 Yi Hua
Yi Hua Xuke Yan
Xuke Yan Liu Liu
Liu Liu Yilong Wang
Yilong Wang Lu Xu
Lu Xu Zhefeng Yuan
Zhefeng Yuan Feng Gao
Feng Gao- Department of Neurology, Children’s Hospital, Zhejiang University School of Medicine, National Clinical Research Center for Child Health, Hangzhou, Zhejiang, China
Objective: To analyze the clinical characteristics and follow-up data of children with different clinical phenotypes of myelin oligodendrocyte glycoprotein antibody-associated disease (MOGAD).
Methods: The basic demographic and clinical features, laboratory and imaging examination results, and follow-up data of 74 Chinese children with different phenotypes of MOGAD were retrospectively reviewed and analyzed.
Results: The male-to-female ratio in this cohort was 1:1.39. The clinical phenotypes of MOGAD included acute disseminated encephalomyelitis (ADEM; n = 37), encephalitis (n = 11), optic neuritis (ON, n = 9), neuromyelitis optica spectrum disorder (NMOSD; n = 9), transverse myelitis (TM; n = 6), leukodystrophy-like manifestations (n = 1), and meningitis (n = 1). The mean age of disease onset was 86 months. The number of leukocytes in the cerebrospinal fluid of patients with ADEM was significantly higher than that in patients with ON but lower than that in patients with TM (p < 0.05). The pathogen detection rate among all patients was 36.5%. Recurrence occurred in 17 patients (23%), with the highest recurrence rate in patients with NMOSD and TM. Patients with recurrence had a significantly higher median age than those without any recurrence (109.00 vs. 82.44 months, p < 0.05). The male-to-female ratio in patients with recurrence was 1:4.67, which differed significantly from that at first onset (p < 0.05).
Conclusion: The most common clinical phenotypes of MOGAD in this cohort were ADEM and encephalitis. Recurrence of MOGAD may be related to age and sex, with a higher recurrence rate observed in females. These findings provide a basis for further exploration of the characteristics of different MOGAD phenotypes.
1. Introduction
Myelin oligodendrocyte glycoprotein (MOG) is a myelin protein that belongs to the immunoglobulin (Ig) superfamily (1). It is exclusively expressed on the surface of myelin sheaths and oligodendrocyte membranes in the central nervous system (CNS) (2). MOG antibody-associated disease (MOGAD) is an idiopathic, rare, immune-mediated inflammatory demyelinating disease of the CNS, which overlaps with other clinical presentations, such as acute disseminated encephalomyelitis (ADEM), neuromyelitis optica spectrum disorder (NMOSD), optic neuritis (ON), and multiple sclerosis (MS) (3). The neuropathological hallmarks of MOGAD consist of MOG-dominant myelin loss, white matter demyelination, intracortical demyelination, etc. (4). Among demyelinating CNS disorders, the relative frequency of MOGAD seems higher in children than in adults (5). It affects both female and male children, with a similar frequency in both genders (6). Recent evidence has suggested that anti-MOG immunoglobulin G (MOG-IgG) may be a pathogenic antibody against MOGAD (7). As a disease with a wide clinical spectrum, the clinical features of MOGAD warrant further investigation. In the present study, we aimed to summarize and analyze the characteristics, treatments, and follow-up data of Chinese children with different clinical phenotypes of MOGAD.
2. Materials and methods
2.1. Study population
In this retrospective study, children who were diagnosed with MOGAD and treated in the Department of Neurology of Children’s Hospital Affiliated to Zhejiang University School of Medicine between March 2017 and February 2021 were qualified for screening. The inclusion criteria were as follows: (1) aged ≤18 years; (2) diagnosed with MOGAD according to the provisional criteria proposed by the International Pediatric Multiple Sclerosis Study Group Criteria (2012) (8) and the International MOGAD panel (9); (3) presented with symptoms of autoimmune encephalitis or fever; (4) tested positive for serum MOG antibodies by cell-based assay; and (5) with complete medical records. Patients were excluded if they (1) presented with another CNS disease, such as cerebral palsy and neonatal brain injury; (2) also had a systemic immune disease, such as lupus erythematosus; (3) were positive for other CNS demyelinating antibodies, such as anti-aquaporin-4 (AQP4) antibody. The experimental protocols were approved by the local ethics committee (approval no. 2021-IRB-161). The parents or guardians of all children were informed of treatment plans and provided written informed consent before laboratory and imaging examinations.
2.2. Data collection
The demographic characteristics and clinical data, including clinical manifestations, laboratory and imaging examination results, treatments, and follow-up data of all patients were collected from the medical records and analyzed retrospectively. All children were tested for serum MOG-IgG using fixed cell-based assay (Euroimmun AG, Germany) with a full-length human MOG as the target antigen and a low positive cut-off value of 1:10. The samples were sent to the V-Medical Laboratory (Hangzhou, China) for analysis. All patients who tested positive for MOG antibodies underwent magnetic resonance imaging (MRI) examinations of the brain and spine, including T1, T2, and fluid-attenuated inversion recovery (FLAIR).
Cerebrospinal fluid (CSF) samples were collected from 71 patients by lumbar puncture. Testing for Epstein–Barr virus (EBV) and herpes simplex virus (HSV) was performed in 55 and 52 patients, respectively. The serum EBV antibody test and mycoplasma pneumoniae antibody test were performed on 60 and 58 patients, respectively.
2.3. Treatments
All patients received methylprednisolone pulse therapy at a dosage of 10–20 mg/kg/day per day for 3–5 days. Concomitant gamma globulin immunomodulatory therapy at a dosage of 1 g/kg/day was administered to 43 patients for 2 days.
The immunosuppressive therapy regime consisted of azathioprine (2 mg/kg/day, divided into two oral doses), methotrexate (20 mg/kg/day, divided into two oral doses), cyclophosphamide (750 mg/m2, administered once every 4 weeks for a total of 4 cycles), and intravenous immunoglobulin (1 g/kg/day, administered for 2 consecutive days).
2.4. Statistical analysis
Data were analyzed using SPSS 20.0 software both before and after the stratification of different clinical phenotypes and treatments. The normality of the data distribution was tested using the Kolmogorov–Smirnov test. Continuous variables with a normal distribution were compared using the Student’s t-test. Categorical variables were compared using the Chi-square test. The test standard was p = 0.05, and p < 0.05 was considered statistically significant.
3. Results
3.1. Basic demographic and clinical characteristics
A total of 74 children qualified for inclusion in this study, including 31 boys and 43 girls, for a male-to-female ratio of 1:1.39. Different clinical phenotypes were observed in patients with MOGAD, including ADEM (n = 37, 50.0%), encephalitis (n = 11, 14.9%), ON (n = 9, 12.1%), neuromyelitis optica spectrum disorder (NMOSD, n = 9, 12.1%), transverse myelitis (TM, n = 6, 8.1%), leukodystrophy-like manifestations (n = 1, 1.4%), and meningitis (n = 1, 1.4%) (Figure 1).
