 Li Shao1†
Li Shao1† Haoyi Wang
Haoyi Wang Ming Qi
Ming Qi Zhaonan Yu
Zhaonan Yu Jing Zhang
Jing Zhang
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
CASE REPORT article
Front. Neurol., 26 July 2023
Sec. Neurogenetics
Volume 14 - 2023 | https://doi.org/10.3389/fneur.2023.1228810
This article is part of the Research TopicCase Reports in Neurogenetics, volume III - 2023View all 20 articles
Background: Ataxia-telangiectasia (A-T) is a multisystem genetic disorder involving ataxia, oculocutaneous telangiectasia, and immunodeficiency caused by biallelic pathogenic variants in the ATM gene. To date, most ATM variants have been reported in the Caucasian population, and few studies have focused on the genotype–phenotype correlation of A-T in the Chinese population. We herein present a Chinese patient with A-T who carries compound heterozygous variants in the ATM gene and conducted a literature review for A-T in China.
Case presentation: A 7-year-old Chinese girl presented with growth retardation, ataxia, medium ocular telangiectasia, cerebellar atrophy, and elevated serum alpha-fetoprotein (AFP) level, which supported the suspicion of A-T. Notably, the serum levels of immunoglobulins were all normal, ruling out immunodeficiency. Exome sequencing and Sanger sequencing revealed two likely pathogenic ATM variants, namely NM_000051.4: c.4195dup (p.Thr1399Asnfs*15) and c.6006 + 1G>T (p.?), which were inherited from her father and mother, respectively. From the Chinese literature review, we found that there was a marked delay in the diagnosis of A-T, and 38.9% (7/18) of A-T patients did not suffer from immunodeficiency in China. No genotype–phenotype correlation was observed in this group of A-T patients.
Conclusion: These results extend the genotype spectrum of A-T in the Chinese population and imply that the diagnosis of A-T in China should be improved.
Ataxia-telangiectasia (A-T; OMIM no. 208900) is a rare multisystem disorder characterized by ataxia, oculocutaneous telangiectasia, global developmental delay, immunodeficiency, radiation hypersensitivity, and/or increased susceptibility to malignancy particularly of lymphoid origin (1–3). This condition is caused by biallelic pathogenic variants in the ATM gene located at chromosome 11q22.3 (1–3). The phenotype of A-T is associated with some degree of preservation of ATM kinase activity. The classical form of A-T is a severe multisystem disorder with an early onset, progressive, and neurodegenerative course due to biallelic loss-of-function variants in the ATM gene, whereas the milder type of A-T, characterized by slow neurological progression and/or later onset and designated “variant A-T”, is caused by biallelic variants with at least one non-truncating variant (missense or splice site variant) resulting in the presence of residual ATM kinase activity (3). The ATM gene harbors 66 exons and encodes a PI3K-family kinase with 3,050 amino acid residues. Hitherto, more than 1,400 unique variants have been identified in the ATM gene (https://www.lovd.nl/ATM).
However, studies investigating ATM variants and A-T in China are scarce (4–9). Herein, we describe a Chinese female patient affected by A-T with compound heterozygous variants in the ATM gene, one of which is novel. Furthermore, we review the literature on the genotype–phenotype spectrum of A-T in the Chinese population.
The proband, a 7-year-old Chinese girl, was referred to our hospital for the evaluation of growth retardation. Her weight and height were below the 3rd percentile, and she exhibited medium ocular telangiectasia since the age of 5. Neurological examinations revealed progressive limb and truncal ataxia, diagnosed on the basis of gait instability, dysarthria, spasticity, and delayed cognitive and motor development. At the last follow-up visit, the 9-year-old patient was not wheelchair-bound. The parents reported that the proband initially developed symptoms of ataxia at the age of 2. Laboratory tests showed that the serum levels of α-fetoprotein (AFP), lactic dehydrogenase (LDH), and carbohydrate antigen 199 (CA199) were significantly elevated, whereas the serum levels of carcinoembryonic antigen (CEA) and immunoglobulins (IgA, IgG, IgM, and IgE) levels were within the normal range (Table 1). Brain magnetic resonance imaging (MRI) unveiled cerebellar atrophy with the enlargement of cerebellar sulci (Figure 1). The parents were both normal, and no relatives were reported to have neurodegenerative diseases except for the grandpa's cousin who presented amentia. A-T was suspected based on the clinical features of ataxia, cerebellar atrophy, ocular telangiectasia, and elevated serum AFP level. Given the absence of immunodeficiency and the conservative wishes of the parents, we decided to follow up with the patient at present. Medical interventions will not be administered until the disease progresses.
