 Hui-Ling Qu
Hui-Ling Qu Xiao-Yu Sun
Xiao-Yu Sun Ying-Jie Dai*
Ying-Jie Dai*
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
CASE REPORT article
Front. Neurol., 24 October 2023
Sec. Neurocritical and Neurohospitalist Care
Volume 14 - 2023 | https://doi.org/10.3389/fneur.2023.1216328
This article is part of the Research TopicCase Reports in Neurocritical and Neurohospitalist Care, volume III - 2023View all 10 articles
Central pontine myelinolysis (CPM) is a heterogeneous nervous system disease of pontine demyelination, usually caused by rapid correction of hyponatremia. In the present study, we report a unique case of a 46-year-old man with a hyperglycemic state complicated with CPM. MRI demonstrated a high signal on T2 and symmetric restricted diffusion in the pontine. In conclusion, the clinical case described confirmed that the hyperosmolar state inherent in hyperglycemia was a likely cause of CPM.
Osmotic demyelination syndrome (ODS) is a rare complication of hyperglycemia and refers to central pontine myelinolysis and extrapontine myelinolysis manifesting with quadriparesis and neurocognitive changes with characteristic changes in magnetic resonance imaging (MRI) (1). The clinical manifestations range from low level of consciousness to dysarthria and then to quadriplegia (2). ODS is usually associated with myelin destruction in the central pontine basement, also known as central pontine myelinolysis (CPM). Here, we report a unique case of a 46-year-old man with a hyperglycemic state complicated with CPM.
A previously healthy 46-year-old man presented with limb weakness and dysarthria for 20 days. There was no history of malnutrition, a known diagnosis of diabetes, alcohol abuse, or smoking. Medical examination showed the patient presented with a body temperature of 36.6°C, respiratory rate of 18 breaths/min, normal resting heart rate of 76 beats/min, and blood pressure of 116/68 mm Hg. No abnormalities were found during heart and lung auscultation, and the patient presented with a soft abdomen and no swelling in either lower limb. A neurological examination revealed that he had mild dysarthria. His muscle tone was normal, with bilateral severe limb weakness and power was grade 2–3 in all of his limbs. Limb reflexes were mildly decreased and plantar flexor and sensation were normal. The blood results showed the following a blood glucose level of 49.7 mmol/L (894.6 mg/dL) and a normal range of 4–7 mmol/L. The hemoglobin A1c was 16.34%, 3-hydroxybutyric acid was 0.5 mmol/L, and he had 1+ ketone (20–29 mg/dL) in his urine. Notably, at presentation, his serum sodium was 128 mmoL/L (128 mEq/L), with a normal range of 133–146 mmol/L, but his serum potassium was markedly low at 2.8 mmol/L (2.8 mEq/L), with a normal range of 3.5–5.0 mmol/L. The calculated serum osmolality was 325 mosm/kg. Normal saline (0.9% NaCl) and insulin therapy (0.1 U/kg/h) were started and provided symptomatic treatment with potassium and sodium supplementation. After 6 h of admission, his blood glucose was 43.4 mmol/L (781.2 mg/dL) and serum sodium was 130 mmoL/L (130 mEq/L), whereas over the first 24 h of admission, his blood glucose reduced to 36.6 mmol/L (658.8 mg/dL), serum sodium increased to 135 mmoL/L (135 mEq/L), and 3-hydroxybutyric acid was 0.3 mmol/L. His blood glucose reduced to 25.6 mmol/L (460.8 mg/dL) over 48 h of admission and his serum sodium increased from 128 to 141 mmol/L (128 to 141 mEq/L). Over the first 48 h of admission, the patient’s serum potassium increased from 2.8 to 4.9 mmol/L (2.8 to 4.9 mEq/L), 3-hydroxybutyric acid was 0.05 mmol/L with negative ketonuria, and the calculated serum osmolality was 327 mOsm/kg. The blood glucose level is shown in Figure 1. In order to exclude peripheral nerve diseases, including acute inflammatory demyelinating polyneuropathy, lumbar puncture and electromyography were performed, and the results showed no abnormalities. The patient gradually became sleepy after admission, and the Glasgow Coma Score fluctuated between 13 and 14. However, there were no significant changes in his limb muscle power assessment. Because of the neurological symptoms, the patient subsequently underwent a cerebral MRI, which demonstrated features of CPM (Figure 2). The patient’s condition became stable and he was discharged to a rehabilitation institution. His blood glucose level was controlled with long-acting and short-acting insulin. In the following 6 months, his symptoms gradually improved and the power returned to grade 5 in all his limbs. The patient gradually resumed his daily activities without any residual symptoms (modified Rankin Scale [mRS] score = 0). Considering the clinical improvement of the patient and the absence of new neurological impairments, no repeated brain imaging was performed.
