 Ling Li
Ling Li- 1Health Management Center, The Third Xiangya Hospital, Central South University, Changsha, China
- 2Center for Experimental Medicine, The Third Xiangya Hospital, Central South University, Changsha, China
- 3Department of Neurology, The Third Xiangya Hospital, Central South University, Changsha, China
- 4Disease Genome Research Center, Central South University, Changsha, China
Genetic epilepsy with febrile seizures plus (GEFSP) is a familial epileptic syndrome that is genetically heterogeneous and inherited in an autosomal dominant form in most cases. To date, at least seven genes have been reported to associate with GEFSP. This study aimed to identify the disease-causing variant in a Chinese Tujia ethnic family with GEFSP by using whole exome sequencing, Sanger sequencing, and in silico prediction. A heterozygous missense variant c.5725A>G (p.T1909A) was identified in the sodium voltage-gated channel alpha subunit 1 gene (SCN1A) coding region. The variant co-segregated with the GEFSP phenotype in this family, and it was predicted as disease-causing by multiple in silico programs, which was proposed as the genetic cause of GEFSP, further genetically diagnosed as GEFSP2. These findings expand the genetic and phenotypic spectrum of GEFSP and should contribute to genetic diagnoses, personalized therapies, and prognoses.
1. Introduction
Genetic epilepsy with febrile seizures plus (GEFSP, also called GEFS+), previously known as generalized epilepsy with febrile seizures plus, was first defined by Scheffer and Berkovic as an inherited epileptic syndrome with genetic and phenotypic heterogeneity (1, 2). It is a familial epilepsy syndrome clinically characterized by various types of seizures, including fever-associated and afebrile seizures (3, 4). GEFSP includes a wide range of subtypes with varying prevalence, of which the estimated prevalence of febrile seizures (FS) is 3–4%, and the overall prevalence is unclear (5, 6). The most common phenotype in the GEFSP pedigrees is FS, which occurs between the age of 3 months and 6 years, followed by febrile seizures plus (FS+), in which episodes with fever persist beyond 6 years or afebrile seizures occur (7). The diagnosis is based on the occurrence of two or more family members that show phenotypes in the spectrum, including FS, FS+, FS/FS+ with various seizures (such as absence, atonic, myoclonic, and partial seizures), and myoclonic-atonic epilepsy, along with Dravet syndrome (8–10). The inheritance patterns include autosomal dominant, autosomal recessive, and complex inheritance in which several genes are involved, accompanied by possibly environmental contributions. In pedigrees with monogenic variants, most of them follow autosomal dominant patterns, and the minority abides by autosomal recessive traits. In some small families, the most common pattern may be complex inheritance (11, 12). To date, at least 11 genetic loci are recorded for GEFSP in the Online Mendelian Inheritance in Man (OMIM, https://www.omim.org/). Variants in the critical regions of at least seven genes, including the sodium voltage-gated channel beta subunit 1 gene (SCN1B), the sodium voltage-gated channel alpha subunit 1 gene (SCN1A), the gamma-aminobutyric acid type A receptor subunit gamma2 gene (GABRG2), the gamma-aminobutyric acid type A receptor subunit delta gene (GABRD), the syntaxin 1B gene (STX1B), the hyperpolarization-activated cyclic nucleotide-gated potassium and sodium channel 1 gene (HCN1), and the hyperpolarization-activated cyclic nucleotide-gated potassium and sodium channel 2 gene (HCN2), were reported to be responsible for GEFSP (13–19). Of these, the SCN1A gene is the most clinically relevant and most frequently reported disease-causing gene for the GEFSP spectrum, and ~11% of reported pedigrees were caused by its variants (20).
In this study, we identified a heterozygous missense variant c.5725A>G (p.T1909A) in the SCN1A gene as the causative variant in a Chinese Tujia ethnic family with GEFSP.
2. Materials and methods
2.1. Participators and clinical evaluations
A non-consanguineous Chinese Tujia ethnic family located in the west of Hunan province was recruited from the Third Xiangya Hospital, Central South University (Changsha, China). The GEFSP diagnosis was based on clinical features, family history, electroencephalography (EEG), and genetic testing. The medical history of the patients was collected. Routine physical examinations and EEG were performed on the proband. All individuals had signed the written informed consent before the peripheral venous blood samples were acquired. The approval of this study was received from the Institutional Review Board of the Third Xiangya Hospital, Central South University, Changsha, China.
2.2. DNA extraction and exome capture
The genomic DNA (gDNA) was extracted from peripheral venous blood samples by using the standard phenol-chloroform extraction method as previously described (21). The gDNA samples of II:1 and III:1 (Figure 1A) were randomly fragmented to 150 bp-250 bp using Covaris technology and prepared for whole exome sequencing (WES). The ends of DNA fragments were repaired, and the “A” base was ligated at the 3′-end of each strand. Adapters were added to both ends of the end-repaired DNA for PCR amplification and further sequencing. The products were then purified and hybridized to the exome array. Hybridized fragments were used for circularization, and non-specific fragments were cleaned out (22). DNA nanoballs were produced by rolling circle amplification. The qualified captured library was loaded on the BGISEQ-500 sequencing platform, and sequencing-derived raw image files were analyzed by BGISEQ-500 base-calling software, performed by the BGI-Shenzhen (Shenzhen, China).
