
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Neurol., 09 June 2023
Sec. Multiple Sclerosis and Neuroimmunology
Volume 14 - 2023 | https://doi.org/10.3389/fneur.2023.1193211
This article is part of the Research TopicMOGAD, Current Knowledge and Future TrendsView all 13 articles
 Milena Trentinaglia1†
Milena Trentinaglia1† Alessandro Dinoto1†
Alessandro Dinoto1† Sara Carta1Vanessa Chiodega1
Sara Carta1Vanessa Chiodega1 Sergio Ferrari1Vincenzo Andreone2
Sergio Ferrari1Vincenzo Andreone2 Giorgia Teresa Maniscalco2,3‡
Giorgia Teresa Maniscalco2,3‡ Sara Mariotto1*‡
Sara Mariotto1*‡Introduction: The association of myelin oligodendrocyte glycoprotein (MOG) antibody associated disease (MOGAD) and tumors has seldom been reported. We aim to investigate the occurrence of tumors in a cohort of patients with MOGAD and to describe their clinical features, in addition to previously reported cases.
Methods: We retrospectively identified patients with MOGAD (i.e., compatible clinical phenotype and positive MOG antibodies analysed with a live cell-based assay) from 1/1/2015 to 1/1/2023 who had a neoplasm diagnosed within 2 years from MOGAD onset. Furthermore, we performed systematic review of literature to identify previously reported cases. Clinical, paraclinical and oncological findings were collected and reported as median (range) or number (percentage).
Results: Two of 150 MOGAD patients (1%) had a concomitant neoplasm in our cohort. Fifteen additional cases were retrieved from literature. Median age was 39 (16–73) years-old, 12 patients were female. ADEM (n = 4;23.5%), encephalomyelitis (n = 3;17.6%), and monolateral optic neuritis (n = 2;11.8%) were the most frequent phenotypes. Median number of treatments was 1 (range 1–4), improvement was reported in 14/17 cases (82.4%). Oncological accompaniments were teratoma (n = 4), CNS (n = 3), melanoma (n = 2), lung (n = 2), hematological (n = 2), ovary (n = 1), breast (n = 1), gastrointestinal (n = 1), and thymic (n = 1) neoplasms. Median time from tumor diagnosis to MOGAD onset was 0 (range − 60 to 20) months. MOG expression in neoplastic tissue was reported in 2/4 patients. Median PNS-CARE score was 3 (range 0–7): 11 patients were classified as “non-PNS,” 5 as “possible PNS,” and 1 as “probable PNS.”
Discussion: Our study confirms that MOG is a low-risk antibody for paraneoplastic neurological syndromes and that the clinical presentation and oncological accompaniments are extremely variable. Most of these patients were classified as non-PNS, whereas only a minority was diagnosed with possible/probable PNS, frequently in association with ovarian teratoma. These findings support the notion that MOGAD is not a paraneoplastic disease.
Paraneoplastic neurological syndromes (PNS) are defined by the presence of specific clinical features in association with cancer and specific autoantibodies (1). Among these autoantibodies, those targeting aquaporin-4 (AQP4) and myelin oligodendrocyte glycoprotein (MOG) are associated with a low risk of cancer, even though MOG or AQP4 expression may be detected on cancer tissue, supporting a paraneoplastic origin in those few reported cases. While the association between neoplasms and AQP4-seropositive neuromyelitis optica spectrum disorder (NMOSD) has been investigated in previous studies (2–4), data on patients affected by MOG antibody-associated disease (MOGAD) (5) are still lacking and limited on few case reports (6). Aim of this study is to report the association between neoplasms and MOGAD in a single-center cohort and, additionally, to provide a systematic evaluation of previously reported cases.
We retrospectively identified patients with MOGAD [i.e with a compatible phenotype and positive MOG-Abs analysed with a live cell-based assay as previously described (7)] and a neoplasm diagnosed within 2 years from disease onset. Clinical, paraclinical, and oncological data were collected and are herein reported. As cancer screening is currently not required in patients with MOGAD, paraneoplastic analyses were performed by treating physicians on an individualized basis according to physical examination or routine laboratory screening. In addition, to identify previously reported cases of paraneoplastic MOGAD, a systematic literature review based on PubMed/Medline database was performed (March 7th, 2023) using the following research queries: (“myelin oligodendrocyte glycoprotein” OR “MOG”OR “MOG-EM” OR “MOGAD”) AND (“cancer” OR “paraneoplastic” OR “tumor” OR “teratoma”) including studies on individual cases. Furthermore, all articles included after abstract screening were cross-referenced. Relevant reported clinical, paraclinical, and oncological findings were collected in an electronic database. PNS Care score was evaluated in each case at clinical presentation, as previously described (1).
A descriptive statistical analysis was performed using median (range) and number (%), as appropriate (IBM SPSS 26) including both data obtained from the retrospective study and the systematic review. For estimating the percentage of cancer occurring within 2 years in patients with MOGAD, we included only cases identified in our cohort excluding those obtained from the systematic review. Finally, patients classified as non-PNS were compared to patients fulfilling the criteria of Probable/Possible/Definite PNS (Fisher’s and U-Mann Whitney tests). Patients gave their informed consent for being included in this study.