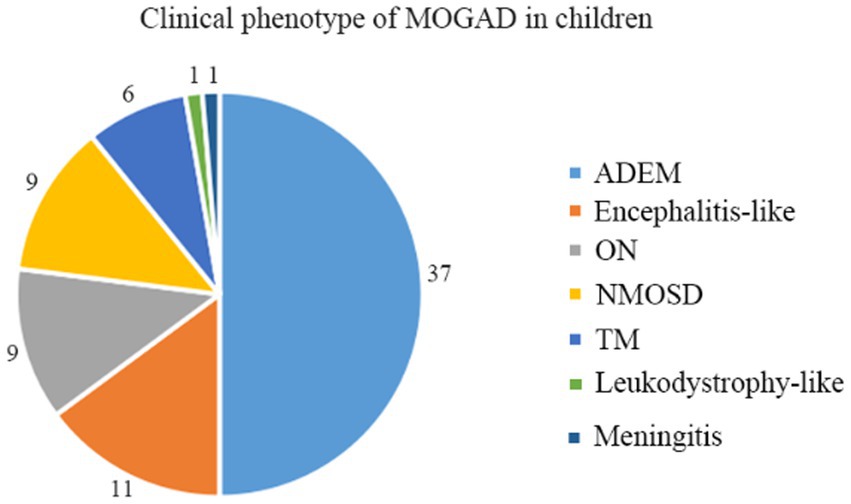
Figure 1. Clinical phenotypes of MOGAD in Chinese children.
The median age of disease onset was 86 months (range, 20–175 months). The clinical phenotype with the lowest mean age of onset was leukodystrophy-like manifestations (43 months), followed by ADEM (66 months) and meningitis (78 months), and the one with the highest mean age of onset was TM (136 months) (Figure 2). The mean age of onset differed significantly between TM and ADEM and between TM and ON (both p < 0.05).
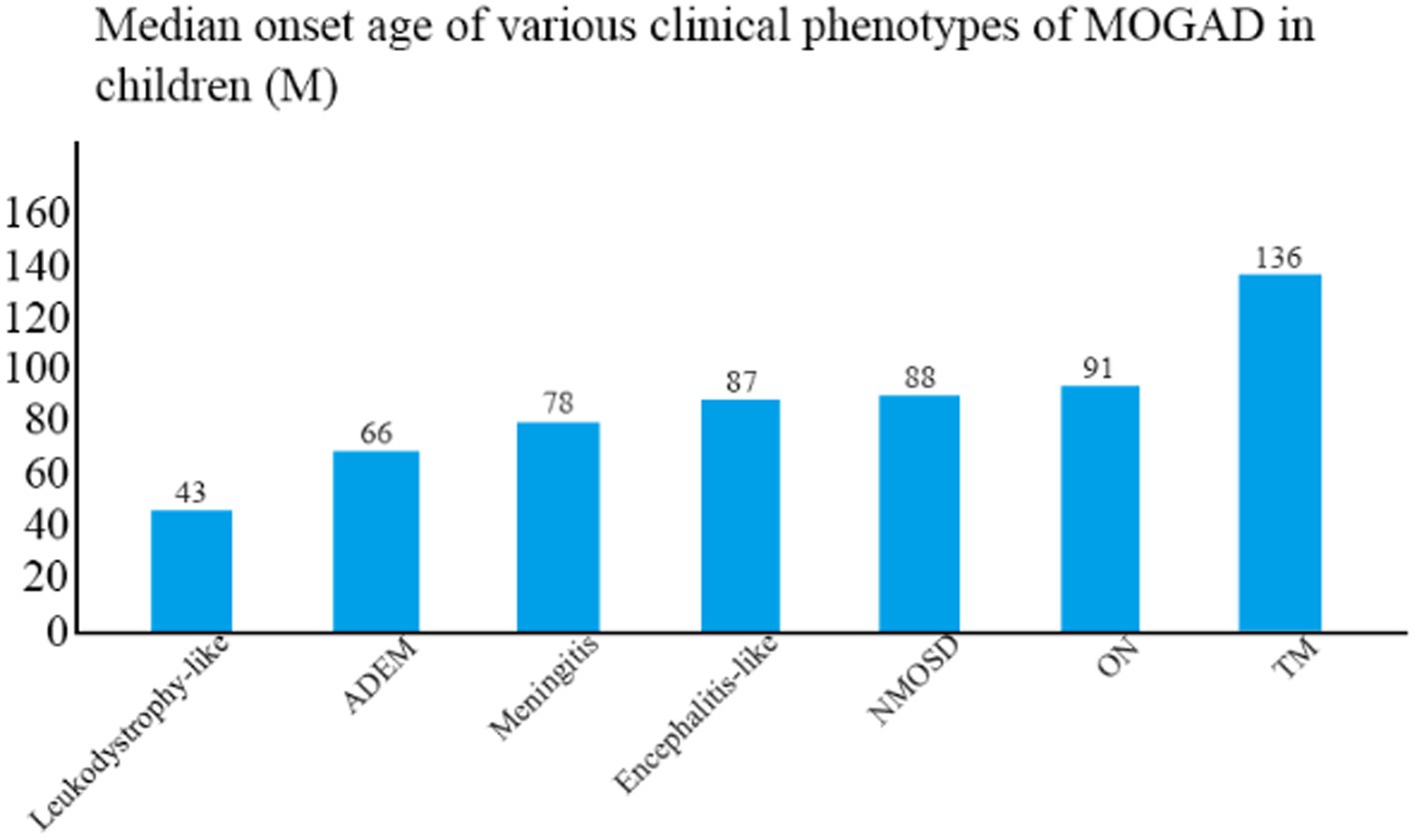
Figure 2. Median age of onset among children with different clinical phenotypes of MOGAD.
The mean hospital stay was 18.6 days for patients with ADEM, 17.9 days for patients with encephalitis, 12.1 days for patients with ON, 14.9 days for patients with NMOSD, 20.5 days for patients with TM, 18 days for patients with leukodystrophy-like manifestations, and 23 days for patients with meningitis (Figure 3). The mean hospital stay of patients with ON differed significantly from those of patients with ADEM, TM, and encephalitis (p < 0.05).
