 Querin Giorgia
Querin Giorgia Marta Gomez Garcia de la Banda
Marta Gomez Garcia de la Banda Piera Smeriglio
Piera Smeriglio- 1APHP, Service de Neuromyologie, Hôpital Pitié-Salpêtrière, Centre Référent pour les Maladies Neuromusculaires Nord/Est/Ile de France, Paris, France
- 2Institut de Myologie, I-Motion Clinical Trials Platform, Paris, France
- 3European Reference Center Network (Euro-NMD ERN), Paris, France
- 4APHP, Pediatric Neurology Department, Hôpital Armand Trousseau, Centre Référent pour les Maladies Neuromusculaires Nord/Est/Ile de France, Paris, France
- 5APHP, Pediatric Neurology and ICU Department, Université Paris Saclay, DMU Santé de l'Enfant et de l'Adolescent, Hôpital Raymond Poincaré, Garches, France
- 6Centre of Research in Myology, Institute of Myology, Sorbonne Université, INSERM, Paris, France
Spinal muscular atrophy (SMA) is a lower motor neuron disease due to biallelic mutations in the SMN1 gene on chromosome 5. It is characterized by progressive muscle weakness of limbs, bulbar and respiratory muscles. The disease is usually classified in four different phenotypes (1–4) according to age at symptoms onset and maximal motor milestones achieved. Recently, three disease modifying treatments have received approval from the Food and Drug Administration (FDA) and the European Medicines Agency (EMA), while several other innovative drugs are under study. New therapies have been game changing, improving survival and life quality for SMA patients. However, they have also intensified the need for accurate biomarkers to monitor disease progression and treatment efficacy. While clinical and neurophysiological biomarkers are well established and helpful in describing disease progression, there is a great need to develop more robust and sensitive circulating biomarkers, such as proteins, nucleic acids, and other small molecules. Used alone or in combination with clinical biomarkers, they will play a critical role in enhancing patients’ stratification for clinical trials and access to approved treatments, as well as in tracking response to therapy, paving the way to the development of individualized therapeutic approaches. In this comprehensive review, we describe the foremost circulating biomarkers of current significance, analyzing existing literature on non-treated and treated patients with a special focus on neurofilaments and circulating miRNA, aiming to identify and examine their role in the follow-up of patients treated with innovative treatments, including gene therapy.
1. Introduction
5q-Spinal muscular atrophy (SMA) is a lower motor neuron (LMN) disease characterized by anterior horn degeneration in the brainstem and spinal cord (SC), muscle weakness and atrophy. It occurs in both pediatric and adult forms with symptoms manifesting from birth (SMA type 0 and 1) to adulthood (SMA type 4) (1). SMA is caused by recessive deletions or loss-of-function mutations in the SMN1 gene (chromosome 5q11.2-q13.3), with an incidence of 1 in 10,000 live births and a carrier frequency of about 1 in 50 (2–5). Complete dysfunction of SMN1 is necessary to determine the SMA phenotype (6–8), with gene deletions accounting for 95% of SMA cases. About 3% of the patients are compound heterozygotes for a deletion and an intragenic point mutation (9). The SMN2 gene, a centromeric paralog of SMN1, is the main genetic modifier of the SMA phenotype and disease severity: multiple copies of SMN2 may be present in the genome (up to 5 or 6 in SMA type 3 and 4), with a higher number inversely correlating with disease severity (10, 11). The SMN2 gene sequence is almost identical to that of SMN1, except for a few nucleotides causing a differential splicing of the gene leading to the production of a truncated and non-functional SMN protein and only to a small amount of residual functional SMN. This event is sufficient to partially compensate for the functional protein loss due to SMN1 mutations.
The SMN protein is highly conserved and widely expressed both in the cytoplasm and the nucleus of cells in all somatic tissues and particularly abundant in motor neurons (12). The complex functions of the SMN protein span from mRNA splicing and snRNP assembly to mRNA trafficking and translation (13). Therefore, loss of SMN expression has important consequences on various aspects of cellular and tissue functions (14–16).
From a clinical point of view, SMA is characterized by irreversible and progressive muscle weakness of axial, limb and bulbar muscles (17, 18). SMA patients exhibit considerable clinical heterogeneity and are traditionally classified in four groups based on the age at onset and maximal motor milestones achieved (with type 1 being the most severe and early onset form, and type 4 being an adult and milder form) (19, 20).
In recent years, significant efforts have been made in the development of new therapies for 5q-SMA (21), resulting in the approval of three disease-modifying treatments by the FDA and EMA. These innovative therapeutic approaches aim either at increasing SMN protein circulating levels by mediating alternative SMN2 splicing (antisense oligonucleotides as nusinersen and small molecules as risdiplam) (22, 23) or at replacing the mutated SMN1 gene (viral vector mediated gene therapy) (24). Other strategies, such as neuroprotective drugs and molecules to enhance muscle strength (e.g., several anti-myostatin drugs) (25, 26) are also under investigation. All these new therapies have transformed the natural history of the disease, leading to improved survival in patients (particularly in type 1) and to the emergence of new phenotypes, with varying neuromuscular prognoses (27, 28). Clinical evolution of treated and non-treated patients is usually performed using neuromuscular evaluations of muscle strength, motor capacities and bulbar and respiratory function. Clinical tools such as SC imaging (29), muscle MRI (30, 31) and neurophysiology (motor unit count) (32, 33) are being explored as possible biomarkers for the follow-up of patients. However, more specific, and sensitive biomarkers capable of assessing the effect of the treatment and of predicting disease trajectories over time are still partially lacking. Moreover, clinical heterogeneity and variability of therapeutic response between treated patients are still a major challenge in SMA. Therefore, there is a growing emphasis on the search for accurate and reliable molecular circulating biomarkers to better classify patients, guide personalized treatment decisions and grant an appropriate post treatment follow-up (34–36).
The objective of this review is to evaluate the latest developments in the rapidly advancing field of molecular biomarkers for SMA, with a particular focus on indicators that have proven useful for both natural history studies and post-treatment investigations. Different putative predictive biomarkers and their role in treated patients are summarized in Table 1.
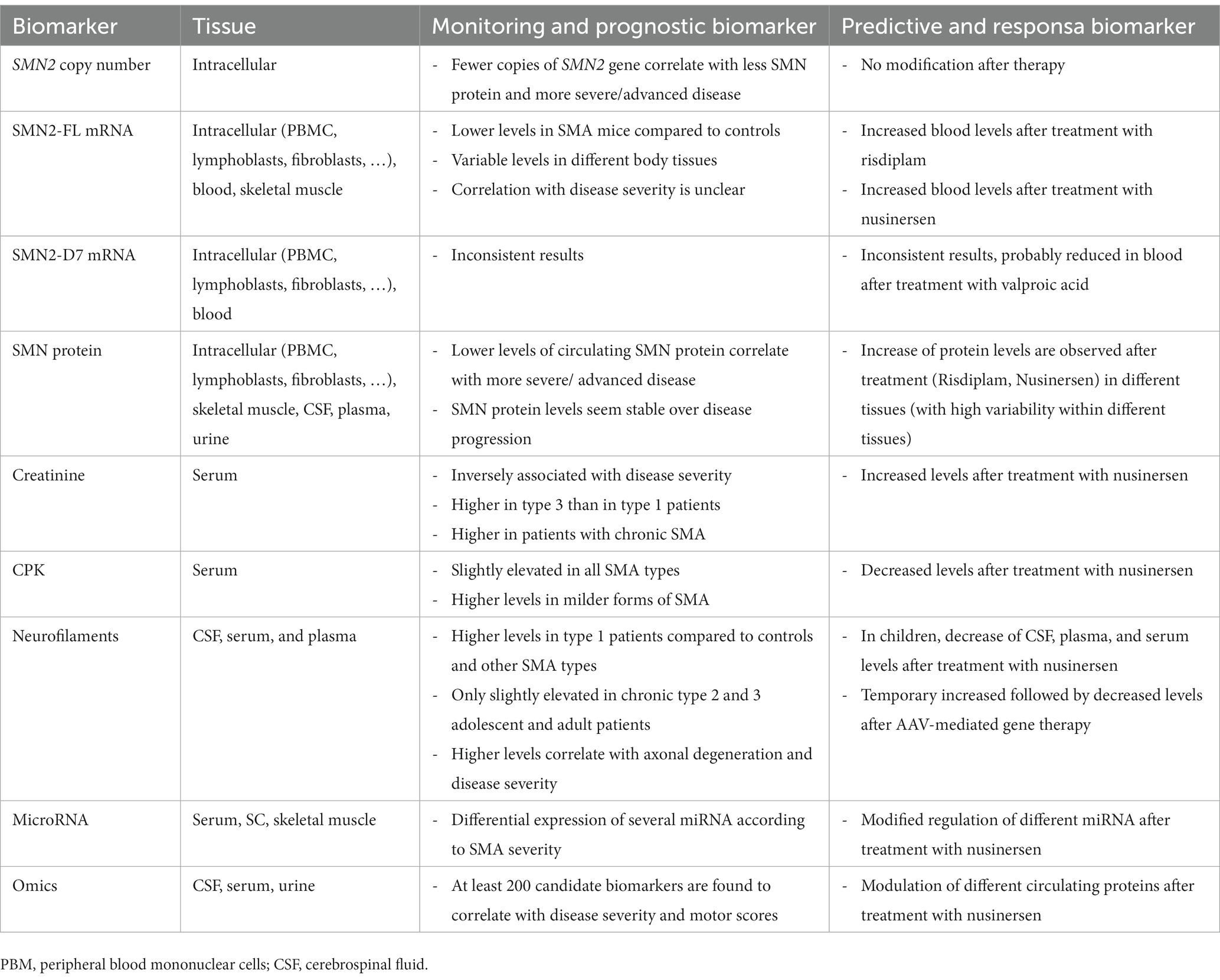
Table 1. Summary of relevant putative biomarkers for untreated and treated SMA patients.
2. Circulating biomarkers in SMA
A biomarker is defined as a characteristic that is objectively measured, serving as an indicator of normal biological processes, pathologic processes, or biological responses to a therapeutic intervention (as per the USA FDA definition). Biomarkers can be identified as: susceptibility/risk, diagnostic, monitoring, prognostic, predictive, response and safety biomarker, diagnostic, prognostic, predictive and pharmacodynamic (37, 38).
Circulating biomarkers are molecules (proteins, antibodies, genetic material…) that can be measured in body fluids such as blood, serum, or cerebrospinal fluid (CSF). Reliable circulating biomarkers should be easy to quantify and possibly correlated to the patients’ clinical function. They should also effectively predict response to therapy, as well as support early preclinical diagnosis, prognosis and follow-up in treated and not treated patients of all ages (39). For each of the summarized biomarkers, we will include the general state-of-the-art knowledge and an additional focus on their relevance in assessing patients’ response to treatment.
