 Lin Cai1,2
†
Lin Cai1,2
† Xiaotao Huang3
†Yan Ye1,2Dailan Yang1,2
Xiaotao Huang3
†Yan Ye1,2Dailan Yang1,2 Linshen Xie4Daigang Fu4Lijun Peng4
Linshen Xie4Daigang Fu4Lijun Peng4 Dingzi Zhou4
Dingzi Zhou4 Juan Liao1,2*
Juan Liao1,2*- 1Department of Gastroenterology, West China School of Public Health and West China Fourth Hospital, Sichuan University, Chengdu, China
- 2Non-communicable Diseases Research Center, West China-PUMC C. C. Chen Institute of Health, Sichuan University, Chengdu, China
- 3Department of Gastroenterology, 903 Hospital, Jiangyou, China
- 4Department of Occupational Disease and Toxicosis, West China School of Public Health and West China Fourth Hospital, Sichuan University, Chengdu, China
Background: Wilson’s disease (WD) is a recessive genetic disorder characterized by copper metabolism dysfunction. It is difficult to obtain an accurate diagnosis due to its variable clinical presentation. This study aimed to describe the clinical characteristics and diagnostic particularities in a series of Chinese WD patients.
Methods: The medical records of 371 patients with WD retrieved from January 2005 to December 2020 were retrospectively reviewed.
Results: The incidence of WD has a male predominance in the adult population. However, the difference in sex distribution is not significant in the pediatric population. Females have an earlier symptom onset than males. The most common initial symptoms were neuropsychiatric manifestations both in the pediatric population (49.7%) and adult population (69.8%), and there was a male predominance (61.8%). Eighty-two percent of patients presented with more than two neurologic symptoms. Fifty-two (14%) patients presented with psychiatric symptoms. The most common WD phenotype was the neuropsychiatric form (48%). The age of onset occurred earlier in patients with the hepatic phenotype than in those with the neuropsychiatric phenotype. Moreover, there was a significant difference in sex distribution regarding phenotype. Females presented with a hepatic phenotype more often than males, and the neuropsychiatric phenotype occurred more frequently in males with an older onset age. Further study showed that the age at onset was a deciding factor for predicting the neuropsychiatric phenotype among the hepatic phenotype. However, sex did not correlate with the phenotype.
Conclusion: Males seem to have a higher disease susceptibility, with symptom onset later than females. Males frequently present with a neuropsychiatric phenotype, while females present with a hepatic phenotype. Age at onset was a deciding factor for predicting the WD phenotype. Further studies focusing on the effect of estrogens on the pathology of WD are suggested.
Introduction
Wilson’s disease (WD) is an inherited autosomal recessive disorder that is caused by pathogenic mutations of ATP7B, which lead to copper metabolism dysfunction. The subsequent gradual aberrant deposition of copper in organs, particularly in the liver and brain, triggers multisystemic symptoms (1). The estimated prevalence of WD is approximately 1/19500, with a higher rate in the Asian and Ashkenazi Jewish populations (2). The clinical presentation and phenotypic characteristics of WD can be highly variable. Patients could present with asymptomatic biochemical abnormalities, hepatitis, steatosis, liver cirrhosis, neurological damage, psychiatric symptoms, and occasionally cardiologic and/or skeletal symptoms. There is no gold standard diagnostic method for establishing the diagnosis of WD. A combination of clinical presentations, abnormal copper metabolism parameters, and/or mutations of the ATP7B gene are necessary to confirm the diagnosis (3).
Wilson’s disease is among a limited number of genetic disorders that can be partly treated or prevented by copper-chelating agents. Early diagnosis and intervention are crucial to minimize the devastating progression of the disease and obtain better outcomes. However, its diagnosis remains a challenge. A delay in diagnosing WD is frequent and may cause irreversible disease deterioration and even fatality. Once the diagnosis is established, lifelong treatment is necessary.
Genetic testing for ATP7B mutations plays an increasing role in the diagnosis of WD. However, the direct molecular genetic diagnosis of WD is difficult at present, as there are more than 500 mutations associated with this condition (4). Additionally, most individuals are compound heterozygotes. Not all suspected patients are able to undergo genetic WD diagnosis or screening, especially those patients in developing countries. The clinical diagnosis mostly relies on both clinical features and biochemical parameters. There is still a lack of reasonably designed clinical trials in China, and most management is based on evidence from other countries. To characterize the patient experience of WD, this study aims to characterize the clinical spectrum and provide a detailed analysis of Chinese WD patients. Moreover, we sought to explore the clinical variants that predict the phenotypes of WD.