Of 185 patients diagnosed with MOGAD from 1/1/2015 to 1/1/2023, two patients out of 150 with available clinical information had a neoplasm within 2 years from MOGAD onset (~1%). These two cases are reported below. Both reported cases resulted negative for additional autoantibodies analysed with commercial and on in-house tissue-based assay (8).
A 59-year-old man presented with acute onset of fever, imbalance, dysarthria, and hallucinations. Brain MRI was unremarkable, while on CSF analysis pleocytosis (645 cells/mm3 N.V. <5) and increased protein levels (477 mg/dL N.V. <45 mg/dL) were observed. Although an extensive infectious and autoimmune screening including antibodies to neuronal and cell surface antigens were negative, the clinical suspicion of brainstem encephalitis led to start treatment with steroids, with complete recovery. Respectively two and 9 months after onset, generalized tonic–clonic seizures occurred and antiepileptic drugs (AED) were commenced. A repeated brain MRI showed multiple T2/FLAIR hyperintense lesions in the supratentorial and infratentorial white matter, with pons contrast enhancement. 4 months later, the patient developed a severe paraparesis with sensory level and sphincter dysfunction. Total body computed tomography (CT) scan was unremarkable while spinal cord MRI revealed multiple T2/FLAIR hyperintensities (at C7-D1, D8-D10, and D11 level) suggestive of mixed short and longitudinally extensive transverse myelitis (LETM). A repeated CSF analysis showed slightly increased cell count (7/mm3), normal protein values (41.8 mg/dL), and negative oligoclonal bands (mirror pattern). An expanded autoimmune screening with live cell-based assay revealed the presence of serum MOG-Abs (titre 1:320). The patient was diagnosed with MOGAD and treated with high dose intravenous steroids followed by slow tapering. Since only a partial response was observed, treatment with Rituximab was commenced. 20 months later, the appearance of a unilateral axillary lymphadenopathy led to a diagnosis of a non-Hogdkin lymphoma and the patient was treated with chemotherapy.
A 64-year-old man while admitted for pneumonia and acute kidney failure developed subacute consciousness impairment requiring orotracheal intubation. On neurological examination he was comatose and unresponsive to pain stimulus with right-sided pyramidal signs. Brain MRI showed diffuse T2/FLAIR hyperintense lesions in the supratentorial and infratentorial white matter involving the brainstem (Figure 1A). EEG revealed generalized theta and delta slowing without epileptic abnormalities. CSF analysis demonstrated mild pleocytosis (10 cells/mm3). An extensive screening for CNS infections, autoimmune/paraneoplastic encephalitis, metabolic encephalopathy, and uremic hemolytic syndrome yielded negative results except for the detection of serum MOG-Abs (titer 1:320) using a live cell-based assay. The patient was diagnosed with MOGAD-related encephalitis and treatment with high dose intravenous steroids followed by oral tapering led to significant neurological improvement. Clinical inspection revealed a skin lesion localized in the right hemithorax suggestive for malignancy. Thus, a biopsy was performed and histological analysis confirmed a locally invasive melanoma (stage pT1b). Total body CT as well as sentinel lymph node biopsy excluded systemic localization. 4 month later the patient was asymptomatic with a normal neurological evalution, MOG-Abs titers were decreased (1:160), and a control MRI showed almost complete resolution of pre-existent lesions (Figure 1B).
