A homozygous missense ALDH18A1 p.Val451Met variant was found in an African-ancestry proband presenting with gait difficulties in adolescence (Table 3). At age 36, the patient manifested, in addition to severe spasticity of the legs, distal amyotrophy, and pes cavus, sensory neuropathy, spasticity of the bulbar region, as well as mild cerebellar signs with nystagmus and past-pointing in the arms. This is the first report of SPG9B in sub-Saharan Africa, with previous reports in European and Japanese populations (53, 54). The majority of these missense variants occur in the L-glutamyl-5-phosphate reductase (G5PR) domain, where the p.Val451Met variant is also located.
Biallelic CYP7B1 p.His401Arg and p.His285Leu variants were detected in a proband with HSP symptom onset in adolescence and accompanied by a substantial proprioceptive loss in his legs (Table 3). The p.His401Arg variant was previously described in a sporadic Taiwanese SPG5A case with another pathogenic CYP7B1 variant (p.Arg112Ter) (55). The second CYP7B1 p.His285Leu variant was previously detected in a homozygous state in an Italian SPG5A proband (56). Biallelic variants in the CYP7B1 gene account for <10% of all AR HSP in European and Asian populations (57).
Among the HSP cases, 52% remained without a genetic diagnosis, irrespective of genetic ancestry, including 11 with HSP and four with spastic ataxia (Figure 1). Searching for structural variants in the WES/WGS data of unsolved cases did not identify any additional disease candidates in the HSP and spastic ataxia genes.
We curated an ALDH18A1 c.809-1G>C splice variant in a proband with gait difficulties since adolescence as a variant of uncertain significance for SPG9B (Table 3) (52). Although this variant is rare in gnomAD, it has a MAF of 0.032% (32/100,000 alleles) in an internal database from the Netherlands (personal communication, E.-J. Kamsteeg). It has been identified in at least three probands by clinical testing (ClinVar SCV001929264.1, SCV001952732.1, and SCV001970262.1) and is classified as likely pathogenic for AR HSP (in each proband, the variant was confirmed in trans with another ALDH18A1 VUS and the parents were unaffected heterozygous carriers). In our proband's WGS data, we have not identified a second candidate ALDH18A1 single nucleotide variant or structural variant consistent with this variant's reported role in SPG9B with autosomal recessive inheritance.
This report describes the mutational profile of inherited neuropathies, spastic paraplegias, and spastic ataxias in a small but first cohort from South Africa comprising 61 probands. Among the European-ancestry cases, despite the small sample size, the profile was similar to that expected for European probands for both the genetic neuropathies and the HSP cases. In our cohort, among the GN group, the notable difference was the lack of CMT1A-PMP22 cases among Black South Africans, whereas this gene accounts for 37% of European cohorts (58). In the HSP group, the difference was the lack of SPG4, SPG7, and SPG11 variants in African-ancestry probands, representing the common AD and AR genotypes in Europeans and Asians (59, 60). The prevalence of HSP genetic subtypes varies by geography, reflecting isolation effects (increased risk of AD-HSP in Norwegian and Sardinian populations attributed to founder effects) (40) and consanguinity (increased risk of AR-HSP in North Africa and Western Asia) (2, 40), neither of which are characteristic of African ancestry subpopulations in South Africa. While referral bias is unlikely to explain this different mutational profile, these findings require validation in a larger sample.
In this cohort, the solved rate among the genetic neuropathies (44%) and the HSP and spastic ataxia group (48%) was similar. The proportion of HSP/spastic ataxia probands achieving a genetic diagnosis was similar to a larger European HSP cohort (49%) (61). However, in this study, the solved rate of GN probands was lower than a larger European GN cohort (~60%), likely due to fewer CMT1 cases, which comprised 55% of that cohort (58).
While the study's aims did not include investigating novel disease genes, 8/28 (29%) resolved probands harbouring a novel disease-causing variant in a known disease gene. This highlights the importance of studying GN and HSP cohorts of diverse and underrepresented ancestries and sharing their pathogenic variants with the wider scientific community via databases such as ClinVar to enhance genetic diagnosis of these conditions worldwide.
