
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Neurol., 18 May 2023
Sec. Neurogenetics
Volume 14 - 2023 | https://doi.org/10.3389/fneur.2023.1170005
This article is part of the Research TopicNeurogenetic Disorders: From the Tests to the ClinicView all 7 articles
 Marlene Ek1,2*Daniel Nilsson1,2,3Martin Engvall1,4Helena Malmgren1,2Håkan Thonberg1,2Maria Pettersson1,2Britt-Marie Anderlid1,2Anna Hammarsjö1,2Hafdis T. Helgadottir1,2Snjolaug Arnardottir5Karin Naess4,6Inger Nennesmo7Martin Paucar1,2Helgi Thor Hjartarson8Rayomand Press9Göran Solders5,9Thomas Sejersen8,10Anna Lindstrand1,2†Malin Kvarnung1,2†
Marlene Ek1,2*Daniel Nilsson1,2,3Martin Engvall1,4Helena Malmgren1,2Håkan Thonberg1,2Maria Pettersson1,2Britt-Marie Anderlid1,2Anna Hammarsjö1,2Hafdis T. Helgadottir1,2Snjolaug Arnardottir5Karin Naess4,6Inger Nennesmo7Martin Paucar1,2Helgi Thor Hjartarson8Rayomand Press9Göran Solders5,9Thomas Sejersen8,10Anna Lindstrand1,2†Malin Kvarnung1,2†Introduction: Neuromuscular disorders (NMDs) have a heterogeneous etiology. A genetic diagnosis is key to personalized healthcare and access to targeted treatment for the affected individuals.
Methods: In this study, 861 patients with NMDs were analyzed with genome sequencing and comprehensive variant calling including single nucleotide variants, small insertions/deletions (SNVs/INDELs), and structural variants (SVs) in a panel of 895 NMD genes, as well as short tandem repeat expansions (STRs) at 28 loci. In addition, for unsolved cases with an unspecific clinical presentation, the analysis of a panel with OMIM disease genes was added.
Results: In the cohort, 27% (232/861) of the patients harbored pathogenic variants, of which STRs and SVs accounted for one-third of the patients (71/232). The variants were found in 107 different NMD genes. Furthermore, 18 pediatric patients harbored pathogenic variants in non-NMD genes.
Discussion: Our results highlight that for children with unspecific hypotonia, a genome-wide analysis rather than a disease-based gene panel should be considered as a diagnostic approach. More importantly, our results clearly show that it is crucial to include STR- and SV-analyses in the diagnostics of patients with neuromuscular disorders.
Neuromuscular disorders (NMDs) encompass a heterogeneous group of disorders that affect the function of peripheral motor nerves, muscles, or neuromuscular junction. Common symptoms include weakness, hypotonia, muscle atrophy, contractures, sensory disturbances, and symptoms from the autonomic nervous system, with onset varying from prenatal to late adulthood. A wider definition of NMDs includes also spastic paraplegia and ataxias. NMDs are caused by constitutional genetic aberrations and/or non-genetic factors.
The number of genes associated to NMDs is continuously increasing and, as of December 2022, 641 genes were listed in the gene table for NMDs, as hosted by Cohen et al. (1) and Dalil Hamroun and Rivier (2). Single nucleotide variants (SNVs)/small insertions-deletions (INDELs) in these genes are the most prevalent type of pathogenic variants, while other variant types such as short tandem repeat expansions (STRs) and structural variants (SVs) are causative in a subset of NMD patients. Given the heterogeneous etiology of NMDs, a clinical diagnostic workup is often challenging. A precise molecular diagnosis is valuable for predicting clinical course, and it is crucial for genetic counseling including carrier testing, prenatal diagnostics, or preimplantation genetic testing. In addition, it is a prerequisite for getting access to targeted treatments and inclusion in clinical trials for novel therapeutics that are gene- and/or mutation-specific.
Several studies of clinical genetic testing in patients with NMDs, using gene panels based on massive parallel sequencing to detect SNVs, have been published with the diagnostic yield varying between 13 and 53% (3–10). However, with this approach, variants other than exonic SNVs/INDELs would go undetected. Recently, efforts in using genome sequencing and bioinformatic pipelines to detect repeat expansions show that this method has a high sensitivity and specificity with an additional diagnostic yield up to 3% in cohorts of various neurological disorders (11, 12).
In this study, we evaluate the clinical utility of genome sequencing with comprehensive variant calling using pipelines for the detection of SNVs/INDELs, STRs, and SVs in a cohort of 861 NMD patients.
In this retrospective study, genetic results were compiled from all 861 referrals for clinical genome sequencing at our center due to suspected neuromuscular disorders including patients with ataxia and/or spastic paraparesis from January 2016 to October 2022. The cohort included patients of all ages from prenatal cases to elderly patients. Genomic DNA was isolated from whole blood using QIAsymphony (QIAGEN, Hilden, Germany) and the QIAsymphony DSP DNA Midi Kit (cat. no. 937255, QIAGEN, Hilden, Germany) according to manufacturer's protocol. For the prenatal cases, DNA was extracted from a chorion villi biopsy or fetal tissue using the EZ1 Advanced XL instrument (QIAGEN, Hilden, Germany) and the EZ1 DNA Tissue Kit (cat. no. 953034, QIAGEN, Hilden, Germany) following manufacturer's protocol. Individuals that were analyzed clinically before December 2019 (n = 455) were previously presented by Stranneheim et al. (13). Over the years, the number of genes in the NMD panel was updated regularly. Furthermore, bioinformatic pipelines were continuously improved (13). Therefore, prior to compiling the results, all negative cases were reanalyzed with the latest NMD gene panel and bioinformatic pipeline as outlined in Figure 1 and described below.