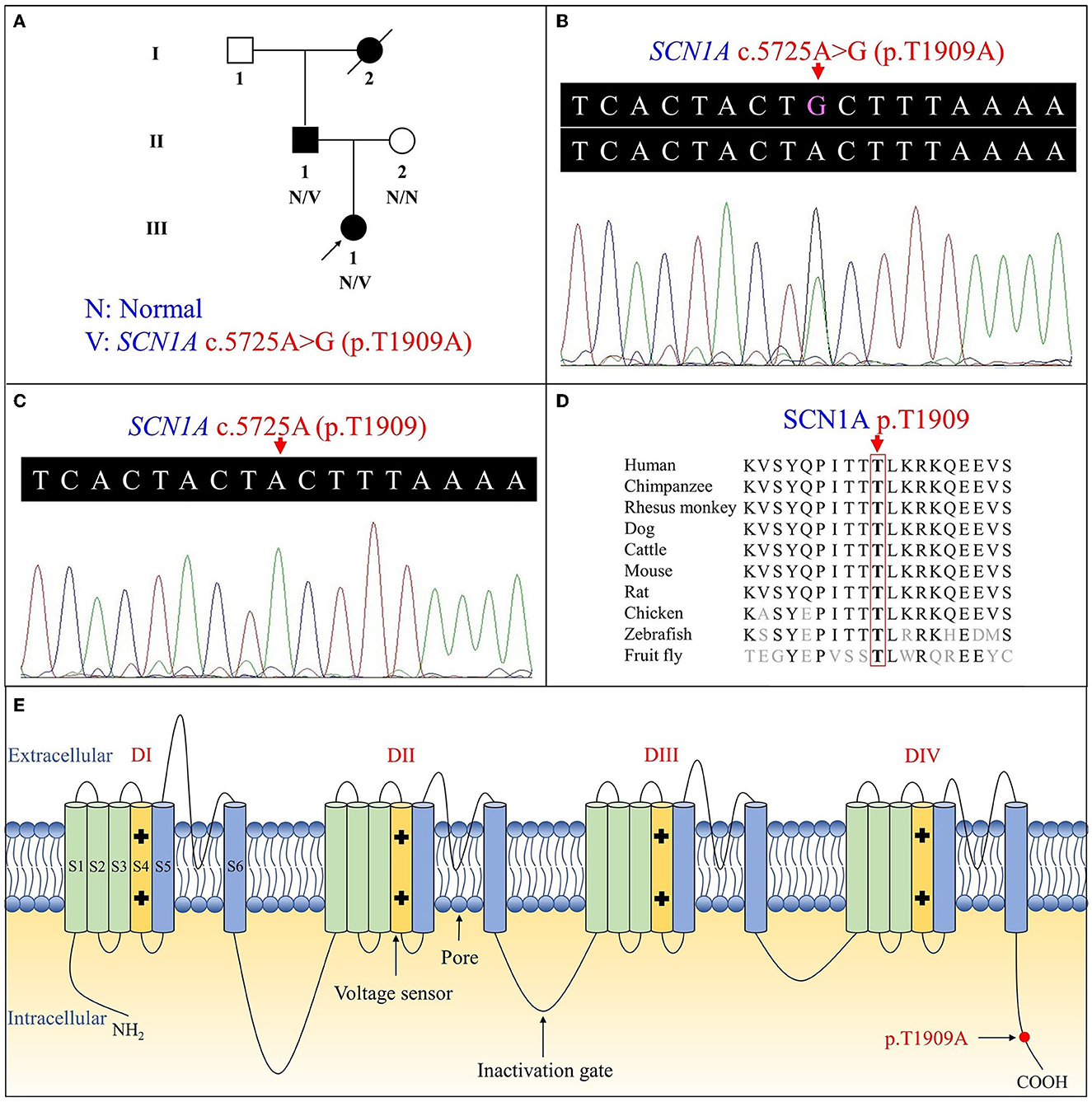
Figure 1. Pedigree figure of the Chinese Tujia ethnic family with genetic epilepsy with febrile seizures plus (GEFSP) and the variant analysis. (A) A pedigree figure of the family. (B) SCN1A sequence of the proband (III:1) with heterozygous c.5725A>G (p.T1909A) variant. (C) Normal SCN1A sequence of the unaffected member (II:2). (D) SCN1A p.T1909 amino acid residue is conserved in multiple species. (E) Schematic structure of SCN1A protein and the location of the p.T1909A variant. S1–S6 represent six alpha-helical transmembrane segments, and DI–DIV represent four homologous domains.
2.3. Read mapping and variant analysis
The clean data were produced by noise-decrease data filtering on raw data and then aligned to the human reference genome (GRCh37/hg19) by Burrows–Wheeler Aligner (BWA, v0.7.15) (23). Picard tools (v2.5.0) were used to label and remove duplicate reads. The Genome Analysis Toolkit (GATK, v3.3.0) was used for local realignment and base quality score recalibration (24, 25). Single nucleotide polymorphisms (SNPs) and insertions-deletions (indels) were called using HaplotypeCaller of GATK and then annotated with the SnpEff tool (26). All candidate variants were screened and analyzed by the public databases, including the Single Nucleotide Polymorphism database (dbSNP, build 141), the 1000 Genomes Project, the National Heart, Lung, and Blood Institute Exome Sequencing Project 6500 (NHLBI ESP6500), the Exome Aggregation Consortium (ExAC), the Genome Aggregation Database (gnomAD), the China Metabolic Analytics Project (ChinaMAP), Human Gene Mutation Database (HGMD), and ClinVar, along with the in-house BGI exome database including 1,943 controls (27–33). The MutationTaster (http://www.mutationtaster.org/), Protein Variation Effect Analyzer (PROVEAN, http://provean.jcvi.org/), Sorting Intolerant from Tolerant (SIFT, http://provean.jcvi.org/), Polymorphism Phenotyping v2 (PolyPhen-2, http://genetics.bwh.harvard.edu/pph2/), and MutationAssessor (http://mutationassessor.org/r3/) were used to obtain the predicted pathogenic effects of variants (34–37). Sanger sequencing was employed to validate the causative variant with an ABI3500 sequencer (Applied Biosystems, Foster City, CA, USA). Primers used for PCR amplification and sequencing were designed and analyzed by the Primer3 program and Primer-BLAST, and the primer sequences for detecting the disease-associated variant are as follows: forward, 5′-GTGACCGGATCCACTGTCTT-3′ and reverse, 5′-GCTTTAAAAGGTGGCGTCTG-3′. The NCBI Basic Local Alignment Search Tool (BLAST, https://blast.ncbi.nlm.nih.gov/Blast.cgi) was used to conduct conservation analysis among multiple species (38). Wild-type and mutant protein structures were predicted by SWISS-MODEL (https://swissmodel.expasy.org/) and displayed by PyMOL software (v2.5.2, Schrödinger, LLC, New York, NY, USA) (39). The American College of Medical Genetics and Genomics (ACMG) guidelines for the sequence variant interpretation were utilized to classify the identified variant (40).