2.1. Genetic based biomarkers
2.1.1. SMN2 copy number
2.1.1.1. Diagnostic and monitoring biomarker
The number of SMN2 copies in an individual is directly linked to their SMN expression level, meaning that a greater number of SMN2 copies is connected to a less severe phenotype (6). The SMN2 copy number, when taken into account alongside factors such as the age at symptom onset and the level of motor skills achieved, functions as an additional criterion for classifying individuals with SMA. Consequently, it can be regarded as a robust indicator of disease severity, applicable to both pediatric and adult patients (40). Despite these aspects, the correlation between SMN2 copy number and patient’s phenotype is not always accurate. There are individuals with 2 copies of SMN2 and less severe symptoms and inversely patients with 3–4 copies and classified as phenotypically very severe (41). These discrepancies have therefore pushed many groups to identify other potential modifiers of clinical phenotype (42). Some of them act as protective modifiers (for example coronin-1C-CORO1C, neurocalcin-NCALD or calcineurin-like EF-hand protein-CHP1) (43–45). Plastin-3 (PSL3) (46, 47), a F-actin-bundling protein localized on the X-chromosome involved in neurotransmitter release and vesicle recycling, is considered to be a positive phenotype modifier of particular interest (48).
2.1.1.2. Predictive biomarkers after treatment
PSL3 overexpression has been shown to improve the clinical phenotype both in animal models of SMA (49) and in human patients (50), with subjects coming from the same family and having the same SMN2 copy number presenting with variable severity according to PSL3 serum levels. Studies on SMA mice have also demonstrated increased efficacy of treatment by nusinersen when combined with overexpression of human PSL3 (51), indicating that PSL3, as well as other modifiers, should be taken into consideration in the perspective of a personalized management of each patient. However, further studies are warranted to confirm the role of PSL3 as a predictive biomarker, while studies performed in patients that measured mRNA found a differential effect depending on age and sex, making the interpretation of the results complex (52).
2.1.2. Survival motor neuron (SMN) mRNA transcript levels
Two different transcripts have been proposed as biomarkers of disease severity: SMN2–full length (SMN2-FL) and SMN transcript lacking exon 7 (SMN-D7) (53–55).
2.1.3. SMN2–full length
2.1.3.1. Monitoring biomarker
Results from different studies have shown conflicting results about the reliability of SMN2-FL transcript as a biomarker of disease severity. Although some studies reported that SMN2-FL mRNA levels are able to differentiate between patients with SMA and healthy controls and that FL-mRNA levels in peripheral blood cells inversely correlate with disease severity especially in type 1 patients (56, 57), they were not able to distinguish between different disease phenotypes (54), while some other studies found no correlation between SMN2-FL expression and disease phenotype at all (55, 58, 59).
2.1.4. SMN transcript lacking exon 7
2.1.4.1. Monitoring biomarker
Different studies have attempted to correlate SMN-D7 expression levels and clinical severity, but it was repeatedly reported that they are globally similar to those of healthy controls both in peripheral blood cells and fibroblasts (54, 55, 58).
2.1.4.2. Response biomarker
Idem, in a study involving patients treated with nusinersen, an increase in SMN-FL levels in extracellular vesicles blood was observed after 14 months of treatment (60). In the first-in-human study with risdiplam, it was shown that FL SMN2 mRNA levels and SMN protein levels increase in whole blood after treatment (56, 57). Studies testing the therapeutic potential of a histone deacetylase inhibitors (valproic acid) on human cell lines, did not show significant changes in SMN-FL levels after treatment (61), while in a corresponding phase 2 trial with valproic acid in children and adolescents SMA patients of all types, SMN2-FL levels were unchanged while SMN-D7 levels were significantly reduced (62). Similar contrasting data were also outlined after clinical trials with phenyl-butyrate (63, 64).
2.1.5. SMN protein levels
SMN protein is ubiquitously expressed and detectable in all cell types, making previous attempts to compare SMN levels in cells in blood and CSF challenging due to tissue-related variations (65).
Some studies (54) show that SMN protein levels are high during the perinatal period in SMA mice and decline rapidly by 3 months, remaining low throughout the mice lifespan (66). Similarly, elevated SMN protein levels in prenatal post-mortem tissues were observed in both SMA patients with 2 SMN2 copies (type 1 and 2 patients) and healthy individuals, with a confirmed decline after birth in healthy new-borns in different disease-related tissues (spinal cord, brain cortex, diaphragm), suggesting the necessity of SMN protein for prenatal development (67, 68).
2.1.5.1. Prognostic biomarker
While a distinct difference in SMN protein levels is evident between SMA mice and healthy controls, in human patients, there is a notable overlap in SMN2 expression across different SMA types, indicating that the clinical presentation is more closely linked to the quantity of SMN2 copies rather than the levels of SMN protein in the blood; however, it’s important to note that the predictive capacity of SMN2 copy number alone is limited in explaining the variability in clinical phenotypes (69). Similar results have been obtained also by Wadmann et al., who describes lower levels of SMN protein in fibroblasts and PBMC of SMA patients compared to healthy controls, without significant differences among SMA types. In this study, they underlined again the relevant differences of SMN protein levels in different tissues, pointing out that, anyway, SMN protein levels in fibroblasts correlate with SMN2 copy number and could be a potential biomarker of disease prognosis (58).
2.1.5.2. Monitoring biomarker
Findings from a large multicentre natural history study of infantile SMA (inclusion <6 months of age) also demonstrated a reduction in SMN protein levels in the blood of SMA infants compared to age-matched healthy infants (57). However, a later study indicated that SMN protein levels remain stable over time despite rapid changes in motor function (70). Additionally, another group reported stability in SMN mRNA and protein levels over a year, with no correlation between levels and either muscle function or structural muscle integrity as assessed by functional clinical scales and quantitative MRI (qMRI) of thigh muscles (71). Furthermore, in a cross-sectional study assessing SMN gene and protein expression in whole blood, there was no difference in SMN expression levels or any other gene expression changes correlating with disease severity between SMA and healthy cohorts (72). Overall, these contrasting results highlight the need for cautious analysis of SMN mRNA and protein levels, considering that systemic levels can be influenced by specific tissue distribution, with peripheral blood levels being globally lower than in motor neurons, age of the patient as well as SMA sub-type.
2.1.5.3. Predictive biomarker
Variable SMN2 mRNA levels and inconsistent SMN protein levels have been noticed among different tissues, such as the spinal cord, central nervous system (CNS) and others, possibly due to limitations in the detection assay (67). Preclinical studies with morpholino antisense oligonucleotides (ASO) showed that a single injection increased SMN protein levels in both SMA and heterozygote mice, but not to the levels of control animals (73). Nevertheless, the systemic delivery and distribution of ASO resulted in a high degree of variability in SMN protein levels among different tissue types, as well as an age-related change in SMN protein levels in all tissues.
In patients treated with risdiplam, an increase of SMN protein in CSF as well as in peripheral blood has been demonstrated as well (74).
Concerning AAV-mediated gene therapy, it has been described that, despite specific CNS tropism of the AAV9 serotype, the vector is potentially able to reach cells all over the body, with high concentrations being found in motor neurons in both animal models and patients. The percentage of circulating SMN protein produced by the transgene is anyway still difficult to estimate (75). Moreover, even if neurons are considered non-dividing cells, implying that the transgene could be express for a long time after delivery, the SMN protein levels of expression in the long term is still under study.
The differences in SMN levels in peripheral tissues and CNS makes it difficult to consider SMN mRNA and protein as relevant biomarkers of the response to therapy, while results proposing it as a pharmacodynamic and target engagement biomarkers seem to me more consistent and encouraging.
2.2. Creatinine and creatine kinase
2.2.1. Monitoring biomarker
Even if CK serum levels are often slightly elevated in all SMA types, they are considered too unspecific to be a reliable diagnostic biomarker of the disease. Nevertheless, taken together with other signs and symptoms of LMN degeneration, high CK serum levels may help orienting the diagnosis of SMA.
2.2.2. Prognostic and predictive biomarker
Some of the first potential prognostic circulating biomarkers proposed as useful in SMA were serum creatinine and CK. Serum creatinine levels are known to be directly correlated to lean muscle mass in healthy individuals. Moreover, serum creatinine is currently under investigation as a circulating biomarker in other MNDs, such as amyotrophic lateral sclerosis (ALS) and spinal-bulbar muscular atrophy (SBMA), where it shows an inverse correlation with the severity of denervation (76–78). In rapidly progressive forms of SMA, creatinine serum levels have been found to be inversely associated with disease severity after adjusting for age and lean mass (34), with higher levels observed in type 3 patients then in type 1. The amount of creatinine declines over time regardless of the SMA type and seems to be a slightly more reactive biomarker than neurophysiological parameters in pre-symptomatic children (79). On the other side, in chronic forms of the disease concerning mostly adolescents and adult patients, creatinine serum levels are considered to be higher than in patients with rapidly progressive forms (79).
CK is an enzyme that catalyzes the reversible transfer of phosphate to creatine, generating phosphocreatine, which serves as a mobilizable energy reserve in skeletal muscle. Increased CK levels have been described especially in milder chronic forms of SMA, including adolescent and adult patients, probably in relation to greater muscle mass (80).
In patients treated with nusinersen, a decrease in CK and a parallel increase in serum creatinine after several months of treatment were observed both in children and adult patients (80, 81). Additionally, serum creatinine and CK levels were higher in responders than in non-responders (82). These findings suggest that serum creatinine and CK levels can be considered easy measurable response biomarkers for assessing response to therapy in SMA.
As anti-myostatin treatments are under study in clinical trials, aiming at directly targeting muscle trophism and strength, the role of creatinine and CK levels, as well as that of other markers of muscle activity, will be of primary interest to provide biological evidence of response to therapy (25).
3. Neurofilament heavy and light chain
Neurofilaments (Nfs) are a group of cytoskeletal proteins belonging to the type IV intermediate filaments family. They consist of three subunits: neurofilament light chain (NfL, 68 kDa), medium chain (NfM, 150 kDa) and heavy chain (NfH, 200 kDa) (83). Nfs are uniquely expressed in neurons, and play a crucial role for axon radial growth and axonal regeneration (84). They are abundant in large, myelinated axons, where they are highly organized into parallel arrays (85). Nfs undergo post-transcriptional regulation processes such as phosphorylation (referred to as p-Nf), which enhances their resistance to degradation (86). However, once they are released following neuronal loss or axonal damage, they can be detected into the CSF, peripheral blood and interstitial fluid (87–89). In light of this background, Nfs have emerged as promising biomarkers for several neurological and neurodegenerative diseases including other motor neuron diseases such as ALS. In ALS, Nfs were demonstrated to be elevated at all disease stages as well as in preclinical carriers of specific gene mutations, suggesting that they could be used as an effective biomarker of disease severity and progression but also of response to future therapies (90–94).
As per existing models of Nfs production and circulation, CSF and blood levels of these proteins are elevated in young, healthy children (95), with a tendency to increase during the early phases of CNS development. Subsequently, both CSF and plasma levels demonstrate a stabilization, followed by a decline in late childhood and adolescence. However, in the context of healthy aging, a slight increase is observed once again. Specifically, within the CSF, the upper reference value for NfL levels shows a 2.5-fold increase between the ages of 20 and 50, and then doubles by the age of 70 (96). This trend could be justified by various factors, including a slowdown in Nfs metabolism, ongoing neural loss due to the aging process, subjects’ body mass index (BMI) and renal function (97).