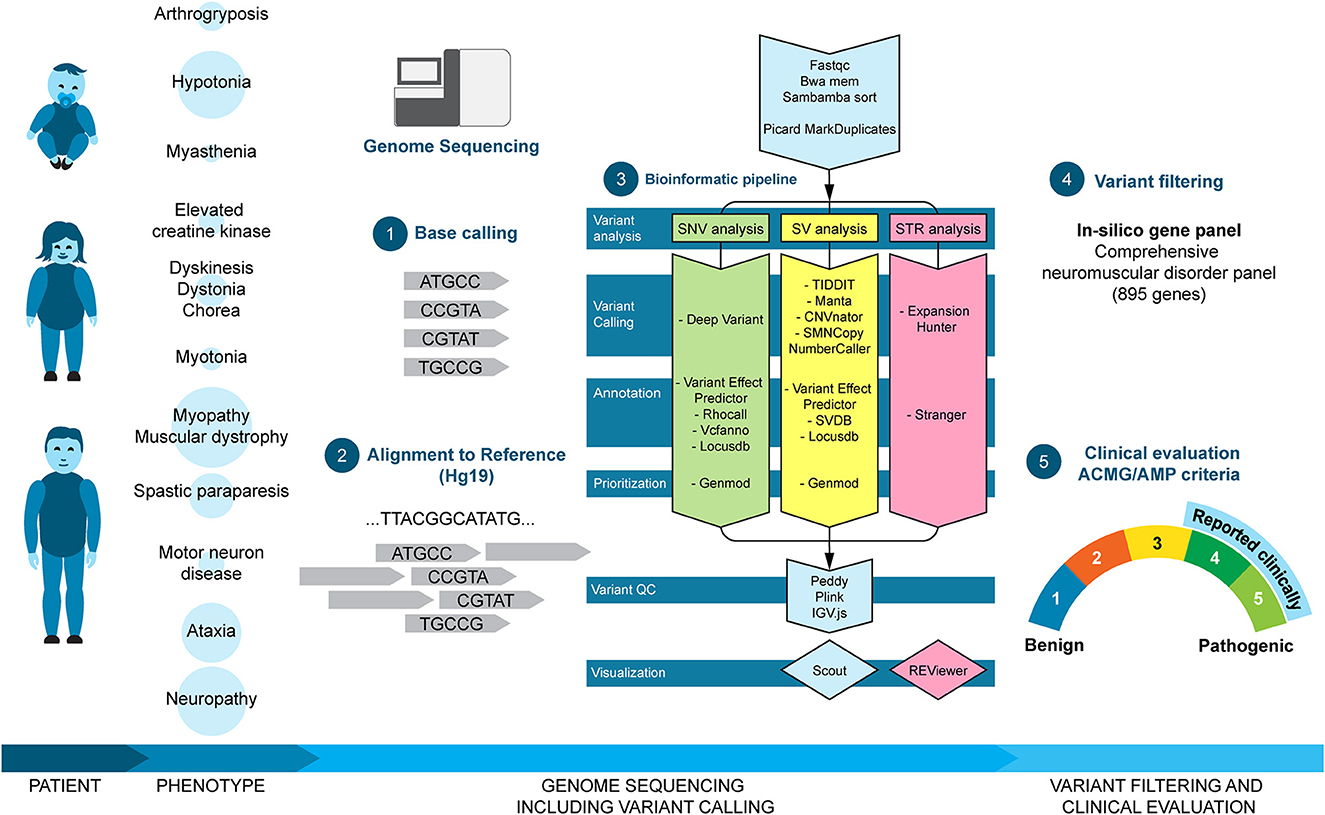
Figure 1. Clinical workflow and bioinformatic pipeline. Patients of different ages were referred for clinical genome sequencing due to neuromuscular disorder phenotypes. The size of the circle behind the phenotype corresponds to the size of the subgroup. Genome sequencing was analyzed using an in-house analysis pipeline including base calling (1), alignment to reference (2), and variant calling and prioritizing (3). Finally, called variants were filtered in silico with the NMD gene panel (4) and classified according to the ACMG/AMP criteria (5). Clinically relevant variants (selected class 3, class 4, and class 5) were reported to the referring doctor.
The workflow of clinical genome sequencing at our center has previously been described in detail (13). For all cases, sequencing reads were mapped to GRCh37 (hg19), and patients were analyzed as singletons. In short, we used an in-house developed analysis pipeline (MIP; https://github.com/Clinical-Genomics/MIP) to detect SNVs, INDELs, and SVs (deletions/duplications and balanced aberrations) in a comprehensive panel of known NMD genes (n = 895) and known pathogenic STR loci (n = 28) (Supplementary Table 1). The panel also included genes associated with ataxia and spastic paraparesis because of a phenotypic overlap and potential clinical difficulty to differentiate between these disorders and pure NMDs. Variants were ranked and visualized in an in-house analysis software (Scout; https://github.com/Clinical-Genomics/scout) (13). Larger SVs (>50 Kb) were visualized in the Cytosure Interpret Software (Oxford Gene Technology) after conversion with a custom program (vcf2cytosure; https://github.com/NBISweden/vcf2cytosure), as described previously (14). SNVs, INDELs, deletions and duplications, and STRs were classified according to the American College of Medical Genetics and Genomics (ACMG)/Association for Molecular Pathology (AMP) guidelines (15) as described previously (16). Class 4 and 5 variants were reported to the referring doctor and are termed as “pathogenic variants.” Moreover, selected class 3 variants, variants of uncertain significance (VUS), were reported either due to a strong and specific overlap between the gene and the patient phenotype or due to compound heterozygosity with a pathogenic variant. STR expansions indicated above normal size (intermediate or pathogenic) by Stranger (https://github.com/Clinical-Genomics/stranger) were inspected using integrative genomics viewer (IGV) for false positive calls. Calls with suspicion of pathogenicity were verified by an alternate method (see Section 2.2). Finally, negative cases with an unspecific or complex phenotype (e.g., hypotonia or additional non-NMD symptoms) and/or a strong suspicion of a genetic disorder (e.g., positive family history) were also screened for known pathogenic variants in a panel including all OMIM disease genes, and reported to the referring doctor, if there was a match between the variant and the patient phenotype (Figure 1). The results were also discussed with the referring doctors, neuropathologists, and neurophysiologists at regular multidisciplinary meetings.
STR expansions detected in the WGS data were verified with a secondary method, either in-house or at an external lab. In-house validated PCR with electrophoresis and a repeat-primed assay (RP-PCR) were used for AR, ATXN2, HTT, and RFC1. Confirmation analysis in-house using commercial kits from Asuragen was performed for C9orf72 (AmplideX® PCR/CE C9orf72 Kit, Asuragen) and DMPK (AmplideX® PCR/CE DMPK Kit, Asuragen). Samples with detected expansions in ATXN7, ATXN8OS, CACNA1A, CNBP, FXN, or PABPN1 were sent to an external lab for PCR with electrophoresis and a repeat-primed assay (RP-PCR) analysis (CENTOGENE, Rostock, Germany).
Small (< 50 Kb) deletions and duplications were verified by MLPA or sequencing (with breakpoint PCR). Large (>50 Kb) deletions and duplications were verified by chromosomal microarray, using a custom 1,80,000 oligonucleotide array, designed with an even distribution across the genome, with ~18 Kb probe spacing (AMADID:031035, Oxford Gene Technology, Begbroke, Oxfordshire, UK). For one individual (case 420), a custom slide with 1 × 1 M oligonucleotides, where probes are designed to cover the exons of 4,645 known disease-causing genes was used (AMADID:068073, Oxford Gene Technology, Begbroke, Oxfordshire, UK). Experiments were performed according to manufacturer's protocol, with some minor changes (14). SNVs with low-quality parameters as well as INDELS were verified by conventional PCR and Sanger sequencing.
