 Jing Zou
Jing Zou Guoping Zhang1
Guoping Zhang1 Hongbin Li
Hongbin Li Antti Mäkitie
Antti Mäkitie
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Neurol., 18 May 2023
Sec. Neuro-Otology
Volume 14 - 2023 | https://doi.org/10.3389/fneur.2023.1159658
This article is part of the Research TopicInflammatory Mechanism, Imaging Diagnostics, and Novel Treatment of Meniere’s DiseaseView all 5 articles
Background: The mechanisms of Meniere's disease (MD) remain largely unknown. The purpose of this study was to identify possible genetic variants associated with immune regulation in MD.
Methods: The whole immune genome of 16 Chinese patients diagnosed with sporadic MD was sequenced using next-generation sequencing.
Results: Definite pathological variants of MEFV (c.1223G>A, c.1105C>T), COL7A1 (c.5287C>T), and ADA (c.445C>T) contributing to the clinical phenotype were found in three patients. Limited and likely pathological variants of TLR3 (c.2228G>A) and RAB27A (c.560G>A) were detected in one patient each. The following definite pathological variants impairing the structure and function of translated proteins were detected in 10 patients, and multigene variants occurred in five patients: PRF1 (c.710C>A), UNC13D (c.1228A>C), COLEC11 (c.169C>T), RAG2 (c.200G>C), BLM (c.1937G>T), RNF31 (c.2533G>A), FAT4 (c.11498A>G), PEPD (c.788A>G), TNFSF12 (c.470G>A), VPS13B (c.11972A>T), TNFRSF13B (c.226G>A), ERCC6L2 (c.4613A>G), TLR3 (c.2228G>A), ADA (c.445C>T), PEPD (c.151G>A), and MOGS (c.2470G>A). The following limited pathological variants impairing the structure and function of translated proteins were detected in five patients, with double gene variants identified in one patient: EXTL3 (c.1396G>A), MTHFD1 (c.2057G>A), FANCA (c.2039T>C), LPIN2 (c.1814C>T), NBAS (c.4049T>C), and FCN3 (c.734G>A).
Conclusion: Patients with sporadic MD carry multiple genetic variants involved in multiple steps of immune regulation, which might render patients susceptible to developing inflammation via both autoimmune and autoinflammation mechanisms upon internal stress.
The mechanisms of Meniere's disease (MD), which is characterized by episodic vertigo, fluctuant hearing loss, tinnitus, and aural fullness, remain largely unknown. Allergy was reported to be associated with MD as early as the 1940's (1, 2), and the autoimmune mechanism was later indicated (3, 4). The excellent therapeutic effect of intratympanic corticosteroids further supports an immune-inflammatory reaction associated with pathological progression (5). Although autoimmunity has been identified in both unilateral and bilateral MD subgroups, the possible immune-inflammatory mechanism has not been evaluated in subgroups according to the classification method (6, 7). High basal levels of IL-1β were detected in the supernatant of 24 out of 113 MD patients (21%), and 20 out of the 24 patients were diagnosed with sporadic MD. Among the 24 patients with high basal levels of IL-1β, 21 (87.5%) showed elevated levels of IL-6, 19 (79.2%) had higher levels of TNF-α, and 17 (70.8%) demonstrated an elevation of IL-1R antagonist levels (8). There is a possibility that the autoinflammatory mechanism was involved in those MD patients with high levels of IL-1β. In contrast to autoimmune diseases that are characterized by the production of specific autoantibodies and the activation of T cells and B cells, autoinflammatory diseases are at the other end and mediated by inflammasomes that further introduce the secretion of active IL-1β (9).
Variants in inflammation-related genes were detected in sporadic MD patients (10). Several clinical subgroups of MD have been reported, but the criteria for genetic variants have not been defined (6, 7). MD patients with a familial history and autoimmunity may have a unique genetic background, and the criteria for MD with autoinflammation have not been defined (10). Zou reported a case of refractory MD demonstrating autoinflammatory characteristics supported by both genotype and associated phenotype and the effects of delivering high-dose steroids to the surface of the intact endolymphatic sac and incus (11). McDermott and Kastner first introduced the term “autoinflammation” in 1999, describing the critical role of impaired membrane TNFR1 clearance and diminished shedding of the potentially antagonistic soluble receptor in autosomal dominant periodic fever syndromes characterized by unexplained fever episodes and severe localized inflammation (12). Six categories of autoinflammatory diseases have been defined based on molecular pathophysiology. However, a mixture of autoinflammation and autoimmunity might be developed during the late stage of diseases possibly mediated through classical dendritic and plasmacytoid dendritic cells-induced differentiation of Th1 and Th17 effector T cell subsets (13). A thorough investigation into the autoinflammatory mechanisms of MD remains warranted.
We aimed to identify possible genetic variants associated with immune regulation, including autoinflammatory and autoimmune mechanisms involved in the occurrence of MD. The whole immune genome of Chinese patients with sporadic MD was, thus, sequenced in the present study.
The protocol was reviewed and approved by the ethics committee of Shanghai Changhai Hospital (CHEC2020-107). Patients who visited the author, JZ, at the Department of Otolaryngology—Head and Neck Surgery, Changhai Hospital, Second Military Medical University, from July 2018 to June 2022, were screened. The inclusion criteria were as follows: (1) diagnosis of definite MD made according to the 2015 criteria of the Barany Society (14) and (2) endolymphatic hydrops (EH) was confirmed using gadolinium enhancement MRI. The exclusion criteria were as follows: vertigo of other origins, such as benign paroxysmal positional vertigo, and diseases of the central nervous system. Since familial MD is a heterogeneous condition, some individuals presenting with a partial syndrome (i.e., episodic vertigo without low-frequency sensorineural hearing loss) fail to meet the diagnostic criteria of MD (15). Familial history ruled out familial clustering of the patients in the current study. Subgroups of unilateral and bilateral MD patients were defined according to previous reports (6, 7). In total, 16 patients diagnosed with definite MD were enrolled (Table 1). The data of two previously reported patients were also included in the current study in order to draw conclusions from accumulated information (11, 16).