3. Results
3.1. Clinical findings
The proband (III:1) was a 22-year-old female with unremarkable spontaneous vaginal delivery and development. Her first seizure occurred at the age of 2 years when she suffered from an upper respiratory tract infection and had a fever of ~38°C. After that, she had recurrent episodes when she caught a fever or felt nervous. She has suffered from attacks of afebrile seizures (AFS) since the age of 14 years. The episodes can manifest as moderate generalized tonic-clonic seizures, with a mean duration of 2–7 min, and focal impaired awareness seizures, with a mean duration of 1–1.5 min. The ambulatory EEG showed interictal epileptiform discharges: bilateral slow waves, sharp waves, and sharp-and-slow wave complexes (41). Irregularly antiepileptic drug lamotrigine treatment not adhering to medical advice was applied, and the efficacy was not good. The self-reported medical history of her father (II:1) revealed fever-associated seizures at 7 months and spontaneous remission before the age of 14 years. Focal impaired awareness seizures began at the age of 7 years, and no AFS were claimed. Detailed features of patients in this family are presented in Table 1.
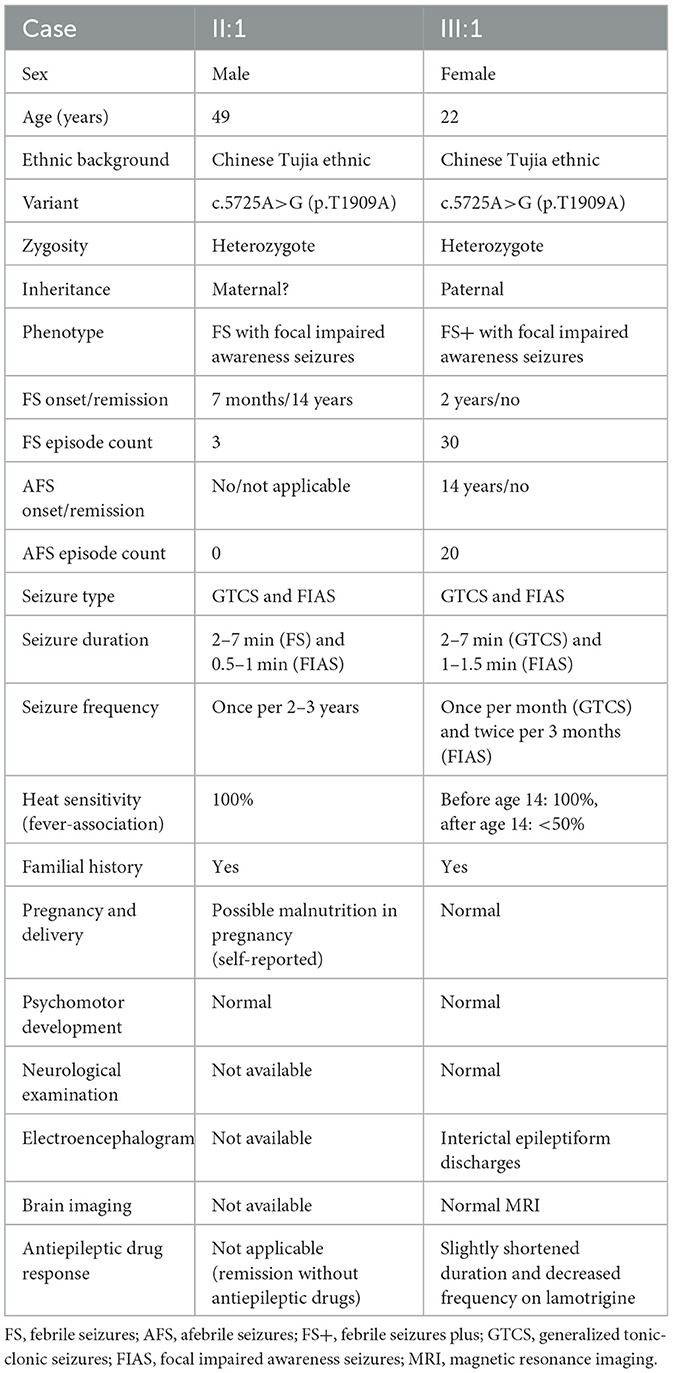
Table 1. Clinical features and genotypes of patients.
3.2. Genetic analysis
Overall, the WES of the two patients generated an average of 203.59 million effective reads. Of these, ~99.95% were mapped to the human reference genome. The target sequence covered 99.68% of bases at ≥10×, and the average sequencing depth was 256.39×. A total of 98,432 SNPs and 17,160 indels were identified in the father (II:1), and ~106,543 SNPs and 18,860 indels were detected in the proband (III:1). A variant filtering scheme, described in recent studies, was applied for detecting the potential disease-associated variant for patients. Common variants registered in dbSNP, 1000 Genomes Project, and NHLBI ESP6500 with an allele frequency of >0.5% were excluded, and damaging variants predicted by in silico tools were reserved. Using the mentioned criteria, only a heterozygous variant of the SCN1A gene (NM_001353948.2), c.5725A>G (p.T1909A), shared by two patients, was judged as the most likely disease-causing variant and predicted to be deleterious by bioinformatics (Table 2). Sanger sequencing corroborated the variant in the patients (II:1 and III:1, Figure 1B) and revealed the absence of the variant in the proband's unaffected mother (II:2, Figure 1C). Conservation analysis showed the high conservation of the mutated p.T1909 residue among 10 species (Figure 1D), and the conformation change caused by the variant was shown in Figure 2. Based on the above evidence, the variant c.5725A>G (p.T1909A) in the SCN1A gene appears to be accountable for the GEFSP in this family. According to ACMG guidelines, the SCN1A gene variant c.5725A>G (p.T1909A) was classified as a “likely pathogenic” (PS1 + PM2 + PP1 + PP3) variant.