The use of population descriptors in this study has only been employed insofar as to guide the analysis of the genomic data (62). African genomes are highly diverse, and African-ancestry individuals from South Africa are not represented in the gnomAD database, the cornerstone of population frequency information when curating variants for rare diseases. Necessarily, our study leveraged allele frequency information from an internal sub-Saharan African ancestry control dataset (n = 1,074 alleles) to guide the curation of candidate variants absent from gnomAD. While this is a substantial continental sample of non-gnomAD ancestry representation, the high levels of diversity and population substructure in Africa will likely require even greater numbers of ancestry-matched controls by country subregion to derive accurate allele frequency information for African subpopulations. This is illustrated by the high regional frequency (MAF > 1%) of the MPV17 p.Gln36Ter variant in South Africa, despite its absence from gnomAD. As genomic research for rare diseases expands in Africa, acknowledging the ongoing importance of case-control study designs and encouraging concerted efforts to share aggregate African frequency data (63) are necessary to avoid misclassification of variant pathogenicity (64). Indeed, the value of studying African genomes to drive the equitable benefit of genomic medicine for rare diseases will not be realised unless African genomic data is shared with the scientific community. This will inform current efforts to evaluate disease-gene validity and define pathogenic disease variants, ultimately guiding gene-based therapy development and clinical trial eligibility.
In this report, we provided confirmatory evidence of phenotype expansion for three disease entities: autosomal recessive CONDSIAS manifesting as hereditary motor neuropathy with hyperreflexia due to a homozygous ADPRS variant; autosomal recessive giant axonal neuropathy presenting as a non-classical early-onset motor and sensory neuropathy due to biallelic GAN variants; and autosomal recessive congenital disorder of glycosylation due to a homozygous RFT1 variant presenting without seizures. These cases were solved through close collaboration between the referring clinician and genomic analysts and highlighted the limitations of using human phenotype ontology terms to direct clinical interpretation of genomic data for genetic diagnosis. Indeed, even beyond phenotype expansion within specific disease entities, as demonstrated by the aforementioned examples, it is increasingly recognised that genetic neuropathies and hereditary spastic paraplegias (grouped separately for this report) may represent a disease spectrum due to their overlapping clinical and genetic features. Therefore, a broad approach to the genetic investigation of these disorders is warranted, while prompt sharing of variant interpretations from Africa will ensure that the ClinGen gene curation expert panel's efforts to assess the evidence of gene-disease relationships for this group of disorders consider global genetic diversity. A case in point is our observation of compound heterozygous MPV17 variants in SA Black probands with CMT2. These case reports, which have been deposited in ClinVar, will now be considered when the genetic evidence for MPV17 variants and CMT2EE is objectively reviewed.
While genetic testing to diagnose NMDs is now the standard of care in developed countries to achieve a definitive molecular diagnosis, the South African probands described in this report were able to access exome and genome sequencing only through their participation in research studies. Although the sequencing data was generated largely by international consortia outside of South Africa, it is noteworthy that the raw data for this report were analysed and interpreted locally as a critical exercise in capacity building, which is essential for the future implementation of local NGS-based genetic testing strategies.
The proportion of our cohort that remains without a genetic diagnosis is similar to other population cohorts, suggesting a common biological, genetic mechanism underlying this missing heritability, such as noncoding genetic variants, complex structural changes, or epigenetic changes (61). Such classes of genetic variation are not adequately examined by short-read approaches, and long-read whole genome sequencing is expected to drive disease gene discovery for these disorders in the future.
This first cohort analysis from a South African neurology clinic using whole exome and whole genome sequencing data suggests that the mutational profile of both genetic neuropathies and hereditary spastic paraplegias (and spastic ataxias) is different in subjects with African ancestry compared to those with European ancestry. This highlights the importance of including genetically diverse cohorts in research programs with the ultimate goal of therapy development.
The datasets presented in this study have been deposited in the ClinVar repository with the following accession numbers: SCV003930375, SCV003930377, SCV003930380, SCV003930346, SCV003852622, SCV003930347, SCV003930343, SCV003930348, SCV003930344, SCV003930345, SCV003852624, SCV003930386, SCV003930388, SCV003930349, SCV003930336, SCV003930350, SCV003930390, SCV003930335, SCV003930338, SCV003930339, SCV003930340, SCV003930361, SCV003930391, SCV003930351, SCV003930362, SCV003930374, SCV003930389, and SCV004024560.
