 Wen-Rong Ge
Wen-Rong Ge Pei-Pei Fu1
Pei-Pei Fu1 Bo Zhang
Bo Zhang Guang Yang
Guang Yang
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
CASE REPORT article
Front. Neurol., 25 April 2023
Sec. Pediatric Neurology
Volume 14 - 2023 | https://doi.org/10.3389/fneur.2023.1163803
This article is part of the Research TopicMalformations of cortical development: clinico-radiological features and genetic causesView all 4 articles
Background: Mutations in the dynein cytoplasmic 1 heavy chain 1 (DYNC1H1) gene are linked to malformations of cortical development (MCD), which may be accompanied by central nervous system (CNS) manifestations. Here, we present the case of a patient with MCD harboring a variant of DYNC1H1 and review the relevant literature to explore genotype-phenotype relationships.
Case presentation: A girl having infantile spasms, was unsuccessfully administered multiple antiseizure medications and developed drug-resistant epilepsy. Brain magnetic resonance imaging (MRI) at 14 months-of-age revealed pachygyria. At 4 years-of-age, the patient exhibited severe developmental delay and mental retardation. A de novo heterozygous mutation (p.Arg292Trp) in the DYNC1H1 gene was identified. A search of multiple databases, including PubMed and Embase, using the search strategy DYNC1H1 AND [malformations of cortical development OR seizure OR intellectual OR clinical symptoms] up to June 2022, identified 129 patients from 43 studies (including the case presented herein). A review of these cases showed that patients with DYNC1H1-related MCD had higher risks of epilepsy (odds ratio [OR] = 33.67, 95% confidence interval [CI] = 11.59, 97.84) and intellectual disability/developmental delay (OR = 52.64, 95% CI = 16.27, 170.38). Patients with the variants in the regions encoding the protein stalk or microtubule-binding domain had the most prevalence of MCD (95%).
Conclusion: MCD, particularly pachygyria, is a common neurodevelopmental disorder in patients with DYNC1H1 mutations. Literature searches reveales that most (95%) patients who carried mutations in the protein stalk or microtubule binding domains exhibited DYNC1H1-related MCD, whereas almost two-thirds of patients (63%) who carried mutations in the tail domain did not display MCD. Patients with DYNC1H1 mutations may experience central nervous system (CNS) manifestations due to MCD.
The dynein cytoplasmic 1 heavy chain 1 (DYNC1H1) gene encodes a large (530 kDa) component of the multisubunit dynein motor complex that mediates retrograde axonal transport and other intracellular processes. These processes include Golgi apparatus regulation, cargo transport, axonal transport, spindle pole organization, organelle motility, and nuclear migration throughout mitosis (1, 2). DYNC1H1 mutations are associated with various clinical manifestations that are typically divided into two major categories: central nervous system (CNS) symptoms (e.g., malformations of cortical development [MCD], intellectual developmental disorder, and developmental and epileptic encephalopathy [DEE]) (3–8); and neuromuscular diseases (e.g., spinal muscular atrophy [SMA] with predominance in the lower extremities and Charcot–Marie–Tooth [CMT], type 20 (1, 2, 9, 10).
The DYNC1H1 protein has four domains: the tail (amino acid [aa] residues 1–1373 and 4222–4646); the ATP-binding AAA motor domain (residues 1868–3168 and 3553–4221); the linker (residues 1374–1867); and the stalk or microtubule-binding domain (MBD; residues 3169–3552) (8). With a gene constraint Z score of 10.97 in the Genome Aggregation Database(gnomAD), DYNC1H1 is known to be exceptionally susceptible to missense mutations (11), which account for most of the DYNC1H1 variants identified to date (11). It remains unclear as to whether any association exists between the location of mutations in the DYNC1H1 gene and the complex and variable clinical symptoms observed in individuals harboring these variants.
MCDs are structural abnormalities that impede normal cortical development, including microcephaly, macrocephaly, brain overgrowth spectrum, focal cortical dysplasia, grey matter heterotopia, lissencephaly, cobblestone malformation, polymicrogyria, radiological spectrum of cobblestone malformation and polymicrogyria, schizencephaly, and dysgyria (12). Patients with MCDs may be asymptomatic or exhibit a range of symptoms, including cognitive impairment, developmental delay, and epilepsy with drug resistance (13). MCD is a common(28%) condition associated with DYNC1H1 mutations (14). However, the association between MCD and clinical symptoms—especially those affecting the CNS—has yet to be comprehensively evaluated in individuals with DYNC1H1 mutations.
In this study, we present the case of a child with MCD owing to a missense mutation in the DYNC1H1 gene and review the literature to explore the association between the location of gene variants and MCD or other clinical symptoms.
The case was a 4-year-old girl and is the only child of unrelated and healthy parents without a family history of neurological disorders. The patient was born at term by spontaneous delivery and exhibited no postnatal abnormalities. The onset of seizures as epileptic spasms began at the age of 5 months. At this time, electroencephalography (EEG) revealed hypsarrhythmia, thus leading to a diagnosis of infantile spasms. Adrenocorticotropic hormone and multiple antiseizure medications, including vigabatrin, failed to control the seizures. A ketogenic diet and vagus nerve stimulation were ineffective at treating the condition, and spasms began when the child was 2 years-of-age (Figures 1A, B). Over time, the type of seizure evolved into absence seizures (confirmed by EEG). The patient exhibits severe developmental delay, with independent sitting at 9 months, and remains unable to walk unassisted. These symptoms were accompanied by severe intellectual disabilities and poor language skills, with a vocabulary consisting of only 10 words.