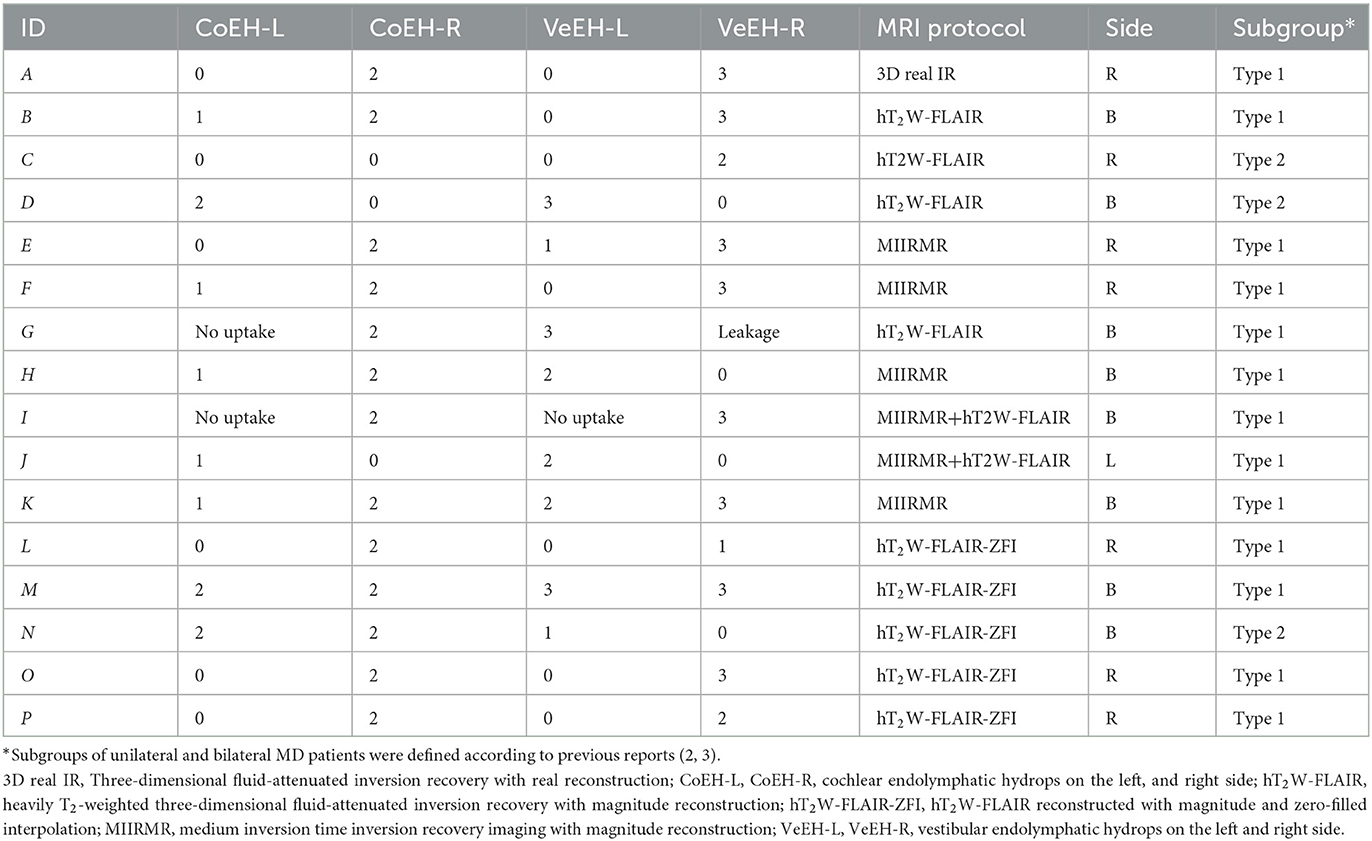
Table 1. EH detected in MD patients using MRI 24 h after intratympanic injection of Gd-DTPA.
Patients were treated mainly with intratympanic dexamethasone (ITDex) and additional high-dose steroid delivery to the surface of the intact endolymphatic sac and incus (ESCS) when they were unsatisfied with ITDex (Table 1). Patient E was treated with endolymphatic sac decompression by another doctor without effect, received ITDex and ESCS without effect, and was finally treated with the triple semicircular canal plugging by another doctor. Patient L was treated by endolymphatic sac shunt surgery in another hospital due to unsatisfactory ITDex and suffered from severe tinnitus after surgery. The following criteria were used for evaluating the outcome of therapy: V2 = no vertigo attack and normal balance, V1 = reduced vertigo, V0 = vertigo remained the same, V(-1) = vertigo became worse; H2 = complete recovery in hearing, H1 = hearing improved >10 dB (mean pure tone at 0.5, 1.0, and 2.0 kHz), H0 = hearing remained the same, and H(-1) = hearing became worse with a mean value >10 dB; T2 = tinnitus disappeared, T1 = reduced tinnitus, T0 = tinnitus remained the same, and T(-1) = tinnitus became worse; F2 = fullness disappeared, F1 = reduced fullness, F0 = fullness remained the same, and F(-1) = fullness became worse.
To detect EH in MD patients, MRI was performed 24 h after intratympanic injection of Gd-DTPA at a previously reported dose using a 3T MR system (MAGNETOM Skyra, Siemens Healthcare, Erlangen, Germany) equipped with a 20-channel Tim 4G head/neck coil (17). Either hT2W-FLAIR, MIIRMR, or hT2W-FLAIR-ZFI was applied for imaging as the protocols were updated three times during the research period (Table 1) (18, 19). The SPACE sequence was also applied to image potential inner ear fibrosis or vestibular schwannoma (17, 18). EH was graded according to previous reports that applied a three-stage method for cochlear hydrops and a four-stage scale for vestibular hydrops based on morphological changes (20, 21). A modification was made in the grading of 4th-grade vestibular hydrops, defining this grade when the bright peripheral rim of the surrounding perilymphatic space is largely absent instead of when the vestibular perilymphatic enhancement becomes invisible.
Genomic DNA was extracted from peripheral blood samples from all patients. Whole exomes including 269, 349, or 423 candidate genes (Table 2) potentially involved in autoinflammatory diseases, autoimmune diseases, and other diseases associated with disorders of immune regulation were selected according to Human Phenotype Ontology (http://human-phenotype-ontology.github.io/) and sequenced using target gene capture and next-generation sequencing using a previously reported protocol (22). Data were analyzed using bwa-0.7.10, samtools-1.0, picard-tools-1.119, bamtools-2.3.0, Genome Analysis TK-3.3.0, and Annovar-2014-11-12. REVEL (Rare Exome Variant Ensemble Learner) software was used to predict the pathogenicity of missense variants based on individual tools regarding impairing the structure and function of translated proteins (23). The following datasets were used as references: 1,000 genomes, NHLBI GO Exome Sequencing Project (ESP6500), the Exome Aggregation Consortium (ExAC), the Exome Aggregation Consortium-East Asian (ExAC-EAS), and the in-house Chinese reference population data of MyGenostics Inc. (Beijing, China) that was archived from 2,273 normal individuals. The clinical significance of sequence variants was interpreted according to the Joint Consensus Recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology published in 2015 (24). The definite pathological variants associated with clinical phenotype and the structure and function of translated proteins as well as variants with autosomal dominant inheritance were further validated using Sanger sequencing.