Subjects and methods
Subjects who had a confirmed diagnosis of WD were included. The diagnosis was based on clinical symptoms, abnormal copper metabolism (decreased level of serum ceruloplasmin, and increased 24 h urine copper excretion), the presence of Kayser-Fleischer rings (KF-rings), and ATP7B mutation analysis if available, according to the Leipzig score (5). The demographic, case history, clinical, laboratory, and brain imaging data and the treatment of WD patients were retrospectively collected in a single center from January 2005 to December 2020.
In accordance with a previous study (5), patients were classified into three disease phenotypes as follows: the hepatic form, which presented with signs of liver injury and/or hepatic symptoms; the neuropsychiatric form, which presented with neuropsychiatric symptoms; and the presymptomatic form.
Gender differences have been reported in the course of liver fibrosis (6), neurodegenerative diseases, and WD (7). In this Southwest China population, we investigated the gender differences in the course and clinical features of WD.
Clinical heterogeneity cannot be explained by genetic determinants. To assess whether the clinical variables were associated with phenotype, the following variants were selected for further analysis: age at onset of disease and gender. The ethics committee of West China Fourth Hospital approved this retrospective analysis (HXSY-EC-2021034). The work was carried out in accordance with the code of ethics of the World Medical Association (Declaration of Helsinki).
Statistical analysis
Statistical analyses were performed with SPSS version 22.0 (IBM, New York, USA). Normally distributed quantitative variables are expressed as the mean ± standard deviation (SD); nonnormally distributed quantitative variables are expressed as the median [IQR]. The relevant parameters were compared using Student’s t test or nonparametric tests, as appropriate. Categorical variables were expressed as counts (percentages) and compared using the χ2 test or Fischer’s exact test when appropriate. A logistic regression analysis was performed to explore the correlation between the clinical parameters and phenotype, and variables with p < 0.05 were then used in the multivariate logistic regression analysis. p-values lower than 0.05 were considered statistically significant.
Results
Patient characteristics
A total of 371 patients who met the criteria for a WD diagnosis were included in the analysis. Demographic and clinical characteristics are summarized in Table 1.