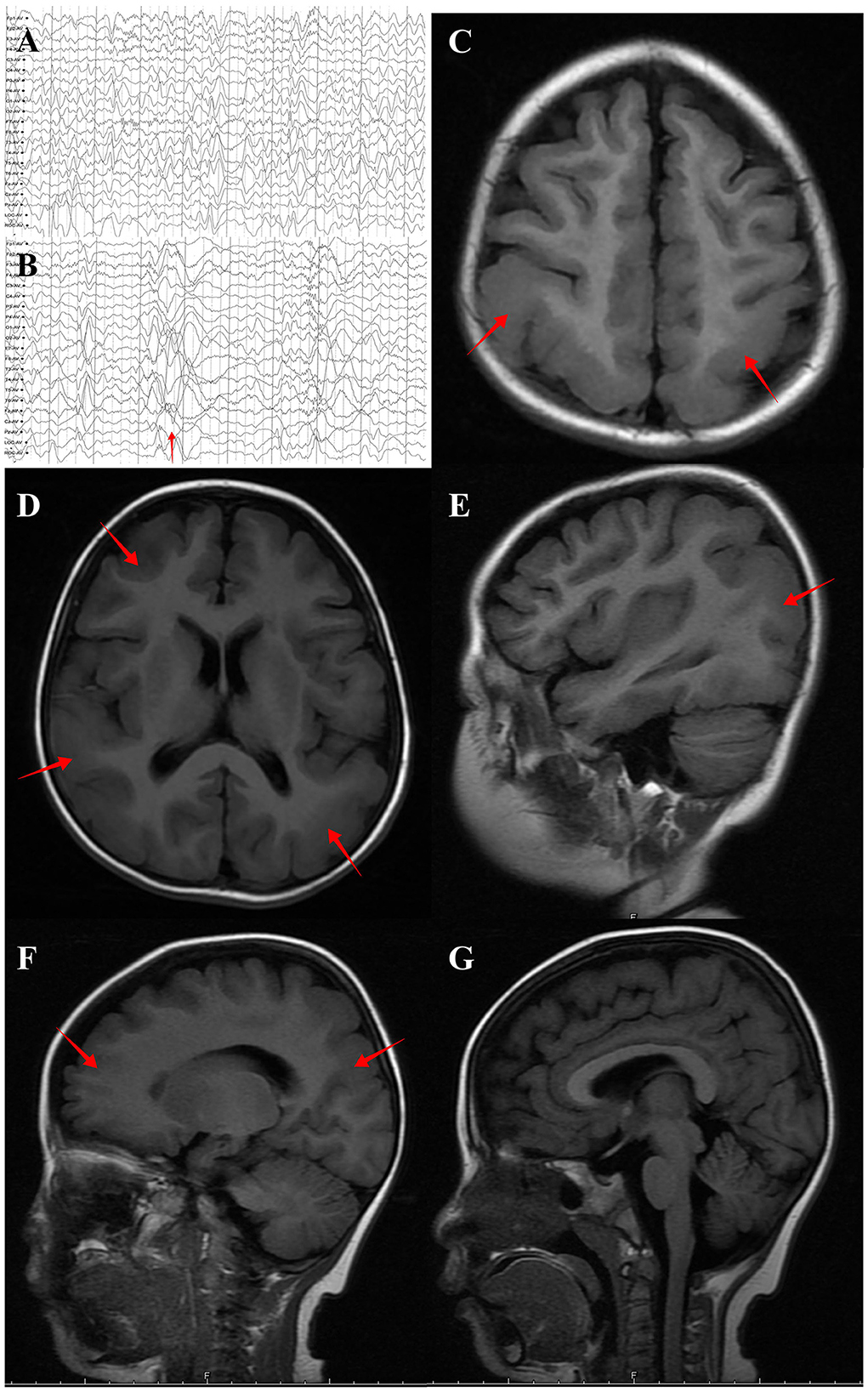
Figure 1. Brain MRI and EEG of the individual with DYNC1H1 variant p.Arg292Trp. (A) Interictal EEG showed multifocal spike-wave complexes at 2 years old. (B) EEG at spasm onset (2 years old) revealed brief, high γ oscillations combined with slow delta waves (red arrow). (C) (Axial plane, T1 flair), (D) (Axial plane, T1 flair), (E) (Sagittal plane, T1 flair), (F) (Sagittal plane, T1 flair) and (G) (Sagittal plane, midline, T1 flair). MRI performed at 4 years old and revealed posterior predominant pachygyria (red arrow).
Brain magnetic resonance imaging (MRI) revealed small, broad, and flat gyri in the bilateral multiple lobes, indicating pachygyria (Figures 1C–F). Genomic DNA of the proband and her parents was extracted from peripheral blood for trio-whole-exome-sequencing (WES), using a method similar to that used in our previous study (15).The results revealed a de novo heterozygous variant (p.Arg292Trp) in the DYNC1H1 gene. In silico analysis using PolyPhen-2 and MutationTaster further indicated that the p.Arg292Trp mutation was “probably damaging,” which was consistent with the PROVEAN server data; the variant was categorized as probably pathogenic according to the American College of Medical Genetics and Genomics guidelines (Supplementary Figure S1). Function- and stability-change predictions were performed as previously described (16, 17). The results showed that this variant caused significant damage to the encoded protein (Figure 2).
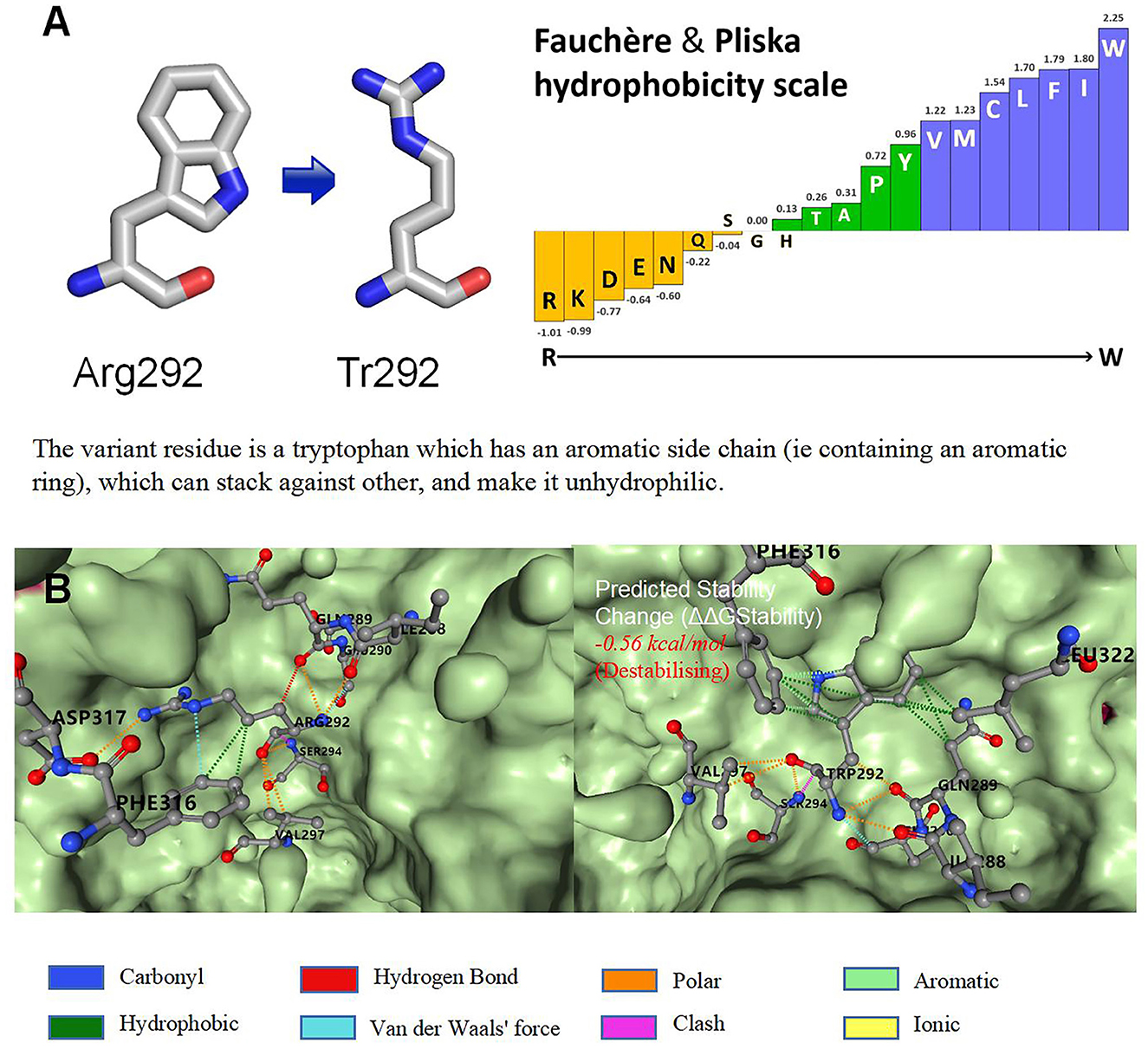
Figure 2. Function and stability prediction of variants. (A) Prediction of p.Arg292Trp function. (B) Prediction of variant stability.