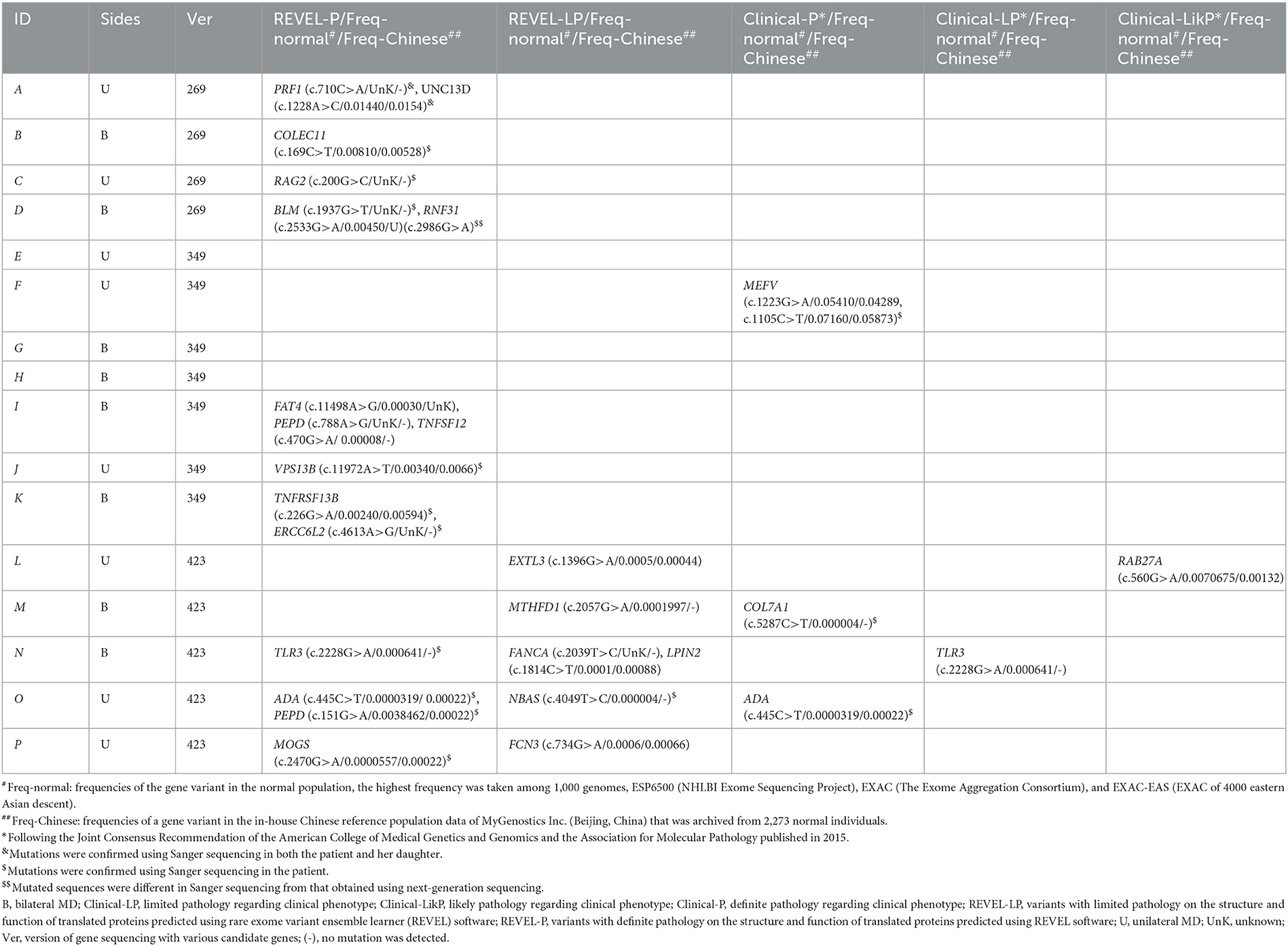
Table 2. Pathogenicity of heterozygote allelic variants in patients with MD detected using targeted next-gene sequencing.
All 16 patients were diagnosed with definite MD; eight patients had bilateral MD, and eight patients had unilateral MD. Various grades of EH were detected in 20 cochleae and 19 vestibules in 24 affected ears, and either cochlear or vestibular EH was detected in 23 affected ears (Figures 1, 2). Mild (grade 1) EH was found in one cochlea and one vestibule each out of seven contralateral ears (Figure 3). EH was absent in one ear of a patient with bilateral MD. Enhancement was not detected in one ear of another patient with bilateral MD (Table 1). There was a significant correlation between cochlear EH and vestibular EH, between cochlear EH and side, and between vestibular EH and side (p < 0.01, Spearman rho).
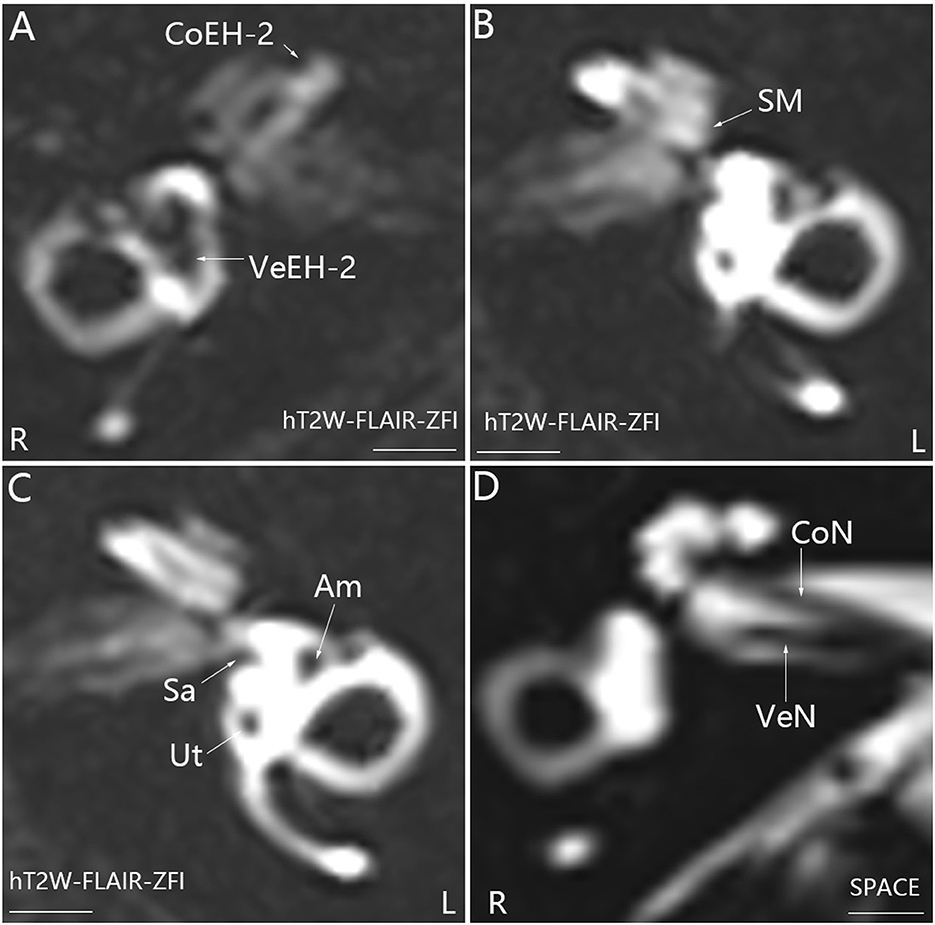
Figure 1. Representative MRI demonstrating EH in the right ear (R) in patient P with definite MD on the right side (A). MR images were taken using the hT2W-FLAIR-ZFI sequence 24 h after intratympanic injection of 0.1 ml of 20-fold-diluted Gd-DTPA that was delivered onto the posterior upper part of the tympanic medial wall on both sides. There was no EH in the left ear (L) (B, C). Neither inner ear fibrosis nor vestibular schwannoma was found on the ipsilateral side imaged using the SPACE sequence (D). Am, ampulla; CoEH-2, grade 2 cochlear EH; CoN, cochlear nerve; Sa, saccule; Ut, utricle; VeEH-2, grade 2 vestibular EH; VeN, vestibular nerve. Scale bar = 3.0 mm.