The studies involving humans were approved by University of Cape Town Human Research Ethics Committee. The studies were conducted in accordance with the local legislation and institutional requirements. Written informed consent for participation in this study was provided by the participants' legal guardians/next of kin. Written informed consent was obtained from the individual(s), and minor(s)' legal guardian/next of kin, for the publication of any potentially identifiable images or data included in this article.
AM analysed the NGS data and wrote the first draft of the manuscript. LW, NF, and KN provided organisational support. JV and GW provided bioinformatics support. CR and MR assisted with data interpretation for ICGNMD cases. MB leads CReATe and assists with data interpretation for CReATe cases. JH, ES, SR, and JW assisted with patient recruitment and clinical phenotyping. JW and JH are ICGNMD PIs and recruited cases from South Africa. MH leads the ICGNMD. MN and JH contributed to the conception, design, and supervision of the study (genomics and clinical phenotyping, respectively). All authors contributed to the manuscript's revision, read, and approved the submitted version.
AM received funding from the UCT Neurology Research Group, the National Research Foundation of South Africa (NRF), and the University of Cape Town. This research was funded in whole or in part by the Wellcome Trust [226519/Z/22/Z] (MN). For the purpose of open access, the author has applied a CC BY public copyright license to any Author Accepted Manuscript version arising from this submission. The costs incurred for storing VCF files in the NHGRI AnVIL were supported by the Google Cloud Research Credits program with the award GCP19980904 (MN). This publication was made possible (in part) by grants from the South African Medical Research Council (SAMRC), the Gabriel Grant Foundation, and the Carnegie Corporation of New York (MN), and the statements made and views expressed are solely the responsibility of the authors. This work was supported by an MRC strategic award to establish an International Centre for Genomic Medicine in Neuromuscular Diseases (ICGNMD) (MR/S005021/1) and the National Institutes of Neurological Diseases and Stroke and the Office of Rare Diseases (U54NS065712, 1UOINS109403-01, and R21TROO3034) (CR and MR). KN was supported by a fellowship from The Guarantors of Brain (UK Charity 1197319), and SR was supported by a fellowship from the ICGNMD.
The authors acknowledge the support of the International Centre for Genomic Medicine in Neuromuscular Diseases (ICGNMD) consortium, which funded the whole exome sequencing of 30 NMD cases (MR/S005021/1). They thank the Clinical Research in ALS and related disorders for Therapeutic Development (CReATe) consortium (U54-NS092091), which funded the whole genome sequencing of five HSP cases, and Joanne Wuu for organizational support. The CReATe Consortium is part of the Rare Diseases Clinical Research Network (RDCRN), an initiative of the Office of Rare Diseases Research (ORDR) and the National Center for Advancing Translational Sciences (NCATS). This consortium is funded through a collaboration between NCATS and the NINDS. The SAHGP dataset was generated by the Southern African Human Genome Programme, a national initiative funded by the Department of Science and Technology of South Africa. This study makes use of data generated by H3Africa. A full list of the investigators who contributed to the data generation is available at https://h3africa.org. The funding for this project comes through the Human Heredity and Health in Africa (H3Africa) Initiative, which is funded by the National Institutes of Health and the Wellcome Trust through the SFA Foundation. The authors acknowledge the use of the Ilifu cloud computing facility (https://www.ilifu.ac.za), a partnership between the University of Cape Town (UCT), the University of the Western Cape, the University of Stellenbosch, Sol Plaatje University, the Cape Peninsula University of Technology, and the South African Radio Astronomy Observatory. The Ilifu facility is supported by contributions from the Inter-University Institute for Data Intensive Astronomy (IDIA—a partnership between UCT, the University of Pretoria, and the University of the Western Cape), the Computational Biology Division at UCT, and the Data Intensive Research Initiative of South Africa (DIRISA).
MR, CR, JV, LW, and MH received a proportion of their salaries and their employer has received payments to support work on the project “An MRC strategic award to establish an International Centre for Genomic Medicine in Neuromuscular Diseases (ICGNMD) MR/S005021/1.” The MRC and UKRI had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
The authors acknowledge the following individuals who assisted in the classification of sequence variants: The Clinical Consult Team at Invitae, J Hoffman (on behalf of GeneDx), E-J Kamsteeg (Radboud University Medical Center), and L Majara, K Sparks, and I Dixon (UCT Neuroscience Institute).