Relevant studies were identified from the existing literature by two independent reviewers (WG and PF, two pediatric neurologists with more than 10 years of experience) by searching MEDLINE, PubMed, Embase, Google, and the China National Knowledge Infrastructure. Most studies on DYNC1H1 have found that most mutations are missense mutations, while mutations that clearly result in protein truncation, such as frameshift or nonsense mutations, are relatively rare (14). At present, the pathogenic mechanism of DYNC1H1 is not fully understood. Animal study has shown that heterozygosity for a null mutation in DYNC1H1 does not cause overt phenotypes in mice (18). In light of this, we only included patients carrying missense mutations in the present study. The search strategy utilized was as follows: DYNC1H1 AND/OR missense AND [malformations of cortical development OR seizure OR intellectual OR clinical symptoms] until June 2022. The search results were restricted to studies in humans that were published in either English or Chinese. To ensure an accurate diagnosis of MCD, we excluded subjects without brain MRI reports.
SPSS v26.0 (IBM, Armonk, NY, USA) and Prism (GraphPad, La Jolla, CA, USA) software were used for statistical analysis. Data were analyzed with the chi-squared test. A logistic regression model was used to identify risk factors for MCD. Differences with p < 0.05 were considered statistically significant.
In total, we identified 129 cases (including the patient reported in the current study) with DYNC1H1 missense variants and neurologic/developmental phenotypes from 43 studies (3–5, 8–10, 14, 19–54). Among them, there were 71 individuals with MCD (55%), 81 (63%) with intellectual disability/developmental delay (ID/DD), and 59 (46%) with epilepsy (Supplementary Table S1; Figure 3A). The proportion of patients with epilepsy was higher in the MCD group than among those without MCD; the same was true for patients with vs. without ID/DD (Table 1). MCD was associated with an elevated risk of epilepsy (odds ratio [OR] = 33.67, 95% confidence interval [CI] = 11.59, 97.84) or ID/DD (OR = 52.64, 95% CI = 16.27, 170.38) (Table 1). In these MCD patients, 58 cases reported the details of their MRI, 45 had pachygyria(one as agyria)(45/58, 78%), and gradient details were available for 35 patients; most of them (30/35, 86%) were posterior predominant, while the remaining five patients were anterior predominant. Of the 45 patients with pachygyria, 41 had epilepsy(41/45, 91%). In addition, 15 patients had hypoplasia of the corpus callosum(15/58, 26%), 11 had polymicrogyria(11/58, 19%) and seven among them were frontal (7/11, 64%) (Supplementary Table S1; Figure 3A). Of the 51 patients for which the time of seizure onset was reported, 37 had an onset in infancy(37/51, 73%). Of the 22 patients with known seizure outcomes, 18 developed drug-resistant epilepsy(18/22, 82%). Patients with pachygyria had a higher proportion of epilepsy and ID/DD when compared to those without MCD (Table 1). Pachygyria was associated with an elevated risk of epilepsy (odds ratio [OR] = 108.65, 95% confidence interval [CI] = 27.43, 430.38) or ID/DD (OR = 44.00, 95% CI = 11.79, 164.17) (Table 1). Compared with patients with other subtype of MCD(n=13), patients with pachygyria had a higher proportion and an elevated risk of epilepsy(OR = 11.96, 95% CI = 2.67, 53.47) (Table 1).