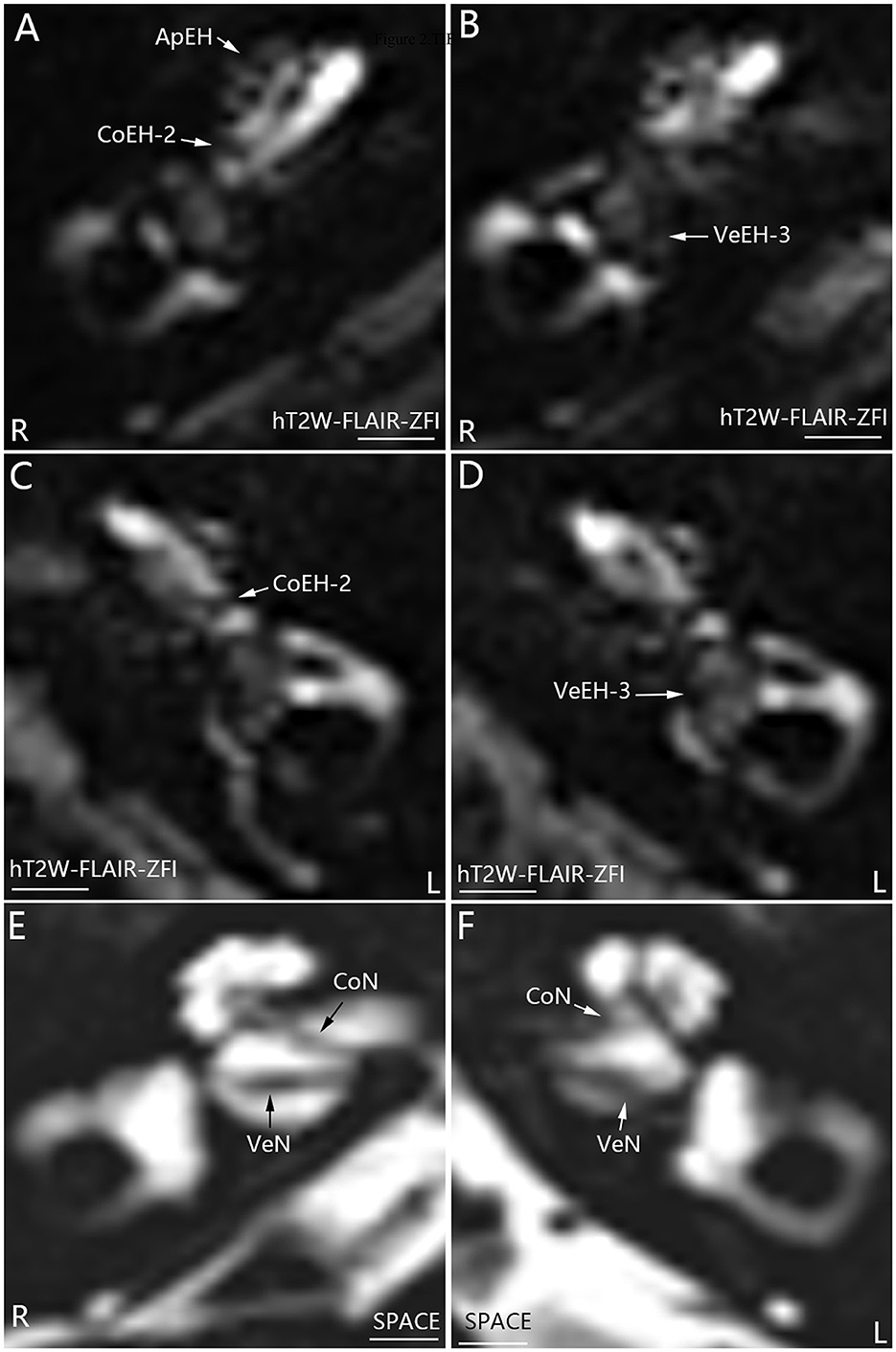
Figure 2. Representative MRI demonstrating EH in both ears in patient M with definite bilateral MD (A–D). MR images were taken using the hT2W-FLAIR-ZFI sequence 24 h after intratympanic injection of 0.1 ml of 20-fold-diluted Gd-DTPA on both sides. Apical EH (ApEH) was also observed in the right ear (R) (A) but not in the left ear (L) (C). Neither inner ear fibrosis nor vestibular schwannoma was observed on either side imaged using the SPACE sequence (E, F). CoEH-2, grade 2 cochlear EH; CoN, cochlear nerve; VeEH-3, grade 3 vestibular EH; VeN, vestibular nerve. Scale bar = 3.0 mm.