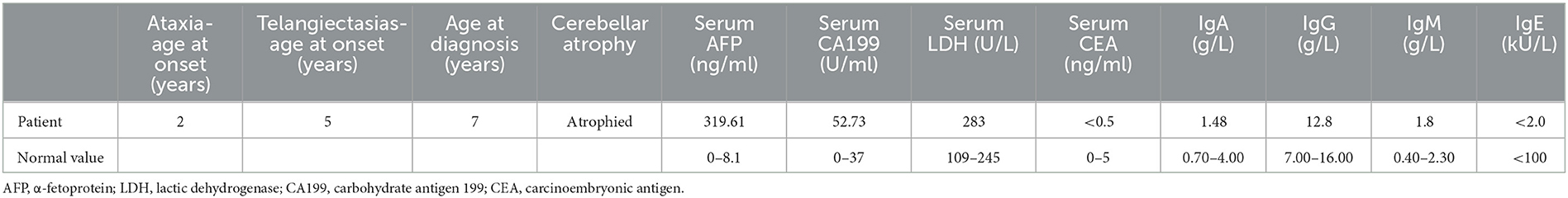
Table 1. Clinical and laboratory features of the patient.
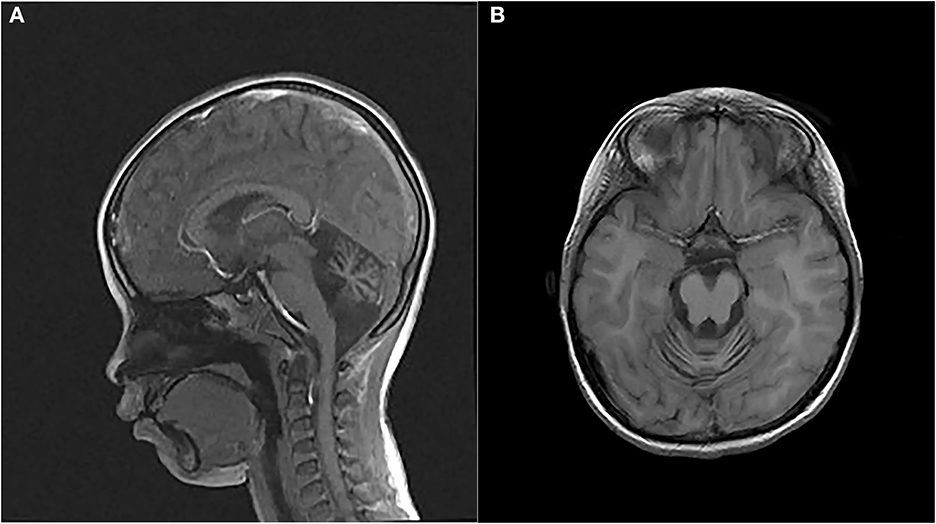
Figure 1. Magnetic resonance images (MRIs) of the patient's brain. (A) Sagittal T2-weighted brain MRI. (B) Axial T1-weighted brain MRI.