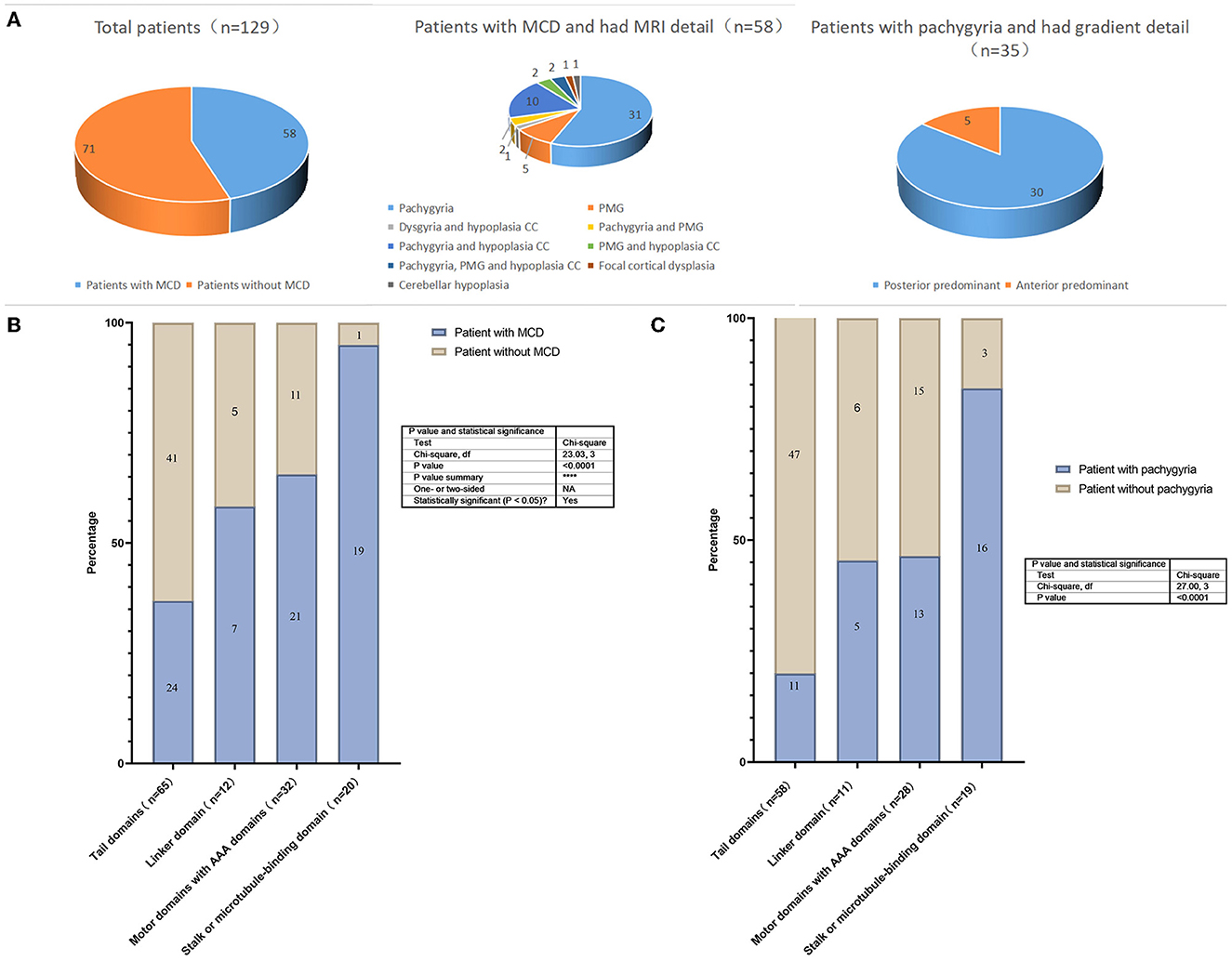
Figure 3. (A) The proportion of malformations of cortical development and subtype in the patients with DYNC1H1 mutations; (B) Prevalence of MCD. Prevalence of different variants in the 4 domains of the DYNC1H1 protein; (C) Prevalence of pachygyria. Prevalence of different variants in the 4 domains of the DYNC1H1 protein. PMG, polymicrogyria; CC, corpus callosum.
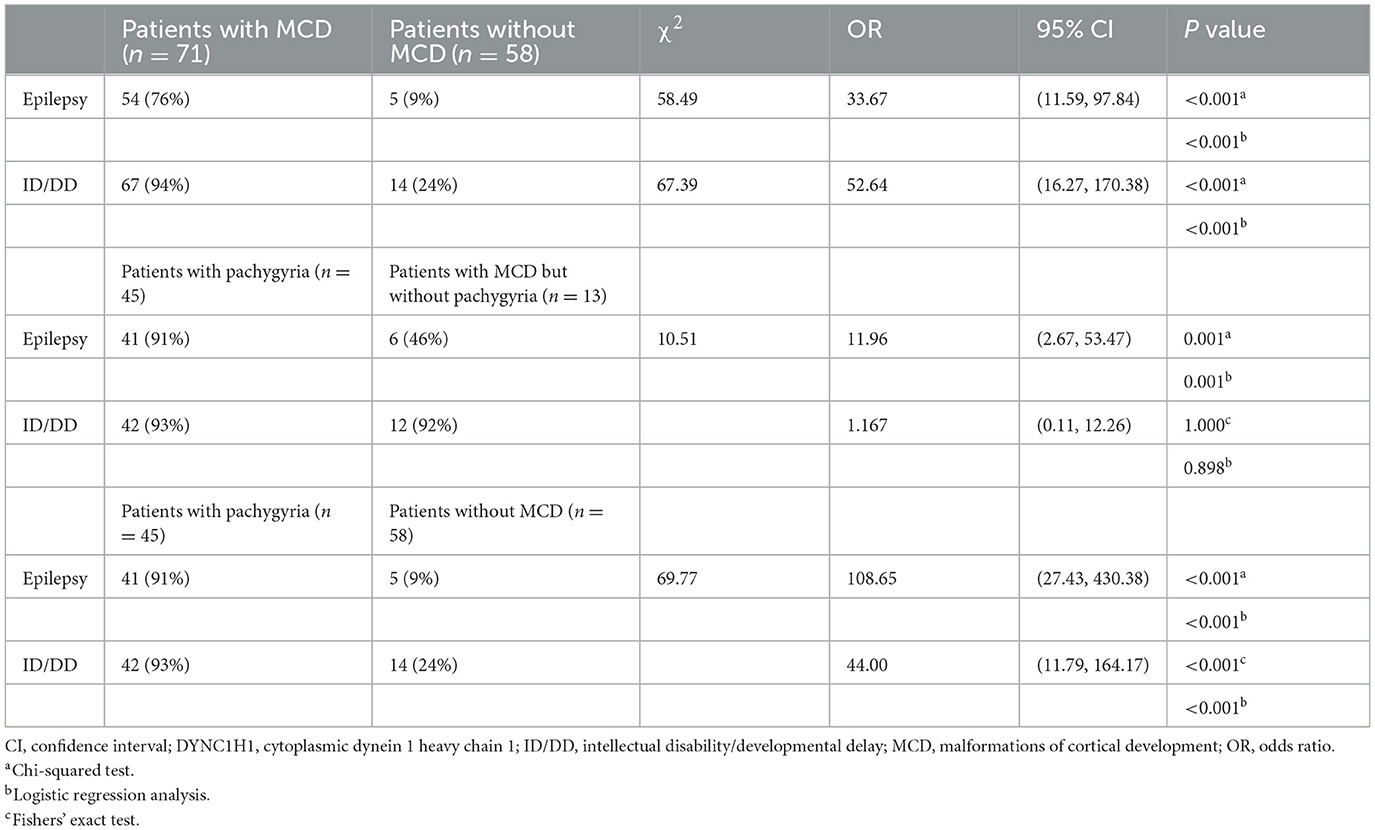
Table 1. Comparison of central nervous system disorders in patients with DYNC1H1 variants with or without malformations of cortical development and with or without pachygyria.