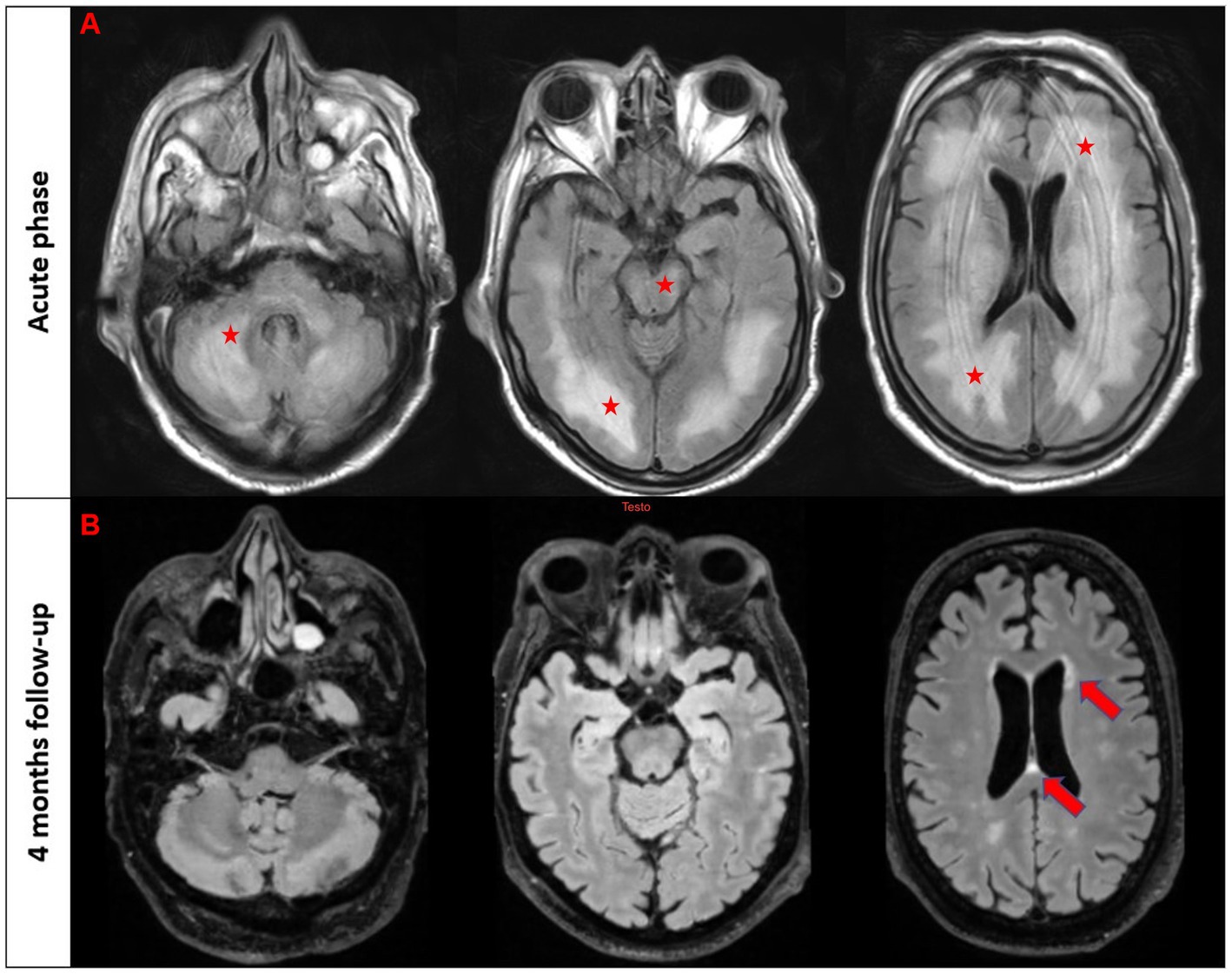
Figure 1. T2/FLAIR imaging in the acute phase (first raw) and follow-up brain MRI after 4 months in patient #2. The first three images from the acute phase reveal a diffuse T2/FLAIR hyperintensity involving bilateral white matter, brainstem, and cerebellum (marked with stars). The follow-up scans demonstrate a nearly complete resolution of these abnormalities with few scattered white matter hyperintensities in the periventricular regions (marked with an arrow).
Of 616 results, 10 studies underwent to full-text evaluation and 8 were included in the final analysis. Furthermore, 7 studies were included through cross-referencing of relevant articles. A total number of 15 studies with 15 cases were included in the final synthesis [(9–23). The PRISMA flow chart is reported in the Supplementary materials.
In the pooled cohort analysis 17 cases were included (Table 1). Median age at onset was 39 years (range 16–73) and 12 patients were female (70.5%). Clinical phenotypes were consistent with: ADEM (n = 4, 23.5%), encephalomyelitis (n = 2, 11.8%), monolateral optic neuritis (n = 2, 11.8%) and one (5.9%) of each of bilateral optic neuritis, encephalitis, encephalomyeloradiculitis, isolated myelitis, myelitis and bilateral optic neuritis, myelitis and bilateral optic neuritis with brainstem involvement, optic neuritis and brainstem syndrome, optic neuritis and meningoencephalitis, brainstem syndrome, encephalitis and myelitis. One patient was also positive for anti-GFAP and anti ITPR−1 antibodies and presented with combined CNS and PNS involvement, which has been previously reported in MOGAD (24, 25). Patients received a median of 1 acute treatment (range 1–4): steroids were administered in 16 cases, intravenous immunoglobulins in 3, plasma exchange in 3, and rituximab in 3. Clinical improvement after immunotherapy was reported in 14 patients (82.4%), whereas no improvement was observed in 3 cases (17.6%). Regarding the oncological accompaniments, associated neoplasms were teratoma (n = 4, in one case with also ganglioneuroma), CNS tumors (n = 3, pituitary macroadenoma, meningioma, and astrocytoma), melanoma (n = 2), lung cancer (n = 2, both adenocarcinoma), hematological malignancies (n = 2, non-Hodgkin lymphoma and cutaneous T cell lymphoma), borderline tumor of the ovary (n = 1), ductal breast carcinoma (n = 1), colon adenocarcinoma (n = 1), and thymic hyperplasia (n = 1). MOG protein expression in the excised tumoral tissue was reported in 2/4 patients (50%). Median time from tumor diagnosis to MOGAD onset was 0 months (range from −60 to +20, with negative values indicating that tumor preceded the onset of the neurological syndrome). Oncological treatment included surgery in 11 cases, chemotherapy in 4, radiotherapy in 1, other treatments in 3 (including hormonal therapy, pembrolizumab, and anti EGFR-TKI). Three patients did not receive any treatment. Median PNS-CARE score was 3 (range 0–7). In particular, 11 patients were classified as “non-PNS,” 5 patients as “possible PNS,” and 1 patient as “probable PNS.” The comparison between patients with non-PNS and with possible and probable PNS did not yield any significant difference, with the exception for a trend favoring the association with ovarian teratoma (p = 0.063), Table 2.