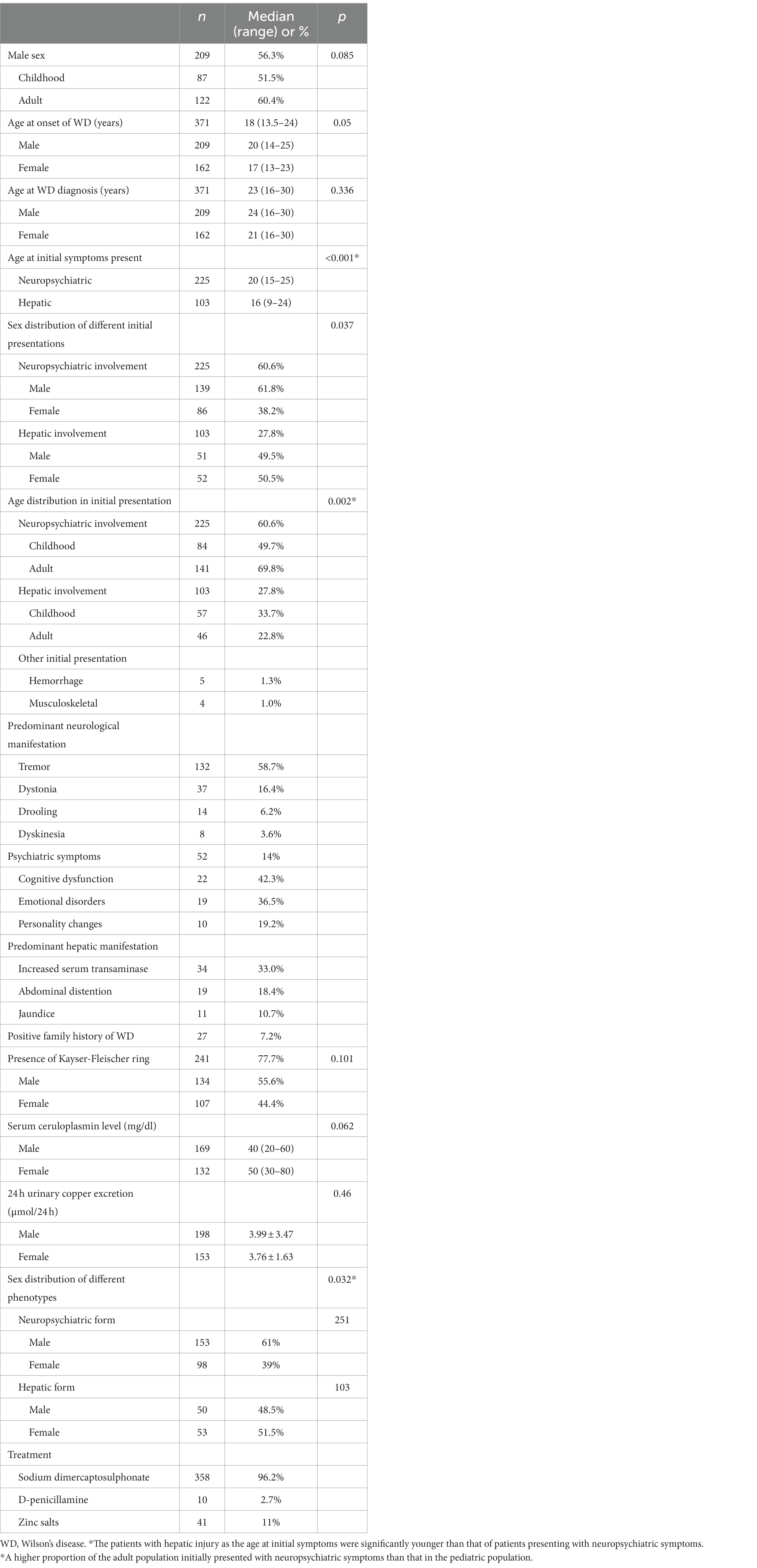
Table 1. Demographic, clinical, and laboratory features of Wilson’s disease (WD) patients.
Regarding the age of onset, 169 (45.6%) subjects were in the pediatric age group (<18 years of age), and 202 (54.4%) were in the adult group. A total of 87.8% of patients were 5 to 35 years of age, and 5.7% were over 40 years of age. The minimum age of onset was 3 years, and the maximum was 54 years. There was a male predominance in adults, with 122 patients (60.4%) being male. In the pediatric population, the gender difference was not obvious, with only 87 (51.5%) being male. However, there was no significant difference in sex distribution between the adult and pediatric populations (p = 0.085). Of the patients studied, 27 patients (7.2%) had a family history of WD, with 26 siblings detected by family screening.
Gender profile in the onset and diagnosis of disease
Regarding the onset of disease, females had an earlier symptom onset than males (Table 1) (p = 0.050). The diagnosis of WD was made at a median age of 23 (range 16–30) years, and there was no statistically significant difference in sex distribution (p = 0.336).
The initial symptoms were neuropsychiatric abnormalities in 225 patients and hepatic abnormalities in 103 patients. The patients with hepatic injury as the initial symptom were 16 (9–24) years of age, which is significantly younger than that of patients presenting with neuropsychiatric symptoms (p < 0.001). Although neuropsychiatric symptoms were also commonly present in childhood (49.7%), a higher proportion of the adult population initially presented with neuropsychiatric symptoms than that in the pediatric population (p = 0.002). Further analyses suggested that a male predominance was observed in patients with neuropsychiatric symptoms compared with those with hepatic symptoms as initial symptoms (p = 0.037). In the hepatic injury group, the sex distribution was nearly equal.
The most common neurological manifestations were tremor (58.7%) and dysarthria (16.4%). In addition, 82% of patients presented with more than two neurologic symptoms. Moreover, 52 (14%) patients presented with psychiatric symptoms. The common manifestations were cognitive dysfunction (42.3%), emotional disorders (36.5%), and personality changes (19.2%). The main hepatic features were increases in the levels of aminotransferases (33.0%) and the incidences of abdominal distention (18.4%) and jaundice (10.7%).