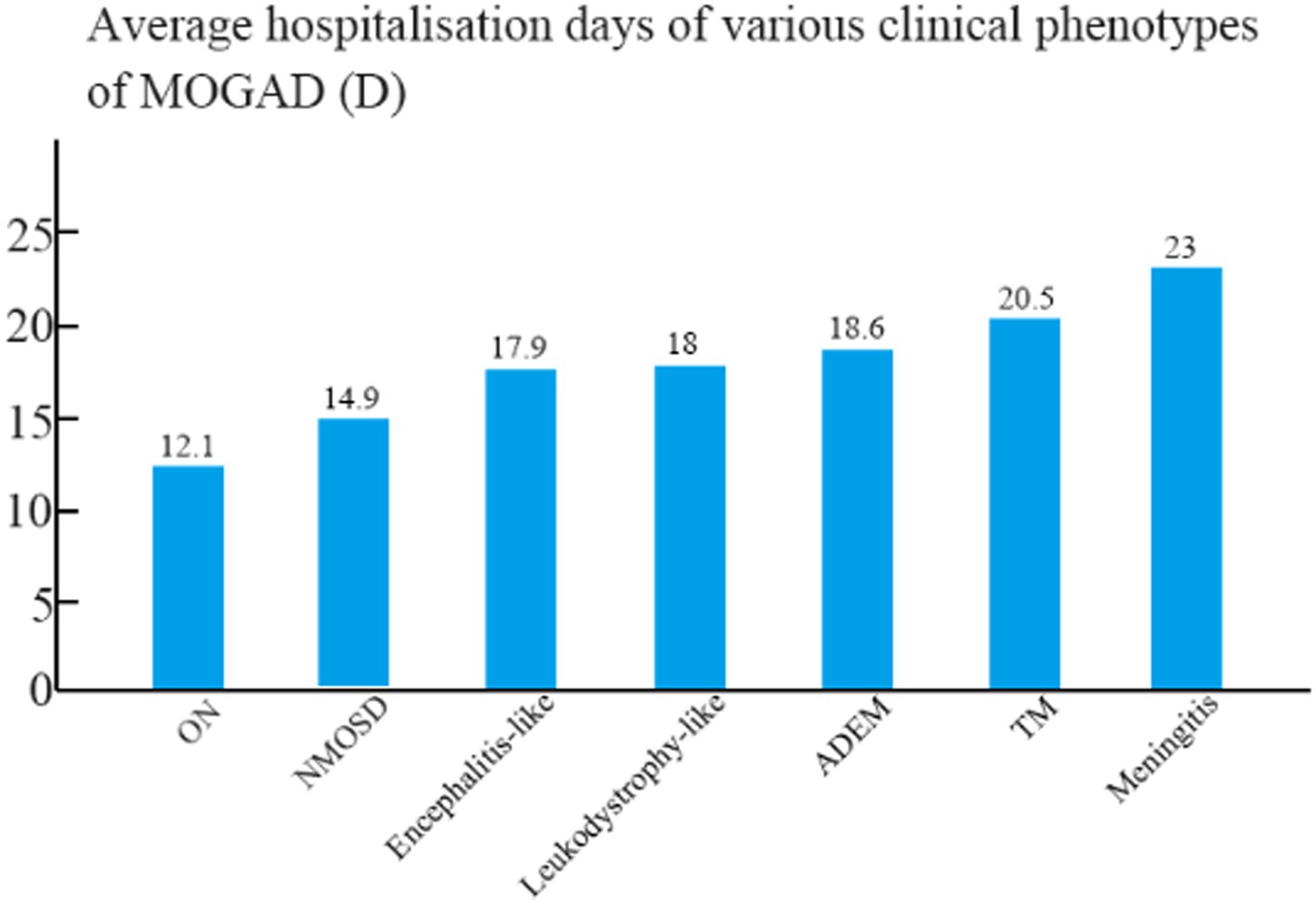
Figure 3. Mean length of hospital stay among children with different clinical phenotypes of MOGAD.
The clinical features observed in the various MOGAD presentations included fever (n = 31), headache (n = 19), dizziness (n = 10), vomiting (n = 11), hypersomnia (n = 10), seizures (n = 14), visual impairment (n = 18), ophthalmalgia (n = 2), muscle weakness (n = 16), dysuria (n = 4), constipation (n = 2), ataxia (n = 5), mood changes (n = 1), facial asymmetry (n = 1), and dysarthria (n = 2).
3.2. Laboratory examination results
All patients were positive for the MOG-IgG antibody. The number of leukocytes in the CSF samples of patients with MOGAD ranged between 2.0 × 106 and 683.0 × 106/L, with an average number of 63.3 × 106/L. The number of leukocytes in patients with ADEM was significantly higher than that in patients with ON but lower than that in patients with TM (p < 0.05). The concentrations of CSF protein fluctuated between 84 and 1,307 mg/L, with a mean value of 295.8 mg/L, and no significant difference was detected among the groups. Among all patients, 3 (5.5%) were positive for EBV; 1 (1.9%) was positive for HSV type II; 9 (15.0%) were positive for serum EBV IgM antibody; and 14 (24.1%) were positive for serum mycoplasma pneumoniae IgM antibody (Table 1). The pathogen detection rate in this cohort was 36.5%. Of the three patients with EBV seropositivity, one exhibited fever but showed no signs of enlarged lymph nodes in the neck, enlarged liver or spleen, or abnormal liver function. There were also no abnormalities in the lymphocyte ratio and lymphocyte morphology. The other two patients did not present with any of these conditions.
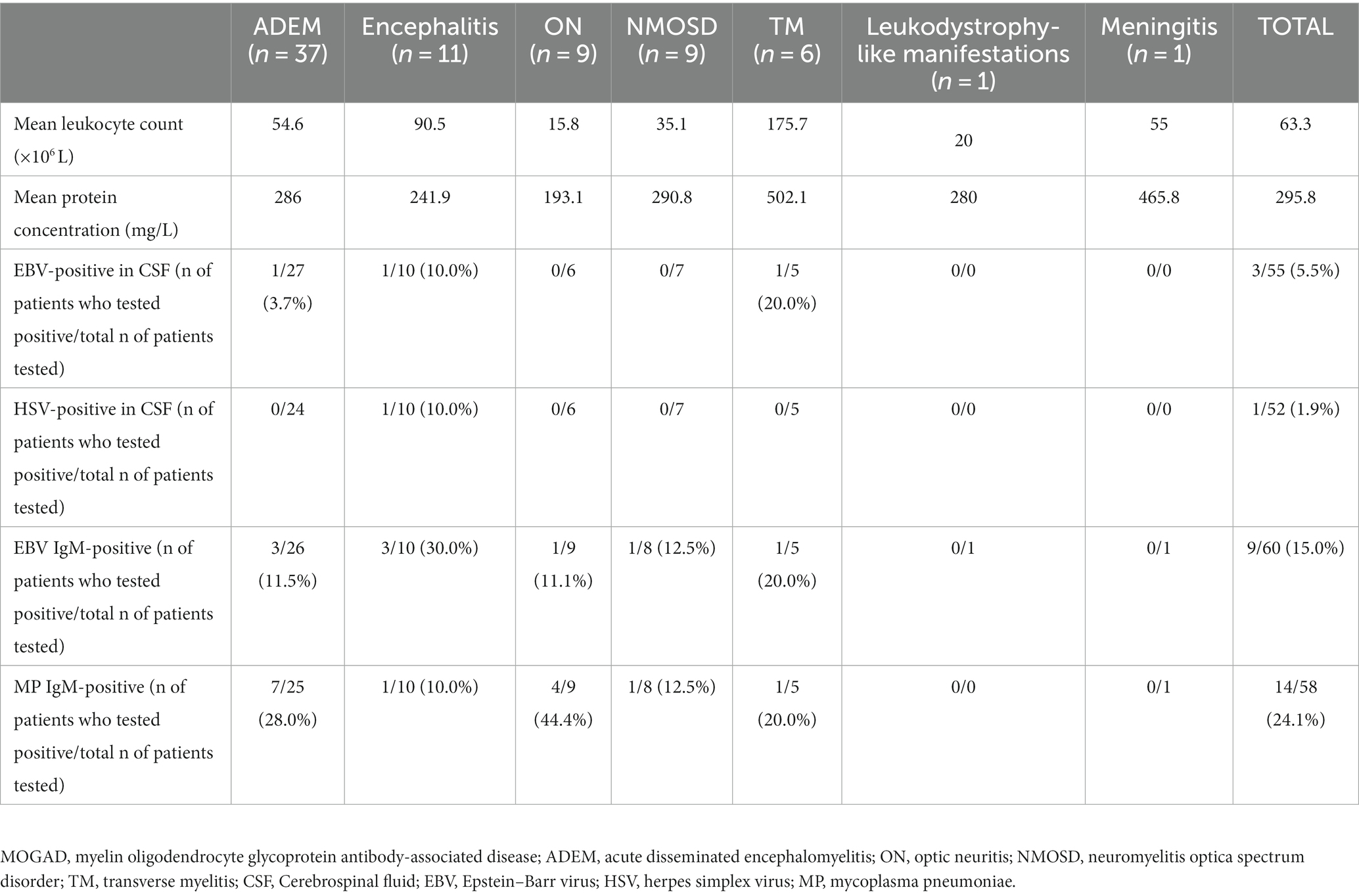
Table 1. Laboratory examination results of patients with different clinical phenotypes of MOGAD.