MR consults for Akcea, Alnylam, Applied Therapeutics, Augustine Therapeutics and Inflectis. MR is an Eidos Therapeutics steering committee member. MH is chair of Muscular Dystrophy UK (MDUK). MB serves on the ALS Association Board of Trustees and holds grants from NIH (R01-NS105479, U01-NS107027, and U54-NS092091) and the Muscular Dystrophy Association (645863), intellectual property from the University of Miami licensed to Biogen (IP-142A), a provisional patent related to determining the onset of amyotrophic lateral sclerosis and consults for Alector, Alexion, Annexon, Arrowhead, Biogen, Cartesian, Denali, Eli Lilly, Horizon, Immunovant, Janssen, Novartis, Roche, Sanofi, Takeda, UCB and UniQure.
The remaining authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fneur.2023.1239725/full#supplementary-material
1. Nel M, Mahungu AC, Monnakgotla N, Botha GR, Mulder NJ, Wu G, et al. Revealing the mutational spectrum in Southern Africans with amyotrophic lateral sclerosis. Neurol Genet. (2022) 8:e654. doi: 10.1212/NXG.0000000000000654
2. Mahungu AC, Monnakgotla N, Nel M, Heckmann JM. A review of the genetic spectrum of hereditary spastic paraplegias, inherited neuropathies and spinal muscular atrophies in Africans. Orphanet J Rare Dis. (2022) 17:1–12. doi: 10.1186/s13023-022-02280-2
3. Rossor AM, Evans MRB, Reilly MM. A practical approach to the genetic neuropathies. Pract Neurol. (2015) 15:187–98. doi: 10.1136/practneurol-2015-001095
4. Sengupta D, Choudhury A, Fortes-Lima C, Aron S, Whitelaw G, Bostoen K, et al. Genetic substructure and complex demographic history of South African Bantu speakers. Nat Commun. (2021) 12:2080. doi: 10.1038/s41467-021-22207-y
5. Choudhury A, Aron S, Botigué LR, Sengupta D, Botha G, Bensellak T, et al. High-depth African genomes inform human migration and health. Nature. (2020) 586:741–8. doi: 10.1038/s41586-020-2859-7
6. Mallick S, Li H, Lipson M, Mathieson I, Gymrek M, Racimo F, et al. The Simons Genome Diversity Project: 300 genomes from 142 diverse populations. Nature. (2016) 538:201–6. doi: 10.1038/nature18964
7. Choudhury A, Ramsay M, Hazelhurst S, Aron S, Bardien S, Botha G, et al. Whole-genome sequencing for an enhanced understanding of genetic variation among South Africans. Nat Commun. (2017) 8:1–12. doi: 10.1038/s41467-017-00663-9
8. Nel M, Mulder N, Europa TA, Heckmann JM. Using whole genome sequencing in an African subphenotype of myasthenia gravis to generate a pathogenetic hypothesis. Front Genet. (2019) 10:136. doi: 10.3389/fgene.2019.00136
9. Nel M, Agenbag GM, Henning F, Cross HM, Esterhuizen A, Heckmann JM. C9orf72 repeat expansions in South Africans with amyotrophic lateral sclerosis. J Neurol Sci. (2019) 401:51–4. doi: 10.1016/j.jns.2019.04.026
10. Schatz MC, Philippakis AA, Afgan E, Banks E, Carey VJ, Carroll RJ, et al. Inverting the model of genomics data sharing with the NHGRI genomic data science analysis, visualization, and informatics lab-space. Cell Genom. (2022) 2:100085. doi: 10.1016/j.xgen.2021.100085
11. Pais LS, Snow H, Weisburd B, Zhang S, Baxter SM, DiTroia S, et al. seqr: a web-based analysis and collaboration tool for rare disease genomics. Hum Mutat. (2022) 43:698–707. doi: 10.1002/humu.24366