Gender profile in other clinical features
A total of 241 (77.7%) patients presented with KF-rings in the cornea. Although there was no statistically significant difference in the sex distribution (p = 0.101), a male predominance (55.6%) was observed in patients presenting with KF-rings (Table 1).
At presentation, 3.3% of patients had normal serum ceruloplasmin levels. Although there were no significant differences, males had higher 24 h urine copper excretion (p = 0.46) and serum ceruloplasmin levels (p = 0.062) than females.
Gender profile in WD phenotype
At presentation, 251 (67.7%) patients had the neuropsychiatric phenotype, and 103 (27.8%) patients had the hepatic phenotype of the disease (Table 1). The differences in the sex distribution between the neuropsychiatric phenotype and the hepatic phenotype were statistically significant (p = 0.032). Females presented more often with a hepatic phenotype than males. Neuropsychiatric presentation occurred more frequently in males. Females had a significantly earlier age of onset than males in the neuropsychiatric phenotype (p = 0.003, Table 2). There were no significant differences in the gender profile regarding the presence of KF-rings, serum ceruloplasmin concentration, and 24-h urinary copper excretion in different WD phenotypes.
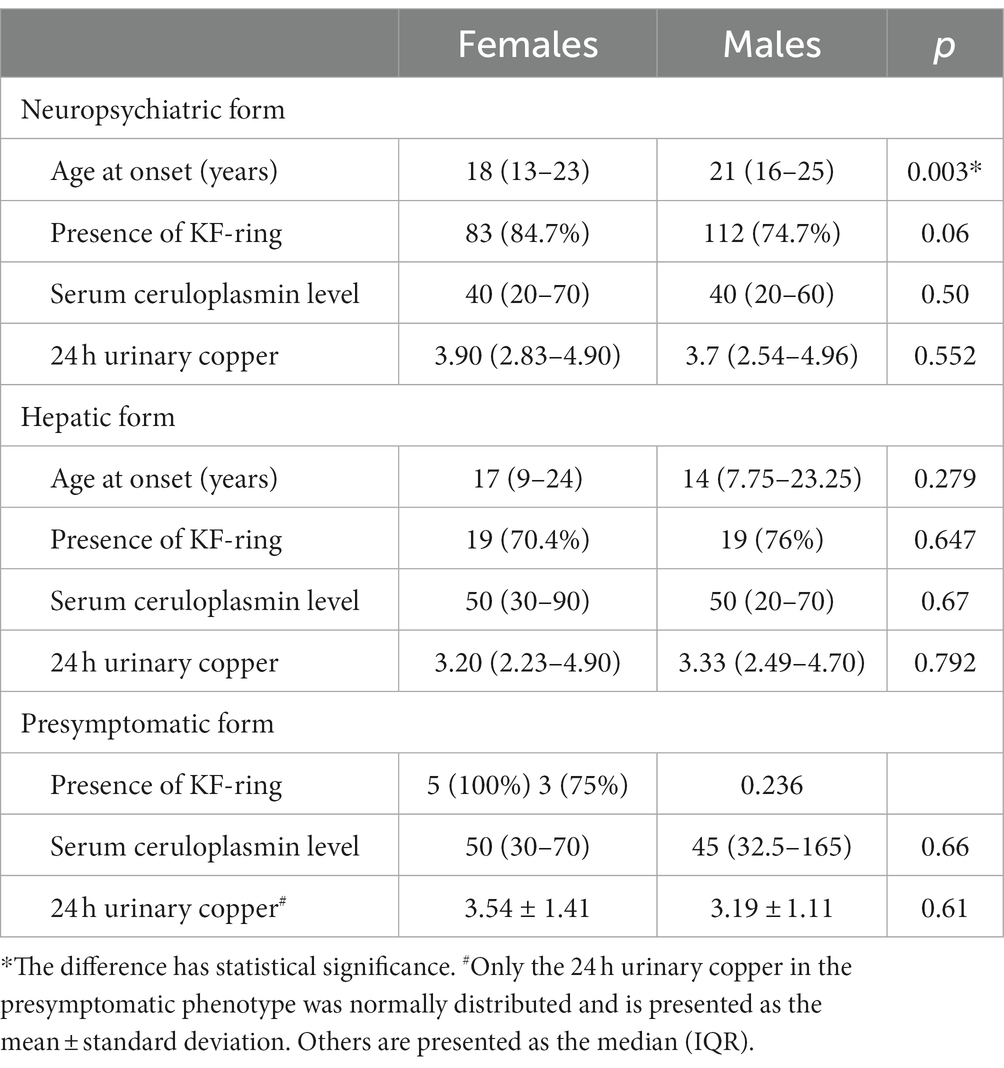
Table 2. The characteristics of different Wilson’s disease phenotypes in females and males.