3.3. Imaging characteristics
All patients underwent MRI scans of the brain and spine. In addition to typical imaging manifestations of ADEM, NMOSD, ON, and TM, one patient with NMOSD presented with area postrema syndrome (Figure 4). Of three children who presented with encephalitis but exhibited no significant abnormalities on cranial MRI in the early stage of the disease, two showed thalamic lesions at 2 weeks after onset, while one exhibited no abnormalities throughout the disease course. One patient who had meningitis and presented with fever as the only clinical manifestation at admission exhibited meningeal enhancement on repeated cranial MRI (Figure 5).
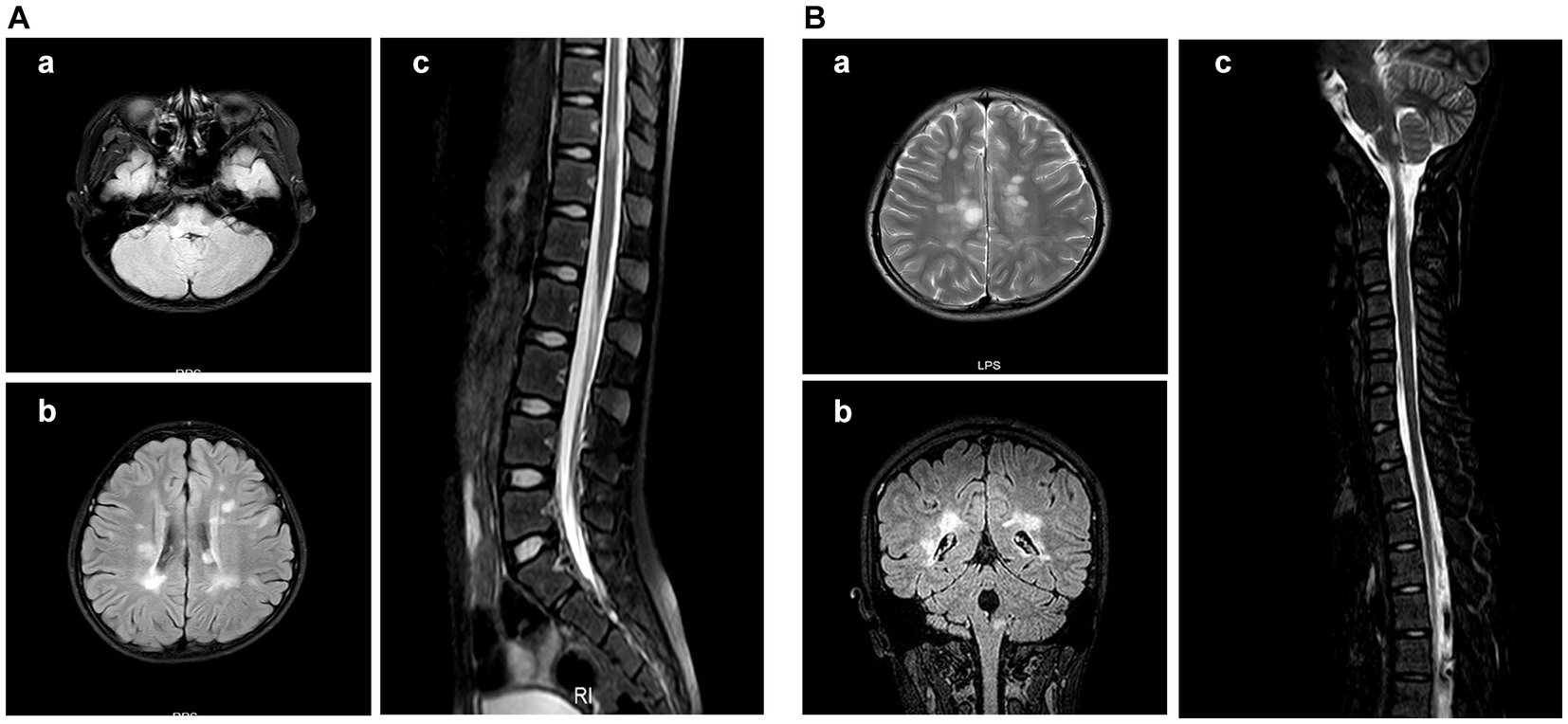
Figure 4. Cranial MRI results of the patient with area postrema syndrome. A female child, aged 133 months, presented with bouts of vomiting and dizziness for over 10 days. (A) Cranial MRI scan at admission. (A-a) Abnormal signal in the right side of the medulla oblongata in the FLAIR sequence. (A-b) Abnormal signal in the bilateral periventricular white matter in the FLAIR sequence. (A-c) Patchy abnormal signal at the T11 level in the FLAIR sequence. (B) Cranial MRI at 30 months after the first MRI scan. The patient complained about headache and had vomited for 2 days. (B-a) The periventricular semioval center of the lateral ventricle in the FLAIR sequence. (B-b) The right medulla oblongata and periventricular white matter in the coronal section of the FLAIR sequence. (B-c) Abnormal signal in the dorsal side of the medulla oblongata in the FLAIR sequence.
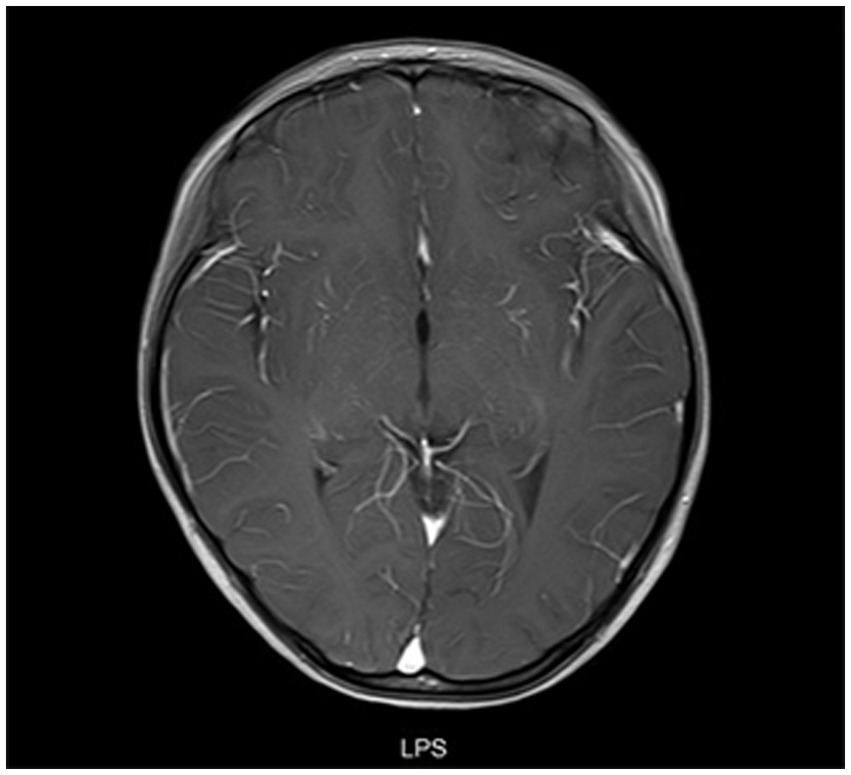
Figure 5. Cranial MRI results of a patient with meningitis who presented with fever. A female child, aged 78 months, was admitted to the hospital with fever for 9 days. At 19 days after admission, cranial enhancement MRI showed that the meninges of the right temporal pole were enhanced and no abnormality was found in the brain parenchyma.