Ethical approval was given by the Regional Ethical Review Authority in Stockholm (ethics permit numbers KS 2012/222-31-3, addendum 1 2023-00178-02). The permit allows for clinical samples to be used for the analysis of scientific importance for clinical development. The approval does not require us to obtain written consent for clinical testing. The research followed the principles of the Helsinki Declaration.
The cohort included 861 patients (472 male and 381 female subjects) between the age of 1 day and 88 years (median age 35 years), as well as eight prenatal cases. Pediatric patients (< 20 years) accounted for one-third of the cases. The distribution of age is shown in Figure 2A. All patients were referred for genome sequencing due to clinical findings compatible with a neuromuscular disorder. Based on clinical findings, the cohort was divided into eleven phenotypic subgroups (myopathy/muscle dystrophy, neuropathy, ataxia, myasthenia, asymptomatic elevation of creatine kinase, unspecified hypotonia, dyskinesia/dystonia/chorea, spastic paraparesis, motor neuron disease, myotonia, and arthrogryposis). The subgroup “motor neuron disease” includes patients with spinal muscular atrophy (SMA) and similar disorders, while it excludes those with a diagnosis of amyotrophic lateral sclerosis. Overall, the most common phenotypic subgroup was myopathy/muscular dystrophy (215/861, 25%). However, among pediatric patients, the most common phenotype was unspecified hypotonia (119/301, 40%). The prevalence of all phenotypes, as well as the relative distribution in pediatric and adult patients, is shown in Figure 2B.
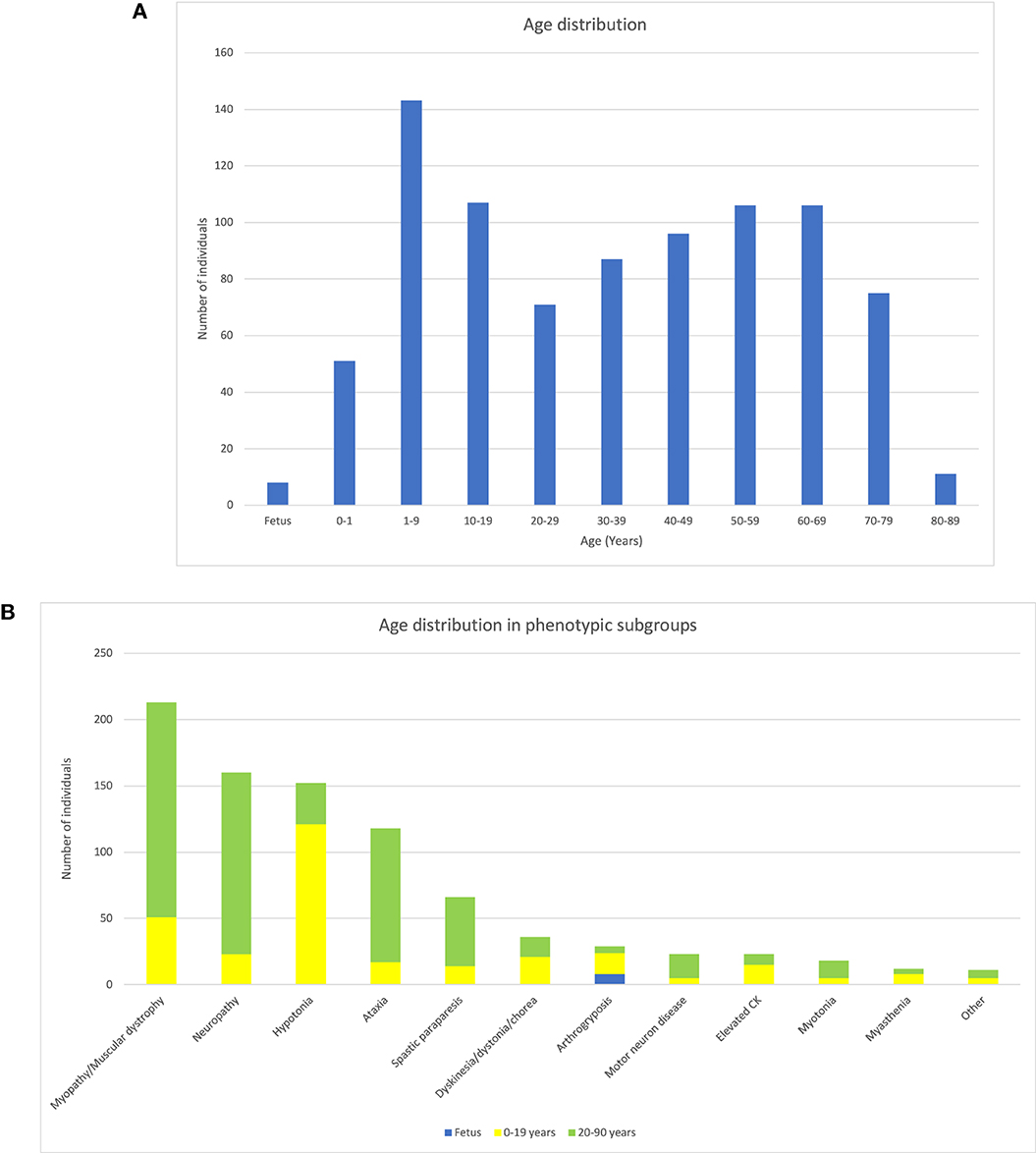
Figure 2. Description of the cohort of NMD patients. (A) Bar chart showing the distribution of patient ages. (B) Bar chart showing the distribution of phenotype subgroups as well as the relation between prenatal cases, pediatric patients, and adult patients in each phenotype group. Patients 0–19 years of age (yellow), ≥20–90 years of age (green), and prenatal cases (blue).
Genome sequencing and analysis of SNV/INDELs in the 895 gene panel detected pathogenic or likely pathogenic variants in 161 out of 861 patients (19%) (Supplementary Table 2). An additional 81 patients (9%) (Supplementary Table 3) harbored variants that were classified as VUSs, while they were still regarded as strong candidates for being causative of the patient's phenotype. The majority of these VUSs were ultra-rare missense variants that were predicted to be damaging and located in genes with a known association with a specific phenotype that matched the patient findings. For clarity, those VUSs are not included when calculating the diagnostic yield.
The genome sequencing data were also analyzed regarding STRs at 28 loci (Supplementary Table 1), with pathogenic repeat expansions detected in 35/861 patients (4%) (Table 1). In these patients, the most prevalent diagnosis was cerebellar ataxia, neuropathy, and vestibular areflexia syndrome (CANVAS, OMIM #614575) due to bi-allelic expansions in RFC1 (13 individuals). Among all individuals with repeat expansions, ataxia was the most common phenotypic finding. However, pathogenic expansions were detected in all phenotypic subgroups, except myasthenia and isolated CK-elevation. Only one pathogenic repeat expansion was found in the pediatric cohort, a 1-year-old girl with progressive hypotonia harbored a pathogenic CAG-repeat in ATXN7, compatible with spinocerebellar ataxia 7 (SCA7, OMIM #164500).