12. Martin AR, Williams E, Foulger RE, Leigh S, Daugherty LC, Niblock O, et al. PanelApp crowdsources expert knowledge to establish consensus diagnostic gene panels. Nat Genet. (2019) 51:1560–5. doi: 10.1038/s41588-019-0528-2
13. Preston CG, Wright MW, Madhavrao R, Harrison SM, Goldstein JL, Luo X, et al. ClinGen variant curation interface: a variant classification platform for the application of evidence criteria from ACMG/AMP guidelines. Genome Med. (2022) 14:1–12. doi: 10.1186/s13073-021-01004-8
14. Richards S, Aziz N, Bale S, Bick D, Das S, Gastier-Foster J, et al. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet Med. (2015) 17:405–24. doi: 10.1038/gim.2015.30
15. Harrison SM, Biesecker LG, Rehm HL. Overview of specifications to the ACMG/AMP variant interpretation guidelines. Curr Protoc Hum Genet. (2019) 103:e93. doi: 10.1002/cphg.93
16. Chunn LM, Nefcy DC, Scouten RW, Tarpey RP, Chauhan G, Lim MS, et al. Mastermind: a comprehensive genomic association search engine for empirical evidence curation and genetic variant interpretation. Front Genet. (2020) 11:577152. doi: 10.3389/fgene.2020.577152
17. Minoche AE, Lundie B, Peters GB, Ohnesorg T, Pinese M, Thomas DM, et al. ClinSV: clinical grade structural and copy number variant detection from whole genome sequencing data. Genome Med. (2021) 13:1–19. doi: 10.1186/s13073-021-00841-x
18. Robinson JT, Thorvaldsdóttir H, Winckler W, Guttman M, Lander ES, Getz G, et al. Integrative genomics viewer. Nat Biotechnol. (2011) 29:24–6. doi: 10.1038/nbt.1754
19. Belyeu JR, Chowdhury M, Brown J, Pedersen BS, Cormier MJ, Quinlan AR, et al. Samplot: a platform for structural variant visual validation and automated filtering. Genome Biol. (2021) 22:161. doi: 10.1186/s13059-021-02380-5
20. Bird TD. Charcot-marie-tooth hereditary neuropathy overview. In: Adam MP, Mirzaa GM, Pagon RA, Wallace SE, Bean LJH, Gripp KW, Amemiya A, editors. GeneReviews®. Seattle, WA: University of Washington (1993–2023).
21. Lawson VH, Graham BV, Flanigan KM. Clinical and electrophysiologic features of CMT2A with mutations in the mitofusin 2 gene. Neurology. (2005) 65:197–204. doi: 10.1212/01.wnl.0000168898.76071.70
22. Ma Y, Sun A, Zhang Y, Fan D, Liu X. The genotype and phenotype features in a large Chinese MFN2 mutation cohort. Front Neurol. (2021) 12:757518. doi: 10.3389/fneur.2021.757518
23. Abati E, Manini A, Velardo D, Del Bo R, Napoli L, Rizzo F, et al. Clinical and genetic features of a cohort of patients with MFN2-related neuropathy. Sci Rep. (2022) 12:6181. doi: 10.1038/s41598-022-10220-0
24. Del Bo R, Moggio M, Rango M, Bonato S, D'Angelo MG, Ghezzi S, et al. Mutated mitofusin 2 presents with intrafamilial variability and brain mitochondrial dysfunction. Neurology. (2008) 71:1959–66. doi: 10.1212/01.wnl.0000327095.32005.a4
25. Sitarz KS, Yu-Wai-Man P, Pyle A, Stewart JD, Rautenstrauss B, Seeman P, et al. MFN2 mutations cause compensatory mitochondrial DNA proliferation. Brain. (2012) 135:e219–e219. doi: 10.1093/brain/aws049
26. Lassuthova P, Rebelo AP, Ravenscroft G, Lamont PJ, Davis MR, Manganelli F, et al. Mutations in ATP1A1 cause dominant Charcot-Marie-Tooth type 2. Am J Hum Genet. (2018) 102:505–14. doi: 10.1016/j.ajhg.2018.01.023
27. Mandich P, Grandis M, Geroldi A, Acquaviva M, Varese A, Gulli R, et al. Gap junction beta 1 (GJB1) gene mutations in Italian patients with X-linked Charcot-Marie-Tooth disease. J Hum Genet. (2008) 53:529–33. doi: 10.1007/s10038-008-0280-4