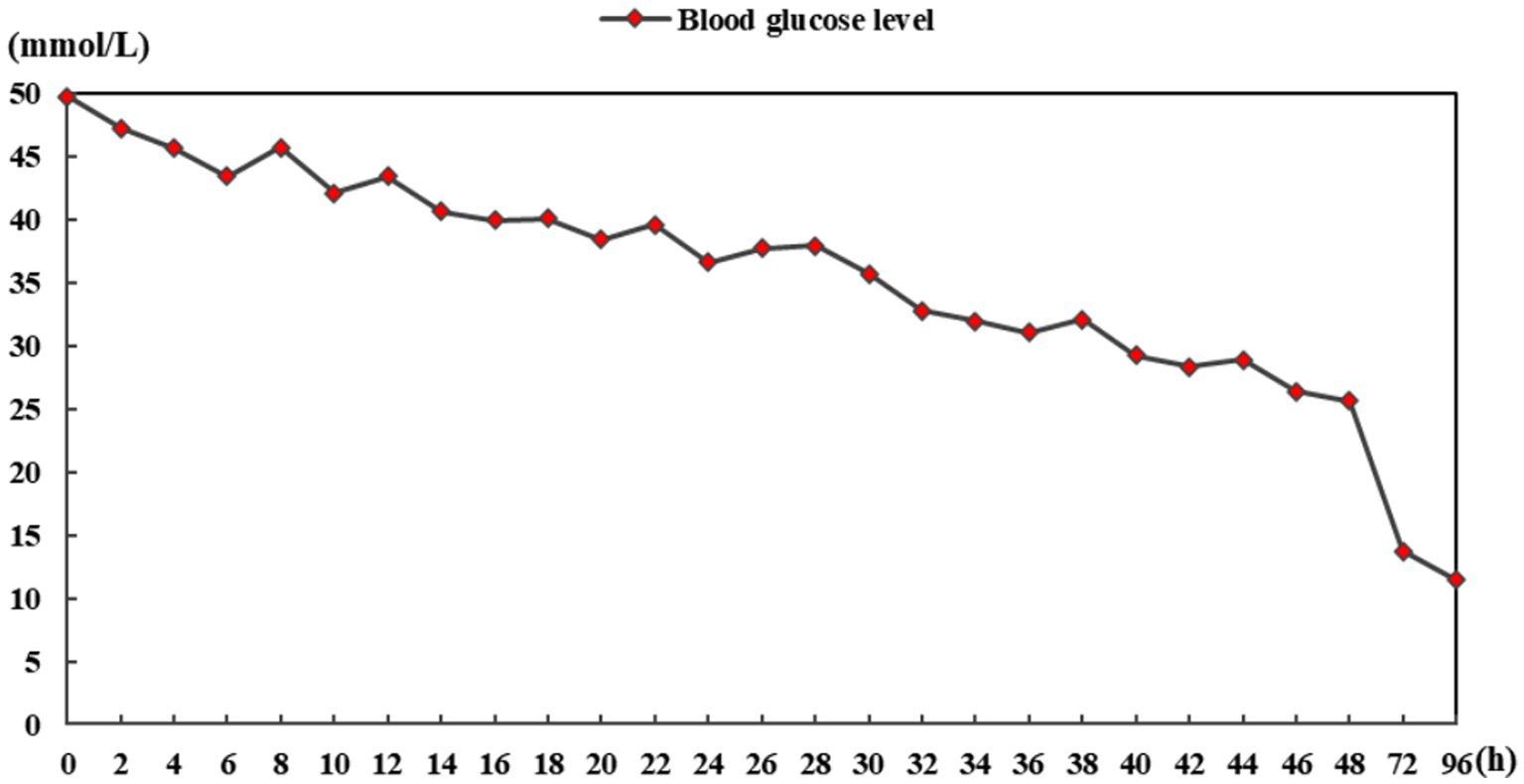
Figure 1. Blood glucose level chart.