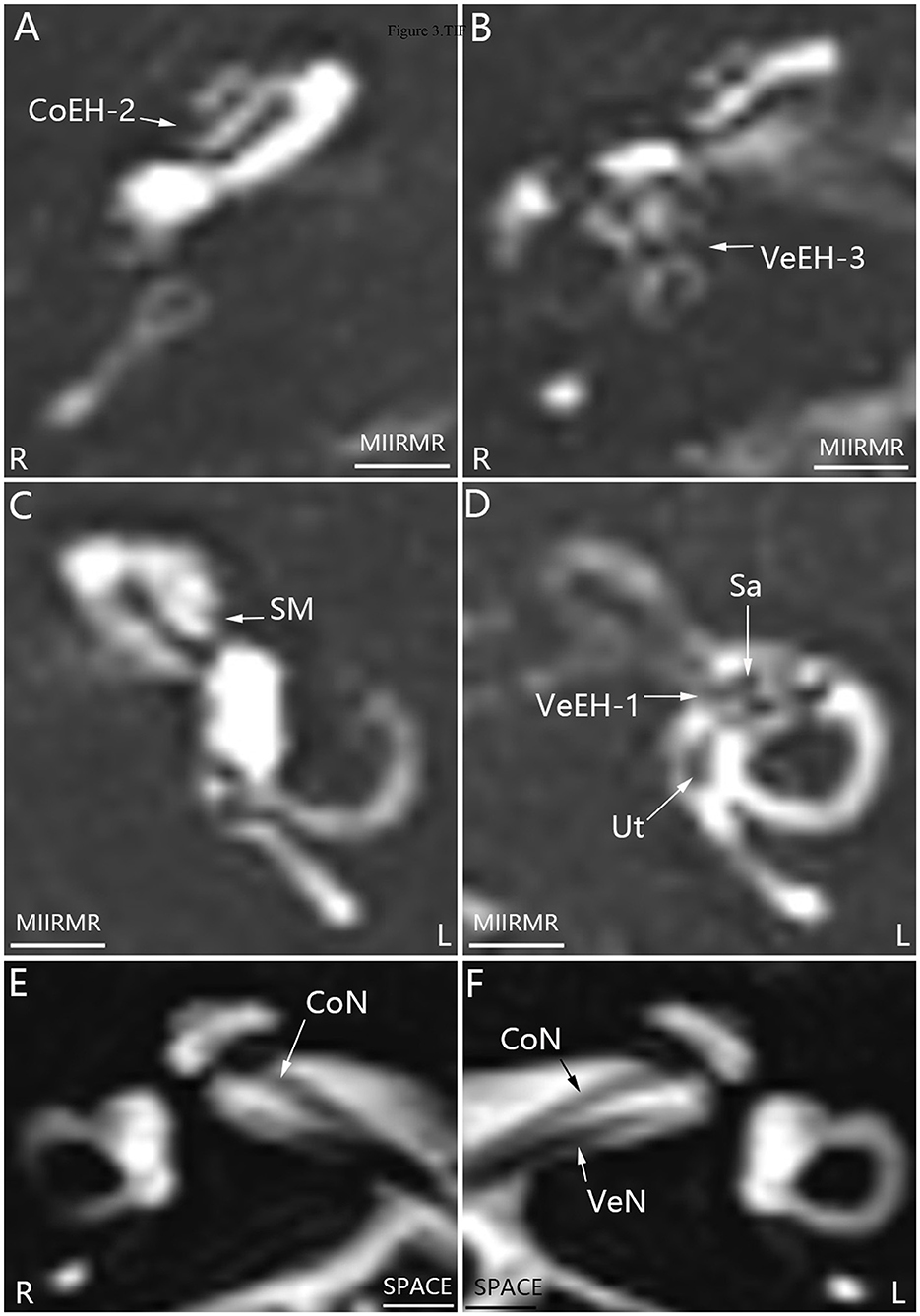
Figure 3. Representative MRI demonstrating EH in both ears in patient E with definite MD on the right side (A, B, D). MR images were taken using the MIIRMR sequence 24 h after intratympanic injection of 0.1 ml of 20-fold-diluted Gd-DTPA on both sides. There was no cochlear EH in the right ear (R) (C). Neither inner ear fibrosis nor vestibular schwannoma was found on the ipsilateral side imaged using the SPACE sequence (E, F). CoEH-2, grade 2 cochlear EH; CoN, cochlear nerve; L, left ear; Sa, saccule; Ut, utricle; VeEH-1 and VeEH-3, grade 1 and 3 vestibular EH; VeN, vestibular nerve. Scale bar = 3.0 mm.
Regarding contribution to clinical phenotype, the following definite pathological variants were found in three out of 16 patients: MEFV (c.1223G > A, c.1105C > T), COL7A1 (c.5287C > T), and ADA (c.445C > T). The likely limited pathological variants TLR3 (c.2228G > A) and RAB27A (c.560G > A) were each detected in one of the 16 patients (Table 2). The patient (M) carrying COL7A1 variation, which causes R mutation in arginine at 1,763 which became a stop codon and terminate the following translation, had bilateral severe to profound hearing loss with the absence of distortion-product otoacoustic emissions but detectable auditory brainstem response. In electrocochleography, action and summating potentials were detected in the left ear but absent in the right ear. ocular vestibular evoked myogenic potentials (oVEMP) was normally recorded in the left ear but absent in the right ear. video head impulse test (vHIT) showed a reduced gain in vestibulo-ocular response with significant corrective saccades in three planes. Caloric testing demonstrated a reduction in the right ear.
Regarding impairments in the structure and function of translated proteins, definite pathological variants were detected in 10 out of 16 patients, and multigene variants occurred in five patients (Table 2). The variants were as follows: PRF1 (c.710C>A), UNC13D (c.1228A>C), COLEC11 (c.169C>T), RAG2 (c.200G>C), BLM (c.1937G>T), RNF31 (c.2533G>A), FAT4 (c.11498A>G), PEPD (c.788A>G), TNFSF12 (c.470G>A), VPS13B (c.11972A>T), TNFRSF13B (c.226G>A), ERCC6L2 (c.4613A>G), TLR3 (c.2228G>A), ADA (c.445C>T), PEPD (c.151G>A), and MOGS (c.2470G>A). Limited pathological variants were detected in five out of 16 patients, and double gene variants appeared in one patient. The variants were as follows: EXTL3 (c.1396G>A), MTHFD1 (c.2057G>A), FANCA (c.2039T>C), LPIN2 (c.1814C>T), NBAS (c.4049T>C), and FCN3 (c.734G>A).
Several variants listed as benign or uncertain heterozygote allelic variants in the currently available database were also detected in all 16 patients (Table 3). Variants with autosomal dominant inheritance were detected in 13 out of 16 patients, and eight patients had multigene involvement. Variants of the MEFV gene were found in six patients, and three patients had variants in multiple loci. Heterozygote variants with autosomal recessive inheritance were detected in 15 out of 16 patients, and 14 patients had multigene variants. A variant with X-linked recessive inheritance, IL2RG (c.1058A>C), was detected in one out of 16 patients. Variants with uncertain inheritance were found in three out of 16 patients, including TIRAP (c.671G>A), POLE (c.504A>T), and TLR4 (c.910T>C).