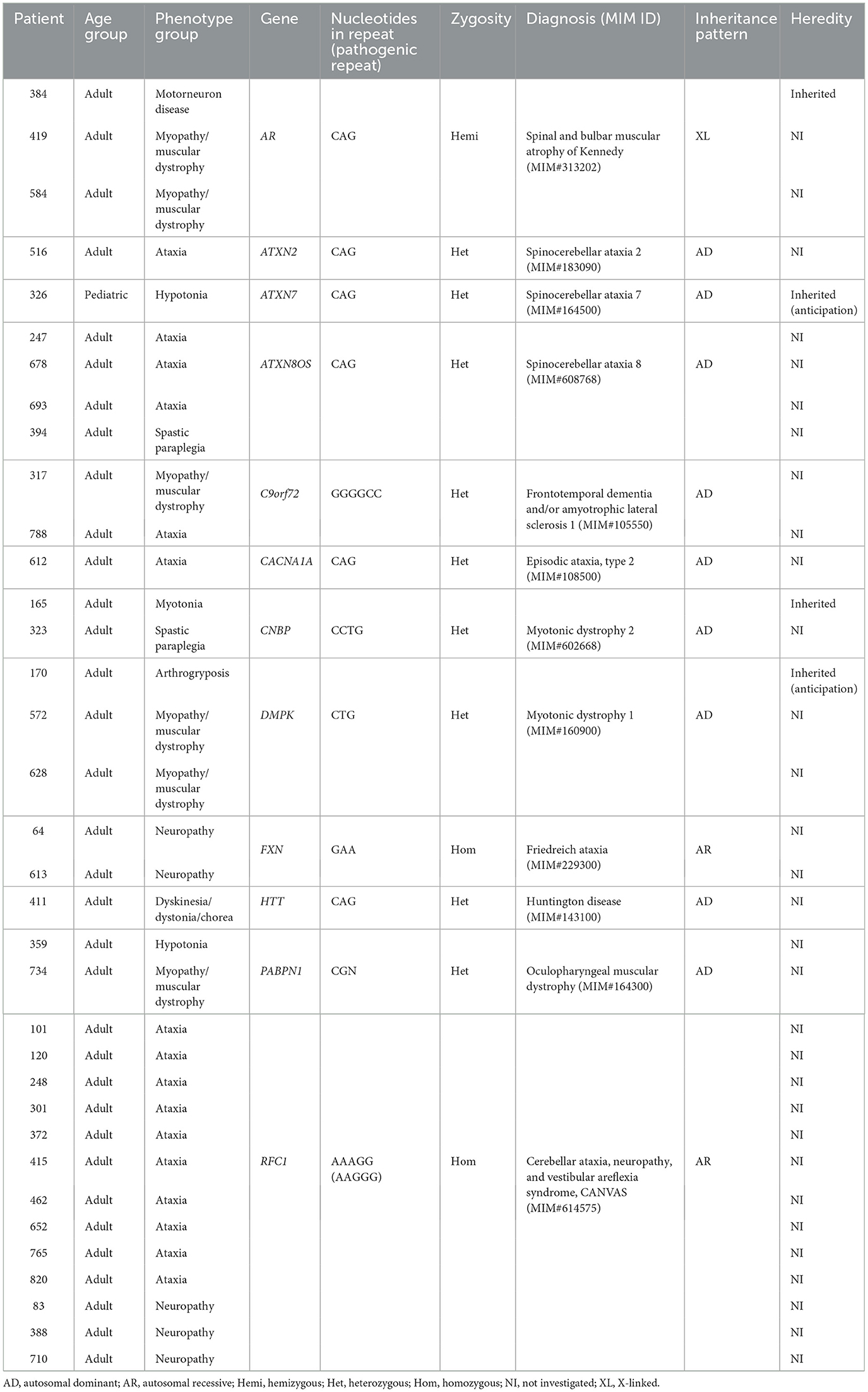
Table 1. Patients with pathogenic expansions of known STR loci.
The analysis of structural variants in the NMD gene panel, including deletion of SMN1 and deletion/duplication of PMP22, identified pathogenic SVs in 36 patients (4%) (Table 2) (sizes ranging from 1.8 Kb to 18.8 Mb). The majority of the variants were intragenic deletions or duplications in DMD (11 individuals). Interestingly, seven of them were female patients with isolated CK-elevation or symptoms such as muscle pain and mild muscle weakness. The second most common gene with pathogenic findings detected by SV-analysis was PMP22, seen in eight patients with Charcot–Marie-Tooth type 1A (CMT1A, OMIM #118220) and one patient with neuropathy, hereditary, with liability to pressure palsies (HNPP, OMIM #162500). Notably, the majority of the CMT1A patients were children. All of them had an affected parent without a previous genetic diagnosis either due to mild symptoms or migration from countries without advanced healthcare. Homozygous deletion of SMN1 was seen in two individuals; a 1-year-old boy and a 52-year-old male, both presented with hypotonia. The number of SMN2 copies in these patients was three and four, respectively, and the phenotypes were classified as SMA type 2 and 4. There were no cases with SMA type 1, likely reflecting that these patients are recognized clinically and confirmed with targeted testing of SMN1, rather than referred for genome sequencing. In total, seven patients harbored rare, non-recurrent SVs in single genes, including CRRPA, DYSF, INF2, and TTBK2 presented in Figure 3. Large SVs, involving several genes, were identified in seven individuals (excluding PMP22) (Table 2).