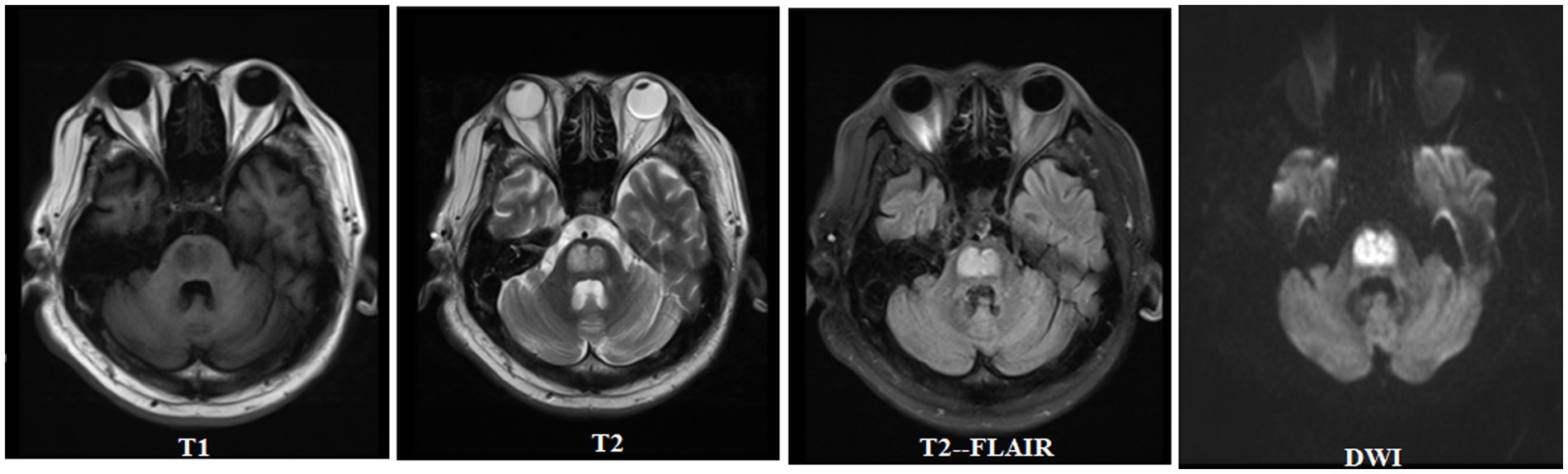
Figure 2. Central pontine myelinolysis in hyperglycemia.
CPM is a heterogeneous nervous system disease of pontine demyelination, usually caused by rapid correction of hyponatremia, and the correction speed exceeds 9 mmol/L/24 h (3, 4). In addition to rapidly correcting hyponatremia, other factors may be chronic alcoholism, hypernatremia, hypokalemia, hypophosphatemia, anorexia nervosa, and diabetes (5). Although rapid correction of hyponatremia is often considered a cause of CPM, considering the speed of initial serum sodium and electrolyte correction, our patient was unlikely to undergo rapid correction. Although the patient reported here presented with a serum sodium growth rate of 6.5 mmol/L/24 h, which did not exceed 9 mmol/L/24 h, we suggest that the inherent hypertonic state of hyperglycemia was more likely the cause of CPM. However, there are limited reports of associations between central pontine myelinolysis and derangement of glucose. It is reported that before the treatment of hyperglycemia and hyperosmolality (6, 7), there were cases of CPM, and our patient’s CPM may also have been caused by hyperosmolality before correction. Several reports have described patients with CPM in association with a hyperosmolar hyperglycemic state or its correction (8–17). Since there was no obvious change in osmotic pressure before and after correction, we consider that the CPM of our patient was related to the hyperosmolar state inherent in hyperglycemia. A diagnosis of CPM secondary to hyperglycemia was made.