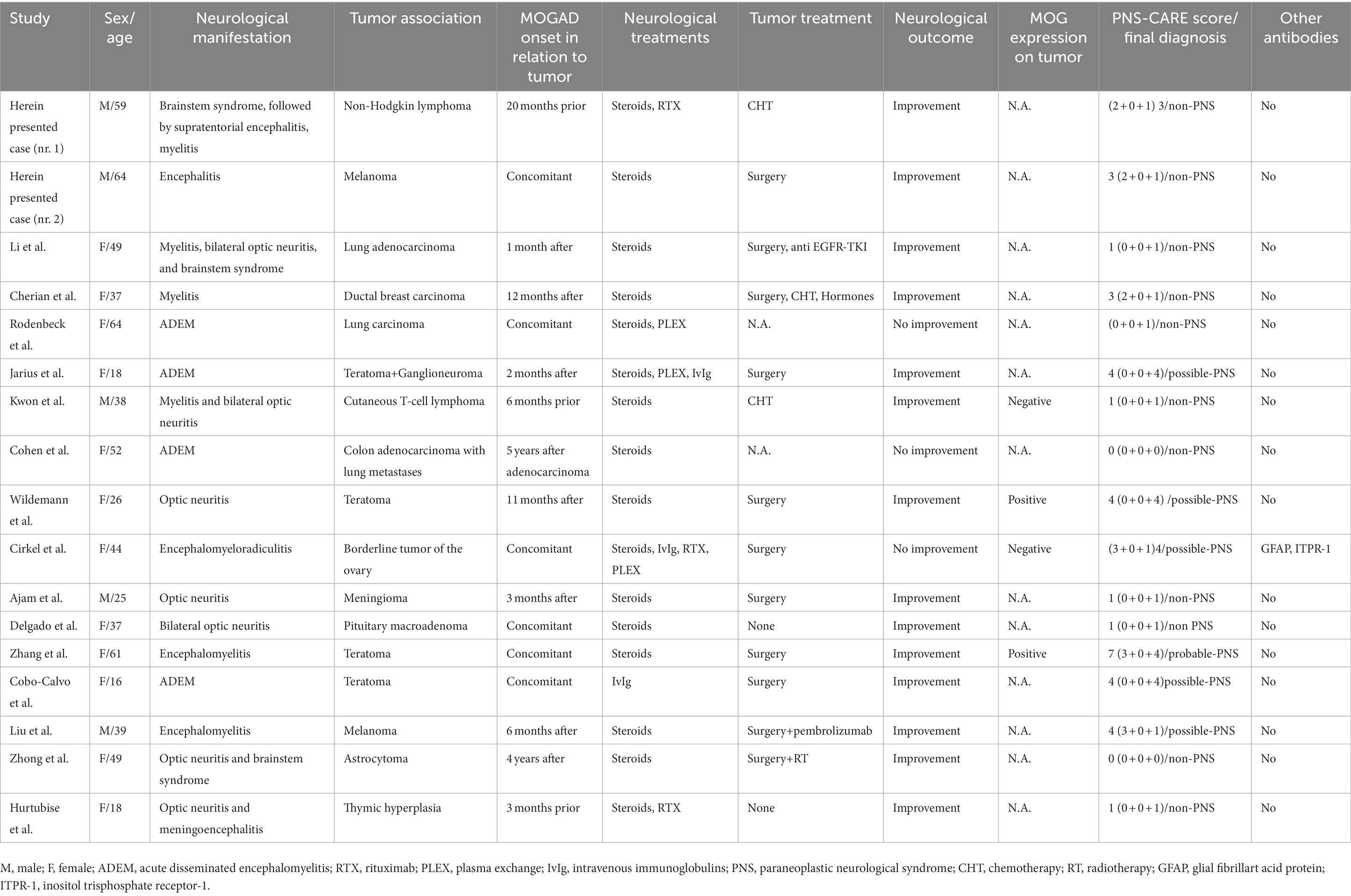
Table 1. Demographic, clinical, and oncological features of the included cohort.