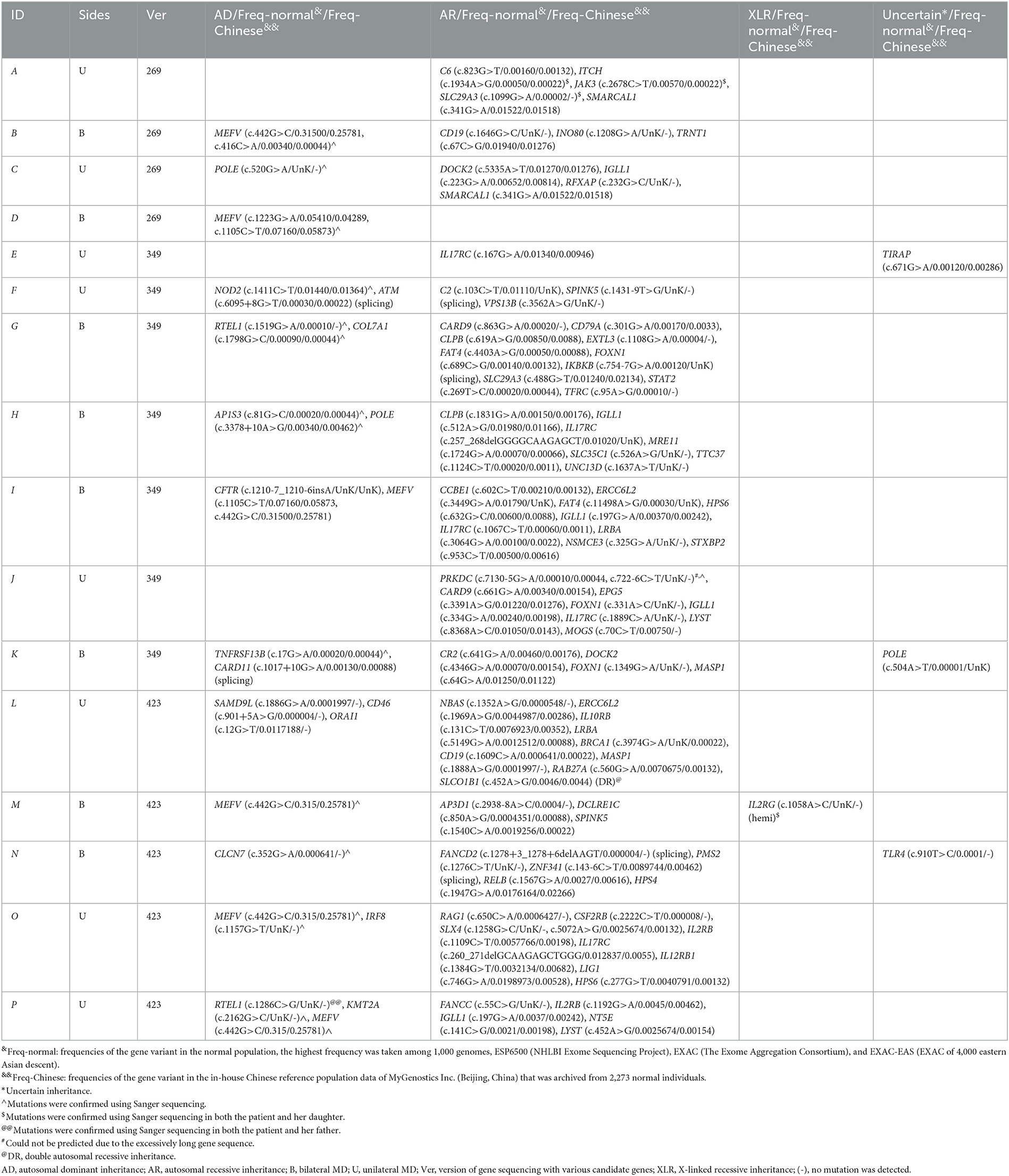
Table 3. Benign/uncertain heterozygote allelic variants in patients with MD detected using targeted next-gene sequencing.
In the present preliminary study, we detected variants in multiple genes involved in the immune response in a small series of Chinese patients with sporadic definite MD. The results indicate that multiple genetic variants exist in most patients with sporadic MD, and the related gene products are involved in the regulation of innate and adaptive immunity.
There is a broad disease spectrum associated with the detected definite pathological variants contributing to clinical phenotypes in human diseases and definite pathological variants with respect to the structure and function of translated proteins. MEFV encodes the protein pyrin, which has been shown to act as a regulator of inflammation mediated by IL-1β. A recent study showed that treatment of cultured human endolymphatic sac epithelial cells with IL-1β reduced epithelial sodium channel expression and the associated current indicating inflammation may disrupt fluid absorption through the endolymphatic sac (25). Mutations in MEFV have been defined as variants specific to autoinflammatory diseases and have also been detected in autoimmune diseases (26, 27). Mutations in COL7A1, which encodes type VII collagen, have been reported to cause dystrophic epidermolysis bullosa, an inherited epidermolysis bullosa that demonstrates activated autoimmunity and inflammatory responses (28). Mutations in COL7A1 were reported to potentially overlap with the TMIE gene that is involved in the mechanoelectrical transduction of cochlear hair cells and detected in a case of bilateral auditory neuropathy (29). It is possible that gene products of TMIE were disrupted by the abnormal stop codon. However, the phenotype of patient M did not fall in the category of auditory neuropathy spectrum disorder that might become atypical at an advanced stage. It is possible that the current patient had a comorbidity of MD and an auditory neuropathy spectrum disorder. ADA is a key enzyme of the purine salvage pathways, and absent or impaired ADA function leads to the accumulation of 2′deoxyadenosine and deoxyadenosine triphosphate, which are toxic metabolites of adenosine, in rapidly proliferating cells such as lymphocytes. This accumulation results in severe combined immunodeficiency. ADA deficiency has also been reported to be associated with sensorineural hearing loss (30). ADA−/− mice developed hearing deficits resulting from extensive damage to cochlear hair cells and impairment of immune cells; these effects were improved by enzyme replacement therapy (31). COLEC11 encodes the CL-K1 component of collectins belonging to a family of pattern recognition molecules with key roles in host defense, tissue homeostasis, and embryogenesis. Mutations in COLEC11 may cause 3M syndrome, which is associated with craniofacial dysmorphism, intellectual disability, and genital, renal, and limb abnormalities (32). RAG2 encodes lymphoid-specific recombinase that initiates V(D)J recombination, a site-specific chromosomal rearrangement process responsible for the immense diversity of TCRs and BCRs, and mutations in RAG2 may cause autoimmune diseases (33). BLM encodes BLM helicase or RECQL3, which is a nucleolar protein essential for the integrity and stability of DNA. Mutations in BLM result in Bloom's syndrome, an autosomal recessive disease with abnormal immunoreaction (34). RNF31 is a linear ubiquitin chain assembly complex component and regulates cell survival by inducing linear ubiquitination of NF-κB signaling components. Mutations in RNF31 may cause immunodeficiency, autoinflammation, amylopectinosis, and lymphangiectasia (35). FAT4 encodes a large protein with extracellular cadherin repeats, EGF-like domains, and laminin G-like domains that play a role in tumor suppression and planar cell polarity. Mutations in FAT4 may cause neuronal defects in addition to the well-known van Maldergem syndrome, characterized by intellectual disability, periventricular heterotopia, characteristic facial features, camptodactyly and syndactyly, small kidneys, osteoporosis, and tracheal anomalies (36). The PEPD gene encodes prolidase, a protease enzyme involved in metal-associated catalytic mechanisms. Mutations in PEPD may cause symptoms such as ulceration and other dermatologic manifestations, telangiectasis, impetigo-like eruptions, lesions, necrotic papules, intellectual disability, respiratory tract infections, and facial dysmorphisms (37). TNFRSF13B encodes the transmembrane activator, calcium-modulator, and cyclophilin ligand interactor involved in class-switch recombination and the maintenance of memory and plasma B cells. Mutations in TNFRSF13B have been identified in common variable immunodeficiency (38). VPS13B encodes the VPS13B protein, which is important for Golgi structure maintenance, and genetic variants have been linked to the autosomal recessive neurodevelopmental disorder Cohen syndrome, which is characterized by intellectual disability, developmental delay, microcephaly, a characteristic facial appearance, progressive retinopathy, myopia, and/or neutropenia (39).
