 Carlo Alberto Cesaroni1†
Carlo Alberto Cesaroni1† Marzia Pollazzon2†
Marzia Pollazzon2† Cecilia Mancini3
Cecilia Mancini3 Susanna Rizzi1Camilla Cappelletti3
Susanna Rizzi1Camilla Cappelletti3 Simone Pizzi3Daniele Frattini1
Simone Pizzi3Daniele Frattini1 Carlotta Spagnoli1Stefano Giuseppe Caraffi2
Carlotta Spagnoli1Stefano Giuseppe Caraffi2 Roberta Zuntini2Gabriele Trimarchi2
Roberta Zuntini2Gabriele Trimarchi2 Marcello Niceta3
Marcello Niceta3 Francesca Clementina Radio3
Francesca Clementina Radio3 Marco Tartaglia3Livia Garavelli2*‡
Marco Tartaglia3Livia Garavelli2*‡ Carlo Fusco1*‡
Carlo Fusco1*‡- 1Child Neurology and Psychiatry Unit, Pediatric Neurophysiology Laboratory, Mother-Child Department, Azienda USL-IRCCS Di Reggio Emilia, Reggio Emilia, Italy
- 2Medical Genetics Unit, Mother-Child Department, Azienda USL-IRCCS of Reggio Emilia, Reggio Emilia, Italy
- 3Molecular Genetics and Functional Genomics Unit, Ospedale Pediatrico Bambino Gesù, IRCCS, Rome, Italy
Objective: We aimed to report on previously unappreciated clinical features associated with FOXP1-related intellectual disability (ID) syndrome, a rare neurodevelopmental disorder characterized by global developmental delay, intellectual disability, and language delay, with or without autistic features.
Methods: We performed whole-exome sequencing (WES) to molecularly characterize an individual presenting with ID, epilepsy, autism spectrum disorder, behavioral problems, and facial dysmorphisms as major features.
Results: WES allowed us to identify a previously unreported de novo splice site variant, c.1429-1G>T (NM_032682.6), in the FOXP1 gene (OMIM*605515) as the causative event underlying the phenotype. Clinical reassessment of the patient and revision of the literature allowed us to refine the phenotype associated with FOXP1 haploinsufficiency, including hyperkinetic movement disorder and flat angiomas as associated features. Interestingly, the patient also has an asymmetric face and choanal atresia and a novel de novo variant of the CHD7 gene.
Conclusion: We suggest that FOXP1-related ID syndrome may also predispose to the development of hyperkinetic movement disorders and flat angiomas. These features could therefore require specific management of this condition.
1. Introduction
The FOXP1 (forkhead box P1) gene (MIM *605515) codifies for a member of the forkhead box family of transcriptional factors playing important roles in the regulation of gene transcription from early development through adulthood (1, 2). The identification of deletions encompassing FOXP1 (3–11) and loss-of-function variants of this gene in individuals with a consistent clinical phenotype (12–20) has led to the conclusion that FOXP1 haploinsufficiency is the molecular basis for the core features of “FOXP1-related intellectual disability (ID) syndrome” (MIM #613670). In addition to ID (mostly mild to moderate), the clinical phenotype of this condition includes a global developmental delay mainly affecting language, autistic spectrum disorder (ASD), behavioral disorders, facial dysmorphisms, ophthalmological anomalies, and cardiac and genitourinary anomalies (21–23). Epilepsy and kidney ailments (unilateral renal agenesis and horseshoe kidney) have been reported in some instances (20, 24). Approximately 100 cases have been described, and in the cases in which the segregation analysis was performed, the disease-causing variant was found to be de novo.
Herein, we report on a patient with a de novo splice site variant in FOXP1, NM_032682.6:c.1429-1G>T, identified by means of whole-exome sequencing (WES) and who has a complex phenotype with features that had not previously been described in this disorder. The boy has a psychomotor delay, ID, epilepsy, ASD, behavioral problems, and dysmorphic features. At birth, he also showed flat angiomas and right choanal atresia. There is no family history of similar features. Moreover, since the age of 9 years, he has been showing a stereotyped hyperkinetic movement disorder not associated with EEG anomalies. While a de novo variant in CHD7, NM_032682.6:c.122T>C, p.(Met41Thr) could explain the right choanal atresia, flat angiomas and the movement disorder are likely to represent new features of this condition that had not previously been characterized.