28. Beijer D, Agnew T, Rack JGM, Prokhorova E, Deconinck T, Ceulemans B, et al. Biallelic ADPRHL2 mutations in complex neuropathy affect ADP ribosylation and DNA damage response. Life Sci Alliance. (2021) 4:e202101057. doi: 10.26508/lsa.202101057
29. Danhauser K, Alhaddad B, Makowski C, Piekutowska-Abramczuk D, Syrbe S, Gomez-Ospina N, et al. Bi-allelic ADPRHL2 mutations cause neurodegeneration with developmental delay, ataxia, and axonal neuropathy. Am J Hum Genet. (2018) 103:817–25. doi: 10.1016/j.ajhg.2018.10.005
30. Mandola AB, Reid B, Sirror R, Brager R, Dent P, Chakroborty P, et al. Ataxia telangiectasia diagnosed on newborn screening–case cohort of 5 years' experience. Front Immunol. (2019) 10:2940. doi: 10.3389/fimmu.2019.02940
31. Stankovic T, Kidd AMJ, Sutcliffe A, McGuire GM, Robinson P, Weber P, et al. ATM mutations and phenotypes in ataxia-telangiectasia families in the british isles: expression of mutant ATM and the risk of leukemia, lymphoma, and breast cancer. Am J Hum Genet. (1998) 62:334–45. doi: 10.1086/301706
32. Aharoni S, Barwick KES, Straussberg R, Harlalka GV, Nevo Y, Chioza BA, et al. Novel homozygous missense mutation in GAN associated with Charcot-Marie-Tooth disease type 2 in a large consanguineous family from Israel. BMC Med Genet. (2016) 17:82. doi: 10.1186/s12881-016-0343-x
33. Lagueny A, Latour P, Vital A, Le Masson G, Rouanet M, Ferrer X, et al. Mild recurrent neuropathy in CMT1B with a novel nonsense mutation in the extracellular domain of the MPZ gene. J Neurol Neurosurg Psychiatry. (2001) 70:232–5. doi: 10.1136/jnnp.70.2.232
34. Subréville M, Bonello-Palot N, Yahiaoui D, Beloribi-Djefaflia S, Fernandes S, Stojkovic T, et al. Genotype–phenotype correlation in French patients with myelin protein zero gene-related inherited neuropathy. Eur J Neurol. (2021) 28:2913–21. doi: 10.1111/ene.14948
35. Meldau S, De Lacy RJ, Riordan GTM, Goddard EA, Pillay K, Fieggen KJ, et al. Identification of a single MPV17 nonsense-associated altered splice variant in 24 South African infants with mitochondrial neurohepatopathy. Clin Genet. (2018) 93:1093–6. doi: 10.1111/cge.13208
36. Stevenson A, Akena D, Stroud RE, Atwoli L, Campbell MM, Chibnik LB, et al. Neuropsychiatric genetics of african populations-psychosis (NeuroGAP-psychosis): a case-control study protocol and GWAS in Ethiopia, Kenya, South Africa and Uganda. BMJ Open. (2019) 9:e025469. doi: 10.1136/bmjopen-2018-025469
37. Pennings M, Schouten MI, van Gaalen J, Meijer RPP, de Bot ST, Kriek M, et al. KIF1A variants are a frequent cause of autosomal dominant hereditary spastic paraplegia. Eur J Hum Genet. (2020) 28:40–9. doi: 10.1038/s41431-019-0497-z
38. Klebe S, Lossos A, Azzedine H, Mundwiller E, Sheffer R, Gaussen M, et al. KIF1A missense mutations in SPG30, an autosomal recessive spastic paraplegia: distinct phenotypes according to the nature of the mutations. Eur J Hum Genet. (2012) 20:645–9. doi: 10.1038/ejhg.2011.261
39. Vaz FM, McDermott JH, Alders M, Wortmann SB, Kölker S, Pras-Raves ML, et al. Mutations in PCYT2 disrupt etherlipid biosynthesis and cause a complex hereditary spastic paraplegia. Brain. (2019) 142:3382–97. doi: 10.1093/brain/awz291