The detected limited and likely pathological variants seemed relevant. TLR3 plays a key role in immune-mediated lupus nephritis by inducing sustained type I interferon activation (40). RAB27A controls the exosome secretion pathway and the terminal transport of lytic granules to immune synapses (41).
Regarding the limited pathological variants detected in five out of the 16 patients, EXTL3 is a member of the exostosin family of glycosyltransferases that regulate glycosylation, a process by which glycans are attached to both proteins and lipids in the ER or Golgi complex. It has been reported that biallelic missense mutations in EXTL3 cause neuroimmunoskeletal dysplasia syndrome, with some individuals presenting with a lack of CD4+ and CD8+ T cells (42). MTHFD1 encodes a trifunctional protein essential for folate metabolism, and the defects are associated with severe combined immunodeficiency (43). FANC encodes FANC proteins that participate in repairing DNA interstrand crosslinks and are important for immunity and organellar homeostasis (44). Variants in the FANC gene cause Fanconi anemia, characterized by congenital abnormalities, bone marrow failure, and cancer predisposition (45). LPIN2 encodes the protein lipin 2, a magnesium-dependent phosphatide phosphatase enzyme that associates with membrane lipids and localizes to organelles. Recessive loss-of-function mutations in LPIN2 are involved in the dysregulation of innate immune responses resulting in systemic inflammation and osteomyelitis, including Majeed syndrome (45). NBAS gene encodes the NBAS protein, which functions as a component of an ER tethering complex involved in retrograde Golgi–ER transport. Variants in NBAS cause a disruption in Golgi-ER transport and formation of large transport vesicles at the ER exit site, induced ER stress and inflammation (46). FCN3 gene encodes the Ficolin-3 protein, which is expressed in the lung and liver and is a recognition molecule in the lectin pathway of the complement system. Variants in FCN3 may cause a deficiency in ficolin-3-dependent complement activation and lead to recurrent infections (47).
Mutations in the abovementioned genes are involved in various diseases, impairing the immune response at various stages, from the level of regulating integrity and stability of DNA, posttranslational modification, and trafficking of proteins and exosome secretion pathway to the final step of the immune response. Although the variants detected in patients with MD in the present study are not identical to those detected in other diseases, these variants are harmful to the gene products and may cause pathological changes in the individual according to current database data. Regarding RNF31 variants involved in linear ubiquitination of NF-κB signaling components, regulatory variants in the NFKB1 gene have been reported to modify hearing outcomes in patients with MD and unilateral sensorineural hearing loss (48). The TLR3 signaling pathway has also been implicated in the innate immune response in the human endolymphatic sac (49). Patient A was also diagnosed with hyperthyroidism and secondary hypothyroidism with negative antibodies against thyroglobulin protein, thyroid peroxidase, and thyroxine receptor, indicating an autoinflammatory condition (11). Patient N has the comorbidity of ankylosing spondylitis, a chronic inflammatory arthritis that may cause inner ear issues. Patient P has the comorbidity of ulcerative colitis, an autoimmune disease that may also be accompanied by MD. We do not expect to discover genetic variants specific to MD because both autoimmunity and autoinflammation may involve in multiple organs. However, individuals carrying the abovementioned genetic variants may be susceptible to developing MD with the appearance of other triggers.
A missense variant in PRKCB was reported to segregate low-frequency sensorineural hearing loss in an autosomal dominant family with MD. PRKCB encodes a serine and threonine-specific protein kinase involved in many different cellular functions, such as neutrophil chemotaxis, melanoma cell growth and proliferation, or induction of apoptosis in endothelial cells (50). Our recent study demonstrated that G-CSF, IL8, and HGF are involved in the development of neutrophil extracellular traps and, through various mechanisms, influence the functions of macrophages, lymphocytes, and dendritic cells, which among others, are key players in the development of EH and MD (51). The behavior patterns of patients with MD showed strong tendencies toward engrossment, self-inhibition, feeling pressed for time, and aggressiveness compared to those of controls that engender day-to-day stress and frustration (52). A recent study on the surgical results of endolymphatic sac drainage surgery and changes in stress-induced plasma arginine-vasopressin levels showed that a gradual plasma vasopressin level elevation in recurrent MD after surgery suggested a link between symptom attacks and internal stress (53). Although endolymphatic sac drainage surgery was reported to be non-specific for MD and the outcome of the surgery might be linked to other biological effects such as shear stress induced by drilling the mastoid (54, 55), the results demonstrating patients without any symptom attacks having significantly low levels of plasma arginine-vasopressin compared to those with symptoms suggest a strong link between symptom attacks and stress (53). It was also reported that acute stress affects endocrine, immune, and metabolic functions in humans, and mood plays a causal role in the modulation of responses to acute stress (56). Individuals carrying the abovementioned genetic variations may be susceptible to developing inflammation and EH under stress conditions.
In addition to variants in immune genes, those in other genes, such as DTNA, FAM136A, SEMA3D, and DPT, relevant to the formation or maintenance of inner ear structures and inner ear ionic homeostasis have been detected in familial MD. Those variations demonstrate genetic heterogeneity in MD (57).
There are obvious limitations in the current study. We were unable to compare our results to a previous report on genetic variants detected in 890 MD patients because different target genes were selected. In the previous report, most of the 69 genes were selected from a list for monogenic sensorineural hearing loss although additional genes were added according to previous results on familial MD among others (58). In the present study, 269–423 target genes were selected by focusing on autoimmunity and autoinflammation. The Chinese individuals may also carry different genetic variants compared with the Spanish group. The small sample size makes it impossible to segregate the phenotype or to perform a burden test to search for the association. A future study using the current targets combined with targets related to the inner ear function based on a large sample selection is needed.
In conclusion, the current preliminary study results suggest that patients with sporadic MD carry multiple genetic variants involved in immune regulation, which might render these patients susceptible to developing inflammation via autoimmune and autoinflammation mechanisms and EH upon stress. Therefore, multiple hits might be involved in triggering the occurrence of MD. However, a large sample size is needed to replicate these findings.
According to national legislation/guidelines, specifically the Administrative Regulations of the People's Republic of China on Human Genetic Resources (http://www.gov.cn/zhengce/content/2019-06/10/content_5398829.htm; http://english.www.gov.cn/policies/latest_releases/2019/06/10/content_281476708945462.htm), no additional raw data is available at this time. Data of this project can be accessed after an approval application to the China National Genebank (CNGB, https://db.cngb.org/cnsa/). Please refer to: https://db.cngb.org/, or email: Q05HQmRiQGNuZ2Iub3Jn for detailed application guidance. The accession code CNP0004038 should be included in the application.