2. Case report
2.1. Medical history
The patient, a 12-year-old boy, is the second child of apparently healthy and unrelated parents. The older sister of the boy is healthy. The second pregnancy of the parents terminated in a miscarriage. Their family history is negative for motor tics. Pregnancy was complicated by gestational diabetes and preeclampsia. The proband was born at 36 weeks of gestation by vaginal delivery. Birth parameters were a weight of 3,200 g (75th−90th percentile), a length of 51 cm (90th−97th percentile), and a head circumference of 36 cm (98th percentile). The Apgar scores were 9 and 9 at 1 and 5 min, respectively. He was admitted to the Neonatal Intensive Care Unit (NICU) for approximately 2 h for cyanosis. During investigations, right choanal atresia was discovered, with subsequent daytime apneas. He underwent surgery for left cryptorchidism at the ages of 3, 4, and 10 years. At 10 years, he underwent permanent Appendix vesicostomy for urinary incontinence. He showed psychomotor delay: walked on tiptoes with help at 17 months; said first words at 4 years of age; and showed partial sphincter control at 4 years, with enuresis. At the age of 12 years, he was able to pronounce sentences but he could not eat on his own. He showed talipes equinovarus with persistent tiptoe walking and had motor impairment with difficulty in independent ambulation. Braces on the lower limbs and wheelchairs for long journeys were introduced.
Motor stereotypies with rocking of the trunk appeared at 7 years. Since the age of 8 years, he has shown recurrent stereotyped hyperkinetic movements, including retrocollis with tongue protrusion and hypertonicity of the arms, that repeat a few times in a less intense fashion and last up to 2 min, which is definable as paroxysmal dystonia (Supplementary Video S1). Sometimes, these episodes were associated with apparent alteration of contact with fixed-gaze focal abnormalities, and bouffées of epileptiform anomalies were recorded. Although not concomitant with these episodes, therapy with valproic acid and clobazam was subsequently introduced, with no benefit. Follow-up routine EEG showed focal paroxysmal anomalies in the phase of reduced alertness and short diffuse bursts of epileptic abnormalities (Figure 1). Since the age of 11 years, a disinhibited conduct of self-stimulation and autoeroticism appeared and required the introduction of risperidone. Hetero-aggressivity and hand stereotypies (previously rubbing and recently clapping) were reported. We also noticed a sensory-seeking behavior with a tendency to seek close physical contact and to smell people, hands, and food.
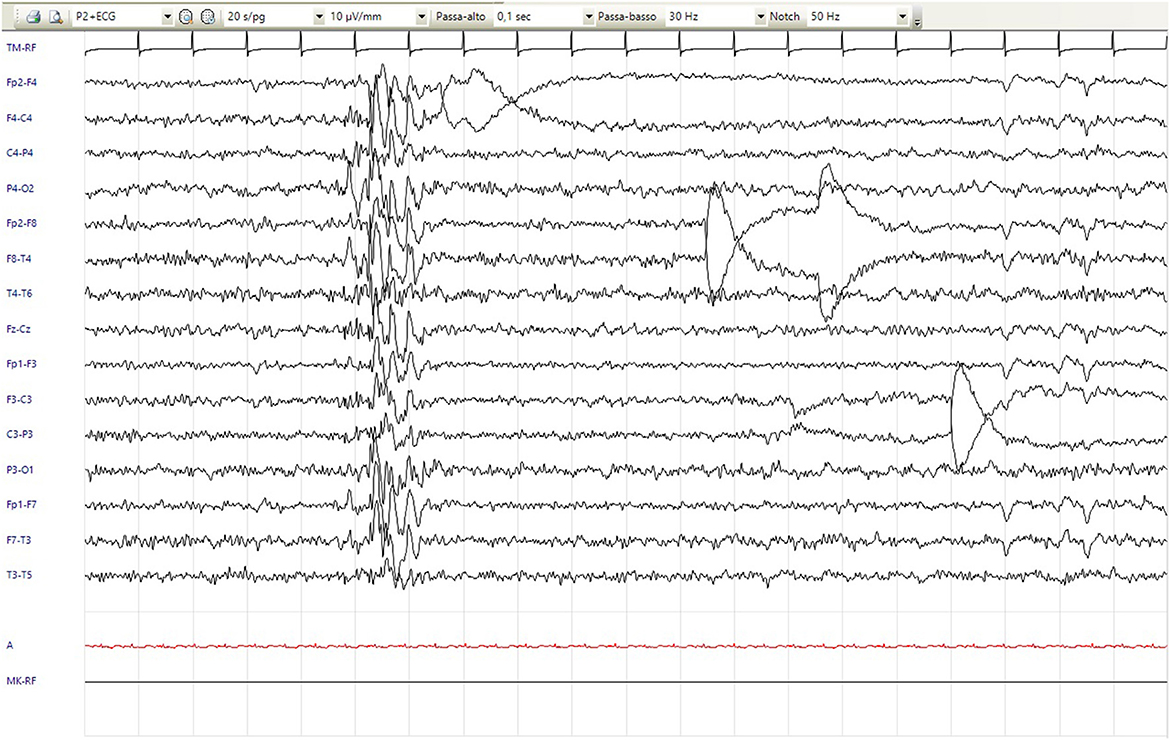
Figure 1. EEG showed short diffuse bursts of epileptic anomalies (20/s/pg, 10 μV/mm, 0,1 s, 30 Hz).