There were 93 mutations identified in the 129 individuals: 44 mutations were located in the tail domain (44/93, 47%), 10 in the linker domain (10/93, 11%), 25 in the motor (AAA) domain (25/93, 27%), and 14 in the stalk/MBD(14/93, 15%). There were statistically significant differences in the prevalence of MCD prevalence according to the domain in which the DYNC1H1 variant was located. The highest prevalence was observed in patients with variants in the stalk/MBD (95%; Figure 3B). The same phenomenon has also been reported in children with pachygyria in that the highest prevalence was observed in patients with variants in the stalk/MBD (84%, Figure 3C).
DYNC1H1-related disorders are heterogeneous and can affect the development and function of the CNS, peripheral nervous system (PNS), or both (14). Historically, more attention has been given to PNS diseases such as SMA or CMT than CNS manifestations. In this study, we reported a case of a DYNC1H1 variant presenting with CNS symptoms, reviewed the literature on DYNC1H1 mutations in MCD, and identified a higher prevalence of MCD than previously reported (14). We suspect that this discrepancy may be because we excluded subjects without brain MRI data. Although DYNC1H1 is widely expressed in different tissues, our multi-databases searches (e.g., Genotype-Tissue Expression, Human Protein Atlas, Functional Annotation of the Mammalian Genome, and ConsensusPathDB) indicated that DYNC1H1 expression may be higher in brain and muscle tissues, especially in the cerebral cortex and skeletal muscles. The DYNC1H1 protein is also involved in neuron-specific processes, specifically retrograde axonal transport. Cytoplasmic DYNC1H1 is an essential component of the cytoplasmic dynamic protein complex and is upregulated during normal development of the nervous system (2, 55). In a previous study, Hoang et al. suspected that the mutations found in DYNC1H1 and associated with disease may have either a dominant-negative or dominant gain-of-function effect (56). Their study showed that these mutations strongly inhibited the gliding of microtubules by dyneins immobilized on surfaces, compromised the activation of processive dynein movement, reduce the run length of processive dynein-dynactin-N-terminal coiled-coil domain of Bicaudal-D2 complexes, and ultimately compromise the expression and motility of the dynein complex in vitro (56). Furthermore, Hoang et al. found that the mutations with the strongest effects on dynein motility were associated with MCD in humans (56). The mutation detected in our patient (p.Arg292Trp) was previously reported by Benson et al. (43), and a previous study reported that a female with this mutation had focal epilepsy (with onset between 13–18 months) and intellectual disability. A similar phenotype was observed in our patient in that she had early-onset epilepsy and severe intellectual disability.
Lissencephaly is a subtype of MCD that is characterized by a thickened cortex and a gyral abnormality ranging from agyria to pachygyria. This condition usually involves the entire brain or large areas of the cerebral hemispheres with an anterior or posterior predominance (12). Many genes have been identified as being associated with lissencephaly, including LIS1, DCX, ARX, CDK5, and DYNC1H1. Various genes may cause the differences in gradients observed. LIS1-related lissencephaly is more likely to result in a posterior-to-anterior gradient, whereas DCX-related lissencephaly is associated with an anterior-to-posterior gradient (57). For DYNC1H1-related MCDs, Scoto et al. (10) demonstrated posterior predominant lesions were the most common manifestations. Our current analysis showed that the most common subtype of DYNC1H1-related MCD was posterior predominant pachygyria, this finding was consistent with the conclusions drawn by Scoto et al. (10). Another subtype, polymicrogyria, is unlike pachygyria in that iappears to be more predominant in the frontal region. MCDs have been previously linked to hypoplasia of the corpus callosum. In this study, we identified 15 MCD patients with corpus callosum hypoplasia; consequently, we consider that this may be one of the characteristic features of DYNC1H1-related MCD.
Besides imaging abnormalities, CNS symptoms have been observed in patients with MCD and the DYNC1H1 mutations. Most individuals with MCD have epilepsy and/or ID/DD. Our literature review found that 76% of individuals with MCDs had recurrent seizures, which is consistent with the rate of 75% reported in a previous study (58). MCDs are an important cause of epilepsy, especially early-stage medically refractory epilepsy (59). In this study, cases in which where clinical details were available indicated that epilepsy had an early onset, and the patient subsequently developed drug resistance. Seizures occur in over 90% of children with lissencephaly (57). Compared to other MCD subtypes with vary widely clinical manifestations, such as PMG and macrocephaly, children with lissencephaly have a relatively higher risk of developing epilepsy (57). In our study, 91% (41/45) of patients with pachygyria/agyria had epilepsy; in addition, the risk and proportions of occurrence were significantly increased when compared to children with other subtypes of MCD, this finding was consistent with a previous report (57). MCD patients may present with developmental delay and cognitive impairment (60); however, according to our literature review, the proportion of ID/DD cases among patients with MCD was alarmingly high. We believe that two factors contribute to this phenomenon: neurological dysfunction caused by the DYNC1H1 mutation, and the cognitive impairment caused by seizure onset. It is important to consider that minor imaging abnormalities may also be involved and could have been overlooked in previous investigations. After comparing the symptoms of patients with and without MCD, we consider that CNS symptoms are more likely to be due to MCD caused by DYNC1H1 variants than the mutation itself.
While the majority of individuals had mutations in the motor domain stalk, patients with mutations in the DYNC1H1 tail domain were more likely to have isolated neuromuscular symptoms such as SMA and CMT (52). Furthermore, patients with mutations in the motor domain (including the AAA domain and MBD or stalk) are more likely to develop MCD (19, 61), which is supported by the results of our literature review. In addition, we found that patients with variants in the stalk/MBD are more likely to develop pachygyria. However, given the large size of the DYNC1H1 protein, and its complex interactions with various other proteins, mutations that affect tertiary structure could also result in alterations to functional outcomes and symptomatology (54). Therefore, the results of this study should be interpreted with caution.
There were some limitations to the current study that need to be considered. First, the nature of the literature review did not allow us to acquire any additional information on the reported cases. Second, to ensure an accurate diagnosis of MCD, we excluded some patients with clinical symptoms but without brain MRI examination, thus reducing the reliability of our results. Third, due to the lack of detailed information for some cranial MRI results, particularly with regards to the gradient of pachygyria, it was not possible to determine the precise proportion of posterior predominant pachygyria in all patients with MCD, even though it is the most commonly observed manifestation, Moreover, the analysis results related to pachygyria must also be treated with caution. Although these findings are of interest at the theoretical level, they require experimental validation in future studies.