To validate the clinical suspicion of A-T, exome sequencing was performed using the peripheral blood from the patient. The procedures of genetic analysis and the filtering condition for variants were described by Shalash et al. (10). We identified compound heterozygous variants in the ATM gene, NM_000051.4 (ATM): c.4195dup and c.6006 + 1G>T. The intronic variant c.6006 + 1G>T (ClinVar Variation ID: 2029577) was expected to disrupt RNA splicing by affecting a donor splice site in intron 40 of the ATM gene, thereby it was classified as pathogenic in the ClinVar database. The variant c.4195dup located at exon 28 led to a frameshift at residue 1399, which was predicted to produce a truncated protein of 1,412 amino acids (p.Thr1399Asnfs*15) lacking the FAT, PI3K/PI4K catalytic, and FATC domain. The CADD and GERP scores were 33 and 4.22, respectively, both of which exceeded the threshold for pathogenicity. Using the MutationTaster tool, both variants were predicted to be disease causing. Familial segregation analysis was verified using Sanger sequencing. The compound heterozygous variants, c.4195dup and c.6006 + 1G>T, were inherited from the unaffected father and mother, respectively (Figure 2). The frameshift variant c.4195dup was not included in the gnomeAD, HGMD, and 1000G databases, thereby confirming its novelty. We did not identify any pathogenic variants in other nervous system-related genes.
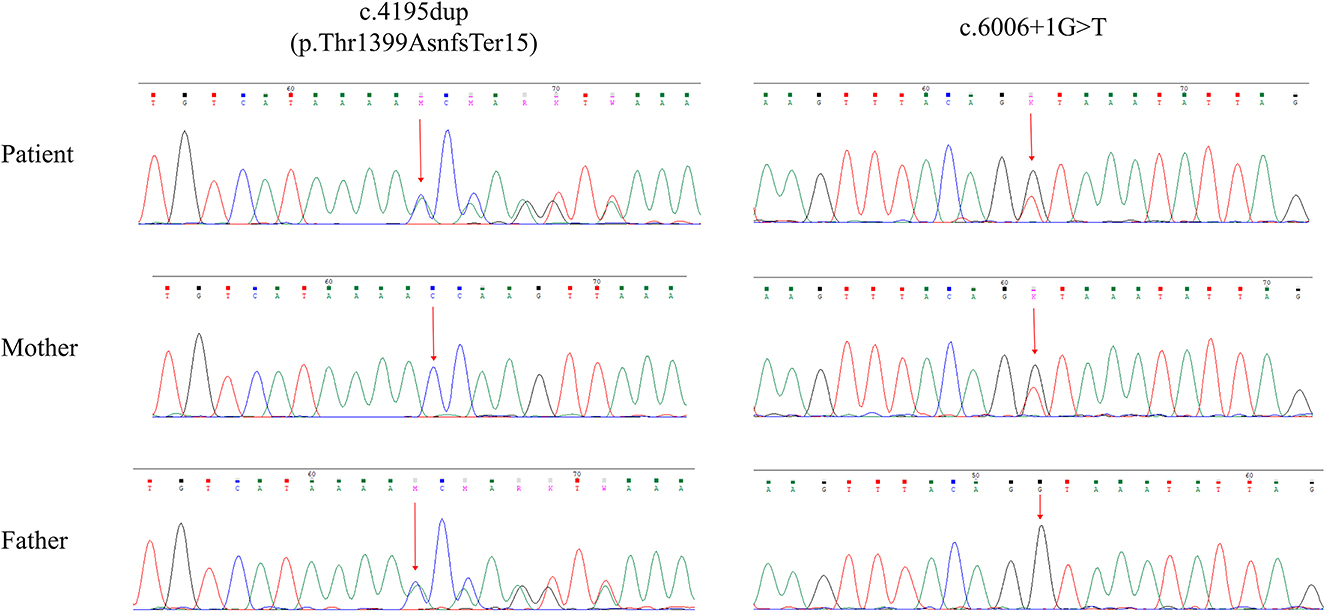
Figure 2. Sanger sequencing validation of the ATM variants of all family members.