2.2. Clinical and instrumental findings
During clinical follow-up, a progressively more elongated face and a more pronounced nasal bridge were noticed (Figure 2). At the last examination (12 years and 6 months), his height was 163.5 cm (>97th percentile), weight was 45 kg (75th−90th percentile), and head circumference was 54 cm (50th−75th percentile). He had a hypomimic and asymmetric face, flat occiput, hypertelorism, periorbital fullness, bilateral ptosis (more pronounced on the left side), epicanthus, exotropia, thick alae nasi, microretrognathia, a thin upper lip, a high-arched palate, small and spaced teeth, poor representation of muscle mass, pectus excavatum, scoliosis, widening of the proximal interphalangeal joints of the hands, flat feet with sandal gap, and II-III toe partial cutaneous syndactyly. He also showed vascular anomalies: visible vessels on the lower eyelids and congenital vascular anomalies (flat angiomas) that disappeared upon pressure, some in the occiput and vertex, and multiple in the back (Figure 2). Abdominal ultrasound, audiometric testing, fundus examination, and electroneurography (ENG) were normal. A small aneurysm at the level of the interatrial septum was detected during the echocardiography. Brain MRI at 6 years revealed cysts of the septum pellucidum and of the cavum vergae (Figure 3A). A brain CT scan performed at 3 years of age revealed right choanal atresia (Figure 3B). The ophthalmological evaluation documented hyperopia and astigmatism.
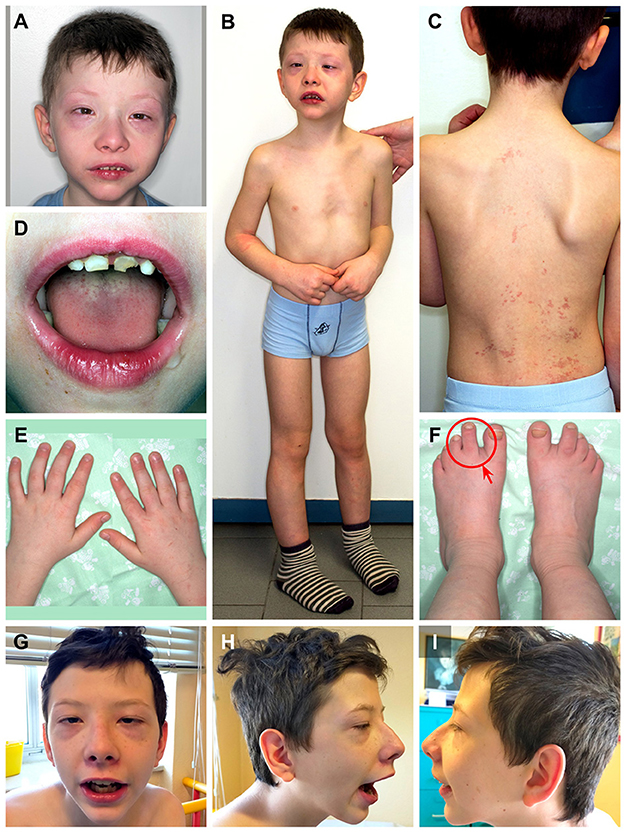
Figure 2. Patient at 9 years and 8 months of age (A-F). (A) Hypertelorism, periorbital fullness, epicanthus, esotropia, thick alae nasi, and microretrognathia; (B) pectus excavatum; (C) scoliosis, congenital vascular anomalies that disappeared upon pressure in the occiput, vertex, and multiple in the back; (D) small and spaced teeth; (E) widening of the proximal interphalangeal joints of the hands; and (F) flat feet with sandal gap and II-III toe partial cutaneous syndactyly (red circle and arrow). Patient at 12 years and 6 months of age (G–I). Note the more pronounced nose and microretrognathia.
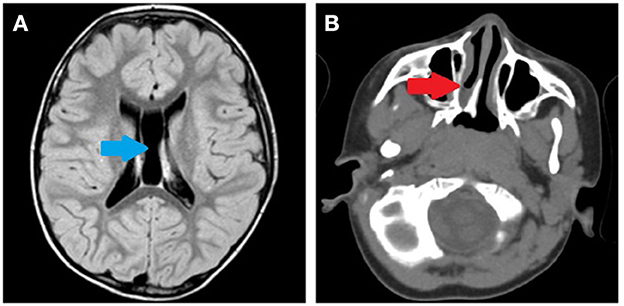
Figure 3. (A) Brain MRI performed at 6 years of age. Cysts of the septum pellucidum and the cavum vergae (blue arrow). (B) Brain CT scan performed at 3 years of age. Right choanal atresia (red arrow).