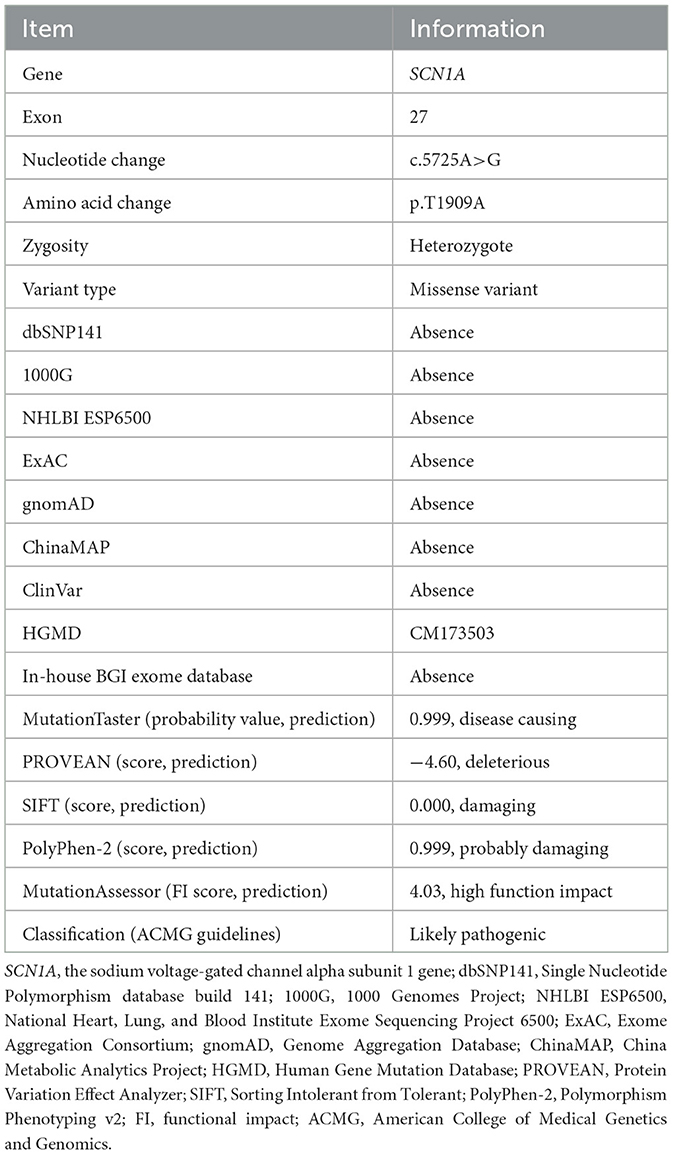
Table 2. Identification of the disease-associated variant in the patients.
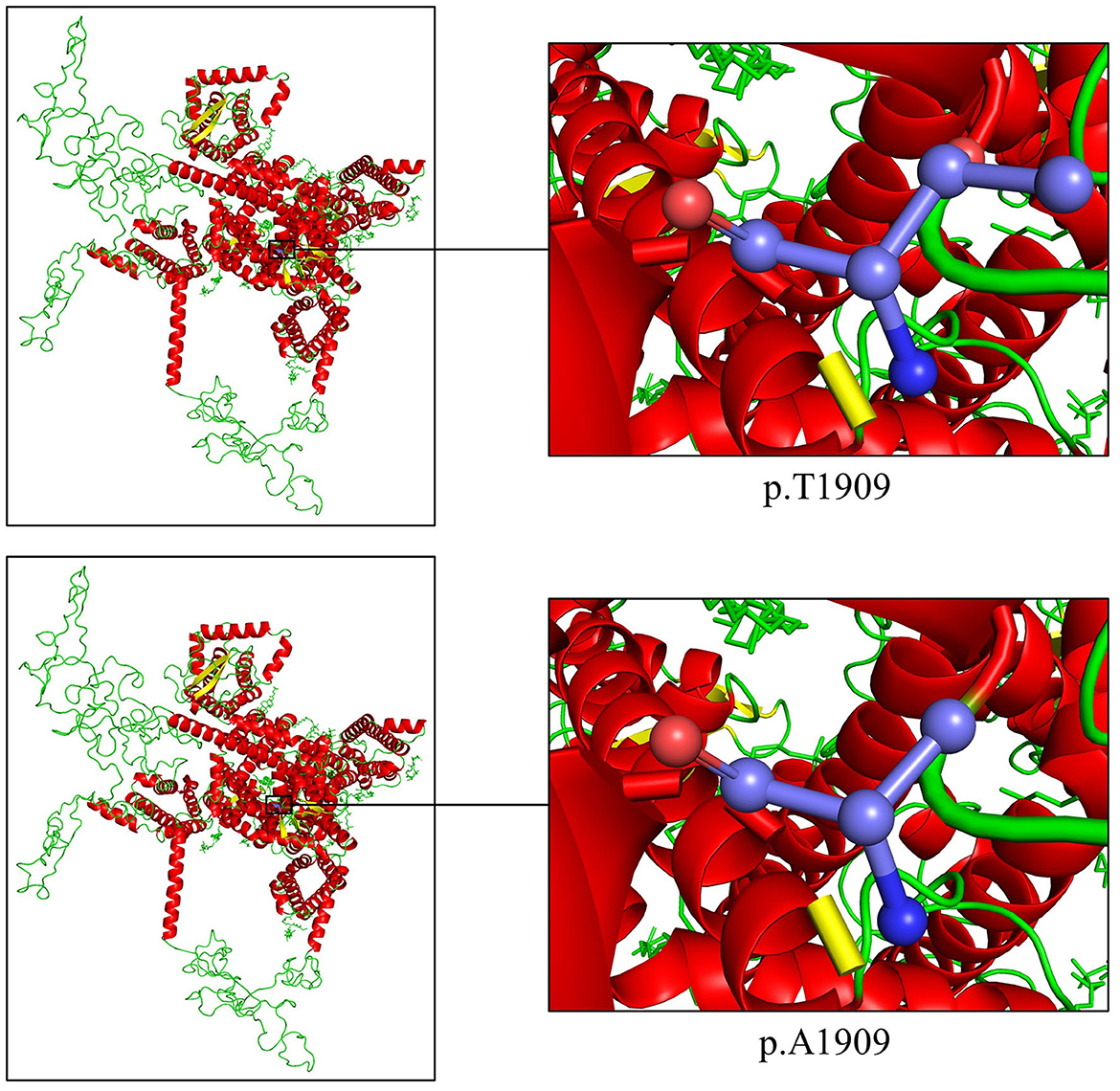
Figure 2. Cartoon model of the SCN1A protein structure by PyMOL 2.5.2 based on the SWISS-MODEL: the threonine and the altered alanine at position 1909 are shown as ball-and-stick models.