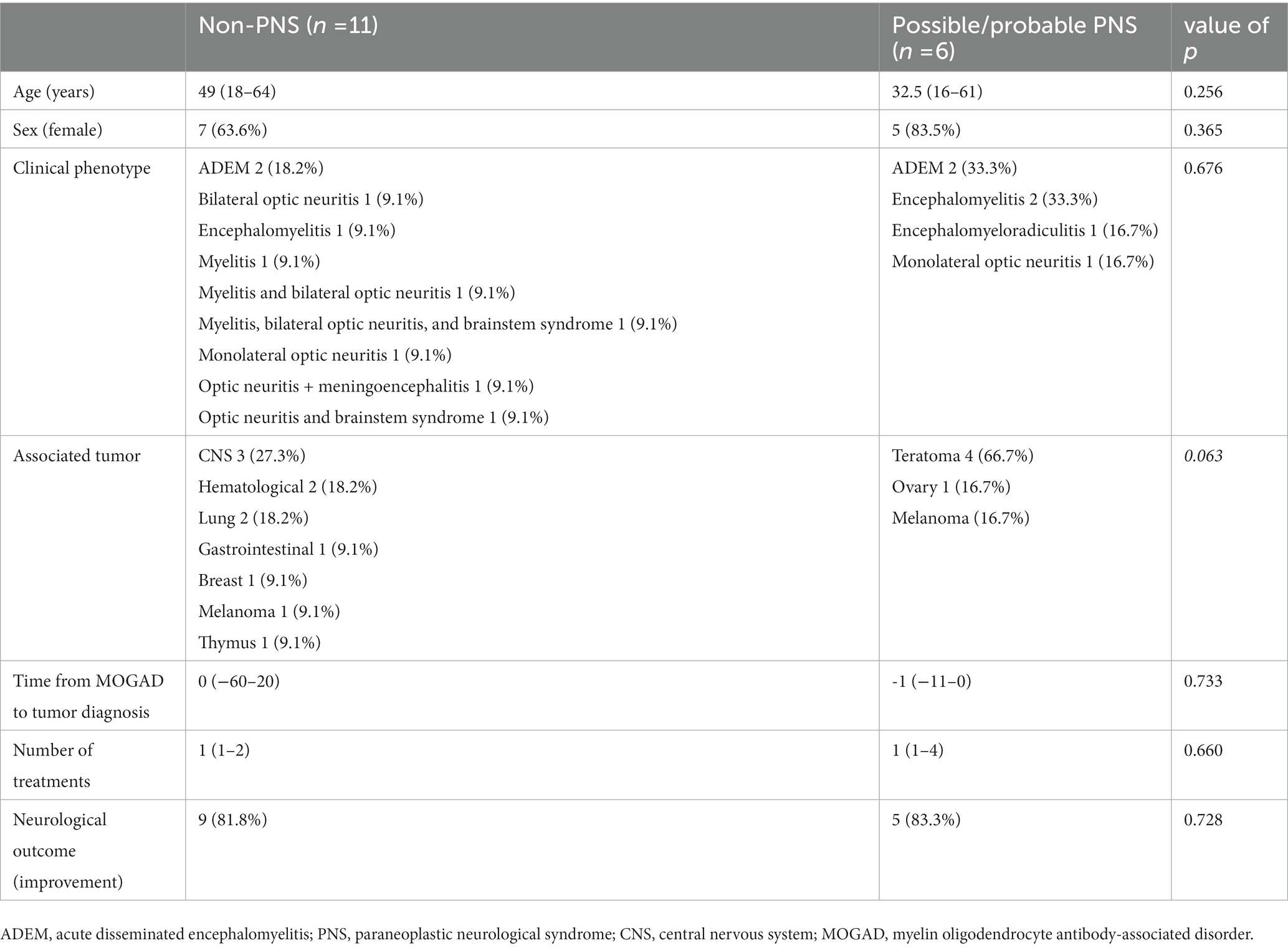
Table 2. Comparison of patients according to the PNS-CARE classification.
Our study supports the weak association between MOGAD and tumors, since: (a) the prevalence of neoplasms in a cohort of patients with MOGAD is low, (b) oncological accompaniments are extremely variable and MOG is usually not expressed in neoplastic tissue, (c) most of reported cases do not fulfill the criteria of PNS; (d) there are no striking features that could distinguish cases with possible/probable PNS from those with non-PNS, with the notable exception for a trend favoring the presence of ovarian teratoma in the first group (Figure 2).