3.1. Monitoring and prognostic biomarker
In children with SMA type 1, plasma levels pNf-H and Nf-L exhibit a substantial increase during the initial months of life when compared to age-matched controls, indicating active and rapid motor neuron degeneration and death at early stages of the disease (83, 98, 99). Additionally, serum Nfs levels are inversely associated with SMN2 copies number, and directly correlate with earlier age of diagnosis and symptom onset and lower baseline motor function (34). Over time, pNf-H levels decline in untreated SMA children, but persistently remain higher compared to healthy controls of the same age, probably due to the active axonal degeneration (99). Recent data have revealed notable fluctuations in pNf-H levels among both SMA patients and mouse models, highlighting the necessity for a comprehensive interpretation of this biomarker’s measurements (100). However, in the context of adolescent and adult patients, the significance of measuring Nfs as informative biomarkers is less clear. Data from published studies indicate that adolescent and adult patients with type 2 and 3 SMA globally show no significant CSF and serum/plasma increase in Nfs compared to age-matched controls, (101–104). A recent paper also suggested that, in a similar cohort of adult SMA type 2 and 3 patients, pNf-H levels in the serum were reduced compared to healthy controls, possibly reflecting motor neuron pool exhaustion as a result of long lasting chronic degeneration (100).
3.2. Response biomarker and surrogate end-point
In pre-symptomatic and symptomatic children, pNf-H has been proposed as a reliable biomarker of response to treatment from the first clinical trial with nusinersen [ENDEAR (22)], which demonstrated a rapid decrease in pNf-H circulating levels within 2 months of intrathecal injections. This decline was also noticeable in the control group, albeit at a slower pace and with less pronounced effect, often followed by stabilization. Intriguingly, treated patients exhibited consistently higher pNf-H levels in both serum and CSF compared to age-matched healthy individuals across all time points despite treatment. Two additional studies of non-ambulatory SMA type 1 infants with 2 SMN2 copies receiving nusinersen therapy further confirmed the rapid decline in Nfs in the CSF, with additional evidence of direct correlation of Nfs levels with clinical presentation, supporting the utility of Nfs as predictive, response and pharmacodynamic biomarkers (105–107).
Among adult patients, a general consensus exists on the fact that these levels do not change after treatment with nusinersen (102, 103) and that a clear correlation with motor improvement cannot be confirmed (108, 109). Interestingly, higher serum Nf-L levels appear to be linked to poorer motor performance, though changes in motor function generally do not correspond with shifts in serum Nf-L. A further confounding factor in adolescent and adult type 2 and 3 patients is that the disease presents slow progression and high clinical heterogeneity. Moreover, treatment availability with nusinersen only started in 2016, implying that adult patients were treated after an extremely variable disease duration, further complicating the possibility to perform reliable statistical analysis of Nfs levels correlation with disease stage and progression (110). However, it is known that younger type 3 patients have a more evident and significant decline of pNf-H and Nf-L levels in the CSF after 3 nusinsersen loading doses (111).
No data are available about Nfs change in patients treated with Risdiplam, in both pediatric and adult patients. Notably, a recent study showed an unexpected increase of Nfs in 7 pre-symptomatic patients SMA infants treated with intravenous gene therapy between 20 and 190 days of age regardless of SMN2 copy number (83). At the same time, infants that were pretreated with nusinersen as a co-therapy within a short interval before their onasemnogene abeparvovec infusion did not show an increase in Nfs levels. One child carrying 2 SMN2 copies, presented incomplete benefit from gene therapy and developed mild symptoms of motor neuron degeneration over time. This individual demonstrated chronically elevated Nfs levels that failed to normalize during the subsequent 18 months after therapy. While the interpretation of these findings should be cautious, one hypothesis could be that nusinersen could have a neuroprotective effect, paving the way for new combination therapy strategies to come.
4. Micro-RNA
MicroRNAs are a class of small (~22 nt) non-protein-coding RNA molecules that can regulate gene expression at a post-transcription level (112, 113). More specifically, miRNA bind to determined sequences in the 30-UTR of target genes, inducing subsequent translational repression and/or decay of target mRNAs (114). Endogenous non-coding RNAs also regulate gene expression via formation of a ribonucleoprotein (RNP) complex with Argonaute (AGO) proteins and complementary base pairing with their target mRNAs (115).
4.1. Pathophysiology and monitoring biomarker
In the latest years of research on microRNAs, their key role in the regulation of muscle and CNS development and regeneration has emerged, placing them within the possible non-invasive biomarkers in neurological and neuromuscular diseases such as SMA (35, 116, 117). Moreover, it has been shown that SMN protein has a critical role in miRNA biogenesis and processing, confirming the relevance of miRNAs in SMA pathogenesis. Their dysregulation is not only a consequence of the disease, but it can contribute to exacerbate its pathological features (118). Given their role in the pathogenesis of SMA, it is possible that miRNA expression changes after disease-modifying treatments, suggesting that they could be a biomarker of response to therapy as well (119). miRNAs are relatively stable in accessible biofluids, such as blood and CSF, and can be detected with accessible and feasible laboratory methods.
Differential expression of different miRNAs (miR-9, miR-206, miR-34, miR-132, miR-225-5p, miR-431, miR-375…) have been reported in SC, skeletal muscle, and serum from SMA and wild-type mice as well as in serum samples from SMA and control patients (114, 119). Mice lacking the miRNA-processing enzyme Dicer selectively in motor neurons display hallmarks of SMA (120). Also, SMN protein has been shown to alter miRNA expression and distribution in neurons. For example, SMN protein down-regulates the expression of miR-9a and, interestingly, miR-9a levels in fibroblasts have shown a positive correlation with SMA severity (121). miR-9 alteration leads to reduced down-regulation of heavy Nfs in motor neurons (114, 121). In fact, neurofilament accumulation has been supported by several studies as one of the more likely causes of selective motor neuron degeneration. The neurons that are most prominently affected by the accumulation of neurofilaments are the largest neurons with the longest axons, such as motor neurons the most vulnerable cells in motor neuron disorders, including SMA (122).
miR-183 is known to be increased in neurites of SMN-deficient neurons. Inhibition of miR-183 expression in the spinal cord of an SMA mouse extends survival and improves motor function of Smn-mutant mice (123).
miR-431, involved in motor neuron neurite length, also plays a role in the SMA motor neuron phenotype. Its expression is namely highly increased in spinal motor neurons and a number of its putative mRNA targets are significantly down-regulated in motor neurons after SMN loss (117).
Another miRNA involved in SMA motor neuron phenotype is miR-375. Besides its role in neurogenesis, miR-375 protects neurons from apoptosis in response to DNA damage. Motor neurons from a SMA patient have shown reduced levels of miR-375, elevated p53 protein levels, and higher susceptibility to DNA damage induced apoptosis (124).
miRNAs could be useful also to better understand and track muscle involvement in SMA. Some miRNAs strictly related to muscle function (including miR-1, miR-133a, miR-206), which are up-regulated by the action of MYOD protein in myoblasts, are reduced in SMA myoblasts since MYOD1 is reduced as well as a result of SMN1 mutations, resulting in impaired metabolism and myotubes formation (116). These results suggest a role of SMN in regulation of myogenesis (125) and propose that specific miRNAs could be effective biomarkers of delayed process in SMA mouse models and patients.
4.2. Predictive and response biomarker
In infants treated with nusinersen, miRNA dosing has been performed on CSF and blood samples, showing how some of them were longitudinally reduced before and after treatment (for example miR-378, known to regulate the balance between myocyte autophagy and apoptosis) (126). miR-378a-3p has been found elevated after treatment especially in patients with more important motor improvement. Similarly, higher baseline levels of miR-142-5p and miR-355-5p seem to be associated with better reaction to nusinersen treatment. On the other side, miR-23a, which has been hypothesized to have a neuroprotective function, has been found in higher concentrations in blood from patients presenting the best response to nusinsersen treatment both at baseline and longitudinally (127).
The myomiRNAs miR133a, 133b, 206 and 1 were demonstrated to be downregulated upon nusinersen treatment in the serum of SMA pediatric patients, with the levls of miR-133a being correlated with motor function improvement. It remains to be established if this miRNA is modulated in adult patients’ treatment (128). In other studies, baseline levels of miR206 negatively correlated with patients’ response (122, 129) suggesting the importance of studying the myomiRNAs before and after therapy. Studies testing a single dose therapeutic morpholino antisense oligomer treatment in SMA mice showed that miR-132 levels (which are known to be pathologically elevated in SMA mice) in the SC, muscle and serum, reversed to the normal levels after treatment, suggesting that miR-132 could be one of the most responsive miRNAs to systemic ASO treatment in the severe mouse model (129).
A very recent study additionally highlighted the importance of miR34 family (miR34a/b/c) in MN functional regulation and has positively correlated this miRNA to the type 1 SMA patients’ response to nusinersen treatment (130). The authors have further demonstrated a therapeutic effect of miR34a administration in pre-symptomatic SMA mice substantiating the importance of this miRNA in the pathophysiology and treatment of SMA.
Altogether, miRNA seem to be encouraging possible biomarkers both of disease severity and response to treatment in SMA patients and animal models. Moreover, they are relatively easy to measure in CSF and blood, permitting iterative sampling in treated patients. However, information about role and function of different miRNA is still under study, and their concentration could change in different tissues as well as with development and aging. Cautions should then be applied in the interpretation of results describing the role of single miRNA especially in treated patients since repetition studies are not available yet and actual results are highly heterogenous. Probably, a combination of different miRNA could hold an interesting value in prediction of disease evolution and response to therapeutic interventions.
5. Omics and other circulating proteins
Over the years, several studies focusing on identifying possible circulating proteins that could represent a reliable biomarker in SMA have been performed. The Biomarkers for SMA (BforSMA) study is an example of this effort. It was a cross-sectional omics study that evaluated blood and urine protein analytes in children (age 2–12 years) with genetically confirmed SMA and age-matched healthy controls (60). A resulting 200 candidate biomarkers were found to correlate with motor scores, and the most significant markers across all outcome measures were plasma protein analytes. The novelty of this study consisted in the use of several unbiased methods (metabolomics, proteomics, and transcriptomics) for the search of biomarkers in a large well-defined SMA patient cohort and age-matched healthy controls. Despite the identification of several putative biomarkers, further validation is needed to confirm these findings.
5.1. Predictive biomarker
Some recent studies have analyzed the role of proteomics as a biomarker in SMA patients treated with nusinersen (131, 132), suggesting that the treatment could modulate the protein profile of the CSF. Further studies are warranted to better define which proteins are the most reactive to treatment both in blood and CSF.
Results from studies in non-treated and treated patients are resumed in Table 2.