4. Discussion
GEFSP is a familial epileptic syndrome with remarkable genetic and phenotypic heterogeneity (42). It was first described in a large Australian family having an unusual concentration of generalized epilepsy and FS (43). Since the SCN1B gene was reported as the first causative gene of GEFSP in 1998, a large number of GEFSP-related variants have been identified in seven pathogenic genes, in which the SCN1A gene is the most frequently reported and most clinically relevant one (44).
In this study, we identified the SCN1A missense variant c.5725A>G (p.T1909A) in two members of the Chinese Tujia ethnic family via WES and Sanger sequencing, which was found to co-segregate with the phenotype in the family. The proband had AFS and uncontrolled seizures in adulthood, and her father had self-limited seizures, in favor of the phenotypic heterogeneity of GEFSP, genetically diagnosed as GEFSP2.
The SCN1A gene, located on chromosome 2q24.3, includes 27 exons, encoding the protein sodium voltage-gated channel alpha subunit 1 (Nav1.1) with 2,009 amino acids (~229 kDa) (45). The Nav1.1 includes four homologous domains (I-IV), each containing six alpha-helical transmembrane segments (S1-S6), in which the S4 is a voltage sensor, and the S5 and S6 form the ion-selective pores (Figure 1E) (46). It is highly expressed in the brain, especially the prefrontal cortex, and forms the voltage-dependent sodium channel with beta subunits as one of the primary sodium channels in the central nervous system (http://biogps.org/), which plays a crucial role in the initiation and propagation of action potentials in neurons (47, 48).
To date, more than 2,584 variants in the SCN1A gene have been recorded, and most are deemed responsible for GEFSP phenotypes (HGMD, http://www.hgmd.cf.ac.uk/ac/index.php). The c.5725A>G variant was located at the exon 27 of the SCN1A gene. It was absent in public databases including dbSNP, the 1000 Genomes Project, NHLBI ESP6500, ExAC, gnomAD, ChinaMAP, ClinVar, and the in-house BGI exome database. The variant was predicted as deleterious or probably damaging by multiple in silico programs including MutationTaster, PROVEAN, SIFT, PolyPhen-2, and MutationAssessor. The threonine at position 1,909 was phylogenetically conserved among varied species from fruit flies to human beings. The polar hydrophilic to non-polar hydrophobic residue change may affect the tertiary structure and impact normal function (49). The p.T1909A variant is located in the cytoplasmic region of the Nav1.1 C-terminal domain, which contains binding sites of interaction proteins and plays an important role in channel inactivation (50).
A de novo c.5725A>G (p.T1909A) variant was reported in a patient with focal epilepsy, recorded in HGMD (CM173503) (44). Moreover, a missense variant involving the same residue, c.5726C>T (p.T1909I, rs121918793), was reported in a 7-month-onset female with severe myoclonic epilepsy of infancy (SMEI, i.e., Dravet syndrome) (51).
Functional studies indicated that different SCN1A variants alter sodium channel properties and functions in different ways and cause distinctive effects on the sodium channel activity, thus affecting the selection of antiepileptic drugs (52). Both gain-of-function (GOF) and loss-of-function (LOF) effects could be responsible for SCN1A-associated epilepsies. In human tsA201 cells with p.R1648C or p.F1661S variant, a small non-inactivating persistent inward current during depolarization was reported to lead to neuron hyperexcitability, which exhibited a GOF effect in the sodium channels (53). GOF-related epilepsy can usually be relieved by commonly prescribed antiepileptic drugs that inhibit sodium channels, such as carbamazepine and phenytoin (54). SCN1A LOF variants mainly impair bipolar GABAergic inhibitory interneurons and lead to diminished inhibition (55). The application of GABA transaminase inhibitors such as valproic acid or GABA receptor-positive allosteric modulators, such as pentobarbital, appears to be effective, while the sodium channel blocker may provoke seizure aggravation (56). In human tsA201 cells, the SMEI-associated p.T1909I variant exhibited a mixture mechanism of GOF and LOF, with increased persistent current and reduction of current density (57). In this study, disease symptom remission and seizure frequency reduction after lamotrigine application in the proband were unsatisfactory. Due to the same biophysical property change (polar hydrophilic to non-polar hydrophobic) of p.T1909I and p.T1909A variants, the same mechanism may be shared. Limited response to medication may be due to the use of a single sodium channel blocking antiepileptic drug. The prescription of GABA transaminase inhibitors or GABA receptor-positive allosteric modulators might be beneficial for the sufferers.
Genetic deficient animal models verified the important role of the SCN1A gene in epilepsy development. The drosophila SCN1A p.K1270T variant knock-in model showed that when the temperature rose, the deactivation threshold for persistent sodium currents reversibly shifted to a more negative voltage, causing sustained depolarizations in GABAergic inhibitory interneurons and leading to reduced inhibitory activity in the brain (58). Heterozygous (Scn1a+/−) mice had spontaneous seizures and occasional deaths beginning on postnatal day 21, attributed to haploinsufficiency of Nav1.1 channels, and homozygous Scn1a knockout (Scn1a−/−) mice developed severe ataxia and seizures and died on postnatal day 15, corresponding to the LOF effect (59). In a bacterial artificial chromosome transgenic SCN1A p.R1648H variant mouse model, experiments showed a delayed recovery of channel inactivation only in inhibitory neurons, suggesting the cell type-dependence of SCN1A mutation and the p.R1648H variant leading to a reduction in inhibitory neurons excitability (60).
5. Conclusion
In summary, a missense variant c.5725A>G (p.T1909A) in the SCN1A gene was identified in a Chinese Tujia ethnic family with GEFSP, further classified as GEFSP2. The different phenotypes of the same variant in the family show the heterogeneity of GEFSP. These findings confirmed the SCN1A-associated GEFSP, which may expand the genetic and phenotypic spectrum of GEFSP and improve genetic diagnoses, personalized therapies, and prognoses. The combination of WES and Sanger sequencing efficiently provided a timely diagnosis and indicated management for those with clinically and/or genetically suspected GEFSP. Additional in vitro studies and the establishment of genetically deficient animal models to explore the functional effect of human SCN1A variants may help to further illuminate potential pathogenic mechanisms.