2.3. Molecular analysis
Genetic works based on CGH array and FMR1 analyses revealed normal results. WES analysis was performed on genomic DNA extracted from peripheral blood leukocytes of the proband and their parents under standard procedures. The capture of the target regions was performed using the SureSelect Human All Exon V7 target enrichment kit, and sequencing was performed on a NovaSeq 6000 platform (Supplementary Table S1). WES raw data were processed using an in-house pipeline based on the GATK Best Practices (25–28). The UCSC GRCh37/hg19 genome assembly was used as a reference for reads alignment by means of the BWA-MEM (bwakit v.0.7.11) algorithm (29). SnpEff v.5.0 (30) and dbNSFP v.4.0 (31) tools were used for variant annotation, including functional impact and in silico prediction of deleteriousness by means of Combined Annotation Dependent Depletion (CADD) v.1.4 (32), Mendelian Clinically Applicable Pathogenicity (M-CAP) v.1.3 (33), and Intervar v.2.0.1 (34). Population frequencies were annotated from the gnomAD database and an in-house database, which included ~2,500 exomes. High-quality SNV and INDELs (GATK v3.8 hard-filtering: QUAL>100) with low-frequency (gnomAD MAF <0.1%, in-house exomes database <1) and within coding exons and splice regions were assessed separately. In the functional impact evaluation, a higher priority was assigned to variants with a CADD score of >20, was tagged as pathogenic/likely pathogenic by InterVar, and relied on Phen2Gene, a tool dedicated to the gene/phenotype associations (35). WES analysis led to the identification of two possibly relevant de novo variants: a pathogenic splice donor site change involving the FOXP1 gene [NM_032682.6:c.1429-1G>T] and a missense variant in CHD7 gene (NM_017780.4:c.122T>C), which predicted to result in the amino acid substitution p.(Met41Thr). Both variants have been submitted to ClinVar (SUB13065401). The FOXP1 variant is absent in the reference population database gnomAD v2.1.1 (accessed on 2023/02/28) and is predicted to abolish the canonical acceptor splice site of exon 17, possibly activating a nearby cryptic acceptor site downstream that would cause a frameshift and generate a premature stop codon. According to the recommendations of the American College of Medical Genetics and Genomics (36, 37), in the absence of experimental data on the effects of the splice site alteration, the variant can be classified as likely pathogenic. The CHD7 variant is reported at an extremely low frequency in the reference population database gnomAD v2.1.1 (accessed on 2023/02/28), predicted damaging by most in silico tools, and can be classified as a variant of uncertain significance (VUS) favoring likely pathogenic, possibly contributing to a subset of the reported clinical features (i.e., asymmetric face and monolateral choanal atresia).
3. Discussion
Our patient had “FOXP1-related intellectual disability (ID) syndrome” with a clinical phenotype fitting the condition with many features that had already been described in previous reports. He presented with mild/moderate ID, a speech delay with echolalia, autism spectrum disorder features, and dysmorphisms. Hyperopia and astigmatism had also been reported. On the contrary, cardiac defects are minor and often restricted to atrial septal defects (38). Notably, Trelles et al. (39) described the complex neurobehavioral profile of the condition. This includes repetitive behaviors and sensory symptoms (e.g., sensory seeking), some of which were also recognized in our patient. Compared to the other described cases, however, the patient also had peculiar characteristics; in particular, he showed hyperkinetic movement disorder not associated with electroencephalographic anomalies and he also had EEG anomalies without clinical manifestations. Therefore, we cannot rule out both a movement disorder and seizures. Among almost 100 individuals with FOXP1-related ID syndrome, seven individuals experienced epilepsy, but no subjects have been reported to exhibit movement disorder (Supplementary Table S2), suggesting another neurological feature that might occur in this condition. A more extended series of cases, however, is required to corroborate this assumption.