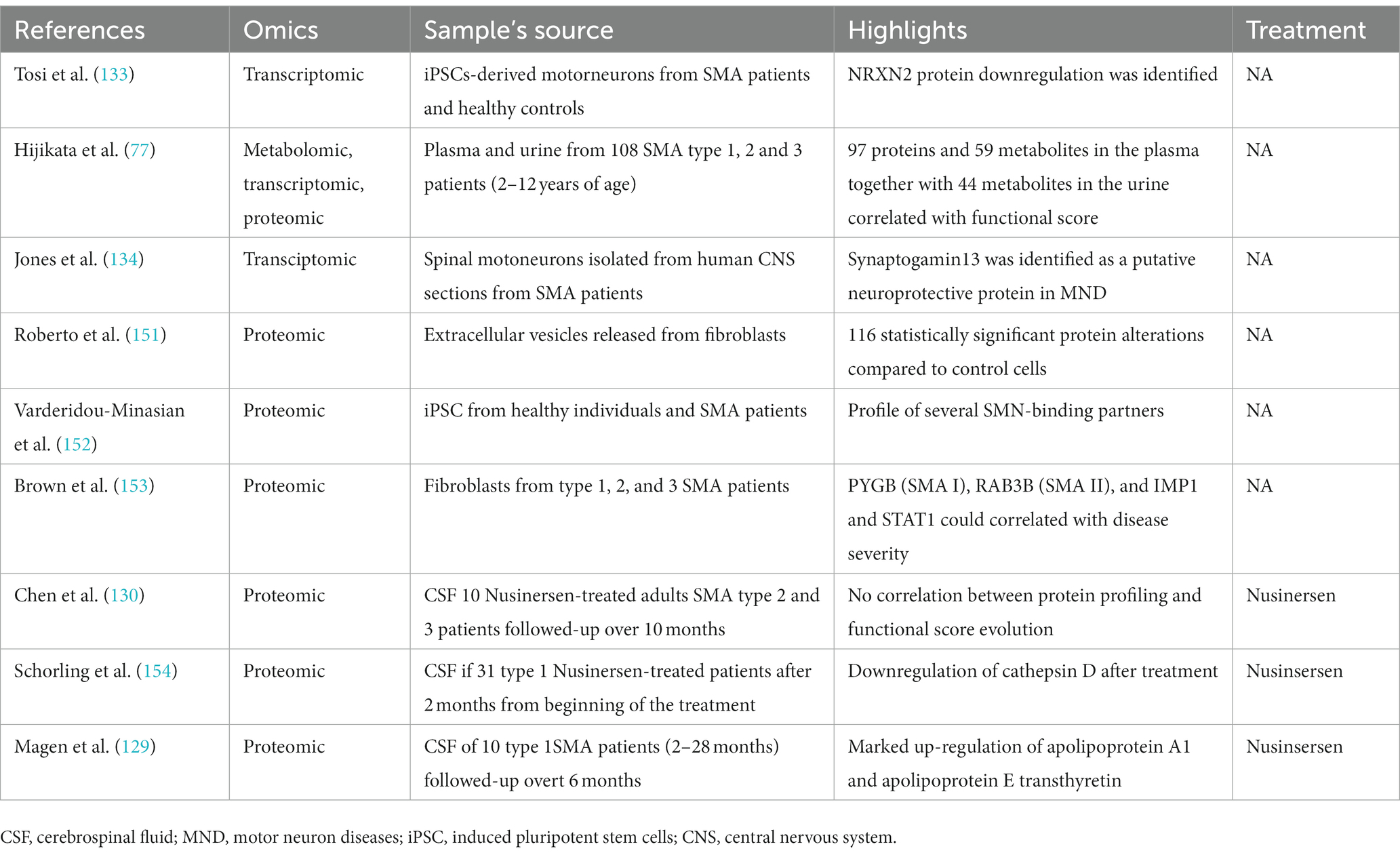
Table 2. Overview of multi-omics approaches used to date to characterize disease progression in non-treated and treated SMA patients.
6. Discussion
SMA is a complex disease characterized by heterogeneous clinical presentation. The recent emergence of several effective drugs and ongoing research into novel therapies highlights the pressing need for reliable biomarkers to guide patient-specific therapeutic strategies. In this context, identifying biomarkers that are accurate but also easily implementable in clinical trials and practice becomes imperative. Furthermore, these biomarkers should be closely associated with therapy response to facilitate informed decisions regarding treatment initiation, cessation, or modification (35, 36). Clinical and functional measurements, as well as imaging and neurophysiology, surely play a relevant role in patients’ classification and in monitoring disease progression (29, 32, 135–139). Nevertheless, circulating biomarkers, including circulating proteins, Nfs and other small molecules such as miRNA, might allow better categorization of patients of all disease sub-types and present great potential to detect significant modifications after treatment, namely in adolescent and adult patients with a relatively stabilized and slowly progressive motor deficit. From a practical point of view, circulating biomarkers are generally easy to access and less operator-dependent (39), allowing repeated measurement over time in treated and non-treated patients.
Among the various putative circulating biomarkers under investigation, Nfs stand out as promising candidates, particularly in children with type 1 and 2 SMA (83, 98, 99). These biomarkers have exhibited favorable responsiveness to treatments such as nusinersen and gene therapy. Their utility extends to monitoring both therapy response and disease progression over time, as they can be assessed in CSF and blood samples. This dual applicability makes neurofilaments valuable tools for tracking disease dynamics and therapeutic efficacy in pediatric SMA patients (83). However, it is important to note that the utility of neurofilaments in adult SMA patients appears limited. In the adult population, alterations in neurofilament levels are only marginal, and the changes following nusinersen therapy are less pronounced and harder to interpret (102, 104, 111). This discrepancy between pediatric and adult populations emphasizes the need for age-specific biomarker considerations when making therapeutic decisions in SMA.
Traditionally, SMN2 copy number has been regarded as the principal genetic modifier in SMA, significantly influencing disease severity (10). Nevertheless, recent research has shed light on the existence of additional positive modifiers, such as PSL3 (46), which are currently under investigation. These emerging modifiers hold the potential to play a role in shaping the phenotype’s severity, necessitating their inclusion in decision-making processes for therapy choices as well as in helping setting patients and families’ expectations. As our understanding of SMA genetics evolves, these modifiers may offer new insights into the disease’s pathogenesis and treatment response.
In contrast to neurofilaments and genetic modifiers, SMN protein and SMN-mRNA appear to be promising predictive and pharmacodynamic biomarkers for systemic treatments in both pediatric and adult SMA patients (55). However, their applicability to intrathecal treatments is limited. This distinction underscores the importance of tailoring biomarker selection to the specific treatment modality employed in SMA patients, further highlighting the complexity of individualized therapy decisions.
In conclusion, the pursuit of effective SMA therapies underscores the critical role of biomarkers in guiding treatment decisions. Neurofilaments emerge as valuable biomarkers in pediatric populations, reflecting both therapy response and disease progression. In contrast, SMN2 copy number and other emerging modifiers like PSL3 are essential considerations for understanding disease severity and therapeutic implications. The distinctions in biomarker utility between pediatric and adult populations, as well as across treatment modalities, emphasize the need for a nuanced and patient-specific approach to SMA management. As research in this field continues to advance, the integration of multifaceted biomarker data will be essential for optimizing therapeutic outcomes in individuals with SMA.
The development of informatic and especially artificial intelligence (AI) tools might also be useful in the categorization of more relevant circulating biomarkers as well as in the development of meaningful biomarkers combinations that could, in the end, be more robust than single biomarkers. Such approach has already been demonstrated to be effective in other pathologies such as cancer or infectious disease (140–142). In this perspective, the development of multimodal tools composed both by clinical and functional and by molecular and circulating biomarkers could improve our ability to predict prognosis and reaction to treatment (36, 137). Composite scores should moreover be adapted to patients’ age, phenotype and to different treatment approaches and have the potential to substantially improve the clinical knowledge and the patients’ care.
Recent approval of three disease-modifying therapies for SMA (143–149) opened the door to great improvements in patient survival and development of new clinical phenotypes. Moreover, it is now well known that treatment must be initiated as early as possible to potentially improve the patient’s response to it. However, phenotypic heterogeneity and the wide spectrum of treatment response still make the decision process and the choice of the most adapted treatment for each patient challenging, adding even more importance to the study and development of new robust biomarkers to guide patient-targeted intervention.
On one side, biomarkers are useful in stratifying patients before therapy administration, helping in the choice between different therapeutic approaches, while, on the other, they are necessary to monitor disease progression under treatment. This is of primary importance in clinical trials to demonstrate the efficacy and safety of new molecules, but also in clinical practice, especially as combination therapies (for example, gene therapy followed by nusinersen or risdiplam), or switch from one treatment to another are proposed to patients with only partial response to a first therapeutic approach (23, 133, 150). With new and different treatments on their ways (for example anti-myostatin drugs), the ability to stratify patients will further help in the decision of when and how to combine therapies.
In this context, individualized treatments choices will gain more importance in the aim of developing a personalized approach specific to each patient. It is in fact nowadays known that response to treatment, but also natural history evolution, can largely differ from one patient to another even in the same family (134) further underlying the need for more precise and specific biomarkers to better characterize patients’ phenotype as well as to predict prognosis with and without treatment.
Author contributions
QG, MG, and PS: Conceptualization, Writing—original draft preparation, Writing—review and editing. QG: Revision and funding acquisition. All authors have read and agreed to the published version of the manuscript. All authors contributed to the article and approved the submitted version.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Mercuri, E, Bertini, E, and Iannaccone, ST. Childhood spinal muscular atrophy: controversies and challenges. Lancet Neurol. (2012) 11:443–52. doi: 10.1016/S1474-4422(12)70061-3
2. Lefebvre, S, Bürglen, L, Reboullet, S, Clermont, O, Burlet, P, Viollet, L, et al. Identification and characterization of a spinal muscular atrophy-determining gene. Cells. (1995) 80:155–65. doi: 10.1016/0092-8674(95)90460-3
3. Brzustowicz, LM, Lehner, T, Castilla, LH, Penchaszadeh, GK, Wilhelmsen, KC, Daniels, R, et al. Genetic mapping of chronic childhood-onset spinal muscular atrophy to chromosome 5q11.2-13. 3. Nature. (1990) 344:540–1. doi: 10.1038/344540a0
4. Wirth, B, Karakaya, M, Kye, MJ, and Mendoza-Ferreira, N. Twenty-five years of spinal muscular atrophy research: from phenotype to genotype to therapy, and what comes Next. Annu Rev Genomics Hum Genet. (2020) 21:231–61. doi: 10.1146/annurev-genom-102319-103602
5. Verhaart, IEC, Robertson, A, Wilson, IJ, Aartsma-Rus, A, Cameron, S, Jones, CC, et al. Prevalence, incidence and carrier frequency of 5q-linked spinal muscular atrophy - a literature review. Orphanet J Rare Dis. (2017) 12:124. doi: 10.1186/s13023-017-0671-8
6. Lunn, MR, and Wang, CH. Spinal muscular atrophy. Lancet. (2008) 371:2120–33. doi: 10.1016/S0140-6736(08)60921-6
7. Schrank, B, Götz, R, Gunnersen, JM, Ure, JM, Toyka, KV, Smith, AG, et al. Inactivation of the survival motor neuron gene, a candidate gene for human spinal muscular atrophy, leads to massive cell death in early mouse embryos. Proc Natl Acad Sci U S A. (1997) 94:9920–5. doi: 10.1073/pnas.94.18.9920
8. Jablonka, S, Rossoll, W, Schrank, B, and Sendtner, M. The role of SMN in spinal muscular atrophy. J Neurol. (2000) 247:I37–42. doi: 10.1007/s004150050555