The brain MRI scans indicated involvement in various locations, including the frontal lobe (n = 35), parietal lobe (n = 35), temporal lobe (n = 21), occipital lobe (n = 21), basal ganglia (n = 21), thalamus (n = 25), brainstem (n = 11), cerebellum (n = 15), corpus callosum (n = 8), and medulla oblongata (n = 4), as well as bilateral involvement of the optic nerve (n = 11) and unilateral involvement of the optic nerve (n = 3). The spinal cord MRI scans revealed affected areas in the cervical cord (n = 19), thoracic cord (n = 21), lumbar and sacral cord (n = 14), and long segment (n = 20).
3.4. Treatments
The shortest mean duration from admission to methylprednisolone therapy was observed in patients with ON and NMOSD (1.22 days and 1.44 days, respectively). The longest mean duration was found in patient with meningitis (19 days), followed by patients with encephalitis (6.54 days). Significant differences in the mean duration from admission to methylprednisolone therapy were observed in patients with NMOSD versus ADEM (p < 0.05), NMOSD versus encephalitis (p < 0.05), ON versus ADEM (p < 0.05), ON versus encephalitis (p < 0.05), and ADEM versus encephalitis (p < 0.05).
Of 74 patients, 15 (20.3%) received immunosuppressive therapy, including 8 patients with ADEM, 5 with NMOSD, 1 with ON, and 1 with TM. Patients were treated monthly with azathioprine (n = 7), mycophenolate mofetil (n = 5), cyclophosphamide (n = 2), and intravenous drip of gamma globulin (n = 2). One patient already taking azathioprine was treated with mycophenolate mofetil due to abnormal liver function. One patient was treated with mycophenolate mofetil due to the poor effect of azathioprine. Recurrence occurred in 13 patients (Table 2).

Table 2. Treatments received by patients with different clinical phenotypes of MOGAD.
3.5. Follow-up
Patients were followed for an average of 11.7 months (range, 60 days to 36 months). During follow-up, recurrence occurred in 17 (23.0%) children (Table 3), including 8 cases with ADEM (6 with multiphasic disseminated encephalomyelitis and 2 with ADEM-NMOSD), 2 with encephalitis (1 with NMOSD and 1 with seizures), 2 with ON (1 with ON-ADEM and 1 with ON-NMOSD), 3 with NMOSD, and 2 with TM (1 with TM-NMOSD and 1 with TM) (Figure 6). Among them, 6 experienced recurrence twice. The clinical features and MRI results of the different MOGAD syndromes are summarized in Supplementary Tables 1, 2, respectively. Representative MR images from patients with MDEM, relapsing NMOSD, TM-NMOSD, and encephalitis are shown in Supplementary Figures 1–4.
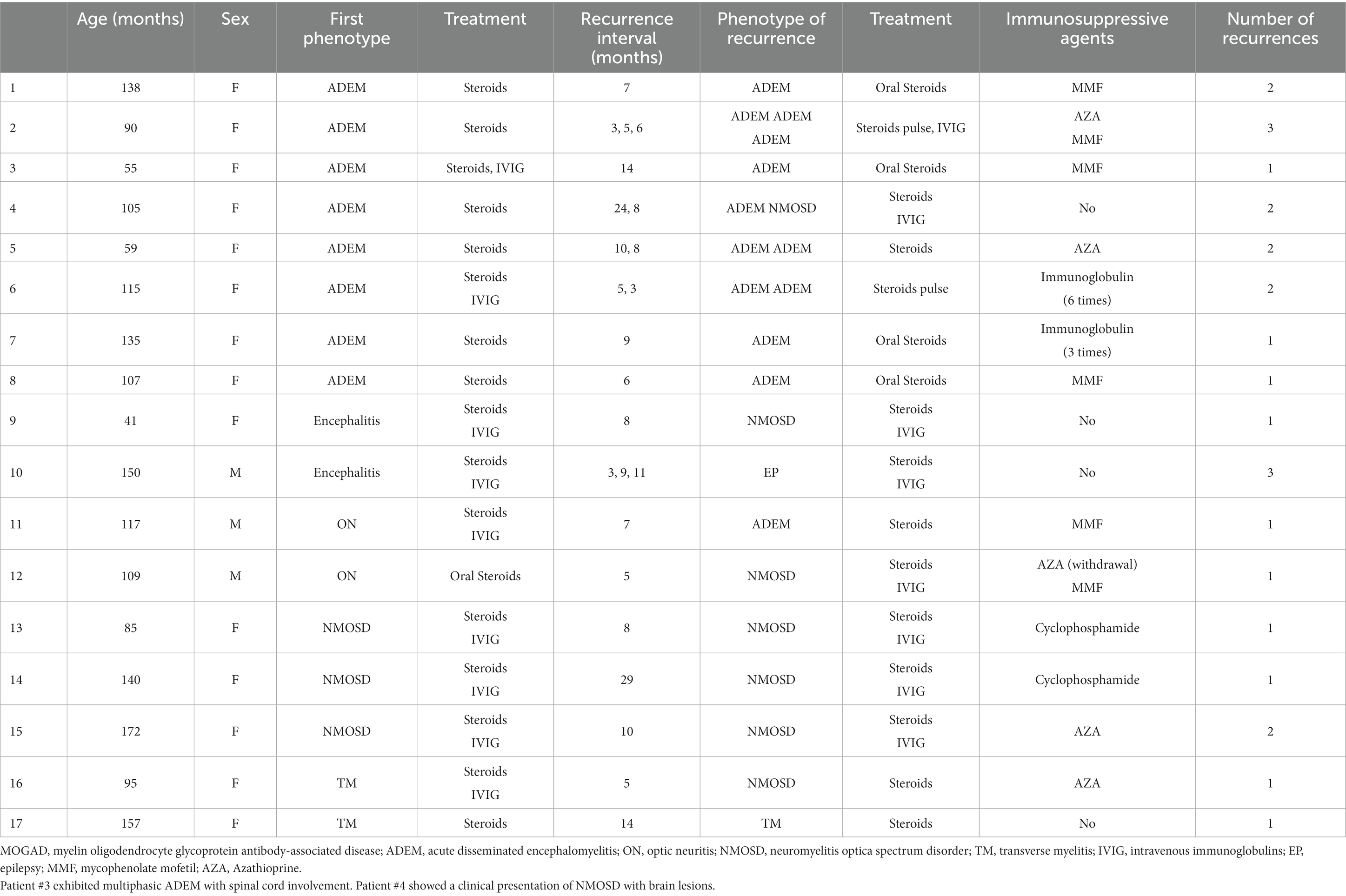
Table 3. Details of recurrence among pediatric patients with different clinical phenotypes of MOGAD.