The FOXP1 transcriptional repressor is a molecular determinant of radial neuronal migration and morphogenesis (40). In particular, Foxp1 is expressed at high levels in a subset of striatal projection neurons of the mouse brain, probably the matrix neurons (41, 42). Foxp1 is crucial for maintaining the cellular composition of the striatum and the proper formation of the striosome-matrix compartments (43). Moreover, gene expression studies in human neural progenitors with altered FOXP1 levels documented a role in regulating pathways involved in striatal neuron identity (44). The expression pattern of Foxp1 mRNA suggests that Foxp1 may play a role in the development and formation of a circuit in the basal ganglia. A similar mechanism is described for Huntington's disease, in which a reduced expression of Foxp1 correlates with the drop of the neuroprotective role in the affected cells (45). Though we have not demonstrated this in our patient, we can speculate that the possible alterations of the mRNA in the basal ganglia could possibly explain his movement disorder (41). In addition, mice heterozygous for a Foxp1 deletion (46) exhibit a reduced mitochondrial membrane potential and complex I activity, as well as decreased expression of the antioxidants superoxide dismutase 2 and glutathione peroxidase 1, resulting in increased oxidative stress and lipid peroxidation. Note that a mitochondrial disorder involving oxidative-stress-related SUCLA2 gene is associated with a hyperkinetic movement disorder (47). The presence of multiple flat angiomas is also peculiar considering that cutaneous vascular anomalies are unexpected in the disorder. Using gene expression profiling, Grundmann et al. (48) showed that FoxP1 induces a specific change in the endothelial transcriptome and stimulates angiogenesis by repressing semaphorin 5B (Sema5B). We, therefore, suggest that the congenital vascular anomalies (flat angiomas) observed in our patient could also belong to the FOXP1-related spectrum.
The present subject also has right choanal atresia and facial asymmetry, which have never been described in the FOXP1-related ID syndrome. We can speculate that these features may be related to the de novo p.(Met41Thr) variant in the CHD7 gene. This gene is usually involved in CHARGE syndrome, and we cannot exclude the contribution of this variant to the complex phenotype of the present subject.
In conclusion, the features that we have described and that distinguish our patient broaden the phenotype of FOXP1-related syndrome and can contribute to defining a more specific follow-up of the patient.
Data availability statement
The datasets presented in this study can be found in online repositories. The names of the repository/repositories and accession number(s) can be found here: https://www.ncbi.nlm.nih.gov/clinvar/, SUB13065401.
Ethics statement
The studies involving human participants were reviewed and approved by the Local Institutional Ethical Committee of the Ospedale Pediatrico Bambino Gesù IRCCS (OPBG), Rome (1702_OPBG_2018). Written informed consent to participate in this study was provided by the participants' legal guardian/next of kin. Written informed consent was obtained from the individual(s), and minor(s)' legal guardian/next of kin, for the publication of any potentially identifiable images or data included in this article.
Author contributions
CAC and MP: conceptualization and writing—original draft preparation. CM, CAC, SP, MN, FCR, MT, SGC, and RZ: genomic analyses. CAC, MP, GT, SR, DF, and CS: clinical data collection and data curation. CAC, MP, SP, FCR, MT, SGC, and RZ: writing—review and editing. LG and CF: supervision. All authors have read and agreed to the published version of the manuscript.
Funding
This project was partially funded by the Italian Ministry of Health (Ricerca 5x1000, CCR-2017-23669081 to MT. RF-2018-12366931, to FR) and Ministero della Ricerca (FOE 2019, to MT).
Acknowledgments
The authors thank the patient's family for their cooperation and their consent to the publication of the photographs. The authors of this publication are members of the European Reference Network on Rare Congenital Malformations and Rare Intellectual Disability ERN-ITHACA [EU Framework Partnership Agreement ID: 3HP-HP-FPA ERN-01-2016/739516]. We also thank the photographers Marco Bonazzi and Luca Valcavi.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher's note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fneur.2023.1207176/full#supplementary-material