9. Wirth, B. An update of the mutation spectrum of the survival motor neuron gene (SMN1) in autosomal recessive spinal muscular atrophy (SMA). Hum Mutat. (2000) 15:228–37. doi: 10.1002/(SICI)1098-1004(200003)15:3<228::AID-HUMU3>3.0.CO;2-9
10. Elsheikh, B, Prior, T, Zhang, X, Miller, R, Kolb, SJ, Moore, D, et al. An analysis of disease severity based on SMN2 copy number in adults with spinal muscular atrophy. Muscle Nerve. (2009) 40:652–6. doi: 10.1002/mus.21350
11. Lefebvre, S, Burlet, P, Liu, Q, Bertrandy, S, Clermont, O, Munnich, A, et al. Correlation between severity and SMN protein level in spinal muscular atrophy. Nat Genet. (1997) 16:265–9. doi: 10.1038/ng0797-265
12. Setola, V, Terao, M, Locatelli, D, Bassanini, S, Garattini, E, and Battaglia, G. Axonal-SMN (a-SMN), a protein isoform of the survival motor neuron gene, is specifically involved in axonogenesis. Proc Natl Acad Sci U S A. (2007) 104:1959–64. doi: 10.1073/pnas.0610660104
13. Singh, RN, Howell, MD, Ottesen, EW, and Singh, NN. Diverse role of survival motor neuron protein. Biochim Biophys Acta. (2017) 1860:299–315. doi: 10.1016/j.bbagrm.2016.12.008
14. Broccolini, A, Engel, WK, and Askanas, V. Localization of survival motor neuron protein in human apoptotic-like and regenerating muscle fibers, and neuromuscular junctions. Neuroreport. (1999) 10:1637–41. doi: 10.1097/00001756-199906030-00003
15. Lanfranco, M, Cacciottolo, R, Borg, RM, Vassallo, N, Juge, F, Bordonné, R, et al. Novel interactors of the Drosophila survival motor neuron (SMN) complex suggest its full conservation. FEBS Lett. (2017) 591:3600–14. doi: 10.1002/1873-3468.12853
16. Otter, S, Grimmler, M, Neuenkirchen, N, Chari, A, Sickmann, A, and Fischer, U. A comprehensive interaction map of the human survival of motor neuron (SMN) complex. J Biol Chem. (2007) 282:5825–33. doi: 10.1074/jbc.M608528200
17. Zerres, K, and Rudnik-Schöneborn, S. 93rd ENMC international workshop: non-5q-spinal muscular atrophies (SMA) - clinical picture (6-8 April 2001, Naarden, the Netherlands). Neuromuscul Disord. (2003) 13:179–83. doi: 10.1016/S0960-8966(02)00211-0
18. Zerres, K, and Rudnik-Schöneborn, S. Natural history in proximal spinal muscular atrophy. Clinical analysis of 445 patients and suggestions for a modification of existing classifications. Arch Neurol. (1995) 52:518–23. doi: 10.1001/archneur.1995.00540290108025
19. Kostova, FV, Williams, VC, Heemskerk, J, Iannaccone, S, Didonato, C, Swoboda, K, et al. Spinal muscular atrophy: classification, diagnosis, management, pathogenesis, and future research directions. J Child Neurol. (2007) 22:926–45. doi: 10.1177/0883073807305662
20. Mercuri, E, Finkel, RS, Muntoni, F, Wirth, B, Montes, J, Main, M, et al. Diagnosis and management of spinal muscular atrophy: part 1: recommendations for diagnosis, rehabilitation, orthopedic and nutritional care. Neuromuscul Disord. (2018) 28:103–15. doi: 10.1016/j.nmd.2017.11.005
21. Faravelli, I, Nizzardo, M, Comi, GP, and Corti, S. Spinal muscular atrophy--recent therapeutic advances for an old challenge. Nat Rev Neurol. (2015) 11:351–9. doi: 10.1038/nrneurol.2015.77
22. Mercuri, E, Darras, BT, Chiriboga, CA, Day, JW, Campbell, C, Connolly, AM, et al. Nusinersen versus sham control in later-onset spinal muscular atrophy. N Engl J Med. (2018) 378:625–35. doi: 10.1056/NEJMoa1710504
23. Erdos, J, and Wild, C. Mid- and long-term (at least 12 months) follow-up of patients with spinal muscular atrophy (SMA) treated with nusinersen, onasemnogene abeparvovec, risdiplam or combination therapies: a systematic review of real-world study data. Eur J Paediatr Neurol. (2022) 39:1–10. doi: 10.1016/j.ejpn.2022.04.006
24. Mendell, JR, Al-Zaidy, S, Shell, R, Arnold, WD, Rodino-Klapac, LR, Prior, TW, et al. Single-dose gene-replacement therapy for spinal muscular atrophy. N Engl J Med. (2017) 377:1713–22. doi: 10.1056/NEJMoa1706198
25. Abati, E, Manini, A, Comi, GP, and Corti, S. Inhibition of myostatin and related signaling pathways for the treatment of muscle atrophy in motor neuron diseases. Cell Mol Life Sci. (2022) 79:374. doi: 10.1007/s00018-022-04408-w
26. Zhou, H, Meng, J, Malerba, A, Catapano, F, Sintusek, P, Jarmin, S, et al. Myostatin inhibition in combination with antisense oligonucleotide therapy improves outcomes in spinal muscular atrophy. J Cachexia Sarcopenia Muscle. (2020) 11:768–82. doi: 10.1002/jcsm.12542
27. Oskoui, M, Levy, G, Garland, CJ, Gray, JM, O’Hagen, J, De Vivo, DC, et al. The changing natural history of spinal muscular atrophy type 1. Neurology. (2007) 69:1931–6. doi: 10.1212/01.wnl.0000290830.40544.b9
28. Finkel, RS, Chiriboga, CA, Vajsar, J, Day, JW, Montes, J, De Vivo, DC, et al. Treatment of infantile-onset spinal muscular atrophy with nusinersen: a phase 2, open-label, dose-escalation study. Lancet. (2016) 388:3017–26. doi: 10.1016/S0140-6736(16)31408-8
29. Querin, G, El Mendili, M-M, Lenglet, T, Behin, A, Stojkovic, T, Salachas, F, et al. The spinal and cerebral profile of adult spinal-muscular atrophy: a multimodal imaging study. Neuroimage Clin. (2019) 21:101618. doi: 10.1016/j.nicl.2018.101618
30. Gallone, A, Mazzi, F, Bonanno, S, Zanin, R, Moscatelli, M, Aquino, D, et al. Muscle quantitative MRI in adult SMA patients on nusinersen treatment: a longitudinal study. Acta Myol. (2022) 41:76–83. doi: 10.36185/2532-1900-074
31. Shimizu-Motohashi, Y, Chiba, E, Mizuno, K, Yajima, H, Ishiyama, A, Takeshita, E, et al. Muscle impairment in MRI affect variability in treatment response to nusinersen in patients with spinal muscular atrophy type 2 and 3: a retrospective cohort study. Brain and Development. (2023) 45:161–70. doi: 10.1016/j.braindev.2022.11.002
32. Günther, R, Neuwirth, C, Koch, JC, Lingor, P, Braun, N, Untucht, R, et al. Motor unit number index (MUNIX) of hand muscles is a disease biomarker for adult spinal muscular atrophy. Clin Neurophysiol. (2019) 130:315–9. doi: 10.1016/j.clinph.2018.11.009
33. Querin, G, Lenglet, T, Debs, R, Stojkovic, T, Behin, A, Salachas, F, et al. The motor unit number index (MUNIX) profile of patients with adult spinal muscular atrophy. Clin Neurophysiol. (2018) 129:2333–40. doi: 10.1016/j.clinph.2018.08.025
34. Pino, MG, Rich, KA, and Kolb, SJ. Update on biomarkers in spinal muscular atrophy. Biomark Insights. (2021) 16:11772719211035643. doi: 10.1177/11772719211035643
35. Kariyawasam, DST, D’Silva, A, Lin, C, Ryan, MM, and Farrar, MA. Biomarkers and the development of a personalized medicine approach in spinal muscular atrophy. Front Neurol. (2019) 10:898. doi: 10.3389/fneur.2019.00898
36. Smeriglio, P, Langard, P, Querin, G, and Biferi, MG. The identification of novel biomarkers is required to improve adult SMA patient stratification. Diagnosis and treatment. J Pers Med. (2020) 10:75. doi: 10.3390/jpm10030075
37. Querin, G, Biferi, MG, and Pradat, P-F. Biomarkers for C9orf7-ALS in symptomatic and pre-symptomatic patients: state-of-the-art in the new era of clinical trials. J Neuromuscul Dis. (2022) 9:25–37. doi: 10.3233/JND-210754
38. Group F-NBW. Glossary In: BEST (biomarkers, endpoint S, and other tools) resource : Food and Drug Administration (US) (2021) Available at:https://www.ncbi.nlm.nih.gov/books/NBK338448/
39. Barp, A, Ferrero, A, Casagrande, S, Morini, R, and Zuccarino, R. Circulating biomarkers in neuromuscular disorders: what is known, what is new. Biomol Ther. (2021) 11:1246. doi: 10.3390/biom11081246
40. Feldkötter, M, Schwarzer, V, Wirth, R, Wienker, TF, and Wirth, B. Quantitative analyses of SMN1 and SMN2 based on real-time light cycler PCR: fast and highly reliable carrier testing and prediction of severity of spinal muscular atrophy. Am J Hum Genet. (2002) 70:358–68. doi: 10.1086/338627
41. Calucho, M, Bernal, S, Alías, L, March, F, Venceslá, A, Rodríguez-Álvarez, FJ, et al. Correlation between SMA type and SMN2 copy number revisited: an analysis of 625 unrelated Spanish patients and a compilation of 2834 reported cases. Neuromuscul Disord. (2018) 28:208–15. doi: 10.1016/j.nmd.2018.01.003
42. Walsh, MB, Janzen, E, Wingrove, E, Hosseinibarkooie, S, Muela, NR, Davidow, L, et al. Genetic modifiers ameliorate endocytic and neuromuscular defects in a model of spinal muscular atrophy. BMC Biol. (2020) 18:127. doi: 10.1186/s12915-020-00845-w
43. Riessland, M, Kaczmarek, A, Schneider, S, Swoboda, KJ, Löhr, H, Bradler, C, et al. Neurocalcin Delta suppression protects against spinal muscular atrophy in humans and across species by restoring impaired endocytosis. Am J Hum Genet. (2017) 100:297–315. doi: 10.1016/j.ajhg.2017.01.005
44. Janzen, E, Mendoza-Ferreira, N, Hosseinibarkooie, S, Schneider, S, Hupperich, K, Tschanz, T, et al. CHP1 reduction ameliorates spinal muscular atrophy pathology by restoring calcineurin activity and endocytosis. Brain. (2018) 141:2343–61. doi: 10.1093/brain/awy167
45. Hosseinibarkooie, S, Peters, M, Torres-Benito, L, Rastetter, RH, Hupperich, K, Hoffmann, A, et al. The power of human protective modifiers: PLS3 and CORO1C unravel impaired endocytosis in spinal muscular atrophy and rescue SMA phenotype. Am J Hum Genet. (2016) 99:647–65. doi: 10.1016/j.ajhg.2016.07.014
46. Wolff, L, Strathmann, EA, Müller, I, Mählich, D, Veltman, C, Niehoff, A, et al. Plastin 3 in health and disease: a matter of balance. Cell Mol Life Sci. (2021) 78:5275–301. doi: 10.1007/s00018-021-03843-5