In conclusion, posterior predominant pachygyria (a subtype of MCD) is a common neurodevelopmental disorder in patients that possess DYNC1H1 mutation. Most patients (95%) who carry mutations in the protein stalk or microtubule binding domains exhibit DYNC1H1-related MCD, whereas almost two-thirds (63%) who carry mutations in the tail domain do not display MCD. In patients with DYNC1H1-related disorders, CNS symptoms tend to be associated with MCD, with seizure onset often occurring in infancy and drug resistance developing over time.
The datasets presented in this article are not readily available because of ethical and privacy restrictions. Requests to access the datasets should be directed to the corresponding authors.
The studies involving human participants were reviewed and approved by the Ethics Committee of the First Medical Center of the People's Liberation Army General Hospital. Written informed consent to participate in this study was provided by the participants' legal guardian/next of kin. Written informed consent was obtained from the minor(s)' legal guardian/next of kin for the publication of any potentially identifiable images or data included in this article.
Y-XD and GY participated in study conception and design. P-PF, W-RG, and BZ acquired the data. W-RG, W-NZ, and BZ analyzed the data. W-RG drafted the initial version of the manuscript. All authors participated in manuscript revision for intellectual content and approved the final published version.
This study was partly financed by the Beijing Natural Science Foundation (No. 7222187), Medical Big Data and Artificial Intelligence Research and Development Project of the Chinese People's Liberation Army General Hospital (No. 2019MBD-004), Epilepsy Research Fund of China Association Against Epilepsy (No. CU-B-2021-11), and Nutrition and Care of Maternal and Child Research Fund Project of Guangzhou Biostime Institute of Nutrition and Care (No. 2021BINCMCF030).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fneur.2023.1163803/full#supplementary-material
1. Kon T, Oyama T, Shimo-Kon R, Imamula K, Shima T, Sutoh K, et al. The 2.8 Å crystal structure of the dynein motor domain. Nature. (2012) 484:345–50. doi: 10.1038/nature10955
2. Schiavo G, Greensmith L, Hafezparast M, Fisher EM. Cytoplasmic dynein heavy chain: the servant of many masters. Trends Neurosci. (2013) 36:641–51. doi: 10.1016/j.tins.2013.08.001
3. Willemsen MH, Vissers LE, Willemsen MA, van Bon BW, Kroes T, de Ligt J, et al. Mutations in DYNC1H1 cause severe intellectual disability with neuronal migration defects. J Med Genet. (2012) 49:179–83. doi: 10.1136/jmedgenet-2011-100542
4. Matsumoto A, Kojima K, Miya F, Miyauchi A, Watanabe K, Iwamoto S, et al. Two cases of DYNC1H1 mutations with intractable epilepsy. Brain Dev. (2021) 43:857–62. doi: 10.1016/j.braindev.2021.05.005
5. Hertecant J, Komara M, Nagi A, Suleiman J, Al-Gazali L, Ali BR. A novel de novo mutation in DYNC1H1 gene underlying malformation of cortical development and cataract. Meta Gene. (2016) 9:124–7. doi: 10.1016/j.mgene.2016.05.004
6. Chung CT, Lee NC, Fan SP, Hung MZ, Lin YH, Chen CH, et al. DYNC1H1 variant associated with epilepsy: Expanding the phenotypic spectrum. Epilepsy Behav Rep. (2023) 21:100580. doi: 10.1016/j.ebr.2022.100580
7. Liu W, Cheng M, Zhu Y, Chen Y, Yang Y, Chen H, et al. DYNC1H1-related epilepsy: Genotype-phenotype correlation. Dev Med Child Neurol. (2023) 65:534–43. doi: 10.1111/dmcn.15414
8. Becker LL, Dafsari HS, Schallner J, Abdin D, Seifert M, Petit F, et al. The clinical-phenotype continuum in DYNC1H1-related disorders-genomic profiling and proposal for a novel classification. J Hum Genet. (2020) 65:1003–17. doi: 10.1038/s10038-020-0803-1
9. Harms MB, Ori-McKenney KM, Scoto M, Tuck EP, Bell S, Ma D, et al. Mutations in the tail domain of DYNC1H1 cause dominant spinal muscular atrophy. Neurology. (2012) 78:1714–20. doi: 10.1212/WNL.0b013e3182556c05
10. Scoto M, Rossor AM, Harms MB, Cirak S, Calissano M, Robb S, et al. Novel mutations expand the clinical spectrum of DYNC1H1-associated spinal muscular atrophy. Neurology. (2015) 84:668–79. doi: 10.1212/WNL.0000000000001269
11. Karczewski KJ, Francioli LC, Tiao G, Cummings BB, Alföldi J, Wang Q, et al. The mutational constraint spectrum quantified from variation in 141,456 humans. Nature. (2020) 581:434–43. doi: 10.1530/ey.17.14.3
12. Severino M, Geraldo AF, Utz N, Tortora D, Pogledic I, Klonowski W, et al. Definitions and classification of malformations of cortical development: practical guidelines. Brain. (2020) 143:2874–94. doi: 10.1093/brain/awaa174
13. Fischer J, Di Donato N. Diagnostic pitfalls in patients with malformations of cortical development. Eur J Paediatr Neurol. (2022) 37:123–8. doi: 10.1016/j.ejpn.2022.01.017
14. Amabile S, Jeffries L, McGrath JM, Ji W, Spencer-Manzon M, Zhang H, et al. DYNC1H1-related disorders: A description of four new unrelated patients and a comprehensive review of previously reported variants. Am J Med Genet A. (2020) 182:2049–57. doi: 10.1002/ajmg.a.61729
15. Liu X, Zhang S, Wan L, Zhang X, Wang H, Zhang H, et al. IQSEC2-related encephalopathy in male children: Novel mutations and phenotypes. Front Mol Neurosci. (2022) 15:984776. doi: 10.3389/fnmol.2022.984776
16. Laskowski RA, Stephenson JD, Sillitoe I, Orengo CA, Thornton JM. VarSite: Disease variants and protein structure. Protein Sci. (2020) 29:111–9. doi: 10.1002/pro.3746
17. Rodrigues C, Pires D, Ascher DB. DynaMut2: Assessing changes in stability and flexibility upon single and multiple point missense mutations. Protein Sci. (2021) 30:60–9. doi: 10.1002/pro.3942
18. Harada A, Takei Y, Kanai Y, Tanaka Y, Nonaka S, Hirokawa N. Golgi vesiculation and lysosome dispersion in cells lacking cytoplasmic dynein. J Cell Biol. (1998) 141:51–9. doi: 10.1083/jcb.141.1.51
19. P oirier K, Lebrun N, Broix L, Tian G, Saillour Y, Boscheron C, et al. Mutations in TUBG1, DYNC1H1, KIF5C and KIF2A cause malformations of cortical development and microcephaly. Nat Genet. (2013) 45:639–47. doi: 10.1038/ng.2613
20. Antoniadi T, Buxton C, Dennis G, Forrester N, Smith D, Lunt P, et al. Application of targeted multi-gene panel testing for the diagnosis of inherited peripheral neuropathy provides a high diagnostic yield with unexpected phenotype-genotype variability. BMC Med Genet. (2015) 16:84. doi: 10.1186/s12881-015-0224-8