Data availability statement
Data of this project can be accessed after an approval application to the China National Genebank (CNGB, https://db.cngb.org/cnsa/). Please refer to https://db.cngb.org/, or email: CNGBdb@cngb.org for detailed application guidance. The accession code CNP0004397 should be included in the application.
Ethics statement
The studies involving human participants were reviewed and approved by the Institutional Review Board of the Third Xiangya Hospital, Central South University, Changsha, China. Written informed consent to participate in this study was provided by the participants' legal guardian/next of kin. Written informed consent was obtained from the individual(s), and minor(s)' legal guardian/next of kin, for the publication of any potentially identifiable images or data included in this article.
Author contributions
LL, LY, and HD conceived and designed this study and wrote the manuscript. WZ, YY, and ZS collected the patient samples and clinical data. LL, LY, WZ, and XD performed the experiments. LL, LY, WZ, XD, and HD analyzed the data. The final version of the manuscript was read and approved by all authors.
Funding
This study was supported by the National Natural Science Foundation of China (Grant Nos. 81800219 and 81873686), the Natural Science Foundation of Hunan Province (Grant Nos. 2020JJ3057, 2020JJ4830, and 2020JJ4865), the Province-level College Students' Innovative Training Plan Program (Grant No. S2021105331043), the Wisdom Accumulation and Talent Cultivation Project of the Third Xiangya Hospital of Central South University (Grant No. YX202109), and the Distinguished Professor of the Lotus Scholars Award Program of Hunan Province, China.
Acknowledgments
We express our deepest gratitude to patients, their families, doctors, and researchers for their cooperation and contribution.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher's note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Scheffer IE, Berkovic SF. Generalized epilepsy with febrile seizures plus. A genetic disorder with heterogeneous clinical phenotypes. Brain. (1997) 120:479–90. doi: 10.1093/brain/120.3.479
2. Zhang YH, Burgess R, Malone JP, Glubb GC, Helbig KL, Vadlamudi L, et al. Genetic epilepsy with febrile seizures plus: refining the spectrum. Neurology. (2017) 89:1210–9. doi: 10.1212/WNL.0000000000004384
3. Gauthier AC, Manganas LN, Mattson RH. A novel inherited SCN1A mutation associated with GEFS+ in benign and encephalopathic epilepsy. J Clin Neurosci. (2017) 40:82–4. doi: 10.1016/j.jocn.2017.02.011
4. Li X, Guo S, Xu S, Chen Z, Wang L, Ding J, et al. Neocortex- and hippocampus-specific deletion of Gabrg2 causes temperature-dependent seizures in mice. Cell Death Dis. (2021) 12:553. doi: 10.1038/s41419-021-03846-x
5. Baulac S, Gourfinkel-An I, Picard F, Rosenberg-Bourgin M, Prud'homme JF, Baulac M, et al. A second locus for familial generalized epilepsy with febrile seizures plus maps to chromosome 2q21-q33. Am J Hum Genet. (1999) 65:1078–85. doi: 10.1086/302593
6. Wu YW, Sullivan J, McDaniel SS, Meisler MH, Walsh EM, Li SX, et al. Incidence of Dravet syndrome in a US population. Pediatrics. (2015) 136:e1310–5. doi: 10.1542/peds.2015-1807
7. Scheffer IE, Nabbout R. SCN1A-related phenotypes: epilepsy and beyond. Epilepsia. (2019) 60 (Suppl. 3):S17–24. doi: 10.1111/epi.16386
8. Scheffer IE, Zhang YH, Jansen FE, Dibbens L. Dravet syndrome or genetic (generalized) epilepsy with febrile seizures plus? Brain Dev. (2009) 31:394–400. doi: 10.1016/j.braindev.2009.01.001
9. Deng H, Zheng W, Song Z. The genetics and molecular biology of fever-associated seizures or epilepsy. Expert Rev Mol Med. (2018) 20:e3. doi: 10.1017/erm.2018.2
10. Ding J, Wang L, Jin Z, Qiang Y, Li W, Wang Y, et al. Do all roads lead to Rome? Genes causing Dravet syndrome and Dravet syndrome-like phenotypes. Front Neurol. (2022) 13:832380. doi: 10.3389/fneur.2022.832380
11. Myers KA, Burgess R, Afawi Z, Damiano JA, Berkovic SF, Hildebrand MS, et al. De novo SCN1A pathogenic variants in the GEFS+ spectrum: not always a familial syndrome. Epilepsia. (2017) 58:e26–30. doi: 10.1111/epi.13649
12. Camfield P, Camfield C. Febrile seizures and genetic epilepsy with febrile seizures plus (GEFS+). Epileptic Disord. (2015) 17:124–33. doi: 10.1684/epd.2015.0737
13. Wallace RH, Wang DW, Singh R, Scheffer IE, George AL Jr, Phillips HA, et al. Febrile seizures and generalized epilepsy associated with a mutation in the Na+-channel beta1 subunit gene SCN1B. Nat Genet. (1998) 19:366–70. doi: 10.1038/1252
14. Escayg A, MacDonald BT, Meisler MH, Baulac S, Huberfeld G, An-Gourfinkel I, et al. Mutations of SCN1A, encoding a neuronal sodium channel, in two families with GEFS+2. Nat Genet. (2000) 24:343–5. doi: 10.1038/74159
15. Baulac S, Huberfeld G, Gourfinkel-An I, Mitropoulou G, Beranger A, Prud'homme JF, et al. First genetic evidence of GABA(A) receptor dysfunction in epilepsy: a mutation in the gamma2-subunit gene. Nat Genet. (2001) 28:46–8. doi: 10.1038/ng0501-46