47. Oprea, GE, Kröber, S, McWhorter, ML, Rossoll, W, Müller, S, Krawczak, M, et al. Plastin 3 is a protective modifier of autosomal recessive spinal muscular atrophy. Science. (2008) 320:524–7. doi: 10.1126/science.1155085
48. Ackermann, B, Kröber, S, Torres-Benito, L, Borgmann, A, Peters, M, Hosseini Barkooie, SM, et al. Plastin 3 ameliorates spinal muscular atrophy via delayed axon pruning and improves neuromuscular junction functionality. Hum Mol Genet. (2013) 22:1328–47. doi: 10.1093/hmg/dds540
49. Kaifer, KA, Villalón, E, Osman, EY, Glascock, JJ, Arnold, LL, Cornelison, DDW, et al. Plastin-3 extends survival and reduces severity in mouse models of spinal muscular atrophy. JCI Insight. (2017) 2:e89970. doi: 10.1172/jci.insight.89970
50. Heesen, L, Peitz, M, Torres-Benito, L, Hölker, I, Hupperich, K, Dobrindt, K, et al. Plastin 3 is upregulated in iPSC-derived motoneurons from asymptomatic SMN1-deleted individuals. Cell Mol Life Sci. (2016) 73:2089–104. doi: 10.1007/s00018-015-2084-y
51. Strathmann, EA, Peters, M, Hosseinibarkooie, S, Rigo, FW, Bennett, CF, Zaworski, PG, et al. Evaluation of potential effects of Plastin 3 overexpression and low-dose SMN-antisense oligonucleotides on putative biomarkers in spinal muscular atrophy mice. PLoS One. (2018) 13:e0203398. doi: 10.1371/journal.pone.0203398
52. Yanyan, C, Yujin, Q, Jinli, B, Yuwei, J, Hong, W, and Fang, S. Correlation of PLS3 expression with disease severity in children with spinal muscular atrophy. J Hum Genet. (2014) 59:24–7. doi: 10.1038/jhg.2013.111
53. Deutsch, L, Osredkar, D, Plavec, J, and Stres, B. Spinal muscular atrophy after Nusinersen therapy: improved physiology in pediatric patients with no significant change in urine, serum, and liquor 1H-NMR metabolomes in comparison to an age-matched. Healthy cohort. Metabolites. (2021) 11:206. doi: 10.3390/metabo11040206
54. Sumner, CJ, Kolb, SJ, Harmison, GG, Jeffries, NO, Schadt, K, Finkel, RS, et al. SMN mRNA and protein levels in peripheral blood: biomarkers for SMA clinical trials. Neurology. (2006) 66:1067–73. doi: 10.1212/01.wnl.0000201929.56928.13
55. Crawford TOPaushkin, SV, Kobayashi, DT, Forrest, SJ, Joyce, CL, Finkel, RS, et al. Evaluation of SMN protein, transcript, and copy number in the biomarkers for spinal muscular atrophy (Bfor SMA) clinical study. PLoS One. (2012) 7:e33572. doi: 10.1371/journal.pone.0033572
56. Tiziano, FD, Pinto, AM, Fiori, S, Lomastro, R, Messina, S, Bruno, C, et al. SMN transcript levels in leukocytes of SMA patients determined by absolute real-time PCR. Eur J Hum Genet. (2010) 18:52–8. doi: 10.1038/ejhg.2009.116
57. Vezain, M, Saugier-Veber, P, Melki, J, Toutain, A, Bieth, E, Husson, M, et al. A sensitive assay for measuring SMN mRNA levels in peripheral blood and in muscle samples of patients affected with spinal muscular atrophy. Eur J Hum Genet. (2007) 15:1054–62. doi: 10.1038/sj.ejhg.5201885
58. Wadman, RI, Stam, M, Jansen, MD, van der Weegen, Y, Wijngaarde, CA, Harschnitz, O, et al. A comparative study of SMN protein and mRNA in blood and fibroblasts in patients with spinal muscular atrophy and healthy controls. PLoS One. (2016) 11:e0167087. doi: 10.1371/journal.pone.0167087
59. Kolb, SJ, Coffey, CS, Yankey, JW, Krosschell, K, Arnold, WD, Rutkove, SB, et al. Baseline results of the neuro NEXT spinal muscular atrophy infant biomarker study. Ann Clin Transl Neurol. (2016) 3:132–45. doi: 10.1002/acn3.283
60. Trifunov, S, Natera-de Benito, D, Carrera-García, L, Codina, A, Expósito-Escudero, J, Ortez, C, et al. Full-length SMN transcript in extracellular vesicles as biomarker in individuals with spinal muscular atrophy type 2 treated with Nusinersen. J Neuromuscul Dis. (2023) 10:653–65. doi: 10.3233/JND-230012
61. Mohseni, J, Zabidi-Hussin, Z, and Sasongko, TH. Histone deacetylase inhibitors as potential treatment for spinal muscular atrophy. Genet Mol Biol. (2013) 36:299–307. doi: 10.1590/S1415-47572013000300001
62. Swoboda, KJ, Scott, CB, Reyna, SP, Prior, TW, LaSalle, B, Sorenson, SL, et al. Phase II open label study of valproic acid in spinal muscular atrophy. PLoS One. (2009) 4:e5268. doi: 10.1371/journal.pone.0005268
63. Andreassi, C, Angelozzi, C, Tiziano, FD, Vitali, T, De Vincenzi, E, Boninsegna, A, et al. Phenylbutyrate increases SMN expression in vitro: relevance for treatment of spinal muscular atrophy. Eur J Hum Genet. (2004) 12:59–65. doi: 10.1038/sj.ejhg.5201102
64. Mercuri, E, Bertini, E, Messina, S, Solari, A, D’Amico, A, Angelozzi, C, et al. Randomized, double-blind, placebo-controlled trial of phenylbutyrate in spinal muscular atrophy. Neurology. (2007) 68:51–5. doi: 10.1212/01.wnl.0000249142.82285.d6
65. Groen, EJN, Perenthaler, E, Courtney, NL, Jordan, CY, Shorrock, HK, van der Hoorn, D, et al. Temporal and tissue-specific variability of SMN protein levels in mouse models of spinal muscular atrophy. Hum Mol Genet. (2018) 27:2851–62. doi: 10.1093/hmg/ddy195
66. Burlet, P, Huber, C, Bertrandy, S, Ludosky, MA, Zwaenepoel, I, Clermont, O, et al. The distribution of SMN protein complex in human fetal tissues and its alteration in spinal muscular atrophy. Hum Mol Genet. (1998) 7:1927–33. doi: 10.1093/hmg/7.12.1927
67. Ramos, DM, d’Ydewalle, C, Gabbeta, V, Dakka, A, Klein, SK, Norris, DA, et al. Age-dependent SMN expression in disease-relevant tissue and implications for SMA treatment. J Clin Invest. (2019) 129:4817–31. doi: 10.1172/JCI124120
68. Zaworski, P, von Herrmann, KM, Taylor, S, Sunshine, SS, McCarthy, K, Risher, N, et al. SMN protein can be reliably measured in whole blood with an Electrochemiluminescence (ECL) immunoassay: implications for clinical trials. PLoS One. (2016) 11:e0150640. doi: 10.1371/journal.pone.0150640
69. Czech, C, Tang, W, Bugawan, T, Mano, C, Horn, C, Iglesias, VA, et al. Biomarker for spinal muscular atrophy: expression of SMN in peripheral blood of SMA patients and healthy controls. PLoS One. (2015) 10:e0139950. doi: 10.1371/journal.pone.0139950
70. Kolb, SJ, Coffey, CS, Yankey, JW, Krosschell, K, Arnold, WD, Rutkove, SB, et al. Natural history of infantile-onset spinal muscular atrophy. Ann Neurol. (2017) 82:883–91. doi: 10.1002/ana.25101
71. Bonati, U, Holiga, Š, Hellbach, N, Risterucci, C, Bergauer, T, Tang, W, et al. Longitudinal characterization of biomarkers for spinal muscular atrophy. Ann Clin Transl Neurol. (2017) 4:292–304. doi: 10.1002/acn3.406
72. Finkel, RS, Crawford, TO, Swoboda, KJ, Kaufmann, P, Juhasz, P, Li, X, et al. Candidate proteins, metabolites and transcripts in the biomarkers for spinal muscular atrophy (Bfor SMA) clinical study. PLoS One. (2012) 7:e35462. doi: 10.1371/journal.pone.0035462
73. Porensky, PN, Mitrpant, C, McGovern, VL, Bevan, AK, Foust, KD, Kaspar, BK, et al. A single administration of morpholino antisense oligomer rescues spinal muscular atrophy in mouse. Hum Mol Genet. (2012) 21:1625–38. doi: 10.1093/hmg/ddr600
74. Poirier, A, Weetall, M, Heinig, K, Bucheli, F, Schoenlein, K, Alsenz, J, et al. Risdiplam distributes and increases SMN protein in both the central nervous system and peripheral organs. Pharmacol Res Perspect. (2018) 6:e00447. doi: 10.1002/prp2.447
75. Burghes, AHM, and Beattie, CE. Spinal muscular atrophy: why do low levels of survival motor neuron protein make motor neurons sick? Nat Rev Neurosci. (2009) 10:597–609. doi: 10.1038/nrn2670
76. Chiò, A, Calvo, A, Bovio, G, Canosa, A, Bertuzzo, D, Galmozzi, F, et al. Amyotrophic lateral sclerosis outcome measures and the role of albumin and creatinine: a population-based study. JAMA Neurol. (2014) 71:1134–42. doi: 10.1001/jamaneurol.2014.1129
77. Hijikata, Y, Hashizume, A, Yamada, S, Inagaki, T, Ito, D, Hirakawa, A, et al. Biomarker-based analysis of preclinical progression in spinal and bulbar muscular atrophy. Neurology. (2018) 90:e1501–9. doi: 10.1212/WNL.0000000000005360
78. Lombardi, V, Querin, G, Ziff, OJ, Zampedri, L, Martinelli, I, Heller, C, et al. Muscle and not neuronal biomarkers correlate with severity in spinal and bulbar muscular atrophy. Neurology. (2019) 92:10.1212/WNL.0000000000007097–e1211. doi: 10.1212/WNL.0000000000007097
79. Alves, CRR, Zhang, R, Johnstone, AJ, Garner, R, Nwe, PH, Siranosian, JJ, et al. Serum creatinine is a biomarker of progressive denervation in spinal muscular atrophy. Neurology. (2020) 94:e921–31. doi: 10.1212/WNL.0000000000008762
80. Rudnik-Schöneborn, S, Lützenrath, S, Borkowska, J, Karwanska, A, Hausmanowa-Petrusewicz, I, and Zerres, K. Analysis of creatine kinase activity in 504 patients with proximal spinal muscular atrophy types I-III from the point of view of progression and severity. Eur Neurol. (1998) 39:154–62. doi: 10.1159/000007926
81. Freigang, M, Wurster, CD, Hagenacker, T, Stolte, B, Weiler, M, Kamm, C, et al. Serum creatine kinase and creatinine in adult spinal muscular atrophy under nusinersen treatment. Ann Clin Transl Neurol. (2021) 8:1049–63. doi: 10.1002/acn3.51340
82. Sarıkaya Uzan, G, Paketçi, C, Günay, Ç, Edem, P, Özsoy, Ö, Hız Kurul, S, et al. The effect of Nusinersen therapy on laboratory parameters of patients with spinal muscular atrophy. Neuropediatrics. (2022) 53:321–9. doi: 10.1055/s-0042-1750719