References
1. OMIM—Online Mendelian Inheritance in Man®. Available online: https://www.omim.org/ (accessed March 22, 2023).
2. Shu W, Yang H, Zhang L, Lu MM, Morrisey EE. Characterization of a new subfamily of winged-helix/forkhead (Fox) genes that are expressed in the lung and act as transcriptional repressors. J Biol Chem. (2001) 276:27488–97. doi: 10.1074/jbc.M100636200
3. Pariani MJ, Spencer A, Graham JM Jr, Rimoin DL. A 785kb deletion of 3p14.1p13, including the FOXP1 gene, associated with speech delay, contractures, hypertonia and blepharophimosis. Eur J Med Genet. (2009) 52:123–127. doi: 10.1016/j.ejmg.2009.03.012
4. Horn, D, Kapeller J, Rivera-Brugues N, Moog U, Lorenz-Depiereux B, Eck S., et al. Identification of FOXP1 deletions in three unrelated patients with mental retardation and significant speech and language deficits. Hum Mutat. (2010) 31:E1851–E1860. doi: 10.1002/humu.21362
5. Carr CW, Moreno-De-Luca D, Parker C, Zimmerman HH, Ledbetter N, Martin CL, et al. Chiari I malformation, delayed gross motor skills, severe speech delay, and epileptiform discharges in a child with FOXP1 haploinsufficiency. Eur J Hum Genet. (2010) 18:1216–20. doi: 10.1038/ejhg.2010.96
6. Tutulan-Cunită AC, Papuc SM, Arghir A, Rötzer KM, Deshpande C, Lungeanu A, et al. 3p interstitial deletion: novel case report and review. J Child Neurol. (2012) 27:1062–6. doi: 10.1177/0883073811431016
7. Palumbo O, D'Agruma L, Minenna AF, Palumbo P, Stallone R, Palladino, et al. 3p141 de novo microdeletion involving the FOXP1 gene in an adult patient with autism, severe speech delay and deficit of motor coordination. Gene. (2013) 516:107–13. doi: 10.1016/j.gene.2012.12.073
8. Dimitrov, B.I, Ogilvie C, Wieczorek D, Wakeling E, Sikkema-Raddatz B, van Ravenswaaij-Arts C.M, et al. 3p14 deletion is a rare contiguous gene syndrome: report of 2 new patients and an overview of 14 patients. Am J Med Genet A. (2015) 167:1223–30. doi: 10.1002/ajmg.a.36556
9. Vuillaume M.L, Cogné B, Jeanne M, Boland A, Ung D.C, Quinquis D, et al. Whole genome sequencing identifies a de novo 2.1 Mb balanced paracentric inversion disrupting FOXP1 and leading to severe intellectual disability. Clin Chim Acta. (2018) 485:218–223. doi: 10.1016/j.cca.2018.06.048
10. Mutlu-Albayrak H, Karaer K. Vocal cord immobility as a cause of aphonia in a child with 3p13p12 deletion syndrome encompassing FOXP1 gene. Int J Pediatr Otorhinolaryngol. (2019) 117:179–81. doi: 10.1016/j.ijporl.2018.11.024
11. Yamamoto-Shimojima K, Okamoto N, Matsumura W, Okazaki T, Yamamoto T. Three Japanese patients with 3p13 microdeletions involving FOXP1. Brain Develop. (2019) 41:257–62. doi: 10.1016/j.braindev.2018.10.016
12. Hamdan FF, Daoud H, Rochefort D, Piton A, Gauthier J, Langlois M, et al. De novo mutations in FOXP1 in cases with intellectual disability, autism, and language impairment. Am J Hum Genet. (2010) 87:671–8. doi: 10.1016/j.ajhg.2010.09.017
13. O'Roak BJ, Deriziotis P, Lee C, Vives L, Schwartz JJ, Girirajan S, et al. Exome sequencing in sporadic autism spectrum disorders identifies severe de novo mutations. Nat Genet. (2011) 43:585–9. doi: 10.1038/ng.835
14. Le Fevre A.K, Taylor S, Malek N.H, Horn D, Carr C.W, Abdul-Rahman O.A, et al. FOXP1 mutations cause intellectual disability and a recognizable phenotype. Am J Med Genet A. (2013) 161A:3166–75. doi: 10.1002/ajmg.a.36174
15. Lozano R, Vino A, Lozano C, Fisher SE, Deriziotis P, A de. novo FOXP1 variant in a patient with autism, intellectual disability and severe speech and language impairment. Eur J Hum Genet. (2015) 23:1702–7. doi: 10.1038/ejhg.2015.66
16. Song H, Makino Y, Noguchi E, Arinami T. A case report of de novo missense FOXP1 mutation in a non-Caucasian patient with global developmental delay and severe speech impairment. Clin Case Rep. (2015) 3:110–3. doi: 10.1002/ccr3.167
17. Myers A, du Souich C, Yang CL, Borovik L, Mwenifumbo J, Rupps R, et al. FOXP1 haploinsufficiency: phenotypes beyond behavior and intellectual disability? Am J Med Genet A. (2017) 173:3172–3181. doi: 10.1002/ajmg.a.38462