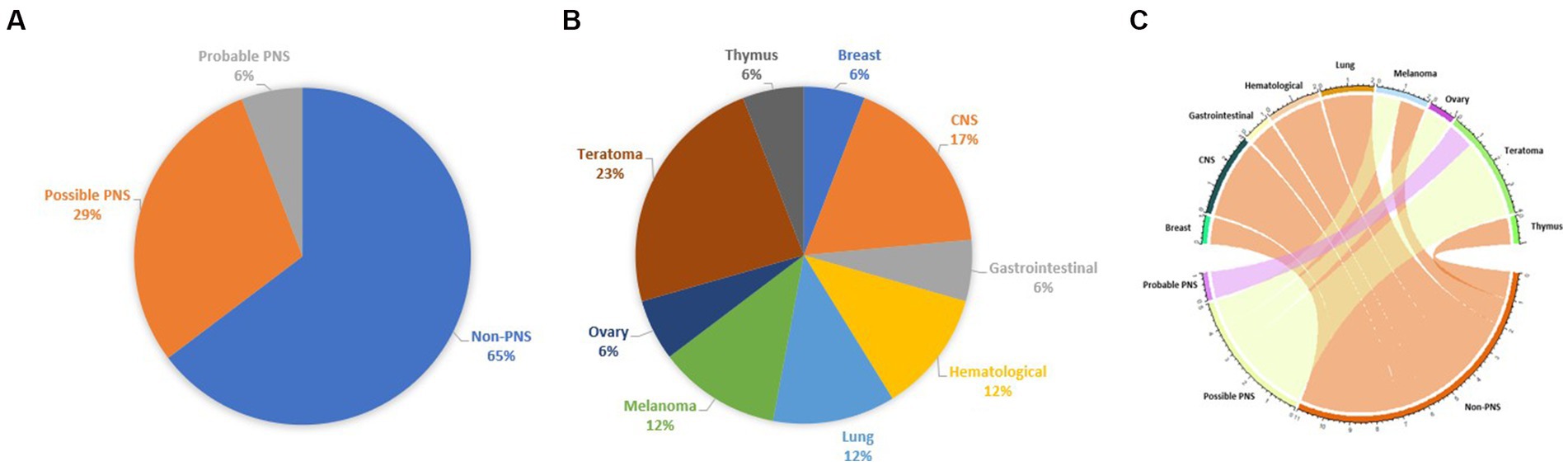
Figure 2. Chart representing the percentage of each tumor in the pooled cohort analysis (A), the fulfillment of PNS diagnostic criteria (B), and a circos plot represents the association between neoplasms and fulfillment of PNS diagnostic criteria (C).
In comparison with a previous study (14) reporting 11.3% of patients with a history of cancer within 12 months from MOGAD onset, with higher rates of prevalence in elderly patients, we observed a significant lower number (1%) of MOGAD cases associated with tumors. In addition, we found a significant lower number of patients with concomitant neoplasms and MOGAD (6.5% vs. 35%). These discrepancies may be explained by the differences in terms of population groups and inclusion criteria.
In support of the absence of a strict association between MOGAD and tumors by systematically reviewing the published literature we observed that neoplasms associated with MOGAD are extremely variable and include even some non-malignant tumors, which mostly fell in the non-PNS group. Even though we did not find an association between MOGAD and a specific cancer, the presence of ovarian teratoma seems to be particularly relevant, as both tumors that expressed MOG protein were teratomas (17, 18), and these data may support a paraneoplastic origin. Furthermore, the presence of teratomas has been associated with different antibody-mediated CNS disorders including AQP4 positive NMOSD (26) and anti-NMDAR encephalitis (27). Consistently, even though no patients were diagnosed with definite PNS, those who had a diagnosis of probable/possible PNS had a statistical trend favoring the presence of an underlying teratoma when compared to non-PNS patients. Consistently, PNS-CARE score was mostly driven by the presence of an underlying teratoma which, alone, would give a score of 4, leading to a diagnosis of possible PNS regardless of the clinical phenotype. This finding could suggest that PNS-CARE scoring system may present some limitations in the setting of a condition characterized by heterogeneous clinical presentations and very specific oncological accompaniments. Of note, one patient without an ovarian teratoma who fulfilled the criteria of possible PNS was receiving treatment with the immune checkpoint inhibitor pembrolizumab, which probably triggered MOGAD. Accordingly, iatrogenic demyelination as an immune-related adverse event of cancer immunotherapy is rare but should not be overlooked.
Finally, we did not find any specific clinical phenotype or demographic feature which may suggest a paraneoplastic trigger and thus should prompt an oncological screening. Furthermore, most of patients improved with immunotherapy only, regardless of neoplasm removal, which is atypical for PNS (28). We previously suggested to perform a paraneoplastic screening regardless of clinical presentation in AQP4 positive NMOSD despite the lack of specific clinical features in suspected paraneoplastic NMOSD, since several cases of paraneoplastic NMOSD have been reported and AQP4 tumor expression can occur even in patients with atypical oncological accompaniments (as non-adenocarcinomas) (2). On the contrary, we do not support tumor screening in MOGAD, since the lack of cancer expression beyond ovarian teratoma and the lack of other suggestive features do not support this extensive screening.
Our study presents several limitations including (a) the small sample size, the absence of a control group, and the unavailability of a systematic and uniform screening of neoplasms in our cohort, (b) the lack of evaluation of MOG expression in many studies, including our cases, and (c) the potential reporting bias favoring the overreporting of patients presenting with MOGAD and neoplasms.
Despite these limitations, our study confirms that MOG is a low-risk antibody for paraneoplastic neurological syndromes and that the clinical presentation and oncological accompaniments are extremely variable. These findings support the notion that MOGAD is not a paraneoplastic disease.