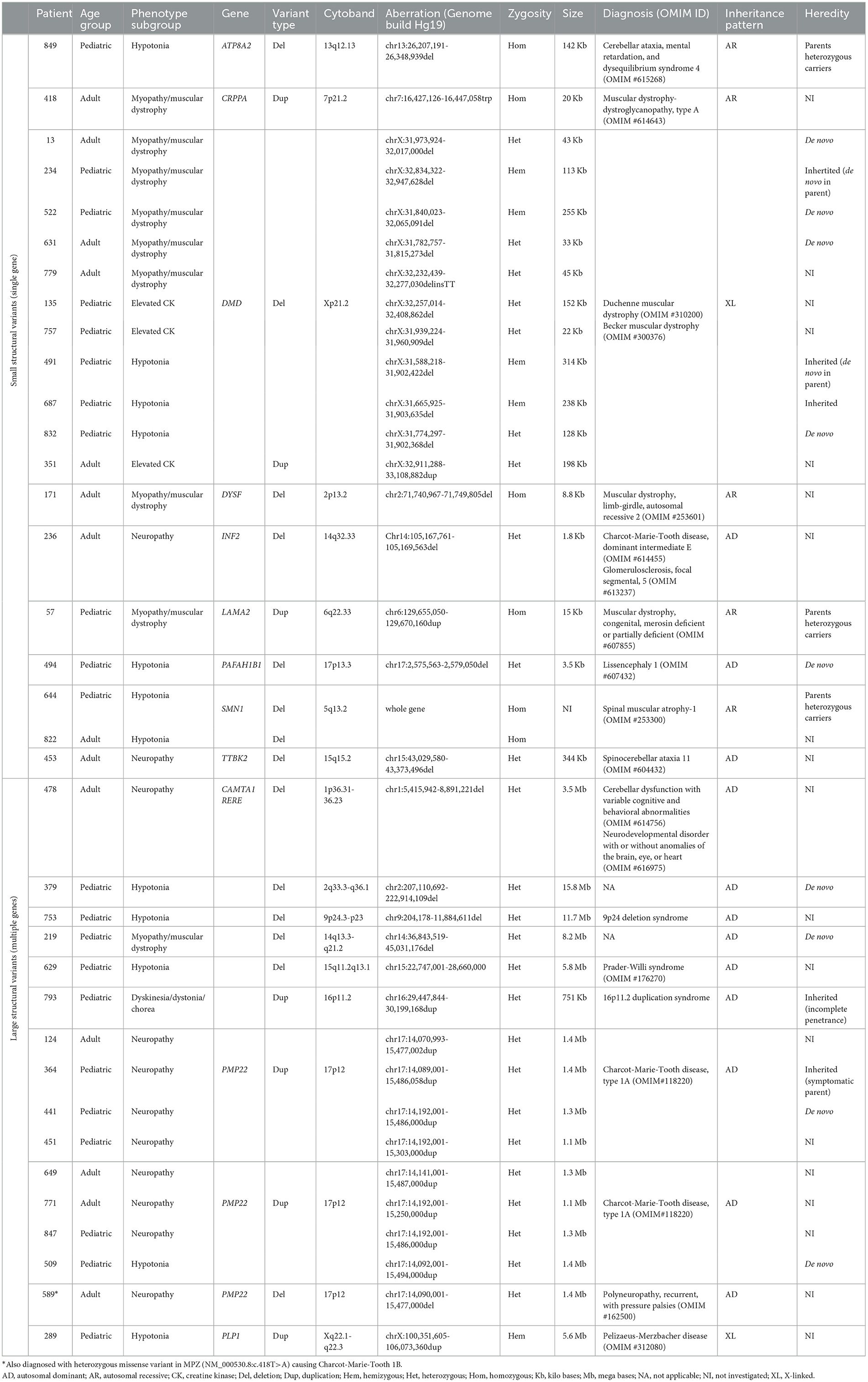
Table 2. Patients with pathogenic and likely pathogenic SVs.
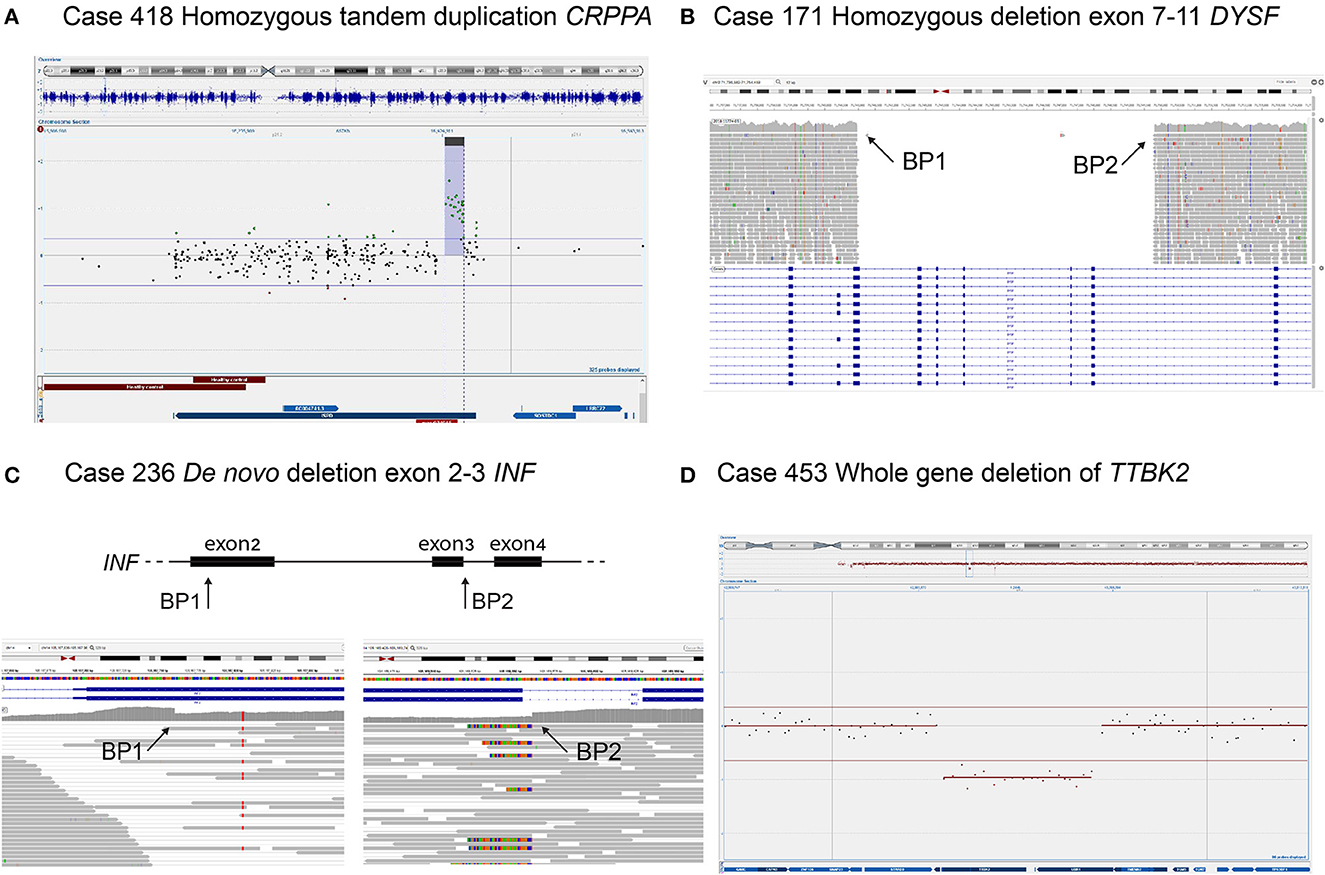
Figure 3. Four individuals with SVs detected in NMD genes. (A) Homozygous tandem duplication of CRRPA in an individual with muscular dystrophy, CK 90, and mild ID. Duplication is shown in copy number variant (CNV) plot from chromosomal microarray with a 1M oligonucleotide slide. (B) Homozygous deletion of exon 7–11 in DYSF in an individual with limb girdle muscular dystrophy and disease onset in their twenties. The deletion is shown in the integrative genomics viewer (IGV) where there are no reads (gray arrows) mapping to the deleted region. (C) De novo deletion of exon 2–3 in INF2 in an individual with the onset of sensory motor neuropathy in early childhood and renal failure before the age of 20 years. The heterozygous deletion is shown in IGV displaying the two breakpoints with a sudden drop and a rise in coverage (arrows), respectively. (D) Whole-gene deletion of TTBK2 in an individual with the onset of ataxia around 40 years of age and sensory-motor neuropathy at nearly 50 years of age. Deletion is shown in CNV plot from chromosomal microarray.