Hyperglycemia and diabetes ketoacidosis are rare causes of CPM, and its physiopathology is uncertain (12). The pathophysiology of CPM may involve the sudden contraction of brain cells, especially oligodendrocytes, and demyelination caused by a rapid increase of serum osmotic pressure due to rapid correction of hyponatremia. Two theories have been proposed to explain oligodendrocyte contraction and myelinolysis, which are sometimes related to the rapid increase of serum osmotic pressure, whether due to the increase of sodium or other reasons, such as hyperglycemia: (i) local inflammatory demyelination caused by blood–brain barrier damage and (ii) oligodendrocyte apoptosis caused by hypertonic stress caused by changes in serum osmotic pressure, which occurs too fast to allow changes in specific molecules (11, 16). Research has shown that long-term episodes of severe hyperglycemia may lead to osmotic stress. Due to the poor control of diabetes, the unstable fluctuation of blood glucose may further lead to an unstable osmotic environment and the inability of normal cell compensation (18). MRI changes related to ODS typically include high signal lesions on T2-weighted MRI. The extension of T2-weighted signals can be explained by demyelination and edema (19).
Brain MRI remains the preferred diagnostic mode for CPM, and as described in the MRI results of this case, CPM may display a high signal on T2 and symmetric restricted diffusion in the pontine (10, 20). Despite reports of asymmetric lesions, pontine lesions are usually symmetrical (21). Our patient displayed the typical radiological and clinical findings of pontine myelinolysis with hyperglycemia, which is a rare phenomenon. Effective specific treatment methods have not yet been determined for the management of ODS cases. Therefore, the current treatment model includes general supportive care and treatment for potential causes, which is everything we do to treat patients. Similarly, there is no clear recommendation for the optimal timing of MRI imaging. The degree of clinical features and/or radiological changes cannot reliably determine the prognosis. The results vary from recovery to near-normal functional levels and death. More than half of ODS cases have recovered well, and the mortality rate of ODS cases has decreased over time. Even with severe neurological manifestations, patients with ODS may have a good prognosis (22).
In conclusion, the clinical case described confirmed that the hyperosmolar state inherent in hyperglycemia was a likely cause of CPM. Clinicians should consider hyperglycemia in patients with diabetes mellitus who develop CPM.
The original contributions presented in the study are included in the article/supplementary material, further inquiries can be directed to the corresponding author.
Written informed consent was obtained from the individual(s) for the publication of any potentially identifiable images or data included in this article.
Y-JD conceived the study. H-LQ and X-YS collected the data and drafted the manuscript. All authors contributed to the article and approved the submitted version.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
1. King, JD, and Rosner, MH. Osmotic demyelination syndrome. Am J Med Sci. (2010) 339:561–7. doi: 10.1097/MAJ.0b013e3181d3cd78
2. Laureno, R, and Karp, BI. Myelinolysis after correction of hyponatremia. Ann Intern Med. (1997) 126:57–62. doi: 10.7326/0003-4819-126-1-199701010-00008
3. Fitts, W, Vogel, AC, and Mateen, FJ. The changing face of osmotic demyelination syndrome: a retrospective, observational cohort study. Neurol Clin Pract. (2021) 11:304–10. doi: 10.1212/CPJ.0000000000000932
4. Rodríguez-Velver, KV, Soto-Garcia, AJ, Zapata-Rivera, MA, Montes-Villarreal, J, Villarreal-Pérez, JZ, and Rodríguez-Gutiérrez, R. Osmotic demyelination syndrome as the initial manifestation of a hyperosmolar hyperglycemic state. Case Rep Neurol Med. (2014) 2014:652523. doi: 10.1155/2014/652523