83. Alves, CRR, Petrillo, M, Spellman, R, Garner, R, Zhang, R, Kiefer, M, et al. Implications of circulating neurofilaments for spinal muscular atrophy treatment early in life: a case series. Mol Ther Methods Clin Dev. (2021) 23:524–38. doi: 10.1016/j.omtm.2021.10.011
84. Yuan, A, Rao, MV, Null, V, and Nixon, RA. Neurofilaments and Neurofilament proteins in health and disease. Cold Spring Harb Perspect Biol. (2017) 9:a018309. doi: 10.1101/cshperspect.a018309
85. Burton, PR, and Wentz, MA. Neurofilaments are prominent in bullfrog olfactory axons but are rarely seen in those of the tiger salamander, Ambystoma tigrinum. J Comp Neurol. (1992) 317:396–406. doi: 10.1002/cne.903170406
86. Goldstein, ME, Sternberger, NH, and Sternberger, LA. Phosphorylation protects neurofilaments against proteolysis. J Neuroimmunol. (1987) 14:149–60. doi: 10.1016/0165-5728(87)90049-x
87. Khalil, M, Teunissen, CE, Otto, M, Piehl, F, Sormani, MP, Gattringer, T, et al. Neurofilaments as biomarkers in neurological disorders. Nat Rev Neurol. (2018) 14:577–89. doi: 10.1038/s41582-018-0058-z
88. Petzold, A. Neurofilament phosphoforms: surrogate markers for axonal injury, degeneration and loss. J Neurol Sci. (2005) 233:183–98. doi: 10.1016/j.jns.2005.03.015
89. Krause, K, Wulf, M, Sommer, P, Barkovits, K, Vorgerd, M, Marcus, K, et al. CSF diagnostics: a potentially valuable tool in neurodegenerative and inflammatory disorders involving motor neurons: a review. Diagnostics (Basel). (2021) 11:1522. doi: 10.3390/diagnostics11091522
90. Weydt, P, Oeckl, P, Huss, A, Müller, K, Volk, AE, Kuhle, J, et al. Neurofilament levels as biomarkers in asymptomatic and symptomatic familial amyotrophic lateral sclerosis. Ann Neurol. (2016) 79:152–8. doi: 10.1002/ana.24552
91. Staffaroni, AM, Quintana, M, Wendelberger, B, Heuer, HW, Russell, LL, Cobigo, Y, et al. Temporal order of clinical and biomarker changes in familial frontotemporal dementia. Nat Med. (2022) 28:2194–206. doi: 10.1038/s41591-022-01942-9
92. Benatar, M, Wuu, J, Andersen, PM, Lombardi, V, and Malaspina, A. Neurofilament light: a candidate biomarker of presymptomatic amyotrophic lateral sclerosis and phenoconversion. Ann Neurol. (2018) 84:130–9. doi: 10.1002/ana.25276
93. Saracino, D, Dorgham, K, Camuzat, A, Rinaldi, D, Rametti-Lacroux, A, Houot, M, et al. Plasma NfL levels and longitudinal change rates in C9orf72 and GRN-associated diseases: from tailored references to clinical applications. J Neurol Neurosurg Psychiatry. (2021) 92:1278–88. doi: 10.1136/jnnp-2021-326914
94. Gaiani, A, Martinelli, I, Bello, L, Querin, G, Puthenparampil, M, Ruggero, S, et al. Diagnostic and prognostic biomarkers in amyotrophic lateral sclerosis: Neurofilament light chain levels in definite subtypes of disease. JAMA. Neurology. (2017) 74:525–32. doi: 10.1001/jamaneurol.2016.5398
95. Paris, A, Bora, P, Parolo, S, Monine, M, Tong, X, Eraly, S, et al. An age-dependent mathematical model of neurofilament trafficking in healthy conditions. CPT Pharmacometrics Syst Pharmacol. (2022) 11:447–57. doi: 10.1002/psp4.12770
96. Beydoun, MA, Noren Hooten, N, Beydoun, HA, Weiss, J, Maldonado, AI, Katzel, LI, et al. Plasma neurofilament light and brain volumetric outcomes among middle-aged urban adults. Neurobiol Aging. (2023) 129:28–40. doi: 10.1016/j.neurobiolaging.2023.04.013
97. Beydoun, MA, Noren Hooten, N, Maldonado, AI, Beydoun, HA, Weiss, J, Evans, MK, et al. BMI and allostatic load are directly associated with longitudinal increase in plasma Neurofilament light among urban middle-aged adults. J Nutr. (2022) 152:535–49. doi: 10.1093/jn/nxab381
98. Darras, BT, Crawford TO, Finkel, RS, Mercuri, E, De Vivo, DC, Oskoui, M, et al. Neurofilament as a potential biomarker for spinal muscular atrophy. Ann Clin Transl Neurol. (2019) 6:932–44. doi: 10.1002/acn3.779
99. Paris, A, Bora, P, Parolo, S, Mac Cannell, D, Monine, M, van der Munnik, N, et al. A pediatric quantitative systems pharmacology model of neurofilament trafficking in spinal muscular atrophy treated with the antisense oligonucleotide nusinersen. CPT Pharmacometrics Syst Pharmacol. (2023) 12:196–206. doi: 10.1002/psp4.12890
100. Spicer, C, Lu, C-H, Catapano, F, Scoto, M, Zaharieva, I, Malaspina, A, et al. The altered expression of neurofilament in mouse models and patients with spinal muscular atrophy. Ann Clin Transl Neurol. (2021) 8:866–76. doi: 10.1002/acn3.51336
101. Glascock, J, Darras, BT, Crawford TO, Sumner, CJ, Kolb, SJ, DiDonato, C, et al. Identifying biomarkers of spinal muscular atrophy for further development. J Neuromuscul Dis. (2023) 10:937–54. doi: 10.3233/JND-230054
102. Wurster, CD, Winter, B, Wollinsky, K, Ludolph, AC, Uzelac, Z, Witzel, S, et al. Intrathecal administration of nusinersen in adolescent and adult SMA type 2 and 3 patients. J Neurol. (2019) 266:183–94. doi: 10.1007/s00415-018-9124-0
103. Wurster, CD, Steinacker, P, Günther, R, Koch, JC, Lingor, P, Uzelac, Z, et al. Neurofilament light chain in serum of adolescent and adult SMA patients under treatment with nusinersen. J Neurol. (2020) 267:36–44. doi: 10.1007/s00415-019-09547-y
104. Totzeck, A, Stolte, B, Kizina, K, Bolz, S, Schlag, M, Thimm, A, et al. Neurofilament heavy chain and tau protein are not elevated in cerebrospinal fluid of adult patients with spinal muscular atrophy during loading with Nusinersen. Int J Mol Sci. (2019) 20:5397. doi: 10.3390/ijms20215397
105. Winter, B, Guenther, R, Ludolph, AC, Hermann, A, Otto, M, and Wurster, CD. Neurofilaments and tau in CSF in an infant with SMA type 1 treated with nusinersen. J Neurol Neurosurg Psychiatry. (2019) 90:1068.2–1068.1069. doi: 10.1136/jnnp-2018-320033
106. Tozawa, T, Kasai, T, Tatebe, H, Shiomi, K, Nishio, H, Tokuda, T, et al. Intrathecal nusinersen treatment after ventriculo-peritoneal shunt placement: a case report focusing on the neurofilament light chain in cerebrospinal fluid. Brain and Development. (2020) 42:311–4. doi: 10.1016/j.braindev.2019.12.006
107. Olsson, B, Alberg, L, Cullen, NC, Michael, E, Wahlgren, L, Kroksmark, A-K, et al. NFL is a marker of treatment response in children with SMA treated with nusinersen. J Neurol. (2019) 266:2129–36. doi: 10.1007/s00415-019-09389-8
108. De Wel, B, De Schaepdryver, M, Poesen, K, and Claeys, KG. Biochemical and clinical biomarkers in adult SMA 3-4 patients treated with nusinersen for 22 months. Ann Clin Transl Neurol. (2022) 9:1241–51. doi: 10.1002/acn3.51625
109. Milella, G, Introna, A, D’Errico, E, Fraddosio, A, Scaglione, G, Morea, A, et al. Cerebrospinal fluid and clinical profiles in adult type 2-3 spinal muscular atrophy patients treated with Nusinersen: an 18-month single-Centre experience. Clin Drug Investig. (2021) 41:775–84. doi: 10.1007/s40261-021-01071-0
110. Rich, KA, Fox, A, Yalvac, M, Heintzman, S, Tellez, M, Bartlett, A, et al. Neurofilament levels in CSF and serum in an adult SMA cohort treated with Nusinersen. J Neuromuscul Dis. (2022) 9:111–9. doi: 10.3233/JND-210735
111. Faravelli, I, Meneri, M, Saccomanno, D, Velardo, D, Abati, E, Gagliardi, D, et al. Nusinersen treatment and cerebrospinal fluid neurofilaments: an explorative study on spinal muscular atrophy type 3 patients. J Cell Mol Med. (2020) 24:3034–9. doi: 10.1111/jcmm.14939
112. Bhatti, GK, Khullar, N, Sidhu, IS, Navik, US, Reddy, AP, Reddy, PH, et al. Emerging role of non-coding RNA in health and disease. Metab Brain Dis. (2021) 36:1119–34. doi: 10.1007/s11011-021-00739-y
113. Bartel, DP, and Chen, C-Z. Micromanagers of gene expression: the potentially widespread influence of metazoan micro RNAs. Nat Rev Genet. (2004) 5:396–400. doi: 10.1038/nrg1328
114. Magri, F, Vanoli, F, and Corti, S. mi RNA in spinal muscular atrophy pathogenesis and therapy. J Cell Mol Med. (2018) 22:755–67. doi: 10.1111/jcmm.13450
115. Slack, FJ, Basson, M, Liu, Z, Ambros, V, Horvitz, HR, and Ruvkun, G. The lin-41 RBCC gene acts in the C. elegans heterochronic pathway between the let-7 regulatory RNA and the LIN-29 transcription factor. Mol Cell. (2000) 5:659–69. doi: 10.1016/s1097-2765(00)80245-2
116. Ikenaka, A, Kitagawa, Y, Yoshida, M, Lin, C-Y, Niwa, A, Nakahata, T, et al. SMN promotes mitochondrial metabolic maturation during myogenesis by regulating the MYOD-mi RNA axis. Life Sci Alliance. (2023) 6:e202201457. doi: 10.26508/lsa.202201457
117. Fenoglio, C, Ridolfi, E, Galimberti, D, and Scarpini, E. An emerging role for long non-coding RNA dysregulation in neurological disorders. Int J Mol Sci. (2013) 14:20427–42. doi: 10.3390/ijms141020427
118. Wertz, MH, Winden, K, Neveu, P, Ng, S-Y, Ercan, E, and Sahin, M. Cell-type-specific mi R-431 dysregulation in a motor neuron model of spinal muscular atrophy. Hum Mol Genet. (2016) 25:2168–81. doi: 10.1093/hmg/ddw084
119. Chen, T-H. Circulating micro RNAs as potential biomarkers and therapeutic targets in spinal muscular atrophy. Ther Adv Neurol Disord. (2020) 13:1756286420979954. doi: 10.1177/1756286420979954
120. Haramati, S, Chapnik, E, Sztainberg, Y, Eilam, R, Zwang, R, Gershoni, N, et al. mi RNA malfunction causes spinal motor neuron disease. Proc Natl Acad Sci U S A. (2010) 107:13111–6. doi: 10.1073/pnas.1006151107
121. Wang, L-T, Chiou, S-S, Liao, Y-M, Jong, Y-J, and Hsu, S-H. Survival of motor neuron protein downregulates mi R-9 expression in patients with spinal muscular atrophy. Kaohsiung J Med Sci. (2014) 30:229–34. doi: 10.1016/j.kjms.2013.12.007