18. Sollis, E, Deriziotis, P, Saitsu, H, Miyake, N, Matsumoto, N, Hoffer M.J.V, et al. Equivalent missense variant in the FOXP2 and FOXP1 transcription factors causes distinct neurodevelopmental disorders. Hum Mutat. (2017) 38:1542–54. doi: 10.1002/humu.23303
19. Johnson T.B, Mechels K, Anderson R.H, Cain J.T, Sturdevant D.A, Braddock S, et al. Characterization of a recurrent missense mutation in the forkhead DNA-binding domain of FOXP1. Sci Rep. (2018) 8:16161. doi: 10.1038/s41598-018-34437-0
20. Urreizti R, Damanti S., Esteve C., Franco-Valls H, Castilla-Vallmanya L., Tonda R., et al. A de novo FOXP1 truncating mutation in a patient originally diagnosed as C syndrome. Sci Rep. (2018) 8:694. doi: 10.1038/s41598-017-19109-9
21. Sollis E, Graham SA, Vino A, Froehlich H, Vreeburg M, Dimitropoulou D, et al. Identification and functional characterization of de novo FOXP1 variants provides novel insights into the etiology of neurodevelopmental disorder. Hum Mol Genet. (2016) 25:546–57. doi: 10.1093/hmg/ddv495
22. Siper PM, De Rubeis S, Trelles MDP, Durkin A, Di Marino D, Muratet F, et al. Prospective investigation of FOXP1 syndrome. Mol Autism. (2017) 8:57. doi: 10.1186/s13229-017-0172-6
23. Lozano R, Gbekie C, Siper PM, Srivastava S, Saland JM, Sethuram S, et al. FOXP1 syndrome: a review of the literature and practice parameters for medical assessment and monitoring. J Neurodev Disord. (2021) 13:18. doi: 10.1186/s11689-021-09358-1
24. Bekheirnia MR, Bekheirnia N, Bainbridge MN, Gu S, Coban Akdemir ZH, Gambin T, et al. Whole-exome sequencing in the molecular diagnosis of individuals with congenital anomalies of the kidney and urinary tract and identification of a new causative gene. Genet Med. (2017) 19:412–20. doi: 10.1038/gim.2016.131
25. Motta M, Pannone L, Pantaleoni F, Bocchinfuso G, Radio FC, Cecchetti S, et al. Enhanced MAPK1 function causes a neurodevelopmental disorder within the RASopathy clinical spectrum. Am J Hum Genet. (2020) 107:499–513. doi: 10.1016/j.ajhg.2020.06.018
26. Radio FC, Pang K, Ciolfi A, Levy MA, Hernández-García A, Pedace L, et al. SPEN haploinsufficiency causes a neurodevelopmental disorder overlapping proximal 1p36 deletion syndrome with an episignature of X chromosomes in females. Am J Hum Genet. (2021) 108:502-516. doi: 10.1016/j.ajhg.2021.01.015
27. Van der Auwera GA, Carneiro MO, Hartl C, Poplin R, Del Angel G, Levy-Moonshine A, et al. From FastQ data to high-confidence variant calls: the genome analysis toolkit best practices pipeline. Curr Protoc Bioinformatics. (2013) 43:11.10.1–33. doi: 10.1002/0471250953.bi1110s43
28. Lin YC, Niceta M, Muto V, Vona B, Pagnamenta AT, Maroofian R, et al. SCUBE3 loss-of-function causes a recognizable recessive developmental disorder due to defective bone morphogenetic protein signaling. Am J Hum Genet. (2021) 108:115–33. doi: 10.1016/j.ajhg.2020.11.015
29. Li H. Aligning sequence reads, clone sequences and assembly contigs with BWA-MEM. arXiv preprint arXiv:1303.3997 (2013). doi: 10.48550/arXiv.1303.3997
30. Cingolani P, Platts A, Wang le L, Coon M, Nguyen T, Wang L, et al. A program for annotating and predicting the effects of single nucleotide polymorphisms, SnpEff: SNPs in the genome of Drosophila melanogaster strain w1118; iso-2; iso-3. Fly. (2012) 6:80–92. doi: 10.4161/fly.19695
31. Liu X, Jian X, Boerwinkle E. dbNSFP v2.0: a database of human non-synonymous SNVs and their functional predictions and annotations. Hum Mutat. (2013) 34:E2393–402. doi: 10.1002/humu.22376
32. Kircher M, Witten DM Jain P, O'Roak BJ, Cooper GM, Shendure J, et al. A general framework for estimating the relative pathogenicity of human genetic variants. Nat Genet. (2014) 46:310–5. doi: 10.1038/ng.2892
33. Jagadeesh KA, Wenger AM, Berger MJ, Guturu H, Stenson PD, Cooper DN, et al. M-CAP eliminates a majority of variants of uncertain significance in clinical exomes at high sensitivity. Nat Genet. (2016) 48:1581–6. doi: 10.1038/ng.3703
34. Li Q, Wang K. InterVar: clinical interpretation of genetic variants by the 2015 ACMG-AMP guidelines. Am J Hum Genet. (2017) 100:267–80. doi: 10.1016/j.ajhg.2017.01.004