The raw data supporting the conclusions of this article will be made available by the authors, without undue reservation.
The studies involving human participants were reviewed and approved by the Ethics Committee of Verona University Hospital. The patients/participants provided their written informed consent to participate in this study.
MT and AD equally contributed and collected data, and wrote the first draft of the manuscript. GM and SM shared the senior co-authorship. All the authors performed a revision to the manuscript, gave the critical intellectual content, contributed to the article, and approved the submitted version.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
The Supplementary material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fneur.2023.1193211/full#supplementary-material
1. Graus, F, Vogrig, A, Muñiz-Castrillo, S, Antoine, JCG, Desestret, V, Dubey, D, et al. Updated diagnostic criteria for paraneoplastic neurologic syndromes. Neurol Neuroimmunol Neuroinflamm. (2021) 8:e1014. doi: 10.1212/NXI.0000000000001014
2. Dinoto, A, Borin, GU, Campana, G, Carta, S, Ferrari, S, and Mariotto, S. Investigating paraneoplastic aquaporin-4-IgG-seropositive neuromyelitis optica spectrum disorder through a data-driven approach. Eur J Neurol. (2022) 29:3466–72. doi: 10.1111/ene.15479
3. Dinoto, A, Bosco, A, Sartori, A, Bratina, A, Bellavita, G, Pasquin, F, et al. Hiccups, severe vomiting and longitudinally extensive transverse myelitis in a patient with prostatic adenocarcinoma and Aquaporin-4 antibodies. J Neuroimmunol. (2021) 352:577488. doi: 10.1016/j.jneuroim.2021.577488
4. Sepúlveda, M, Sola-Valls, N, Escudero, D, Rojc, B, Barón, M, Hernández-Echebarría, L, et al. Clinical profile of patients with paraneoplastic neuromyelitis optica spectrum disorder and aquaporin-4 antibodies. Mult Scler J. (2018) 24:1753–9. doi: 10.1177/1352458517731914
5. Sechi, E, Cacciaguerra, L, Chen, JJ, Mariotto, S, Fadda, G, Dinoto, A, et al. Myelin oligodendrocyte glycoprotein antibody-associated disease (MOGAD): a review of clinical and MRI features, diagnosis, and management. Front Neurol. (2022) 13:885218. doi: 10.3389/fneur.2022.885218
6. Molazadeh, N, Bose, G, Lotan, I, and Levy, M. Autoimmune diseases and cancers overlapping with myelin oligodendrocyte glycoprotein antibody-associated disease (MOGAD): a systematic review. Mult Scler J Exp Transl Clin. (2022) 8:20552173221128170. doi: 10.1177/20552173221128170
7. Mariotto, S, Ferrari, S, Monaco, S, Benedetti, MD, Schanda, K, Alberti, D, et al. Clinical spectrum and IgG subclass analysis of anti-myelin oligodendrocyte glycoprotein antibody-associated syndromes: a multicenter study. J Neurol. (2017) 264:2420–30. doi: 10.1007/s00415-017-8635-4
8. Dinoto, A, McKeon, A, Vattemi, G, Carta, S, Ferrari, S, and Mariotto, S. Neuronal intermediate filament paraneoplastic autoimmunity complicating avelumab therapy of Merkel cell carcinoma. J Neuroimmunol. (2022) 368:577882. doi: 10.1016/j.jneuroim.2022.577882
9. Cherian, A, Shetty, SC, Divya, KP, Prabhakaran, PK, Pavuluri, H, and Paramasivan, NK. MOG antibody-positive paraneoplastic myelopathy in breast carcinoma: the new culprit. Acta Neurol Belg. (2022) 122:1603–5. doi: 10.1007/s13760-021-01602-8
10. Cirkel, A, Wandinger, K-P, Ditz, C, Leppert, J, Hanker, L, Cirkel, C, et al. Paraneoplastic encephalomyeloradiculits with multiple autoantibodies against ITPR-1, GFAP and MOG: case report and literature review. Neurol Res Pract. (2021) 3:48. doi: 10.1186/s42466-021-00145-w
11. Cobo-Calvo, Á, Ruiz, A, D’Indy, H, Poulat, AL, Carneiro, M, Philippe, N, et al. MOG antibody-related disorders: common features and uncommon presentations. J Neurol. (2017) 264:1945–55. doi: 10.1007/s00415-017-8583-z
12. Hurtubise, B, Frohman, EM, Galetta, S, Balcer, LJ, Frohman, TC, Lisak, RP, et al. MOG antibody-associated disease and thymic hyperplasia: from the national multiple sclerosis Society case conference proceedings. Neurol Neuroimmunol Neuroinflamm. (2023) 10:e200077. doi: 10.1212/NXI.0000000000200077