21. Chan S, van Alfen N, Thuestad IJ, Ip J, Chan AO, Mak C, et al. A recurrent de novo DYNC1H1 tail domain mutation causes spinal muscular atrophy with lower extremity predominance, learning difficulties and mild brain abnormality. Neuromuscul Disord. (2018) 28:750–6. doi: 10.1016/j.nmd.2018.07.002
22. Peeters K, Chamova T, Jordanova A. Clinical and genetic diversity of SMN1-negative proximal spinal muscular atrophies. Brain. (2014) 137:2879–96. doi: 10.1093/brain/awu169
23. Di Donato N, Timms AE, Aldinger KA, Mirzaa GM, Bennett JT, Collins S, et al. Analysis of 17 genes detects mutations in 81% of 811 patients with lissencephaly. Genet Med. (2018) 20:1354–64. doi: 10.1038/gim.2018.8
24. Weedon MN, Hastings R, Caswell R, Xie W, Paszkiewicz K, Antoniadi T, et al. Exome sequencing identifies a DYNC1H1 mutation in a large pedigree with dominant axonal Charcot-Marie-Tooth disease. Am J Hum Genet. (2011) 89:308–12. doi: 10.1016/j.ajhg.2011.07.002
25. Tsurusaki Y, Saitoh S, Tomizawa K, Sudo A, Asahina N, Shiraishi H, et al. A DYNC1H1 mutation causes a dominant spinal muscular atrophy with lower extremity predominance. Neurogenetics. (2012) 13:327–32. doi: 10.1007/s10048-012-0337-6
26. Jamuar SS, Lam AT, Kircher M, D'Gama AM, Wang J, Barry BJ, et al. Somatic mutations in cerebral cortical malformations. N Engl J Med. (2014) 371:733–43. doi: 10.1056/NEJMoa1314432
27. Punetha J, Monges S, Franchi ME, Hoffman EP, Cirak S, Tesi-Rocha C. Exome sequencing identifies dync1h1 variant associated with vertebral abnormality and spinal muscular atrophy with lower extremity predominance. Pediatr Neurol. (2015) 52:239–44. doi: 10.1016/j.pediatrneurol.2014.09.003
28. Laššuthová P, Šafka BroŽková D, Krutová M, Neupauerová J, Haberlová J, Mazanec R, et al. Improving diagnosis of inherited peripheral neuropathies through gene panel analysis. Orphanet J Rare Dis. (2016) 11:118. doi: 10.1186/s13023-016-0500-5
29. Li JT, Dong SQ, Zhu DQ, Yang WB, Qian T, Liu XN, et al. Expanding the phenotypic and genetic spectrum of neuromuscular diseases caused by dync1h1 mutations. Front Neurol. (2022) 13:943324. doi: 10.3389/fneur.2022.943324
30. Ding D, Chen Z, Li K, Long Z, Ye W, Tang Z, et al. Identification of a de novo DYNC1H1 mutation via WES according to published guidelines. Sci Rep. (2016) 6:20423. doi: 10.1038/srep20423
31. Niu Q, Wang X, Shi M, Jin Q. A novel DYNC1H1 mutation causing spinal muscular atrophy with lower extremity predominance. Neurol Genet. (2015) 1:e20. doi: 10.1212/NXG.0000000000000017
32. Mahjani B, De Rubeis S, Gustavsson Mahjani C, Mulhern M, Xu X, Klei L, et al. Prevalence and phenotypic impact of rare potentially damaging variants in autism spectrum disorder. Mol Autism. (2021) 12:65. doi: 10.1186/s13229-021-00465-3
33. Strickland AV, Schabhüttl M, Offenbacher H, Synofzik M, Hauser NS, Brunner-Krainz M, et al. Mutation screen reveals novel variants and expands the phenotypes associated with DYNC1H1. J Neurol. (2015) 262:2124–34. doi: 10.1007/s00415-015-7727-2
34. Chen Y, Xu Y, Li G, Li N, Yu T, Yao RE, et al. Exome Sequencing Identifies De Novo DYNC1H1 Mutations Associated With Distal Spinal Muscular Atrophy and Malformations of Cortical Development. J Child Neurol. (2017) 32:379–86. doi: 10.1177/0883073816683083
35. Fiorillo C, Moro F, Yi J, Weil S, Brisca G, Astrea G, et al. Novel dynein DYNC1H1 neck and motor domain mutations link distal spinal muscular atrophy and abnormal cortical development. Hum Mutat. (2014) 35:298–302. doi: 10.1002/humu.22491
36. Farwell KD, Shahmirzadi L, El-Khechen D, Powis Z, Chao EC, Tippin Davis B, et al. Enhanced utility of family-centered diagnostic exome sequencing with inheritance model-based analysis: results from 500 unselected families with undiagnosed genetic conditions. Genet Med. (2015) 17:578–86. doi: 10.1038/gim.2014.154
37. Palmer EE, Schofield D, Shrestha R, Kandula T, Macintosh R, Lawson JA, et al. Integrating exome sequencing into a diagnostic pathway for epileptic encephalopathy: Evidence of clinical utility and cost effectiveness. Mol Genet Genomic Med. (2018) 6:186–99. doi: 10.1002/mgg3.355
38. Zhu X, Petrovski S, Xie P, Ruzzo EK, Lu YF, McSweeney KM, et al. Whole-exome sequencing in undiagnosed genetic diseases: interpreting 119 trios. Genet Med. (2015) 17:774–81. doi: 10.1038/gim.2014.191
39. Gelineau-Morel R, Lukacs M, Weaver KN, Hufnagel RB, Gilbert DL, Stottmann RW. Congenital Cataracts and Gut Dysmotility in a DYNC1H1 Dyneinopathy Patient. Genes (Basel). (2016) 7. doi: 10.3390/genes7100085
40. Hayashi S, Uehara DT, Tanimoto K, Mizuno S, Chinen Y, Fukumura S, et al. Comprehensive investigation of CASK mutations and other genetic etiologies in 41 patients with intellectual disability and microcephaly with pontine and cerebellar hypoplasia (MICPCH). PLoS One. (2017) 12:e0181791. doi: 10.1371/journal.pone.0181791
41. Rochtus A, Olson HE, Smith L, Keith LG, El Achkar C, Taylor A, et al. Genetic diagnoses in epilepsy: The impact of dynamic exome analysis in a pediatric cohort. Epilepsia. (2020) 61:249–58. doi: 10.1111/epi.16427
42. Friedman D, Kannan K, Faustin A, Shroff S, Thomas C, Heguy A, et al. Cardiac arrhythmia and neuroexcitability gene variants in resected brain tissue from patients with sudden unexpected death in epilepsy (SUDEP). NPJ Genom Med. (2018) 3:9. doi: 10.1038/s41525-018-0048-5
43. Su T, Yan Y, Hu Q, Liu Y, Xu S. De novo DYNC1H1 mutation causes infantile developmental and epileptic encephalopathy with brain malformations. Mol Genet Genomic Med. (2022) 10:e1874. doi: 10.1002/mgg3.1874
44. Benson KA, White M, Allen NM, Byrne S, Carton R, Comerford E, et al. A comparison of genomic diagnostics in adults and children with epilepsy and comorbid intellectual disability. Eur J Hum Genet. (2020) 28:1066–77. doi: 10.1038/s41431-020-0610-3
45. Helbig KL, Farwell Hagman KD, Shinde DN, Mroske C, Powis Z, Li S, et al. Diagnostic exome sequencing provides a molecular diagnosis for a significant proportion of patients with epilepsy. Genet Med. (2016) 18:898–905. doi: 10.1038/gim.2015.186
46. Hou MF, Fan JJ, Zou QP, Fu R, He JJ, Liu J. A de novo mutation of the DYNC1H1-related infantile spasms in a fmaily and literature review. Chin J Appl Clin Pediatr. (2019). 34:1022–4.