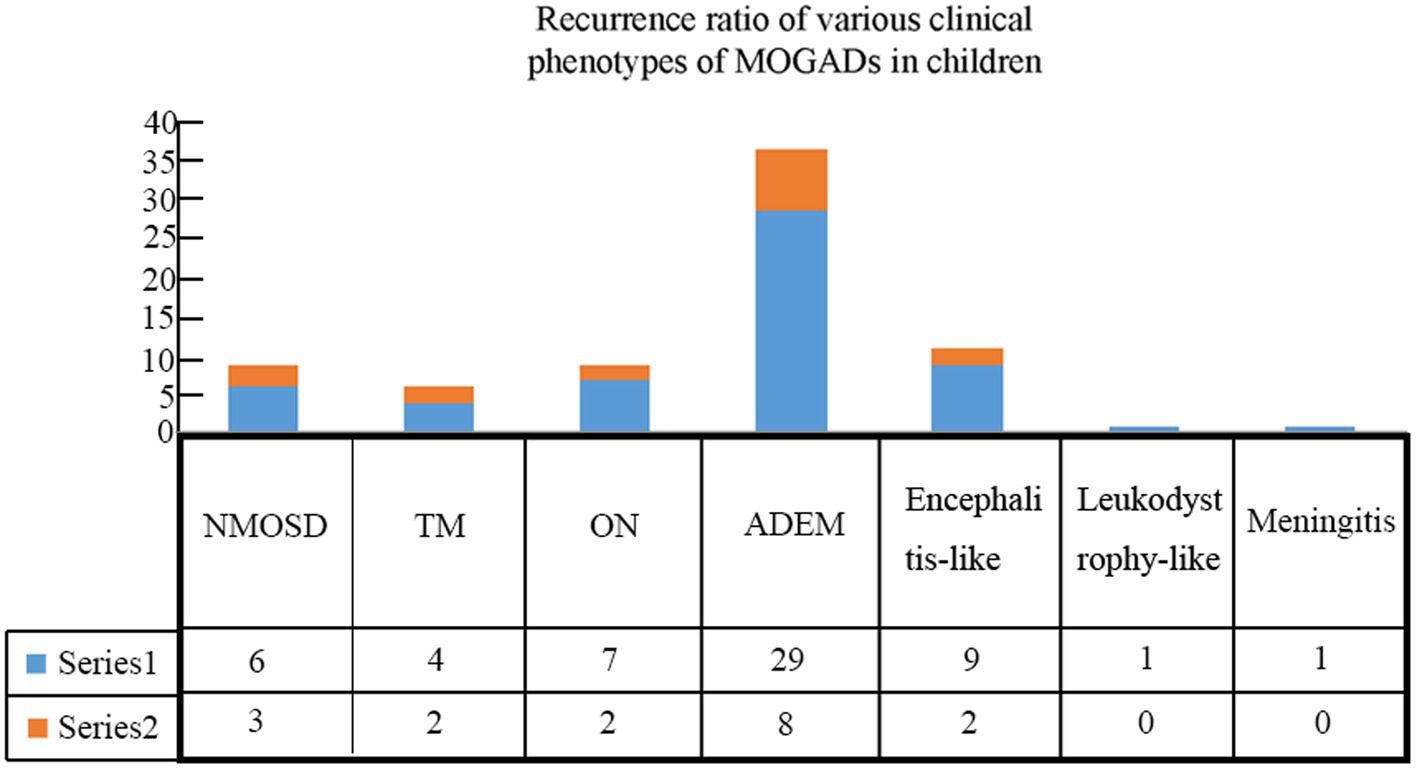
Figure 6. Recurrence rates among children with different clinical phenotypes of MOGAD.
The median age of patients with recurrence was 109 months, which was significantly higher than that of patients with no recurrence (82.5 months) (p < 0.05). The male-to-female ratio in patients with recurrence was 1:4.67, which was significantly lower than that in patients at first onset (p < 0.05). We further divided 74 patients into four groups according to their serum MOG levels: 1:10, 1:32, 1:100, and 1:320. Pairwise comparison analysis showed no significant difference in the recurrence rate among the four groups (p < 0.05) (Table 4). The average interval to the first recurrence was 9.8 months. All patients with recurrence were re-treated with hormone or immunosuppressive therapy, and the symptoms were relieved in the acute phase. Thirteen patients with recurrence were given immunosuppressants. Of them, a second recurrence occurred in 4 (30.8%) patients after treatment.
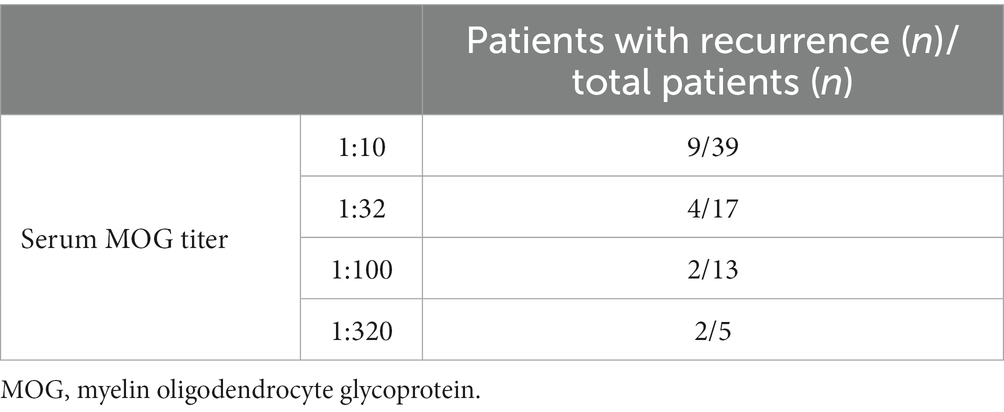
Table 4. Recurrence rates among patients with different serum MOG titers.