Finally, genome-wide screening for pathogenic variants affecting genes not included in the NMD gene panel resulted in diagnostic findings in an additional 18 patients (Supplementary Table 4). Most patients in this group had early-onset hypotonia and, over time, the majority developed additional symptoms such as intellectual disability (ID) or epilepsy (EP). A recurrent diagnosis in this group was Coffin–Siris syndrome (CSS1, OMIM #135900; CSS2, OMIM #614607) (four individuals). Patients with pathogenic variants in genes outside the NMD gene panel are not included when calculating the diagnostic yield.
Altogether, the diagnostic yield increased from 19% (161/861 patients), with SNV/INDEL analysis in the NMD gene panel, to 27% (232/861 patients) with the addition of STR- and SV-analyses (Figure 4A). The total diagnostic yield was the same in pediatric patients compared to adults. However, the prevalence of pathogenic SVs and STRs differed between the groups, with STRs being more frequent among adult patients, while SVs were more prevalent among pediatric patients (Figures 4B, C).
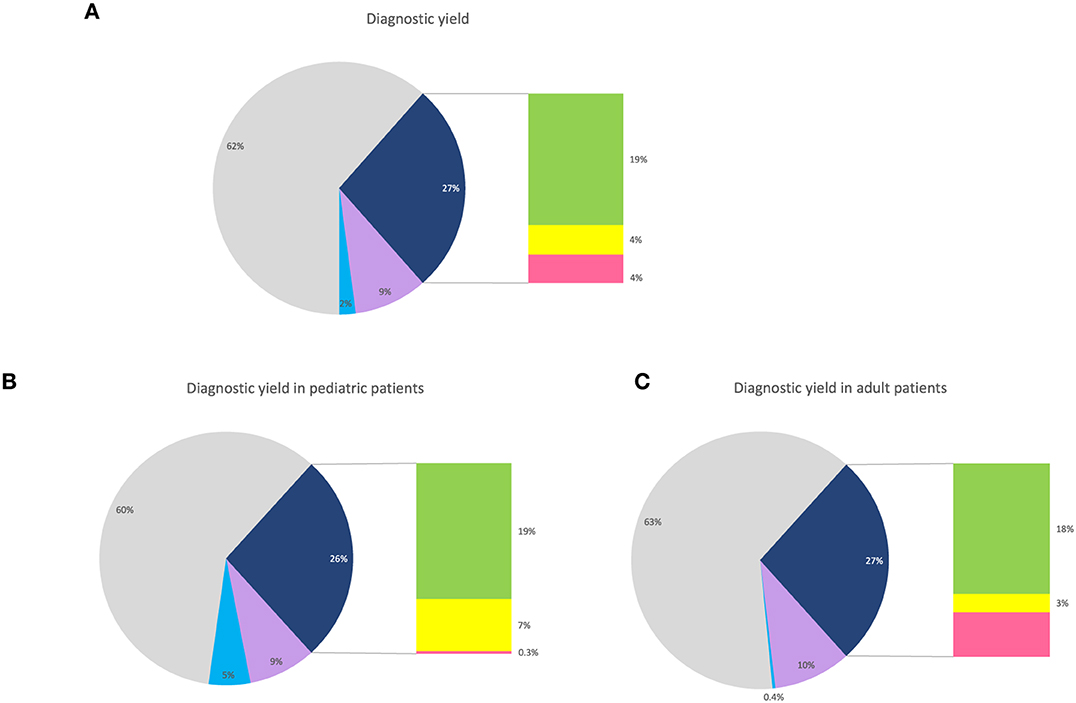
Figure 4. Diagnostic yield. (A) Pie chart showing the percentage of all patients with pathogenic or likely pathogenic findings in NMD genes (dark blue), with a bar chart showing the distribution of SNVs/INDELs (green), SVs (yellow), and STRs (pink). For visualization, patients with VUS in NMD genes (purple) and pathogenic findings in non-NMD genes (light blue). (B) Pie chart showing the percentage of pediatric patients with pathogenic or likely pathogenic findings in NMD genes (dark blue), with a bar chart showing the distribution of SNVs/INDELs (green), SVs (yellow), and STRs (pink). For visualization, patients with VUS in NMD genes (purple) and pathogenic findings in non-NMD genes (light blue). (C) Pie chart showing the percentage of adult patients with pathogenic or likely pathogenic findings in NMD genes (dark blue), with a bar chart showing the distribution of SNVs/INDELs (green), SVs (yellow), and STRs (pink). For visualization, patients with VUS in NMD genes (purple) and pathogenic findings in non-NMD genes (light blue).
In the phenotypic subgroups, the highest overall yield was seen in those with asymptomatic elevation of creatine kinase (12/23, 52%) and those with arthrogryposis (12/30, 40%) (Figure 5). Looking at the yield added by STR- and SV-analysis, in the different phenotype groups, the largest increase in yield by STR-analysis was seen in those with ataxia, while the largest increase in yield by SV-analysis was seen in those with hypotonia (Figure 5).