47. Kolbjer S, Martin DA, Pettersson M, Dahlin M, Anderlid BM. Lissencephaly in an epilepsy cohort: Molecular, radiological and clinical aspects. Eur J Paediatr Neurol. (2021) 30:71–81. doi: 10.1016/j.ejpn.2020.12.011
48. Viollet LM, Swoboda KJ, Mao R, Best H, Ha Y, Toutain A, et al. A novel pathogenic variant in DYNC1H1 causes various upper and lower motor neuron anomalies. Eur J Med Genet. (2020) 63:104063. doi: 10.1016/j.ejmg.2020.104063
49. Fernández Perrone AL, Moreno Fernández P, Álvarez S, Fernández-Jaén A. DYNC1H1 de novo mutation, spinal muscular atrophy and attention problems. Neurologia (Engl Ed). (2022) 37:406–9. doi: 10.1016/j.nrl.2021.08.001
50. Derksen A, Mirchi A, Tran LT, Cao-Lei L, Oskoui M, Srour M, et al. A novel de novo variant in dync1h1 causes spinal muscular atrophy lower extremity predominant in identical twins: a case report. Child Neurol Open. (2021) 8:2329048X211027438. doi: 10.1177/2329048X211027438
51. Yang H, Gong P, Jiao X, Niu Y, Zhou Q, Zhang Y, et al. De Novo variants in the dync1h1 gene associated with infantile spasms. Front Neurol. (2021) 12:733178. doi: 10.3389/fneur.2021.733178
52. Beecroft SJ, McLean CA, Delatycki MB, Koshy K, Yiu E, Haliloglu G, et al. Expanding the phenotypic spectrum associated with mutations of DYNC1H1. Neuromuscul Disord. (2017) 27:607–15. doi: 10.1016/j.nmd.2017.04.011
53. Retterer K, Juusola J, Cho MT, Vitazka P, Millan F, Gibellini F, et al. Clinical application of whole-exome sequencing across clinical indications. Genet Med. (2016) 18:696–704. doi: 10.1038/gim.2015.148
54. Lin Z, Liu Z, Li X, Li F, Hu Y, Chen B, et al. Whole-exome sequencing identifies a novel de novo mutation in DYNC1H1 in epileptic encephalopathies. Sci Rep. (2017) 7:258. doi: 10.1038/s41598-017-00208-6
55. Eschbach J, Dupuis L. Cytoplasmic dynein in neurodegeneration. Pharmacol Ther. (2011) 130:348–63. doi: 10.1016/j.pharmthera.2011.03.004
56. Hoang HT, Schlager MA, Carter AP, Bullock SL. DYNC1H1 mutations associated with neurological diseases compromise processivity of dynein-dynactin-cargo adaptor complexes. Proc Natl Acad Sci U S A. (2017) 114:E1597–1597E1606. doi: 10.1073/pnas.1620141114
57. Parrini E, Conti V, Dobyns WB, Guerrini R. Genetic basis of brain malformations. Mol Syndromol. (2016) 7:220–33. doi: 10.1159/000448639
58. Leventer RJ, Phelan EM, Coleman LT, Kean MJ, Jackson GD, Harvey AS. Clinical and imaging features of cortical malformations in childhood. Neurology. (1999) 53:715–22. doi: 10.1212/WNL.53.4.715
59. D'Gama AM, Poduri A. Precision therapy for epilepsy related to brain malformations. Neurotherapeutics. (2021) 18:1548–63. doi: 10.1007/s13311-021-01122-6
60. Chang BS, Walsh CA. Mapping form and function in the human brain: the emerging field of functional neuroimaging in cortical malformations. Epilepsy Behav. (2003) 4:618–25. doi: 10.1016/j.yebeh.2003.09.006
Keywords: DYNC1H1 gene, malformations of cortical development, variant, microtubule-binding domain, case report
Citation: Ge W-R, Fu P-P, Zhang W-N, Zhang B, Ding Y-X and Yang G (2023) Case report: Genotype and phenotype of DYNC1H1-related malformations of cortical development: a case report and literature review. Front. Neurol. 14:1163803. doi: 10.3389/fneur.2023.1163803
Received: 11 February 2023; Accepted: 11 April 2023;
Published: 25 April 2023.
Edited by:
Mariasavina Severino, Giannina Gaslini Institute (IRCCS), ItalyReviewed by:
Ana Filipa Geraldo, Centro Hospitalar de Vila Nova de Gaia, PortugalCopyright © 2023 Ge, Fu, Zhang, Zhang, Ding and Yang. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Guang Yang, eWFuZ2czMDFAMTI2LmNvbQ==; Ying-Xue Ding, ZC15aW5neHVlQDEyNi5jb20=
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.