40. Vander Stichele G, Durr A, Yoon G, Schüle R, Blackstone C, Esposito G, et al. An integrated modelling methodology for estimating global incidence and prevalence of hereditary spastic paraplegia subtypes SPG4, SPG7, SPG11, and SPG15. BMC Neurol. (2022) 22:115. doi: 10.1186/s12883-022-02595-4
41. Tesson C, Koht J, Stevanin G. Delving into the complexity of hereditary spastic paraplegias: how unexpected phenotypes and inheritance modes are revolutionizing their nosology. Hum Genet. (2015) 134:511–38. doi: 10.1007/s00439-015-1536-7
42. Namekawa M, Muriel M-P, Janer A, Latouche M, Dauphin A, Debeir T, et al. Mutations in the SPG3A gene encoding the GTPase atlastin interfere with vesicle trafficking in the ER/Golgi interface and Golgi morphogenesis. Mol Cell Neurosci. (2007) 35:1–13. doi: 10.1016/j.mcn.2007.01.012
43. Zhao G, Liu X. Clinical features and genotype-phenotype correlation analysis in patients with ATL1 mutations: a literature reanalysis. Transl Neurodegener. (2017) 6:9. doi: 10.1186/s40035-017-0079-3
44. Roxburgh RH, Marquis-Nicholson R, Ashton F, George AM, Lea RA, Eccles D, et al. The pAla510Val mutation in the SPG7 (paraplegin) gene is the most common mutation causing adult onset neurogenetic disease in patients of British ancestry. J Neurol. (2013) 260:1286–94. doi: 10.1007/s00415-012-6792-z
45. Stevanin G, Santorelli FM, Azzedine H, Coutinho P, Chomilier J, Denora PS, et al. Mutations in SPG11, encoding spatacsin, are a major cause of spastic paraplegia with thin corpus callosum. Nat Genet. (2007) 39:366–72. doi: 10.1038/ng1980
46. Pereira MC, Loureiro JL, Pinto-Basto J, Brandão E, Lopes AM, Neves G, et al. Alu elements mediate large SPG11 gene rearrangements: further spatacsin mutations. Genet Med. (2012) 14:143–51. doi: 10.1038/gim.2011.7
47. Paisan-Ruiz C, Dogu O, Yilmaz A, Houlden H, Singleton A. SPG11 mutations are common in familial cases of complicated hereditary spastic paraplegia. Neurology. (2008) 70(Issue 16, Part 2):1384–9. doi: 10.1212/01.wnl.0000294327.66106.3d
48. Lynch DS, Koutsis G, Tucci A, Panas M, Baklou M, Breza M, et al. Hereditary spastic paraplegia in Greece: characterisation of a previously unexplored population using next-generation sequencing. Eur J Hum Genet. (2016) 24:857–63. doi: 10.1038/ejhg.2015.200
49. Li C, Yan Q, Duan F, Zhao C, Zhang Z, Du Y, et al. Mild cognitive impairment in novel SPG11 mutation-related sporadic hereditary spastic paraplegia with thin corpus callosum: case series. BMC Neurol. (2021) 21:12. doi: 10.1186/s12883-020-02040-4
50. Ondruskova N, Vesela K, Hansikova H, Magner M, Zeman J, Honzik T. RFT1-CDG in adult siblings with novel mutations. Mol Genet Metab. (2012) 107:760–2. doi: 10.1016/j.ymgme.2012.10.002
51. Quelhas D, Jaeken J, Fortuna A, Azevedo L, Bandeira A, Matthijs G, et al. RFT1-CDG: absence of epilepsy and deafness in two patients with novel pathogenic variants. JIMD Rep. (2019) 43:111–6. doi: 10.1007/8904_2018_112
52. Coutelier M, Goizet C, Durr A, Habarou F, Morais S, Dionne-Laporte A, et al. Alteration of ornithine metabolism leads to dominant and recessive hereditary spastic paraplegia. Brain. (2015) 138:2191–205. doi: 10.1093/brain/awv143
53. Kalmár T, Maróti Z, Zimmermann A, Sztriha L. Tremor as an early sign of hereditary spastic paraplegia due to mutations in ALDH18A1. Brain Dev. (2021) 43:144–51. doi: 10.1016/j.braindev.2020.07.015