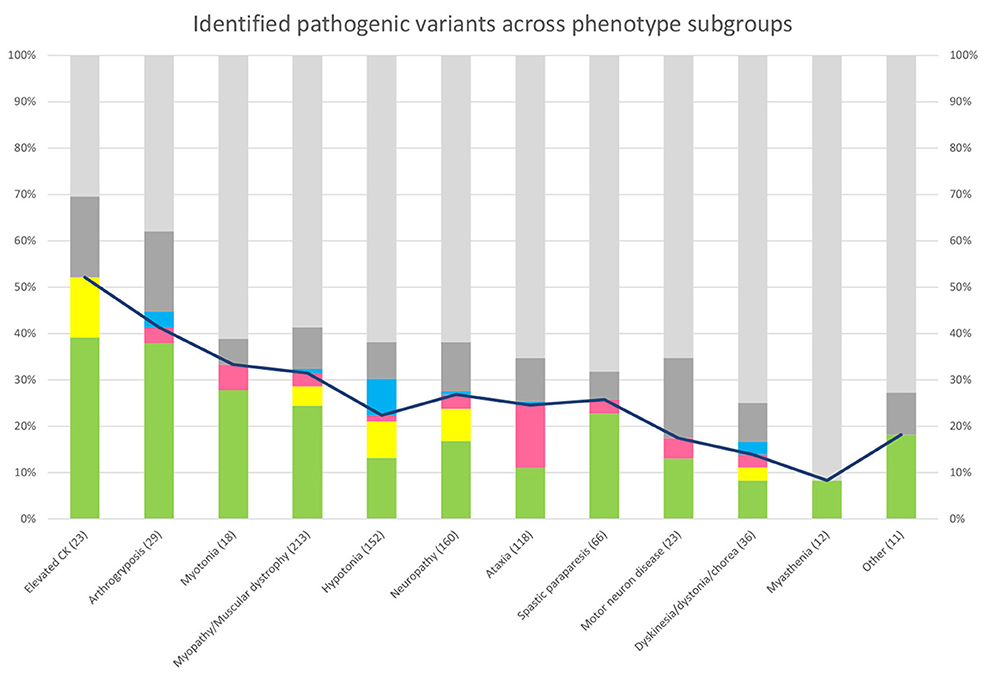
Figure 5. Genetic findings across phenotype subgroups. Bar chart showing the percentage of patients with pathogenic or likely pathogenic SNVs/INDELs (green), SVs (yellow), and STRs (pink). For visualization, patients with pathogenic findings in non-NMD genes (blue) and VUS in NMD genes (dark gray). Line chart showing the diagnostic yield in each phenotypic subgroup.
Despite the proven heterogeneity among NMDs, a few genes were recurrently affected in our cohort: DMD (n = 14), RFC1 (n = 13), RYR1 (n = 9), PMP22 (n = 9), FKRP (n = 8), CAPN3 (n = 7), COL6A1 (n = 7), and DYSF (n = 5). In total, causative variants were detected in 107 genes (among the 232 patients with pathogenic findings) in the NMD gene panel (Supplementary Table 2).
For all patients in the cohort with an established molecular diagnosis, the most common inheritance pattern was autosomal recessive (45%), followed by autosomal dominant (43%) and X-linked (11%). The remaining 1% of diagnoses had a digenic or mitochondrial inheritance pattern. In pediatric patients, inheritance from the parents was examined. Parental samples were analyzed for 35 of the 50 patients harboring pathogenic variants in genes associated with autosomal dominant disorders. This resulted in 26 variants found to be de novo, and nine were inherited from either a healthy parent (n = 4) or a symptomatic parent (n = 5). The variants that were inherited from a healthy parent were all associated with disorders known to have reduced penetrance or anticipation. For patients with bi-allelic autosomal recessive variants, 23 of the 32 families were followed up, revealing 21 individuals with both parents being healthy heterozygous carriers. For the additional two individuals, one carried a paternally inherited variant in combination with a de novo variant, and one individual had a father affected by the same disorder as the child and compound heterozygous for the same variants in ANO5 as the child. In the latter case, the mother carried a relatively common founder mutation in ANO5. The parents were unrelated. For the variants with X-linked inheritance, maternal samples for 12 of 14 variants were investigated, eight were found to be de novo, and four inherited from the mother (Table 2; Supplementary Table 2). The individuals who inherited the variant were all boys and thereby hemizygous for the pathogenic variant.
In this study, 861 patients with neuromuscular disorders were analyzed with genome sequencing and comprehensive variant calling. The overall diagnostic yield for pathogenic or likely pathogenic variants was 27% (232/861 patients). Previous studies have shown a diagnostic yield for SNVs/INDELs in NMD patients, varying between 13 and 53% (3–10). One explanation for the variable figures is differences in calculating yield. In our study, we report only pathogenic and likely pathogenic variants according to ACMG classification (15), while many studies with higher yield have included variants of unknown significance. Furthermore, inclusion criteria affect outcome. In the present study, inclusion criteria were set to reflect the clinical landscape, with the inclusion of all patients referred for genome sequencing. This may have affected outcome as not all patients had an a priori strong suspicion of a genetic disorder.
Remarkably, out of all pathogenic and likely pathogenic NMD variants in our cohort, one-third of them were repeat expansions or deletions/duplications.
By applying STR- and SV- callers to the genome sequencing data, 71 patients received a molecular diagnosis. Some of these patients might have reached an etiological diagnosis through subsequent targeted genetic tests. However, requests for targeted tests are often based on thorough clinical investigations, such as muscle biopsies, neurophysiological assessments, and MRI of muscles and/or the CNS. This approach is cost- and time-consuming, and it may lead to a delay in a definite diagnosis for the patient. Furthermore, for most of the patients with pathogenic STRs and SVs, the specific diagnosis was difficult to pinpoint prior to genetic testing despite a meticulous clinical workup.
In our cohort, as described above, the major diagnostic advantage of genome sequencing over targeted gene panels or exome sequencing was the potential of adding bioinformatic tools for STR- and SV-analysis. In addition, genome sequencing offered the possibility to analyze deep intronic variants, exemplified by two de novo variants in COL6A1 (c.930 + 189C > T) and one homozygous variant in DYSF (c.5785-824C > T), detected in the cohort. Moreover, genome sequencing (like exome sequencing) also offers the possibility to analyze genes not included in the primary gene list. The NMD gene list applied in this study is a broad panel based on the expanded definition of neuromuscular disorders to include also genes associated with ataxia and spastic paraparesis. Despite using a comprehensive gene list, a fraction of the patients with a presumed neuromuscular disorder were diagnosed with genetic disorders due to variants in genes outside the panel. The vast majority of these patients were children with hypotonia or movement disorders, in whom pathogenic variants in genes associated with ID- or EP-syndromes were detected. These results illustrate the difficulty in applying gene panels based on disease groups and favor an even broader gene panel, particularly for children with unspecific phenotypes such as hypotonia.
In summary, this study of genome sequencing and comprehensive variant calling in 861 patients with NMDs is the largest study to date. The main finding of the study shows that one-third of the patients with pathogenic findings in the NMD gene panel have a repeat expansion or a deletion/duplication as the cause of their phenotype. This finding leads us to conclude that it is crucial to include bioinformatic pipelines for STR- and SV-analysis in massive parallel sequencing diagnostics for patients with neuromuscular disorders.
The datasets presented in this article are not readily available because of ethical and privacy restrictions. Requests to access the datasets should be directed to the corresponding author.
The studies involving human participants were reviewed and approved by the Regional Ethical Review Authority in Stockholm, Sweden. Written informed consent from the participants' legal guardian/next of kin was not required to participate in this study in accordance with the national legislation and the institutional requirements.