16. Dibbens LM, Feng HJ, Richards MC, Harkin LA, Hodgson BL, Scott D, et al. GABRD encoding a protein for extra- or peri-synaptic GABAA receptors is a susceptibility locus for generalized epilepsies. Hum Mol Genet. (2004) 13:1315–9. doi: 10.1093/hmg/ddh146
17. Schubert J, Siekierska A, Langlois M, May P, Huneau C, Becker F, et al. Mutations in STX1B, encoding a presynaptic protein, cause fever-associated epilepsy syndromes. Nat Genet. (2014) 46:1327–32. doi: 10.1038/ng.3130
18. Bonzanni M, DiFrancesco JC, Milanesi R, Campostrini G, Castellotti B, Bucchi A, et al. A novel de novo HCN1 loss-of-function mutation in genetic generalized epilepsy causing increased neuronal excitability. Neurobiol Dis. (2018) 118:55–63. doi: 10.1016/j.nbd.2018.06.012
19. Dibbens LM, Reid CA, Hodgson B, Thomas EA, Phillips AM, Gazina E, et al. Augmented currents of an HCN2 variant in patients with febrile seizure syndromes. Ann Neurol. (2010) 67:542–6. doi: 10.1002/ana.21909
20. Mahoney K, Moore SJ, Buckley D, Alam M, Parfrey P, Penney S, et al. Variable neurologic phenotype in a GEFS+ family with a novel mutation in SCN1A. Seizure. (2009) 18:492–7. doi: 10.1016/j.seizure.2009.04.009
21. Chen X, Deng S, Xia H, Yuan L, Xu H, Tang S, et al. Identification of a CCDC114 variant in a Han-Chinese patient with situs inversus. Exp Ther Med. (2020) 20:3336–42. doi: 10.3892/etm.2020.9059
22. Deng S, Wu S, Xia H, Xiong W, Deng X, Liao J, et al. Identification of a frame shift mutation in the CCDC151 gene in a Han-Chinese family with Kartagener syndrome. Biosci Rep. (2020) 40:BSR20192510. doi: 10.1042/BSR20192510
23. Li H, Durbin R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics. (2009) 25:1754–60. doi: 10.1093/bioinformatics/btp324
24. Van der Auwera GA, Carneiro MO, Hartl C, Poplin R, Del Angel G, Levy-Moonshine A, et al. From FastQ data to high confidence variant calls: the Genome Analysis Toolkit best practices pipeline. Curr Protoc Bioinformatics. (2013) 43:11.10.1–33. doi: 10.1002/0471250953.bi1110s43
25. Yuan L, Xu H, Yuan J, Deng X, Xiong W, Yang Z, et al. A novel FN1 variant associated with familial hematuria: TBMN? Clin Biochem. (2016) 49:816–20. doi: 10.1016/j.clinbiochem.2016.01.026
26. Cingolani P, Platts A, Wang LL, Coon M, Nguyen T, Wang L, et al. A program for annotating and predicting the effects of single nucleotide polymorphisms, SnpEff: SNPs in the genome of Drosophila melanogaster strain w1118; iso-2; iso-3. Fly. (2012) 6:80–92. doi: 10.4161/fly.19695
27. Sherry ST, Ward MH, Kholodov M, Baker J, Phan L, Smigielski EM, et al. dbSNP: the NCBI database of genetic variation. Nucleic Acids Res. (2001) 29:308–11. doi: 10.1093/nar/29.1.308
28. Landrum MJ, Chitipiralla S, Brown GR, Chen C, Gu B, Hart J, et al. ClinVar: improvements to accessing data. Nucleic Acids Res. (2020) 48:D835–44. doi: 10.1093/nar/gkz972
29. Karczewski KJ, Francioli LC, Tiao G, Cummings BB, Alföldi J, Wang Q, et al. The mutational constraint spectrum quantified from variation in 141,456 humans. Nature. (2020) 581:434–43. doi: 10.1038/s41586-020-2308-7
30. Tennessen JA, Bigham AW, O'Connor TD, Fu W, Kenny EE, Gravel S, et al. Evolution and functional impact of rare coding variation from deep sequencing of human exomes. Science. (2012) 337:64–9. doi: 10.1126/science.1219240
31. Li L, Huang P, Sun X, Wang S, Xu M, Liu S, et al. The ChinaMAP reference panel for the accurate genotype imputation in Chinese populations. Cell Res. (2021) 31:1308–10. doi: 10.1038/s41422-021-00564-z
32. 1000 Genomes Project Consortium, Auton A, Brooks LD, Durbin RM, Garrison EP, Kang HM, et al. A global reference for human genetic variation. Nature. (2015) 526:68–74. doi: 10.1038/nature15393
33. Yuan M, Guo Y, Xia H, Xu H, Deng H, Yuan L. Novel SCN5A and GPD1L variants identified in two unrelated Han-Chinese patients with clinically suspected Brugada syndrome. Front Cardiovasc Med. (2021) 8:758903. doi: 10.3389/fcvm.2021.758903
34. Schwarz JM, Cooper DN, Schuelke M, Seelow D. MutationTaster2: mutation prediction for the deep-sequencing age. Nat Methods. (2014) 11:361–2. doi: 10.1038/nmeth.2890
35. Choi Y, Chan AP, PROVEAN web server: a tool to predict the functional effect of amino acid substitutions and indels. Bioinformatics. (2015) 31:2745–7. doi: 10.1093/bioinformatics/btv195
36. Adzhubei IA, Schmidt S, Peshkin L, Ramensky VE, Gerasimova A, Bork P, et al. A method and server for predicting damaging missense mutations. Nat Methods. (2010) 7:248–9. doi: 10.1038/nmeth0410-248
37. Reva B, Antipin Y, Sander C. Predicting the functional impact of protein mutations: application to cancer genomics. Nucleic Acids Res. (2011) 39:e118. doi: 10.1093/nar/gkr407
38. Altschul SF, Gish W, Miller W, Myers EW, Lipman DJ. Basic local alignment search tool. J Mol Biol. (1990) 215:403–10. doi: 10.1016/S0022-2836(05)80360-2
39. Waterhouse A, Bertoni M, Bienert S, Studer G, Tauriello G, Gumienny R, et al. SWISS-MODEL: homology modelling of protein structures and complexes. Nucleic Acids Res. (2018) 46:W296–303. doi: 10.1093/nar/gky427