54. Wei Q, Dong H-L, Pan L-Y, Chen C-X, Yan Y-T, Wang R-M, et al. Clinical features and genetic spectrum in Chinese patients with recessive hereditary spastic paraplegia. Transl Neurodegener. (2019) 8:19. doi: 10.1186/s40035-019-0157-9
55. Lan M-Y, Yeh T-H, Chang Y-Y, Kuo H-C, Sun HS, Lai S-C, et al. Clinical and genetic analysis of Taiwanese patients with hereditary spastic paraplegia type 5. Eur J Neurol. (2015) 22:211–4. doi: 10.1111/ene.12407
56. Arnoldi A, Crimella C, Tenderini E, Martinuzzi A, D'Angelo M, Musumeci O, et al. Clinical phenotype variability in patients with hereditary spastic paraplegia type 5 associated with CYP7B1 mutations. Clin Genet. (2012) 81:150–7. doi: 10.1111/j.1399-0004.2011.01624.x
57. Chou C, Soong B, Lin K, Tsai Y, Jih K, Liao Y, et al. Clinical characteristics of Taiwanese patients with Hereditary spastic paraplegia type 5. Ann Clin Transl Neurol. (2020) 7:486–96. doi: 10.1002/acn3.51019
58. Fridman V, Bundy B, Reilly MM, Pareyson D, Bacon C, Burns J, et al. CMT subtypes and disease burden in patients enrolled in the Inherited Neuropathies Consortium natural history study: a cross-sectional analysis. J Neurol Neurosurg Psychiatry. (2015) 86:873–8. doi: 10.1136/jnnp-2014-308826
59. Meyyazhagan A, Orlacchio A. Hereditary spastic paraplegia: an update. Int J Mol Sci. (2022) 23:1697. doi: 10.3390/ijms23031697
60. Wang J, Fang F, Ding C, Li J, Wu Y, Zhang W, et al. Clinical and genetic spectrum of hereditary spastic paraplegia in Chinese children. Dev Med Child Neurol. (2023) 65:416–23. doi: 10.1111/dmcn.15385
61. Kara E, Tucci A, Manzoni C, Lynch DS, Elpidorou M, Bettencourt C, et al. Genetic and phenotypic characterization of complex hereditary spastic paraplegia. Brain. (2016) 139:1904–18. doi: 10.1093/brain/aww111
62. Using Population Descriptors in Genetics and Genomics Research. Washington, DC: National Academies Press (2023).
63. Lumaka A, Carstens N, Devriendt K, Krause A, Kulohoma B, Kumuthini J, et al. Increasing African genomic data generation and sharing to resolve rare and undiagnosed diseases in Africa: a call-to-action by the H3Africa rare diseases working group. Orphanet J Rare Dis. (2022) 17:230. doi: 10.1186/s13023-022-02391-w
Keywords: whole exome sequencing, whole genome sequencing, Charcot-Marie-Tooth disease, hereditary spastic paraplegia, African, equity, diversity and inclusion
Citation: Mahungu AC, Steyn E, Floudiotis N, Wilson LA, Vandrovcova J, Reilly MM, Record CJ, Benatar M, Wu G, Raga S, Wilmshurst JM, Naidu K, Hanna M, Nel M and Heckmann JM (2023) The mutational profile in a South African cohort with inherited neuropathies and spastic paraplegia. Front. Neurol. 14:1239725. doi: 10.3389/fneur.2023.1239725
Received: 13 June 2023; Accepted: 02 August 2023;
Published: 29 August 2023.
Edited by:
Daniele Orsucci, Azienda Usl Toscana Nord Ovest, ItalyReviewed by:
Corrado Italo Angelini, University of Padua, ItalyCopyright © 2023 Mahungu, Steyn, Floudiotis, Wilson, Vandrovcova, Reilly, Record, Benatar, Wu, Raga, Wilmshurst, Naidu, Hanna, Nel and Heckmann. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Jeannine M. Heckmann, amVhbmluZS5oZWNrbWFubkB1Y3QuYWMuemE=; Melissa Nel, bWVsaXNzYS5uZWxAdWN0LmFjLnph
†These authors have contributed equally to this work and share senior authorship
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.