35. Zhao M, Havrilla JM, Fang L, Chen Y, Peng J, Liu C, et al. Phen2Gene: rapid phenotype-driven gene prioritization for rare diseases. NAR Genom Bioinform. (2020) 2:lqaa032. doi: 10.1093/nargab/lqaa032
36. Richards S, Aziz N, Bale S, Bick D, Das S, Gastier-Foster J, et al. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American college of medical genetics and genomics and the association for molecular pathology. Genet Med. (2015) 17:405–24. doi: 10.1038/gim.2015.30
37. Tavtigian SV, Harrison SM, Boucher KM, Biesecker LG. Fitting a naturally scaled point system to the ACMG/AMP variant classification guidelines. Hum Mutat. (2020) 41:1734–7. doi: 10.1002/humu.24088
38. Meerschaut I, Rochefort D, Revencu N, Petre J, Corsello C, Rouleau GA, et al. FOXP1-related intellectual disability syndrome: a recognisable entity. J Med Genet. (2017) 54:613–623. doi: 10.1136/jmedgenet-2017-104579
39. Trelles MP, Levy T, Lerman B, Siper P, Lozano R, Halpern D, et al. Individuals with FOXP1 syndrome present with a complex neurobehavioral profile with high rates of ADHD, anxiety, repetitive behaviors, and sensory symptoms. Mol Autism. (2021) 12:61. doi: 10.1186/s13229-021-00469-z
40. Li X, Xiao J, Fröhlich H, Tu X, Li L, Xu Y, et al. Foxp1 regulates cortical radial migration and neuronal morphogenesis in developing cerebral cortex. PLoS ONE. (2015) 10:e0127671. doi: 10.1371/journal.pone.0127671
41. Tamura S, Morikawa Y, Iwanishi H, Hisaoka T, Senba E. Foxp1 gene expression in projection neurons of the mouse striatum. Neuroscience. (2004) 124:261–7. doi: 10.1016/j.neuroscience.2003.11.036
42. Fong WL, Kuo HY, Wu HL, Chen SY, Liu FC. Differential and overlapping pattern of Foxp1 and Foxp2 expression in the striatum of adult mouse brain. Neuroscience. (2018) 388:214–23. doi: 10.1016/j.neuroscience.2018.07.017
43. Anderson AG, Kulkarni A, Harper M, Konopka G. Single-cell analysis of Foxp1-Driven mechanisms essential for striatal development. Cell Rep. (2020) 30:3051–7. doi: 10.1016/j.celrep.2020.02.030
44. Araujo DJ, Anderson AG, Berto S, Runnels W, Harper M, Ammanuel S, et al. FoxP1 orchestration of ASD-relevant signaling pathways in the striatum. Genes Dev. (2015) 29:2081–96. doi: 10.1101/gad.267989.115
45. Louis Sam Titus ASC, Yusuff T, Cassar M, Thomas E, Kretzschmar D, D'Mello SR, et al. Reduced Expression of Foxp1 as a Contributing Factor in Huntington's Disease. J Neurosci. (2017) 37:6575–87. doi: 10.1523/JNEUROSCI.3612-16.2017
46. Wang J, Fröhlich H, Torres FB, Silva RL, Poschet G, Agarwal A, et al. Mitochondrial dysfunction and oxidative stress contribute to cognitive and motor impairment in FOXP1 syndrome. Proc Natl Acad Sci U S A. (2022) 119:e2112852119. doi: 10.1073/pnas.2112852119
47. Ghaoui R, Sue CM. Movement disorders in mitochondrial disease. J Neurol. (2018) 265:1230–40. doi: 10.1007/s00415-017-8722-6
Keywords: autism, epilepsy, movement disorder, angiomas, choanal atresia, FOXP1
Citation: Cesaroni CA, Pollazzon M, Mancini C, Rizzi S, Cappelletti C, Pizzi S, Frattini D, Spagnoli C, Caraffi SG, Zuntini R, Trimarchi G, Niceta M, Radio FC, Tartaglia M, Garavelli L and Fusco C (2023) Case report: Expanding the phenotype of FOXP1-related intellectual disability syndrome and hyperkinetic movement disorder in differential diagnosis with epileptic seizures. Front. Neurol. 14:1207176. doi: 10.3389/fneur.2023.1207176
Received: 17 April 2023; Accepted: 23 June 2023;
Published: 14 July 2023.
Edited by:
Laura Cif, Université de Montpellier, FranceReviewed by:
Inge Anita Meijer, Montreal University, CanadaGudrun Rappold, Heidelberg University, Germany
Copyright © 2023 Cesaroni, Pollazzon, Mancini, Rizzi, Cappelletti, Pizzi, Frattini, Spagnoli, Caraffi, Zuntini, Trimarchi, Niceta, Radio, Tartaglia, Garavelli and Fusco. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Livia Garavelli, bGl2aWEuZ2FyYXZlbGxpQGF1c2wucmUuaXQ=; Carlo Fusco, Y2FybG8uZnVzY29AYXVzbC5yZS5pdA==
†These authors have contributed equally to this work and share first authorship
‡These authors share last authorship