Seventeen Chinese cases with A-T from six studies were collected to reveal the genotypic and phenotypic features of A-T in China (Table 2) (4–9). Combined with the case in this article, we found that there was a delay of ≥5 years in diagnosis since the onset of clinical presentations in most cases. With regard to the ATM genotype, compound heterozygosity is the major allelic type in China possibly due to the legal prohibition of consanguineous marriage in our country. Most cases had at least one ATM truncated (frameshift or non-sense) variant, which is in agreement with the classical clinical signs of A-T. Seven cases did not present immunodeficiency, which is one of the cardinal clinical indications in A-T. No genotype–phenotype correlation was found in this group of A-T patients.
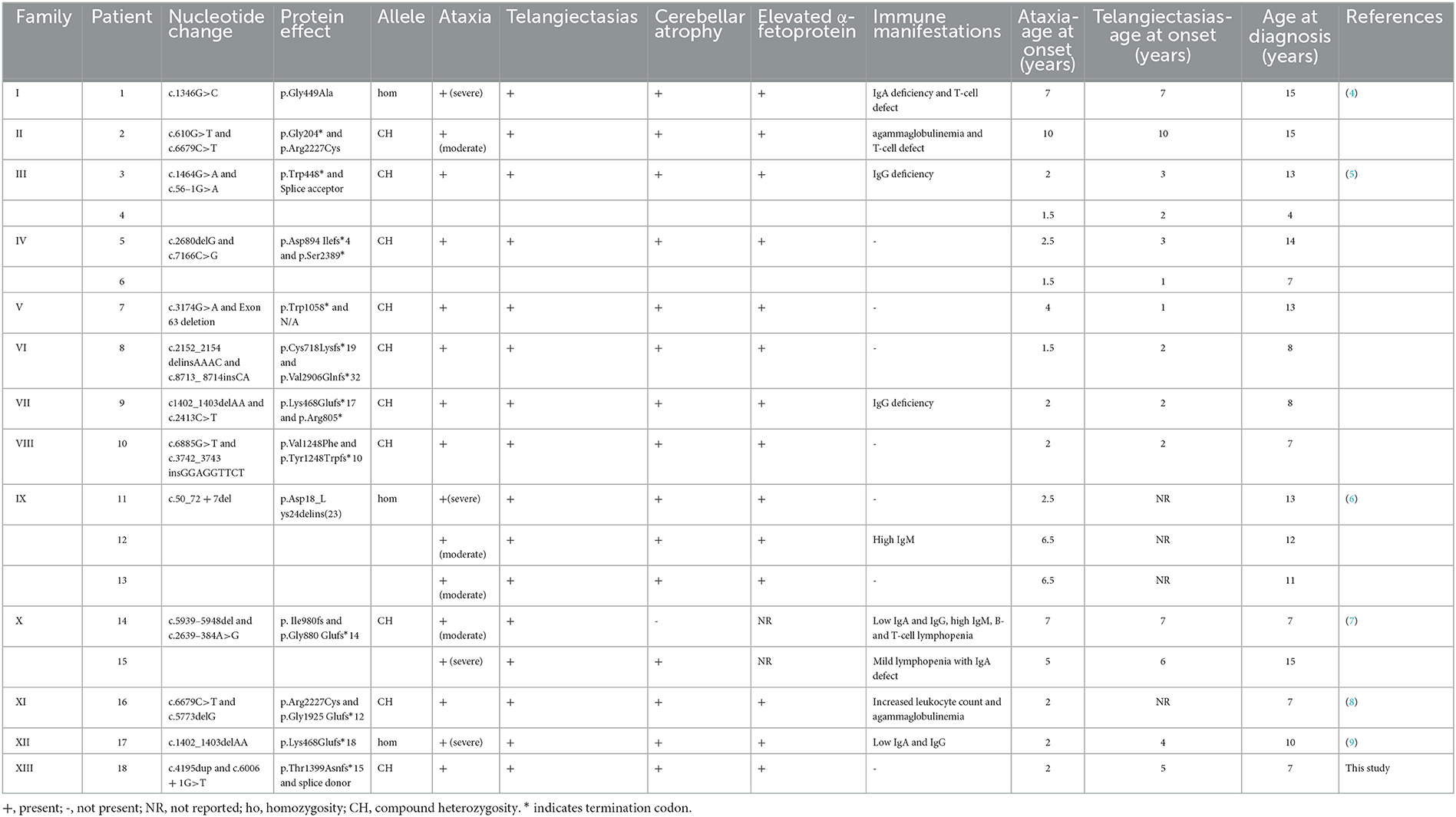
Table 2. Literature review for Chinese patients with ataxia-telangiectasia.