122. Catapano, F, Zaharieva, I, Scoto, M, Marrosu, E, Morgan, J, Muntoni, F, et al. Altered levels of Micro RNA-9, −206, and −132 in spinal muscular atrophy and their response to antisense oligonucleotide therapy. Mol Ther Nucleic Acids. (2016) 5:e331. doi: 10.1038/mtna.2016.47
123. Kye, MJ, and Gonçalves, ICG. The role of mi RNA in motor neuron disease. Front Cell Neurosci. (2014) 8:15. doi: 10.3389/fncel.2014.00015
124. Bhinge, A, Namboori, SC, Bithell, A, Soldati, C, Buckley, NJ, and Stanton, LW. MiR-375 is essential for human spinal motor neuron development and may be involved in motor neuron degeneration. Stem Cells. (2016) 34:124–34. doi: 10.1002/stem.2233
125. Abiusi, E, Infante, P, Cagnoli, C, Lospinoso Severini, L, Pane, M, Coratti, G, et al. SMA-mi Rs (mi R-181a-5p, −324-5p, and -451a) are overexpressed in spinal muscular atrophy skeletal muscle and serum samples. elife. (2021) 10:e68054. doi: 10.7554/eLife.68054
126. D’Silva, AM, Kariyawasam, D, Venkat, P, Mayoh, C, and Farrar, MA. Identification of novel CSF-derived mi RNAs in treated Paediatric onset spinal muscular atrophy: an exploratory study. Pharmaceutics. (2023) 15:170. doi: 10.3390/pharmaceutics15010170
127. Zaharieva, IT, Scoto, M, Aragon-Gawinska, K, Ridout, D, Doreste, B, Servais, L, et al. Response of plasma micro RNAs to nusinersen treatment in patients with SMA. Ann Clin Transl Neurol. (2022) 9:1011–26. doi: 10.1002/acn3.51579
128. Bonanno, S, Marcuzzo, S, Malacarne, C, Giagnorio, E, Masson, R, Zanin, R, et al. Circulating Myomi Rs as potential biomarkers to monitor response to Nusinersen in pediatric SMA patients. Biomedicine. (2020) 8:21. doi: 10.3390/biomedicines8020021
129. Magen, I, Aharoni, S, Yacovzada, NS, Tokatly Latzer, I, Alves, CRR, Sagi, L, et al. Muscle micro RNAs in the cerebrospinal fluid predict clinical response to nusinersen therapy in type II and type III spinal muscular atrophy patients. Eur J Neurol. (2022) 29:2420–30. doi: 10.1111/ene.15382
130. Chen, T-H, Chang, S-H, Wu, Y-F, Yen, Y-P, Hsu, F-Y, Chen, Y-C, et al. MiR34 contributes to spinal muscular atrophy and AAV9-mediated delivery of MiR34a ameliorates the motor deficits in SMA mice. Mol Ther Nucleic Acids. (2023) 32:144–60. doi: 10.1016/j.omtn.2023.03.005
131. Bianchi, L, Sframeli, M, Vantaggiato, L, Vita, GL, Ciranni, A, Polito, F, et al. Nusinersen modulates proteomics profiles of cerebrospinal fluid in spinal muscular atrophy type 1 patients. Int J Mol Sci. (2021) 22:4329. doi: 10.3390/ijms22094329
132. Kessler, T, Latzer, P, Schmid, D, Warnken, U, Saffari, A, Ziegler, A, et al. Cerebrospinal fluid proteomic profiling in nusinersen-treated patients with spinal muscular atrophy. J Neurochem. (2020) 153:650–61. doi: 10.1111/jnc.14953
133. Tosi, M, Catteruccia, M, Cherchi, C, Mizzoni, I, and D’Amico, A. Switching therapies: safety profile of Onasemnogene abeparvovec-xioi in a SMA1 patient previously treated with Risdiplam. Acta Myol. (2022) 41:117–20. doi: 10.36185/2532-1900-077
134. Jones, CC, Cook, SF, Jarecki, J, Belter, L, Reyna, SP, Staropoli, J, et al. Spinal muscular atrophy (SMA) subtype concordance in siblings: findings from the Cure SMA cohort. J Neuromuscul Dis. (2020) 7:33–40. doi: 10.3233/JND-190399
135. El Mendili, M-M, Lenglet, T, Stojkovic, T, Behin, A, Guimarães-Costa, R, Salachas, F, et al. Cervical spinal cord atrophy profile in adult SMN1-linked SMA. PLoS One. (2016) 11:e0152439. doi: 10.1371/journal.pone.0152439
136. Seferian, AM, Moraux, A, Annoussamy, M, Canal, A, Decostre, V, Diebate, O, et al. Upper limb strength and function changes during a one-year follow-up in non-ambulant patients with Duchenne muscular dystrophy: an observational multicenter trial. PLoS One. (2015) 10:e0113999. doi: 10.1371/journal.pone.0113999
137. Querin, G, Lenglet, T, Debs, R, Stojkovic, T, Behin, A, Salachas, F, et al. Development of new outcome measures for adult SMA type III and IV: a multimodal longitudinal study. J Neurol. (2021) 268:1792–802. doi: 10.1007/s00415-020-10332-5
138. Chabanon, A, Seferian, AM, Daron, A, Péréon, Y, Cances, C, Vuillerot, C, et al. Prospective and longitudinal natural history study of patients with type 2 and 3 spinal muscular atrophy: baseline data Nat his-SMA study. PLoS One. (2018) 13:e0201004. doi: 10.1371/journal.pone.0201004
139. Durmus, H, Yilmaz, R, Gulsen-Parman, Y, Oflazer-Serdaroglu, P, Cuttini, M, Dursun, M, et al. Muscle magnetic resonance imaging in spinal muscular atrophy type 3: selective and progressive involvement. Muscle Nerve. (2017) 55:651–6. doi: 10.1002/mus.25385
140. Besser, J, Carleton, HA, Gerner-Smidt, P, Lindsey, RL, and Trees, E. Next-generation sequencing technologies and their application to the study and control of bacterial infections. Clin Microbiol Infect. (2018) 24:335–41. doi: 10.1016/j.cmi.2017.10.013
141. Allicock, OM, Guo, C, Uhlemann, A-C, Whittier, S, Chauhan, LV, Garcia, J, et al. Bac cap Seq: a platform for diagnosis and characterization of bacterial infections. MBio. (2018) 9:e02007–18. doi: 10.1128/mBio.02007-18
142. Malone, ER, Oliva, M, Sabatini, PJB, Stockley, TL, and Siu, LL. Molecular profiling for precision cancer therapies. Genome Med. (2020) 12:8. doi: 10.1186/s13073-019-0703-1
143. Finkel, RS, Mercuri, E, Darras, BT, Connolly, AM, Kuntz, NL, Kirschner, J, et al. Nusinersen versus sham control in infantile-onset spinal muscular atrophy. N Engl J Med. (2017) 377:1723–32. doi: 10.1056/NEJMoa1702752
144. Mercuri, E, and Sansone, V. Nusinersen in adults with spinal muscular atrophy: new challenges. Lancet Neurol. (2020) 19:283–4. doi: 10.1016/S1474-4422(20)30068-5
145. Mercuri, E, Muntoni, F, Baranello, G, Masson, R, Boespflug-Tanguy, O, Bruno, C, et al. Onasemnogene abeparvovec gene therapy for symptomatic infantile-onset spinal muscular atrophy type 1 (STR1VE-EU): an open-label, single-arm, multicentre, phase 3 trial. Lancet Neurol. (2021) 20:832–41. doi: 10.1016/S1474-4422(21)00251-9
146. Strauss, KA, Farrar, MA, Muntoni, F, Saito, K, Mendell, JR, Servais, L, et al. Onasemnogene abeparvovec for presymptomatic infants with two copies of SMN2 at risk for spinal muscular atrophy type 1: the phase III SPR1NT trial. Nat Med. (2022) 28:1381–9. doi: 10.1038/s41591-022-01866-4
147. Masson, R, Mazurkiewicz-Bełdzińska, M, Rose, K, Servais, L, Xiong, H, Zanoteli, E, et al. Safety and efficacy of risdiplam in patients with type 1 spinal muscular atrophy (FIREFISH part 2): secondary analyses from an open-label trial. Lancet Neurol. (2022) 21:1110–9. doi: 10.1016/S1474-4422(22)00339-8
148. Oskoui, M, Day, JW, Deconinck, N, Mazzone, ES, Nascimento, A, Saito, K, et al. Two-year efficacy and safety of risdiplam in patients with type 2 or non-ambulant type 3 spinal muscular atrophy (SMA). J Neurol. (2023) 270:2531–46. doi: 10.1007/s00415-023-11560-1
149. Pane, M, Berti, B, Capasso, A, Coratti, G, Varone, A, D’Amico, A, et al. Onasemnogene abeparvovec in spinal muscular atrophy: predictors of efficacy and safety in naïve patients with spinal muscular atrophy and following switch from other therapies. EClinicalMedicine. (2023) 59:101997. doi: 10.1016/j.eclinm.2023.101997
150. Chiriboga, CA, Bruno, C, Duong, T, Fischer, D, Mercuri, E, Kirschner, J, et al. Risdiplam in patients previously treated with other therapies for spinal muscular atrophy: an interim analysis from the JEWELFISH study. Neurol Ther. (2023) 12:543–57. doi: 10.1007/s40120-023-00444-1
151. Roberto, J, Poulin, KL, Parks, RJ, and Vacratsis, PO. Label-free quantitative proteomic analysis of extracellular vesicles released from fibroblasts derived from patients with spinal muscular atrophy. Proteomics. (2021) 21:e2000301. doi: 10.1002/pmic.202000301
152. Varderidou-Minasian, S, Verheijen, BM, Harschnitz, O, Kling, S, Karst, H, van der Pol, WL, et al. Spinal Muscular Atrophy Patient iPSC-Derived Motor Neurons Display Altered Proteomes at Early Stages of Differentiation. ACS Omega. (2021) 6:35375–35388. doi: 10.1021/acsomega.1c04688
153. Brown, SJ, Kline, RA, Synowsky, SA, Shirran, SL, Holt, I, Sillence, KA, et al. The Proteome Signatures of Fibroblasts from Patients with Severe, Intermediate and Mild Spinal Muscular Atrophy Show Limited Overlap. Cells (2022) 11:2624. doi: 10.3390/cells11172624
154. Schorling, DC, K.lbel, H, Hentschel, A, Pechmann, A, Meyer, N, Wirth, B, et al. Cathepsin D as biomarker in cerebrospinal fluid of nusinersen-treated patients with spinal muscular atrophy. Eur J Neurol. (2022) 29:2084–2096. doi: 10.1111/ene.15331
Glossary
Keywords: SMA, circulating biomarkers, individualized medicine, neurofilaments, miRNA, innovative treatments, gene therapy
Citation: Giorgia Q, Gomez Garcia de la Banda M and Smeriglio P (2023) Role of circulating biomarkers in spinal muscular atrophy: insights from a new treatment era. Front. Neurol. 14:1226969. doi: 10.3389/fneur.2023.1226969
Edited by:
Angela Lucia Berardinelli, Neurological Institute Foundation Casimiro Mondino (IRCCS), ItalyReviewed by:
Tim Hagenacker, Essen University Hospital, GermanyNilo Riva, San Raffaele Hospital (IRCCS), Italy
Olivier Binda, University of Ottawa, Canada
Juan F. Vázquez Costa, Hospital Universitario y Politécnico de La Fe, Spain
Copyright © 2023 Giorgia, Gomez Garcia de la Banda and Smeriglio. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Querin Giorgia, Zy5xdWVyaW5AaW5zdGl0dXQtbXlvbG9naWUub3Jn
†These authors have contributed equally to this work and share first authorship