For disease phenotype prediction, both univariate and multivariate logistic regression were performed. As shown in Table 3, only age at onset was a deciding factor when predicting hepatic phenotype and neuropsychiatric phenotype. Early onset age is inclined toward the hepatic phenotype rather than the neuropsychiatric phenotype. There was no significant correlation between sex distribution and the WD phenotype in multivariate logistic regression.
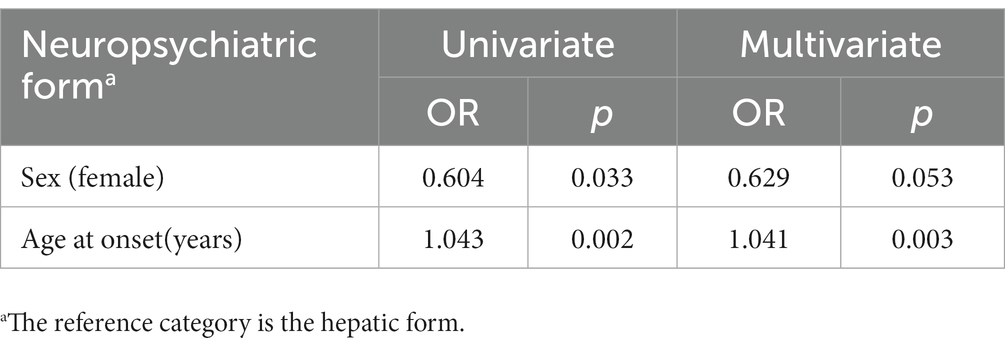
Table 3. Univariate and multivariate logistic regression of factors associated with the neuropsychiatric phenotype and hepatic phenotype.
Discussion
There are five main findings in our study. ① There was a male predominance in the prevalence of WD in the adult population. However, the gender difference was not significant in childhood. ② Females have an earlier symptom onset than males, but there is no significant gender difference regarding the age of diagnosis. ③ The most common initial symptoms were neuropsychiatric manifestations, with a significant male predominance. The tendency becomes more apparent in adults. Hepatic injury is present at an earlier age of onset than neuropsychiatric symptoms. ④ The most common WD phenotype was the neuropsychiatric form. There was a significant gender difference. Females presented more often with a hepatic phenotype than males. Neuropsychiatric presentation occurred more frequently in males. ⑤ Earlier age at onset was a deciding factor for predicting the hepatic phenotype rather than the neuropsychiatric phenotype.
WD is a highly heterogeneous disease. There is still a lack of sufficient knowledge on the natural history of untreated patients with WD. It is well known that WD trends toward a young age of onset, as most diagnoses are made between the ages of 5 and 35 years. Our results demonstrated that the incidence of age distribution in the population was very similar to that in a previous report (8). In our study, there was a male predominance in the prevalence of this condition in adults. Interestingly, no significant gender difference was observed in childhood. These findings indicate that sex hormones may play a role in the pathogenesis of WD, with estrogens exerting antioxidant, neurotrophic, and anti-inflammatory effects (9).
There was a gender difference regarding disease presentation in WD patients. In accordance with a previous study (10), the neurologic manifestations start later and are more common in males. Hepatic presentation was more common among females. A possible explanation is that estrogens exert a protective role in the brain. Gromadzka et al. demonstrated that iron metabolism differences may be associated with sex-related clinical heterogeneity (11). Further studies are needed to elucidate the underlying mechanisms.
Interestingly, the occurrence of neurological symptoms in childhood is higher than that in in previous studies (12). The discrepancies could possibly be explained by the median age of children in our study being older than 10 years. Neurological symptoms are more prevalent in older children (13).