Patient #10 experienced recurrent seizures characterized by frequent and intense episodes, which were controlled after immunomodulatory treatment, with a seizure-free period lasting several months. The main manifestation of the recurrence was seizures without fever or encephalopathy. Multiple cranial MRI showed no notable abnormalities. The initial presentation of this patient was fever and convulsions. The CSF white blood cell (WBC) count was 240 × 106/L, with 64% monocytes. Cranial MRI did not reveal any significant abnormalities, but the electroencephalogram (EEG) indicated diffuse δ activity. The treatment consisted of methylprednisolone, intravenous immunoglobulin infusion, and oral sodium valproate. At the follow-up examination, the CSF WBC count was 32 × 106/L, with 90% monocytes. The first follow-up examination was conducted after a 3-month interval, and the patient experienced recurrent seizures and status epilepticus again, without fever or signs of encephalopathy. The CSF WBC count was 36 × 106/L, and the EEG showed increased δ activity in the left hemisphere and epileptic discharges in the left frontal lobe. Cranial and spinal MRI showed no abnormalities. The patient was treated with methylprednisolone, in addition to combination therapy with oxcarbazepine and sodium valproate to control the seizures. Subsequently, in the fourth month, occasional seizures occurred, and the CSF WBC count was 4 × 106/L. The EEG indicated a basic rhythm with slow waves (≤8 Hz), and increased θ and δ activity was observed in the left hemisphere. Lacosamide was added to the treatment regimen for anti-seizure therapy, and seizure control was achieved. The second recurrence occurred 9 months after the previous one, with seizures again, but without fever or signs of encephalopathy. The CSF WBC count was 45 × 106/L, and the EEG showed a basic rhythm with slow waves (≤8 Hz), as well as increased θ and δ activity in the left hemisphere. Cranial MRI was normal. The treatment included methylprednisolone, intravenous immunoglobulin infusion, and mycophenolate mofetil as immunomodulatory therapy, in addition to continued oral sodium valproate, oxcarbazepine, and lacosamide for anti-seizure therapy. Seizure control was achieved. The third recurrence occurred 11 months after the previous one, with seizures again, but without fever or signs of encephalopathy. The CSF WBC count was 97 × 106/L, and the EEG showed a basic rhythm with slow waves (≤8 Hz), as well as increased θ and δ activity in the left hemisphere. Cranial MRI was normal. Methylprednisolone, mycophenolate mofetil, sodium valproate, oxcarbazepine, and lacosamide were used for anti-seizure therapy. The child remained seizure-free for nearly 1 year.
4. Discussion
Pediatric patients with MOGAD exhibit a wide range of monophasic and recurrent disorders (10). In this study, the most common clinical phenotype of MOGAD was ADEM, accounting for 50% of the children. This finding is consistent with the study reported by the E.U. Pediatric MOG Consortium, which showed that 46% of pediatric patients with MOGAD presented with ADEM (11). The second most common phenotype was encephalitis (14.9%), with a higher prevalence rate in this cohort than in the above-mentioned E.U. study (11), which may be explained by a higher detection rate of the MOG antibody in our study. Thus, to achieve early diagnosis, testing for both autoimmune encephalitis antibodies and CNS demyelinating antibodies should be conducted in children with encephalitis. Patients with TM had the highest median age of disease onset compared to those with other clinical phenotypes, suggesting that elder children were prone to have lesions in the spinal cord. The shortest hospital stay was observed in patients with ON, possibly because steroid pulse therapy was administered to them immediately after admission. Children with meningitis had the longest hospital stay, which may be related to the lack of neurological signs and symptoms, which leads to the late diagnosis of these patients.
The CSF leukocyte count was elevated in all clinical phenotypes of MOGAD in comparison with the healthy range, while no significant increase in the CSF protein concentration was observed. Atypical changes in imaging examinations are often observed in patients with MOGAD. In a previous cohort study of 56 children positive for MOG antibodies, 8 (14%) exhibited symptoms of area postrema syndrome (APS), with 7 presenting both ADEM and APS, and 1 displaying APS alone (12). In a multicenter study of 274 adult patients with positive AQP4 antibodies and 107 adult patients positive for MOG antibodies in South Korea, 41 (14.9%) AQP4-positive patients had the first symptom of APS, and 47 (17.2%) exhibited APS during the disease, while no patient positive for MOG antibodies had the first symptoms of APS, and only 2 (1.9%) patients exhibited APS symptoms during the disease (13). Venkatesan et al. (14) found that 9% of patients with MOG-related encephalitis phenotype showed normal cranial MRI results (8), which was consistent with our study, in which 1 out of 11 (9.1%) patients with encephalitis exhibited no cranial MRI abnormalities throughout the disease course. The above data suggest that patients with MRI-negative encephalitis may have MOGAD and should be observed for demyelination during follow-up. The cranial MRI scans of a meningitis patient with fever as the only clinical manifestation showed meningeal enhancement without parenchymal changes, suggesting consistent absence of demyelinating changes in this patient.
The incidence of EBV positivity in other patients, such as those with fever, was much lower compared to that in patients with MOGAD. Previous studies have indicated a potential link between viral infection and the development of MOGAD (15, 16). Despite the absence of evidence for intracellular EBV infection in these specific patients, we believe it is important to consider the possibility of EBV infection being associated with MOGAD and to further investigate this correlation. In addition, patient #10 experienced recurrent seizures. Although the cranial MRI did not show any notable abnormalities, there was a decrease in the cerebrospinal fluid leukocyte count, which later increased. In addition, the serum MOG antibody titers at a dilution of 1:10 were positive in multiple tests, and the seizure symptoms were improved following immunomodulatory therapy. MOG encephalitis occurred 22 days after HSV encephalitis. Therefore, we considered that the seizures were associated with MOG positivity.
Methylprednisolone pulse therapy and immunoglobulin immunomodulatory therapy are efficient first-line treatments for MOGAD, especially in the acute phase (17). In this study, all patients received methylprednisolone pulse therapy and 43 (58.1%) also were given concomitant immunomodulatory therapy with gamma globulin. Treatment with methylprednisolone pulse therapy is associated with significantly decreased visual acuity, reduced risk of disability, and improved early diagnosis in patients with ON and NMOSD. Patients with encephalitis received methylprednisolone pulse therapy after 6.54 days of admission, which was significantly different compared to patients with NMOSD, ON, and ADEM. Therefore, the recognition of MOGAD patients with encephalitis needs to be improved. Among 15 patients who were given immunosuppressive therapy, there were 2 with monophasic disease. Both patients were diagnosed with the NMOSD phenotype, presenting with acute onset and significant visual impairment. Despite two courses of high-dose steroid therapy and intravenous immunoglobulin treatment, they experienced limited improvement in visual acuity. Considering the potentially disabling nature of the disease and the anticipated long-term use of steroids, immunosuppressive therapy was prescribed. The highest recurrence rates in this study were observed in patients with NMOSD (33%) and TM (33%), which is consistent with the findings of Waters et al. (18), who showed that the recurrence of MOGAD was associated with persistent positive antibodies and patients who remained seropositive during follow-up had a higher risk of recurrence than those who converted to seronegative status (38% vs. 13%). In the present study, we found no association between high serum MOG antibody titers at the beginning of the disease course and the rate of disease recurrence.
5. Limitations
The sample size for each clinical phenotype was small, and all patients were from the same hospital. Additionally, the follow-up period was short for some patients. Future investigations with a larger sample size and longer follow-up are needed.
6. Conclusion
This study showed that ADEM and encephalitis are the most common clinical phenotypes of MOGAD in Chinese pediatric patients. The age of onset, hospital stay, number of leukocytes in the CSF, and recurrence rate significantly differed in children with different MOGAD phenotypes. Moreover, atypical MRI changes were found in some MOGAD cases. These findings contribute to a better understanding of the characteristics, treatment options, and recurrence rates of each clinical phenotype of MOGAD and may provide insight for improving the diagnosis and treatment of these diseases.
Data availability statement
The raw data supporting the conclusions of this article will be made available by the authors, without undue reservation.
Ethics statement
The studies involving humans were approved by the Department of Neurology of Children’s Hospital Affiliated to Zhejiang University School of Medicine. The studies were conducted in accordance with the local legislation and institutional requirements. Written informed consent for participation in this study was provided by the participants’ legal guardians/next of kin.