40. Richards S, Aziz N, Bale S, Bick D, Das S, Gastier-Foster J, et al. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet Med. (2015) 17:405–24. doi: 10.1038/gim.2015.30
41. Cheng D, Yan X, Xu K, Zhou X, Chen Q. The effect of interictal epileptiform discharges on cognitive and academic performance in children with idiopathic epilepsy. BMC Neurol. (2020) 20:233. doi: 10.1186/s12883-020-01807-z
42. Peycheva V, Ivanova N, Kamenarova K, Panova M, Pacheva I, Ivanov I, et al. SCN1A mutation spectrum in a cohort of Bulgarian patients with GEFS+ phenotype. Turk J Pediatr. (2020) 62:711–25. doi: 10.24953/turkjped.2020.05.002
43. Myers KA, Scheffer IE, Berkovic SF, ILAE Genetics Commission. Genetic literacy series: genetic epilepsy with febrile seizures plus. Epileptic Disord. (2018) 20:232–8. doi: 10.1684/epd.2018.0985
44. Cetica V, Chiari S, Mei D, Parrini E, Grisotto L, Marini C, et al. Clinical and genetic factors predicting Dravet syndrome in infants with SCN1A mutations. Neurology. (2017) 88:1037–44. doi: 10.1212/WNL.0000000000003716
45. Plummer NW, Meisler MH. Evolution and diversity of mammalian sodium channel genes. Genomics. (1999) 57:323–31. doi: 10.1006/geno.1998.5735
46. Catterall WA. From ionic currents to molecular mechanisms: the structure and function of voltage-gated sodium channels. Neuron. (2000) 26:13–25. doi: 10.1016/S0896-6273(00)81133-2
47. Catterall WA, Kalume F, Oakley JC. Nav1.1 channels and epilepsy. J Physiol. (2010) 588:1849–59. doi: 10.1113/jphysiol.2010.187484
48. Alekov AK, Rahman MM, Mitrovic N, Lehmann-Horn F, Lerche H. Enhanced inactivation and acceleration of activation of the sodium channel associated with epilepsy in man. Eur J Neurosci. (2001) 13:2171–6. doi: 10.1046/j.0953-816x.2001.01590.x
49. Lu Q, Guo Y, Yi J, Deng X, Yang Z, Yuan X, et al. Identification of an ND4 mutation in Leber hereditary optic neuropathy. Optom Vis Sci. (2017) 94:1090–4. doi: 10.1097/OPX.0000000000001147
50. Deschênes I, Trottier E, Chahine M. Implication of the C-terminal region of the alpha-subunit of voltage-gated sodium channels in fast inactivation. J Membr Biol. (2001) 183:103–14. doi: 10.1007/s00232-001-0058-5
51. Ohmori I, Ouchida M, Ohtsuka Y, Oka E, Shimizu K. Significant correlation of the SCN1A mutations and severe myoclonic epilepsy in infancy. Biochem Biophys Res Commun. (2002) 295:17–23. doi: 10.1016/S0006-291X(02)00617-4
52. Ragsdale DS. How do mutant Nav1.1 sodium channels cause epilepsy? Brain Res Rev. (2008) 58:149–59. doi: 10.1016/j.brainresrev.2008.01.003
53. Rhodes TH, Lossin C, Vanoye CG, Wang DW, George AL Jr. Noninactivating voltage-gated sodium channels in severe myoclonic epilepsy of infancy. Proc Natl Acad Sci U S A. (2004) 101:11147–52. doi: 10.1073/pnas.0402482101
54. Mantegazza M, Curia G, Biagini G, Ragsdale DS, Avoli M. Voltage-gated sodium channels as therapeutic targets in epilepsy and other neurological disorders. Lancet Neurol. (2010) 9:413–24. doi: 10.1016/S1474-4422(10)70059-4
55. Martin MS, Dutt K, Papale LA, Dubé CM, Dutton SB, de Haan G, et al. Altered function of the SCN1A voltage-gated sodium channel leads to gamma-aminobutyric acid-ergic (GABAergic) interneuron abnormalities. J Biol Chem. (2010) 285:9823–34. doi: 10.1074/jbc.M109.078568
56. Hayashi K, Ueshima S, Ouchida M, Mashimo T, Nishiki T, Sendo T, et al. Therapy for hyperthermia-induced seizures in Scn1a mutant rats. Epilepsia. (2011) 52:1010–7. doi: 10.1111/j.1528-1167.2011.03046.x
57. Ohmori I, Kahlig KM, Rhodes TH, Wang DW, George AL Jr. Nonfunctional SCN1A is common in severe myoclonic epilepsy of infancy. Epilepsia. (2006) 47:1636–42. doi: 10.1111/j.1528-1167.2006.00643.x
58. Sun L, Gilligan J, Staber C, Schutte RJ, Nguyen V, O'Dowd DK, et al. A knock-in model of human epilepsy in Drosophila reveals a novel cellular mechanism associated with heat-induced seizure. J Neurosci. (2012) 32:14145–55. doi: 10.1523/JNEUROSCI.2932-12.2012
59. Yu FH, Mantegazza M, Westenbroek RE, Robbins CA, Kalume F, Burton KA, et al. Reduced sodium current in GABAergic interneurons in a mouse model of severe myoclonic epilepsy in infancy. Nat Neurosci. (2006) 9:1142–9. doi: 10.1038/nn1754
Keywords: genetic epilepsy with febrile seizures plus, SCN1A, whole exome sequencing, missense variant, genetic diagnoses
Citation: Li L, Yuan L, Zheng W, Yang Y, Deng X, Song Z and Deng H (2023) An SCN1A gene missense variant in a Chinese Tujia ethnic family with genetic epilepsy with febrile seizures plus. Front. Neurol. 14:1229569. doi: 10.3389/fneur.2023.1229569
Received: 26 May 2023; Accepted: 30 June 2023;
Published: 27 July 2023.
Edited by:
Tao Sun, Ningxia Medical University, ChinaReviewed by:
Jiangwei Ding, First Affiliated Hospital of Zhengzhou University, ChinaJianxiang Liao, Shenzhen Children's Hospital, China
Copyright © 2023 Li, Yuan, Zheng, Yang, Deng, Song and Deng. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Hao Deng, hdeng008@163.com
†These authors have contributed equally to this work