Psychiatric symptoms are nonspecific, and the clinical features are variable. That may cause difficulties in diagnosis. In this study, the occurrence of psychiatric manifestations was lower than that in a previous study (14), and a male predominance was observed. In fact, neuropsychiatric WD is less well appreciated in childhood. Unfortunately, there is still a lack of dedicated scales to assess the psychiatric symptoms of WD. In this study, the diagnosis of the psychiatric form of WD is based on psychiatric signs and symptoms, psychiatric examination. It is difficult to assess whether psychiatric symptoms occurred as a result of central nervous system involvement since psychiatric symptoms may not occur together with neurologic symptoms. Further studies are needed to explore this question in newly diagnosed cases with psychopathologic assessment. There is also a need to elaborate a validate evaluation scale, with a multidisciplinary team involving hepatologists, neurologists, and psychiatrists to assess the psychiatric profile of WD.
In our study, both hepatic and neurological WD may initially display subtle symptoms that are easily overlooked. Thus, diagnostic vigilance should be proposed when faced with any liver disease and/or neurological disease with unknown etiological factors. Moreover, careful history-taking and review of laboratory data and imaging are necessary.
ATP7B gene mutations are the pathophysiological basis of WD (15), but the correlation between genotype and disease phenotype still lacks evidence. Ferenci et al. demonstrated no correlations between genotype and phenotype regarding initial symptomatic manifestation (16). Both siblings and monozygotic twins commonly have different WD phenotypes, and the same gene mutations display clinical variability among isolated populations (17–19). Moreover, several reports describe that clinical phenotypes are influenced by age (20) and sex (16). In our study, only the age of onset was a deciding factor for distinguishing the neuropsychiatric phenotype from the hepatic phenotype. Further studies are needed to explore the specific role of epigenetic factors and sex hormones and their mechanisms.
The length of this study, including the that of the follow-up, is insufficient. Second, we did not measure the level of estrogen. Third, due to the retrospective design of this study, the mechanisms underlying the sex differences remain unclear.
Conclusion
In summary, the data obtained in a cohort of 371 patients with WD clearly show gender differences in the clinical features of WD, and the age of onset was a deciding factor for disease phenotype prediction.
Data availability statement
The raw data supporting the conclusions of this article will be made available by the authors, without undue reservation.
Ethics statement
The studies involving human participants were reviewed and approved by the Ethics Committee of West China Fourth Hospital. Written informed consent from the participants was not required to participate in this study in accordance with the national legislation and the institutional requirements.
Author contributions
LC and XH implemented the data analysis, interpreted the results, and drafted this manuscript. JL provided critical revision of the manuscript for important intellectual content. LC, YY, and XH did the preliminary analysis. DY collated the results. LX, DF, LP, DZ, and JL revised the manuscript. All authors contributed to the conception and design of the research and read and approved the final manuscript.
Acknowledgments
The authors would like to thank the patients for their involvement in the present study.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The handling editor HS declared a shared parent affiliation with the author's LC, YY, DY, LX, DF, LP, DZ, and JL at the time of review.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. EASL Clinical Practice Guidelines . Wilson's disease. J Hepatol. (2012) 56:671–85. doi: 10.1016/j.jhep.2011.11.007
2. Wallace, DF, and Dooley, JS. ATP7B variant penetrance explains differences between genetic and clinical prevalence estimates for Wilson disease. Hum Genet. (2020) 139:1065–75. doi: 10.1007/s00439-020-02161-3