5. Chaudhary, A, Chaudhary, A, Yadav, RS, Shrestha, Y, and Shah, R. Pediatric osmotic demyelination syndrome in a case of type 1 diabetes mellitus with diabetic ketoacidosis. Clin Case Rep. (2022) 10:e05584. doi: 10.1002/ccr3.5584
6. Bline, K, Singh, D, Poeppelman, R, Lo, W, and O’Brien, N. Extrapontine Myelinolysis and microhemorrhages: rare finding in pediatric diabetic ketoacidosis. Pediatr Neurol. (2018) 89:68–70. doi: 10.1016/j.pediatrneurol.2018.09.006
7. Saini, M, Mamauag, MJ, and Singh, R. Central pontine myelinolysis: a rare presentation secondary to hyperglycaemia. Singap Med J. (2015) 56:e71–3. doi: 10.11622/smedj.2015065
8. Bonkowsky, JL, and Filloux, FM. Extrapontine myelinolysis in a pediatric case of diabetic ketoacidosis and cerebral edema. J Child Neurol. (2003) 18:144–7. doi: 10.1177/08830738030180021201
9. Burns, JD, Kosa, SC, and Wijdicks, EFM. Central pontine myelinolysis in a patient with hyperosmolar hyperglycemia and consistently normal serum sodium. Neurocrit Care. (2009) 11:251–4. doi: 10.1007/s12028-009-9241-9
10. Gonzalez Caldito, N, Karim, N, and Gebreyohanns, M. Teaching NeuroImage: central pontine Myelinolysis in diabetic ketoacidosis. Neurology. (2021) 97:e1971–2. doi: 10.1212/WNL.0000000000012301
11. Guerrero, WR, Dababneh, H, and Nadeau, SE. Hemiparesis, encephalopathy, and extrapontine osmotic myelinolysis in the setting of hyperosmolar hyperglycemia. J Clin Neurosci. (2013) 20:894–6. doi: 10.1016/j.jocn.2012.05.045
12. Matías-Guiu, JA, Molino, AM, Jorquera, M, Jiménez, R, and Ruiz-Yagüe, M. Pontine and extrapontine myelinolysis secondary to glycemic fluctuation. Neurologia. (2016) 31:345–7. doi: 10.1016/j.nrl.2014.06.005
13. McComb, RD, Pfeiffer, RF, Casey, JH, Wolcott, G, and Till, DJ. Lateral pontine and extrapontine myelinolysis associated with hypernatremia and hyperglycemia. Clin Neuropathol. (1989) 8:284–8.
14. McKee, AC, Winkelman, MD, and Banker, BQ. Central pontine myelinolysis in severely burned patients: relationship to serum hyperosmolality. Neurology. (1988) 38:1211–7. doi: 10.1212/WNL.38.8.1211
15. O’Malley, G, Moran, C, Draman, MS, King, T, Smith, D, Thompson, CJ, et al. Central pontine myelinolysis complicating treatment of the hyperglycaemic hyperosmolar state. Ann Clin Biochem. (2008) 45:440–3. doi: 10.1258/acb.2008.007171
16. Sharma, C, Kumawat, BL, Panchal, M, and Shah, M. Osmotic demyelination syndrome in type 1 diabetes in the absence of dyselectrolytaemia: an overlooked complication? BMJ Case Rep. (2017) 2017:bcr2016219148. doi: 10.1136/bcr-2016-219148
17. Sivaswamy, L, and Karia, S. Extrapontine myelinolysis in a 4 year old with diabetic ketoacidosis. Eur J Paediatr Neurol. (2007) 11:389–93. doi: 10.1016/j.ejpn.2007.02.017
18. Donnelly, H, Connor, S, and Quirk, J. Central pontine myelinolysis secondary to hyperglycaemia. Pract Neurol. (2016) 16:493–5. doi: 10.1136/practneurol-2016-001389
19. Murase, T, Sugimura, Y, Takefuji, S, Oiso, Y, and Murata, Y. Mechanisms and therapy of osmotic demyelination. Am J Med. (2006) 119:S69–73. doi: 10.1016/j.amjmed.2006.05.010
20. Ruzek, KA, Campeau, NG, and Miller, GM. Early diagnosis of central pontine myelinolysis with diffusion-weighted imaging. AJNR Am J Neuroradiol. (2004) 25:210–3.
21. Bansal, LR. Therapeutic effect of steroids in osmotic demyelination of infancy. Child Neurol Open. (2018) 5:2329048X1877057. doi: 10.1177/2329048X18770576
Keywords: central pontine myelinolysis, hyponatremia, hyperosmolar, osmotic demyelination syndrome, blood glucose
Citation: Qu H-L, Sun X-Y and Dai Y-J (2023) Central pontine myelinolysis: a rare finding in hyperosmolar hyperglycemia. Front. Neurol. 14:1216328. doi: 10.3389/fneur.2023.1216328
Edited by:
Barak Bar, University of Wisconsin-Madison, United StatesReviewed by:
Jerome Gnanaraj, Johns Hopkins University, United StatesCopyright © 2023 Qu, Sun and Dai. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Ying-Jie Dai, MTUwMDk4ODgzMzNAMTYzLmNvbQ==
†These authors have contributed equally to this work
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.