Author contributions
YH wrote the main manuscript text. XY prepared Tables 1–4. LL prepared Figures 1–6. YW and LX statistics. PJ and ZY specialist in neurology. FG concept and design. All authors reviewed the manuscript and approved the final draft before submission.
Funding
This study is supported by Key Research and Development Plan of Zhejiang Province (2020C03038); National Natural Science Foundation of China (Grant No. 82001232); Zhejiang Provincial Natural Science Foundation of China (Grant No. LQ19H090002); STI2030-Major Projects+2021ZD 0201700.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary material
The Supplementary material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fneur.2023.1188323/full#supplementary-material
References
1. Delarasse, C, Della Gaspera, B, Lu, CW, Lachapelle, F, Gelot, A, Rodriguez, D, et al. Complex alternative splicing of the myelin oligodendrocyte glycoprotein gene is unique to human and non-human Primates. J Neurochem. (2006) 98:1707–17. doi: 10.1111/j.1471-4159.2006.04053.x
2. Peschl, P, Bradl, M, Höftberger, R, Berger, T, and Reindl, M. Myelin oligodendrocyte glycoprotein: deciphering a target in inflammatory demyelinating diseases. Front Immunol. (2017) 8:529. doi: 10.3389/fimmu.2017.00529
3. Reindl, M, and Waters, P. Myelin oligodendrocyte glycoprotein antibodies in neurological disease. Nat Rev Neurol. (2019) 15:89–2. doi: 10.1038/s41582-018-0112-x
4. Höftberger, R, Guo, Y, Flanagan, EP, Lopez-Chiriboga, AS, Endmayr, V, Hochmeister, S, et al. The pathology of central nervous system inflammatory demyelinating disease accompanying myelin oligodendrocyte glycoprotein autoantibody. Acta Neuropathol. (2020) 139:875–2. doi: 10.1007/s00401-020-02132-y
5. Sechi, E, Cacciaguerra, L, Chen, JJ, Mariotto, S, Fadda, G, Dinoto, A, et al. Myelin oligodendrocyte glycoprotein antibody-associated disease (Mogad): a review of clinical and Mri features, diagnosis, and management. Front Neurol. (2022) 13:885218. doi: 10.3389/fneur.2022.885218
6. Jurynczyk, M, Messina, S, Woodhall, MR, Raza, N, Everett, R, Roca-Fernandez, A, et al. Clinical presentation and prognosis in Mog-antibody disease: a UK study. Brain. (2017) 140:3128–38. doi: 10.1093/brain/awx276
7. Jarius, S, Paul, F, Aktas, O, Asgari, N, Dale, RC, de Seze, J, et al. Mog encephalomyelitis: international recommendations on diagnosis and antibody testing. J Neuroinflammation. (2018) 15:134. doi: 10.1186/s12974-018-1144-2
8. Krupp, LB, Tardieu, M, Amato, MP, Banwell, B, Chitnis, T, Dale, RC, et al. International pediatric multiple sclerosis study group criteria for pediatric multiple sclerosis and immune-mediated central nervous system demyelinating disorders: revisions to the 2007 definitions. Mult Scler. (2013) 19:1261–7. doi: 10.1177/1352458513484547
9. Banwell, B, Bennett, JL, Marignier, R, Kim, HJ, Brilot, F, Flanagan, EP, et al. Diagnosis of myelin oligodendrocyte glycoprotein antibody-associated disease: international Mogad panel proposed criteria. Lancet Neurol. (2023) 22:268–2. doi: 10.1016/s1474-4422(22)00431-8
10. Hennes, EM, Baumann, M, Lechner, C, and Rostásy, K. Mog Spectrum disorders and role of Mog-antibodies in clinical practice. Neuropediatrics. (2018) 49:003–11. doi: 10.1055/s-0037-1604404
11. Bruijstens, AL, Lechner, C, Flet-Berliac, L, Deiva, K, Neuteboom, RF, Hemingway, C, et al. Paediatric Mog consortium consensus: part 1 - classification of clinical phenotypes of Paediatric myelin oligodendrocyte glycoprotein antibody-associated disorders. Eur J Paediatr Neurol. (2020) 29:2–13. doi: 10.1016/j.ejpn.2020.10.006
12. Kunchok, A, Krecke, KN, Flanagan, EP, Jitprapaikulsan, J, Lopez-Chiriboga, AS, Chen, JJ, et al. Does area Postrema syndrome occur in myelin oligodendrocyte glycoprotein-igg-associated disorders (Mogad)? Neurology. (2020) 94:85–8. doi: 10.1212/wnl.0000000000008786
13. Hyun, JW, Kwon, YN, Kim, SM, Lee, HL, Jeong, WK, Lee, HJ, et al. Value of area Postrema syndrome in differentiating adults with Aqp4 vs Mog antibodies. Front Neurol. (2020) 11:396. doi: 10.3389/fneur.2020.00396
14. Venkatesan, A, Tunkel, AR, Bloch, KC, Lauring, AS, Sejvar, J, Bitnun, A, et al. Case definitions, diagnostic algorithms, and priorities in encephalitis: consensus statement of the international encephalitis consortium. Clin Infect Dis. (2013) 57:1114–28. doi: 10.1093/cid/cit458
15. Burrer, R, Buchmeier, MJ, Wolfe, T, Ting, JP, Feuer, R, Iglesias, A, et al. Exacerbated pathology of viral encephalitis in mice with central nervous system-specific autoantibodies. Am J Pathol. (2007) 170:557–6. doi: 10.2353/ajpath.2007.060893
16. Nakamura, Y, Nakajima, H, Tani, H, Hosokawa, T, Ishida, S, Kimura, F, et al. Anti-Mog antibody-positive Adem following infectious mononucleosis due to a primary Ebv infection: a case report. BMC Neurol. (2017) 17:76. doi: 10.1186/s12883-017-0858-6
17. Bruijstens, AL, Wendel, EM, Lechner, C, Bartels, F, Finke, C, Breu, M, et al. Paediatric Mog consortium consensus: part 5 - treatment of Paediatric myelin oligodendrocyte glycoprotein antibody-associated disorders. Eur J Paediatr Neurol. (2020) 29:41–53. doi: 10.1016/j.ejpn.2020.10.005
Keywords: myelin oligodendrocyte glycoprotein, myelin oligodendrocyte glycoprotein antibody-associated disease, clinical phenotype, diagnosis, treatment
Citation: Hua Y, Yan X, Liu L, Wang Y, Xu L, Jiang P, Yuan Z and Gao F (2023) Phenotypic characteristics of myelin oligodendrocyte glycoprotein antibody-associated disease in children: a single-center, retrospective study. Front. Neurol. 14:1188323. doi: 10.3389/fneur.2023.1188323
Edited by:
Silvia Noemi Tenembaum, Garrahan Hospital, ArgentinaReviewed by:
Terrence Thomas, KK Women's and Children's Hospital, SingaporeChao Zhang, Tianjin Medical University General Hospital, China
Copyright © 2023 Hua, Yan, Liu, Wang, Xu, Jiang, Yuan and Gao. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Feng Gao, ZXBpbGVwc3lAemp1LmVkdS5jbg==
†These authors have contributed equally to this work