13. Jarius, S, Kleiter, I, Ruprecht, K, Asgari, N, Pitarokoili, K, Borisow, N, et al. MOG-IgG in NMO and related disorders: a multicenter study of 50 patients. Part 3: brainstem involvement – frequency, presentation and outcome. J Neuroinflammation. (2016) 13:281. doi: 10.1186/s12974-016-0719-z
14. Kwon, YN, Koh, J, Jeon, YK, Sung, JJ, Park, SH, and Kim, SM. A case of MOG encephalomyelitis with T- cell lymphoma. Mult Scler Relat Disord. (2020) 41:102038. doi: 10.1016/j.msard.2020.102038
15. Li, K, Zhan, Y, and Shen, X. Multiple intracranial lesions with lung adenocarcinoma: a rare case of MOG-IgG-associated encephalomyelitis. Mult Scler Relat Disord. (2020) 42:102064. doi: 10.1016/j.msard.2020.102064
16. Liu, Q, Wang, B, and Zhao, W. MOG-IgG-associated demyelination induced by pembrolizumab treatment in a patient with malignant melanoma. Neurology. (2022) 98:501–2. doi: 10.1212/WNL.0000000000200055
17. Wildemann, B, Jarius, S, Franz, J, Ruprecht, K, Reindl, M, and Stadelmann, C. MOG-expressing teratoma followed by MOG-IgG-positive optic neuritis. Acta Neuropathol. (2021) 141:127–31. doi: 10.1007/s00401-020-02236-5
18. Zhang, XY, An, DM, and Liu, L. MOG antibody disease with ovarian teratoma: a case report and review of the literature. J Neuroimmunol. (2022) 367:577858. doi: 10.1016/j.jneuroim.2022.577858
19. Zhong, G, Zhang, J, Liu, X, Yang, S, and Gu, H. Astrocytoma with myelin oligodendrocyte glycoprotein antibody associated encephalomyelitis: a case report. Medicine. (2022) 101:e31003. doi: 10.1097/MD.0000000000031003
20. Ajam, Y, and Li, X. Anti-myelin oligodendrocyte glycoprotein (MOG) antibody-associated bilateral optic neuritis: to treat or not to treat?. 145th Annual Meeting American Neurological Association. Ann Neurol. (2020).
21. Cohn, SJ, Macaron, G, Mahajan, KR, et al. MOG-antibody related disease presenting with a fulminant CNS demyelinating syndrome in an adult ACTRIMS Forum. Mult Scler J. (2020).
22. Delgado, S, Dionísio, J, Figueiredo, C, et al. Anti-MOG bilateral optic neuritis and suspicion of a compressive pituitary macroadenoma: the relevance of semiology. Eur J Neurol. (2021).
23. Rodenbeck, K, Crane, P, and Sitzmanno, A. Myelopathy in the setting of malignancy: the importance of a thoughtful approach to autoantibody testing based on clinical presentation (P1295). Neurology. (2021).
24. Dinoto, A, Licciardi, NM, Reindl, M, Chiodega, V, Schanda, K, Carta, S, et al. Peripheral neuropathy and MOG-IgG: a clinical and neuropathological retrospective study. Mult Scler Relat Disord. (2022) 68:104214. doi: 10.1016/j.msard.2022.104214
25. Rinaldi, S, Davies, A, Fehmi, J, Beadnall, HN, Wang, J, Hardy, TA, et al. Overlapping central and peripheral nervous system syndromes in MOG antibody-associated disorders. Neurol Neuroimmunol Neuroinflamm. (2021) 8:e924. doi: 10.1212/NXI.0000000000000924
26. Bernard-Valnet, R, Cobo-Calvo, A, Siegfried, A, Marasescu, R, Bonnan, M, Ballan, G, et al. Paraneoplastic neuromyelitis optica and ovarian teratoma: a case series. Mult Scler Relat Disord. (2019) 31:97–100. doi: 10.1016/j.msard.2019.03.031
27. Dalmau, J, Lancaster, E, Martinez-Hernandez, E, Rosenfeld, MR, and Balice-Gordon, R. Clinical experience and laboratory investigations in patients with anti-NMDAR encephalitis. Lancet Neurol. (2011) 10:63–74. doi: 10.1016/S1474-4422(10)70253-2
Keywords: myelin oligodendrocyte glycoprotein antibody-associated disease (MOGAD), paraneoplastic neurological syndrome, tumor, cancer, immune checkpoint inhibitors
Citation: Trentinaglia M, Dinoto A, Carta S, Chiodega V, Ferrari S, Andreone V, Maniscalco GT and Mariotto S (2023) Investigating the association between neoplasms and MOG antibody-associated disease. Front. Neurol. 14:1193211. doi: 10.3389/fneur.2023.1193211
Edited by:
Sasitorn Siritho, Bumrungrad International Hospital, ThailandReviewed by:
Masako Kinoshita, National Hospital Organization Utano National Hospital, JapanCopyright © 2023 Trentinaglia, Dinoto, Carta, Chiodega, Ferrari, Andreone, Maniscalco and Mariotto. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Sara Mariotto, c2FyYS5tYXJpb3R0b0B1bml2ci5pdA==
†These authors have contributed equally to this work
‡These authors share senior authorship
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.