We report a Chinese patient affected by A-T with compound heterozygous ATM genotype; one of the identified variants is unreported. In China, patients are suspected of A-T only when classical clinical signs, namely ataxia and oculocutaneous telangiectasia are present. However, the onset age of ataxia and oculocutaneous telangiectasia is sometimes different. Some manifestations may even occur prior to ataxia and oculocutaneous telangiectasia, such as recurrent infections of unknown origins (7). These factors, combined with the extremely low prevalence of A-T at 1:40,000–1:300,000 (3), collectively lead to a delay in the diagnosis of A-T in China. According to the Chinese literature review (Table 2), it takes more than 5 years in China to complete the diagnosis of A-T since the onset of symptoms; therefore, great efforts are still warranted to ameliorate the diagnosis of this rare genetic disorder. Devaney et al. (11) reported the presentation and diagnostic delay in A-T, which further consolidated our conclusions.
The early measurement of serum AFP is an easily detectable and reliable biological hallmark of A-T (3). As expected, the serum AFP level is elevated in all reported Chinese patients with A-T (Table 2), except for two patients whose AFP levels were not reported (7). Immunodeficiency is also an important indicator for A-T, which is associated with ATM protein dysfunction in immunoglobulin class-switch recombination (CSR), V(D)J recombination, and B- and T-cell homeostasis (12, 13). It has been reported that immunodeficiency is present in approximately two-thirds of A-T patients (1, 3). The Chinese literature review (Table 2) shows that 38.9% (7/18) of Chinese patients with A-T, including the one in our case, have normal immune status at the time of diagnosis, which is in agreement with the literature data. For these patients who have no signs of immunodeficiency, elevated serum AFP levels and neurological manifestations are the most suggestive.
In conclusion, our findings extend the genotype spectrum of A-T in the Chinese population and suggest that the diagnosis of A-T in China should be improved in clinical practice. The limitations of our study are the small number of patients and a lack of functional investigation.
The datasets presented in this article are not readily available because of ethical and privacy restrictions. Requests to access the datasets should be directed to the corresponding authors.
The studies involving human participants were reviewed and approved by the Ethics Committee of Jinhua Maternity and Child Health Care Hospital. Written informed consent to participate in this study was provided by the participants' legal guardian/next of kin. Written informed consent was obtained from the individual(s) and minor(s)' legal guardian/next of kin, for the publication of any potentially identifiable images or data included in this article. Written informed consent was obtained from the participant/patient(s) for the publication of this case report.