Conceptualizing was done by AL, MK, and TS. The contribution of samples and phenotyping was done by GS, HHj, IN, KN, MEn, MPa, RP, and SA. Data curation was done by AL, AH, B-MA, DN, HM, HT, HHe, MEk, MK, and MPe. Formal analysis was done by AL, MEk, and MK. Funding acquisition and project administration was done by AL and MK. Visualization and writing of the first draft were done by MEk and MK. Writing—review and editing were done by AL, AH, B-MA, DN, GS, HM, HT, HHe, HHj, IN, KN, MEn, MEk, MK, MPa, MPe, RP, SA, and TS. All authors contributed to the article and approved the submitted version.
This research was funded by Swedish Research Council, grant number 2019-02078; the Swedish Brain Foundation, grant number FO2022-0256; the Stockholm Regional Council (ALF funding); and the Swedish Rare Diseases Research Foundation (Sällsyntafonden).
Karolinska membership of EURO-NMD and ERN-ITHACA is gratefully acknowledged.
AL has received honoraria from Illumina Inc., and has been an advisor for Oxford Nanopore Technologies, both unrelated to the content in this article.
The remaining authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fneur.2023.1170005/full#supplementary-material
1. Cohen E, Bonne G, Rivier F, Hamroun D. The 2022 version of the gene table of neuromuscular disorders (nuclear genome). Neuromuscul Disord. (2021) 31:1313–57. doi: 10.1016/j.nmd.2021.11.004
2. Dalil Hamroun GB, Rivier R. Gene Table of Neuromuscular Disorders. (2023). Available from: https://www.musclegenetable.fr (accessed November 2, 2022).
3. Tsang MHY, Chiu ATG, Kwong BMH, Liang R, Yu MHC, Yeung KS, et al. Diagnostic value of whole-exome sequencing in Chinese pediatric-onset neuromuscular patients. Mol Genet Genomic Med. (2020) 8:e1205. doi: 10.1002/mgg3.1205
4. Haskell GT, Adams MC, Fan Z, Amin K, Guzman Badillo RJ, Zhou L, et al. Diagnostic utility of exome sequencing in the evaluation of neuromuscular disorders. Neurol Genet. (2018) 4:e212. doi: 10.1212/NXG.0000000000000212
5. Laquerriere A, Jaber D, Abiusi E, Maluenda J, Mejlachowicz D, Vivanti A, et al. Phenotypic spectrum and genomics of undiagnosed arthrogryposis multiplex congenita. J Med Genet. (2022) 59:559–67. doi: 10.1136/jmedgenet-2020-107595
6. Topf A, Johnson K, Bates A, Phillips L, Chao KR, England EM, et al. Sequential targeted exome sequencing of 1001 patients affected by unexplained limb-girdle weakness. Genet Med. (2020) 22:1478–88. doi: 10.1038/s41436-020-0840-3
7. Reddy HM, Cho KA, Lek M, Estrella E, Valkanas E, Jones MD, et al. The sensitivity of exome sequencing in identifying pathogenic mutations for LGMD in the United States. J Hum Genet. (2017) 62:243–52. doi: 10.1038/jhg.2016.116
8. Ghaoui R, Cooper ST, Lek M, Jones K, Corbett A, Reddel SW, et al. Use of whole-exome sequencing for diagnosis of limb-girdle muscular dystrophy: outcomes and lessons learned. JAMA Neurol. (2015) 72:1424–32. doi: 10.1001/jamaneurol.2015.2274
9. Harris E, Topf A, Barresi R, Hudson J, Powell H, Tellez J, et al. Exome sequences versus sequential gene testing in the UK highly specialised service for limb girdle muscular dystrophy. Orphanet J Rare Dis. (2017) 12:151. doi: 10.1186/s13023-017-0699-9
10. Benkirane M, Marelli C, Guissart C, Roubertie A, Ollagnon E, Choumert A, et al. High rate of hypomorphic variants as the cause of inherited ataxia and related diseases: study of a cohort of 366 families. Genet Med. (2021) 23:2160–70. doi: 10.1038/s41436-021-01250-6
11. Rajan-Babu IS, Peng JJ, Chiu R, Study I, Study C, Li C, et al. Genome-wide sequencing as a first-tier screening test for short tandem repeat expansions. Genome Med. (2021) 13:126. doi: 10.1186/s13073-021-00961-4
12. Ibañez K, Polke J, Hagelstrom RT, Dolzhenko E, Pasko D, Thomas ERA, et al. Whole genome sequencing for the diagnosis of neurological repeat expansion disorders in the UK: a retrospective diagnostic accuracy and prospective clinical validation study. Lancet Neurol. (2022) 21:234–45. doi: 10.1016/S1474-4422(21)00462-2
13. Stranneheim H, Lagerstedt-Robinson K, Magnusson M, Kvarnung M, Nilsson D, Lesko N, et al. Integration of whole genome sequencing into a healthcare setting: high diagnostic rates across multiple clinical entities in 3219 rare disease patients. Genome Med. (2021) 13:40. doi: 10.1186/s13073-021-00855-5
14. Lindstrand A, Eisfeldt J, Pettersson M, Carvalho CMB, Kvarnung M, Grigelioniene G, et al. From cytogenetics to cytogenomics: whole-genome sequencing as a first-line test comprehensively captures the diverse spectrum of disease-causing genetic variation underlying intellectual disability. Genome Med. (2019) 11:68. doi: 10.1186/s13073-019-0675-1
15. Richards S, Aziz N, Bale S, Bick D, Das S, Gastier-Foster J, et al. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet Med. (2015) 17:405–24. doi: 10.1038/gim.2015.30
Keywords: neuromuscular disorders, genome sequencing, single nucleotide variant, structural variant, repeat expansion, ataxia
Citation: Ek M, Nilsson D, Engvall M, Malmgren H, Thonberg H, Pettersson M, Anderlid B-M, Hammarsjö A, Helgadottir HT, Arnardottir S, Naess K, Nennesmo I, Paucar M, Hjartarson HT, Press R, Solders G, Sejersen T, Lindstrand A and Kvarnung M (2023) Genome sequencing with comprehensive variant calling identifies structural variants and repeat expansions in a large fraction of individuals with ataxia and/or neuromuscular disorders. Front. Neurol. 14:1170005. doi: 10.3389/fneur.2023.1170005
Received: 20 February 2023; Accepted: 21 April 2023;
Published: 18 May 2023.
Edited by:
Bo Yuan, Baylor College of Medicine, United StatesReviewed by:
Henry Houlden, University College London, United KingdomCopyright © 2023 Ek, Nilsson, Engvall, Malmgren, Thonberg, Pettersson, Anderlid, Hammarsjö, Helgadottir, Arnardottir, Naess, Nennesmo, Paucar, Hjartarson, Press, Solders, Sejersen, Lindstrand and Kvarnung. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Marlene Ek, marlene.ek.2@ki.se
†These authors share last authorship
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.