3. Członkowska, A, Litwin, T, Dusek, P, Ferenci, P, Lutsenko, S, Medici, V, et al. Wilson disease. Nat Rev Dis Primers. (2018) 4:21. doi: 10.1038/s41572-018-0018-3
4. Ferenci, P . Wilson's disease. Clin Gastroenterol Hepatol. (2005) 3:726–33. doi: 10.1016/s1542-3565(05)00484-2
5. Ferenci, P, Caca, K, Loudianos, G, Mieli-Vergani, G, Tanner, S, Sternlieb, I, et al. Diagnosis and phenotypic classification of Wilson disease. Liver Int. (2003) 23:139–42. doi: 10.1034/j.1600-0676.2003.00824.x
6. Rigamonti, C, Andorno, S, Maduli, E, Capelli, F, Boldorini, R, and Sartori, M. Gender and liver fibrosis in chronic hepatitis: the role of iron status. Aliment Pharmacol Ther. (2005) 21:1445–51. doi: 10.1111/j.1365-2036.2005.02517.x
7. Li, X, Feng, Z, Tang, W, Yu, X, Qian, Y, Liu, B, et al. Sex differences in clinical characteristics and brain MRI change in patients with Wilson's disease in a Chinese population. Front Physiol. (2018) 9:1429. doi: 10.3389/fphys.2018.01429
8. Poujois, A, and Woimant, F. Challenges in the diagnosis of Wilson disease. Ann Trans Med. (2019) 7:S67. doi: 10.21037/atm.2019.02.10
9. Członkowska, A, Ciesielska, A, Gromadzka, G, and Kurkowska-Jastrzebska, I. Gender differences in neurological disease: role of estrogens and cytokines. Endocrine. (2006) 29:243–56. doi: 10.1385/endo:29:2:243
10. Litwin, T, Gromadzka, G, and Członkowska, A. Gender differences in Wilson's disease. J Neurol Sci. (2012) 312:31–5. doi: 10.1016/j.jns.2011.08.028
11. Gromadzka, G, Wierzbicka, D, Litwin, T, and Przybyłkowski, A. Difference in iron metabolism may partly explain sex-related variability in the manifestation of Wilson's disease. J Trace Elem Med Biol. (2020) 62:126637. doi: 10.1016/j.jtemb.2020.126637
12. Fernando, M, van Mourik, I, Wassmer, E, and Kelly, D. Wilson disease in children and adolescents. Arch Dis Child. (2020) 105:499–505. doi: 10.1136/archdischild-2018-315705
13. Socha, P, Janczyk, W, Dhawan, A, Baumann, U, D'Antiga, L, Tanner, S, et al. Wilson's disease in children: a position paper by the Hepatology Committee of the European Society for Paediatric gastroenterology, Hepatology and nutrition. J Pediatr Gastroenterol Nutr. (2018) 66:334–44. doi: 10.1097/mpg.0000000000001787
14. Zimbrean, PC, and Schilsky, ML. Psychiatric aspects of Wilson disease: a review. Gen Hosp Psychiatry. (2014) 36:53–62. doi: 10.1016/j.genhosppsych.2013.08.007
15. Yuan, XZ, Yang, RM, and Wang, XP. Management perspective of Wilson's disease: early diagnosis and individualized therapy. Curr Neuropharmacol. (2021) 19:465–85. doi: 10.2174/1570159x18666200429233517
16. Ferenci, P, Stremmel, W, Członkowska, A, Szalay, F, Viveiros, A, Stättermayer, AF, et al. Age and sex but not ATP7B genotype effectively influence the clinical phenotype of Wilson disease. Hepatology. (2019) 69:1464–76. doi: 10.1002/hep.30280
17. Członkowska, A, Gromadzka, G, and Chabik, G. Monozygotic female twins discordant for phenotype of Wilson's disease. Move Disord. (2009) 24:1066–9. doi: 10.1002/mds.22474
18. Chabik, G, Litwin, T, and Członkowska, A. Concordance rates of Wilson's disease phenotype among siblings. J Inherit Metab Dis. (2014) 37:131–5. doi: 10.1007/s10545-013-9625-z
19. Kegley, KM, Sellers, MA, Ferber, MJ, Johnson, MW, Joelson, DW, and Shrestha, R. Fulminant Wilson's disease requiring liver transplantation in one monozygotic twin despite identical genetic mutation. Am J Transplant Off J Am Soc Transplant Am Soc Transplant Surg. (2010) 10:1325–9. doi: 10.1111/j.1600-6143.2010.03071.x
Keywords: Wilson’s disease, gender difference, clinical characteristics, phenotype, age of onset
Citation: Cai L, Huang X, Ye Y, Yang D, Xie L, Fu D, Peng L, Zhou D and Liao J (2023) Role of gender and age in features of Wilson’s disease. Front. Neurol. 14:1176946. doi: 10.3389/fneur.2023.1176946
Edited by:
Huifang Shang, Sichuan University, ChinaReviewed by:
Barbara Garavaglia, IRCCS Carlo Besta Neurological Institute Foundation, ItalyValentina Medici, University of California, Davis, United States
Copyright © 2023 Cai, Huang, Ye, Yang, Xie, Fu, Peng, Zhou and Liao. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Juan Liao, anVhbmxpYW9Ac2N1LmVkdS5jbg==
†These authors have contributed equally to this work and share first authorship