JX, LS, and JZ were the patient's physicians. HW and MQ reviewed the literature and contributed to manuscript drafting. HW and ZY performed the mutation analysis. JX and JZ conceptualized and designed the study, coordinated and supervised data collection, and critically reviewed the manuscript for important intellectual content. HW, ZY, and JZ were responsible for the revision of the manuscript for important intellectual content. All authors contributed to the article and approved the submitted version.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
1. Rothblum-Oviatt C, Wright J, Lefton-Greif MA, McGrath-Morrow SA, Crawford TO, Lederman HM. Ataxia telangiectasia: a review. Orphanet J Rare Dis. (2016) 11:159. doi: 10.1186/s13023-016-0543-7
2. Amirifar P, Ranjouri MR, Yazdani R, Abolhassani H, Aghamohammadi A. Ataxia-telangiectasia: a review of clinical features and molecular pathology. Pediatr Allergy Immunol. (2019) 30:277–88. doi: 10.1111/pai.13020
3. Amirifar P, Ranjouri MR, Lavin M, Abolhassani H, Yazdani R, Aghamohammadi A. Ataxia-telangiectasia: epidemiology, pathogenesis, clinical phenotype, diagnosis, prognosis and management. Expert Rev Clin Immunol. (2020) 16:859–71. doi: 10.1080/1744666X.2020.1810570
4. Jiang H, Tang B, Xia K, Hu Z, Shen L, Tang J, et al. Mutation analysis of the ATM gene in two Chinese patients with ataxia telangiectasia. J Neurol Sci. (2006) 241:1–6. doi: 10.1016/j.jns.2005.09.001
5. Huang Y, Yang L, Wang J, Yang F, Xiao Y, Xia R, et al. Twelve novel Atm mutations identified in Chinese ataxia telangiectasia patients. Neuromolecular Med. (2013) 15:536–40. doi: 10.1007/s12017-013-8240-3
6. Chen W, Liu S, Hu H, Chen G, Zhu S, Jia B, et al. Novel homozygous ataxia-telangiectasia (A-T) mutated gene mutation identified in a Chinese pedigree with A-T. Mol Med Rep. (2019) 20:1655–62. doi: 10.3892/mmr.2019.10402
7. Gu C, Wang H, Shu J, Zheng J, Li D, Cai C, et al. RNA sequencing combining with whole exome sequencing reveals a compound heterozygous variant in ATM in a girl with atypical ataxia-telangiectasia. Clin Chim Acta. (2021) 523:6–9. doi: 10.1016/j.cca.2021.08.026
8. Li XL, Wang YL. Ataxia-telangiectasia complicated with Hodgkin's lymphoma: A case report. World J Clin Cases. (2020) 8:2387–91. doi: 10.12998/wjcc.v8.i11.2387
9. Zhang L, Jia Y, Qi X, Li M, Wang S, Wang Y. Trihexyphenidyl for treatment of dystonia in ataxia telangiectasia: a case report. Childs Nerv Syst. (2020) 36:873–5. doi: 10.1007/s00381-019-04399-3
10. Shalash AS, Rösler TW, Salama M, Pendziwiat M, Müller SH, Hopfner F, et al. Evidence for pathogenicity of variant ATM Val1729Leu in a family with ataxia telangiectasia. Neurogenetics. (2021) 22:143–7. doi: 10.1007/s10048-021-00639-4
11. Devaney R, Pasalodos S, Suri M, Bush A, Bhatt JM. Ataxia telangiectasia: presentation and diagnostic delay. Arch Dis Child. (2017) 102:328–30. doi: 10.1136/archdischild-2016-310477
12. Bakkenist CJ, Kastan MB, DNA. Damage activates ATM through intermolecular autophosphorylation and dimer dissociation. Nature. (2003) 421:499–506. doi: 10.1038/nature01368
13. Driessen GJ, Ijspeert H, Weemaes CM, Haraldsson Á, Trip M, Warris A, et al. Antibody deficiency in patients with ataxia telangiectasia is caused by disturbed B- and T-cell homeostasis and reduced immune repertoire diversity. J Allergy Clin Immunol. (2013) 131:1367–75. doi: 10.1016/j.jaci.2013.01.053
Keywords: ataxia-telangiectasia, ATM gene, cerebellar atrophy, case report, literature review
Citation: Shao L, Wang H, Xu J, Qi M, Yu Z and Zhang J (2023) Ataxia-telangiectasia in China: a case report of a novel ATM variant and literature review. Front. Neurol. 14:1228810. doi: 10.3389/fneur.2023.1228810
Received: 25 May 2023; Accepted: 04 July 2023;
Published: 26 July 2023.
Edited by:
Félix Javier Jiménez-Jiménez, Hospital Universitario del Sureste, SpainReviewed by:
Pedro J. Garcia-Ruiz, University Hospital Fundación Jiménez Díaz, SpainCopyright © 2023 Shao, Wang, Xu, Qi, Yu and Zhang. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Jing Zhang, emhhbmdqMDk2QHllYWgubmV0; Zhaonan Yu, eXV6aGFvbmFuODcwMjEzQDE2My5jb20=
†These authors have contributed equally to this work
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.