The studies involving human participants were reviewed and approved by the Ethical Committee of Shanghai Changhai Hospital (CHEC2020-107). The patients/participants provided their written informed consent to participate in this study.
JZ made substantial contributions to the conception and design of the work, acquisition, analysis and interpretation of data, and drafting the work. GZ, HL, and ZZ participated in clinical work and acquisition of data. QZ performed the audiological measurements and participated in acquisition of data. IP and AM participated in drafting the work. All authors contributed to the article and approved the submitted version.
This study was supported by the National Natural Science Foundation of China (81771006) and the Ministry of Health Fund Industry of China (201202005).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
1. Williams HL. The syndrome of physical or intrinsic allergy of the head; vasomotor rhinitis and the vasodilating pain syndrome, their relationship to myalgia and endolymphatic hydrops (Meniere's disease) and the reasons for including all four as the syndrome of physical allergy of the head. Proc Staff Meet Mayo Clin. (1946) 21:58–64.
2. Kjaer M. Meniere's disease and allergy. Acta Otolaryngol. (1947) 35:390–403. doi: 10.3109/00016484709124231
3. Brookes GB. Circulating immune complexes in Meniere's disease. Arch Otolaryngol Head Neck Surg. (1986) 112:536–40. doi: 10.1001/archotol.1986.03780050060010
4. Riente L, Bongiorni F, Nacci A, Migliorini P, Segnini G, Delle Sedie A, et al. Antibodies to inner ear antigens in Meniere's disease. Clin Exp Immunol. (2004) 135:159–63. doi: 10.1111/j.1365-2249.2004.02317.x
5. Patel M, Agarwal K, Arshad Q, Hariri M, Rea P, Seemungal BM, et al. Intratympanic methylprednisolone versus gentamicin in patients with unilateral Meniere's disease: A randomised, double-blind, comparative effectiveness trial. Lancet. (2016) 388:2753–62. doi: 10.1016/S0140-6736(16)31461-1
6. Frejo L, Soto-Varela A, Santos-Perez S, Aran I, Batuecas-Caletrio A, Perez-Guillen V, et al. Clinical subgroups in bilateral Meniere disease. Front Neurol. (2016) 7:182. doi: 10.3389/fneur.2016.00182
7. Frejo L, Martin-Sanz E, Teggi R, Trinidad G, Soto-Varela A, Santos-Perez S, et al. Extended phenotype and clinical subgroups in unilateral Meniere disease: A cross-sectional study with cluster analysis. Clin Otolaryngol. (2017) 42:1172–80. doi: 10.1111/coa.12844
8. Frejo L, Gallego-Martinez A, Requena T, Martin-Sanz E, Amor-Dorado JC, Soto-Varela A, et al. Proinflammatory cytokines and response to molds in mononuclear cells of patients with Meniere disease. Sci Rep. (2018) 8:5974. doi: 10.1038/s41598-018-23911-4
9. Krainer J, Siebenhandl S, Weinhausel A. Systemic autoinflammatory diseases. J Autoimmun. (2020) 109:102421. doi: 10.1016/j.jaut.2020.102421
10. Gallego-Martinez A, Lopez-Escamez JA. Genetic architecture of Meniere's disease. Hear Res. (2020) 397:107872. doi: 10.1016/j.heares.2019.107872
11. Zou J. Autoinflammatory characteristics and short-term effects of delivering high-dose steroids to the surface of the intact endolymphatic sac and incus in refractory Meniere's disease. J Otol. (2019) 14:40–50. doi: 10.1016/j.joto.2019.01.001
12. McDermott MF, Aksentijevich I, Galon J, McDermott EM, Ogunkolade BW, Centola M, et al. Germline mutations in the extracellular domains of the 55 kDa TNF receptor, TNFR1, define a family of dominantly inherited autoinflammatory syndromes. Cell. (1999) 97:133–44. doi: 10.1016/S0092-8674(00)80721-7
13. Hedrich CM. Shaping the spectrum—From autoinflammation to autoimmunity. Clin Immunol. (2016) 165:21–8. doi: 10.1016/j.clim.2016.03.002
14. Lopez-Escamez JA, Carey J, Chung WH, Goebel JA, Magnusson M, Mandala M, et al. Diagnostic criteria for Meniere's disease. J Vestib Res. (2015) 25:1–7. doi: 10.3233/VES-150549
15. Requena T, Espinosa-Sanchez JM, Cabrera S, Trinidad G, Soto-Varela A, Santos-Perez S, et al. Familial clustering and genetic heterogeneity in Meniere's disease. Clin Genet. (2014) 85:245–52. doi: 10.1111/cge.12150
16. Zou J, Zhao Z, Zhang G, Zhang Q, Pyykko I. MEFV. IRF8, ADA, PEPD, and NBAS gene variants and elevated serum cytokines in a patient with unilateral sporadic Meniere's disease and vascular congestion over the endolymphatic sac. J Otol. (2022) 17:175–81. doi: 10.1016/j.joto.2022.03.001
17. Zou J, Wang Z, Chen YK, Zhang GP, Lu JP, Zheng HL. Optimization of delivering minimum Gd-DTPA at the posterior upper point on tympanic medial wall and hT2W-3D-FLAIR sequence for detecting endolymphatic hydrops. Zhonghua Er Bi Yan Hou Tou Jing Wai Ke Za Zhi. (2018) 53:931–8. doi: 10.3760/cma.j.issn.1673-0860.2018.12.011
18. Zou J, Wang Z, Chen Y, Zhang G, Chen L, Lu J, et al. Detection of endolymphatic hydrops in Meniere's disease in 8 minutes using MIIRMR and a 20-channel coil after targeted gadolinium delivery. World J Otorhinolaryngol Head Neck Surg. (2019) 5:180–7. doi: 10.1016/j.wjorl.2019.04.001
19. Zou J, Chen L, Li H, Zhang G, Pyykko I, Lu J. High-quality imaging of endolymphatic hydrops acquired in 7 minutes using sensitive hT(2)W-3D-FLAIR reconstructed with magnitude and zero-filled interpolation. Eur Arch Otorhinolaryngol. (2022) 279:2279–90. doi: 10.1007/s00405-021-06912-4
20. Bernaerts A, Vanspauwen R, Blaivie C, van Dinther J, Zarowski A, Wuyts FL, et al. The value of four stage vestibular hydrops grading and asymmetric perilymphatic enhancement in the diagnosis of Meniere's disease on MRI. Neuroradiology. (2019) 61:421–9. doi: 10.1007/s00234-019-02155-7
21. Barath K, Schuknecht B, Naldi AM, Schrepfer T, Bockisch CJ, Hegemann SC. Detection and grading of endolymphatic hydrops in Meniere disease using MR imaging. Am J Neuroradiol. (2014) 35:1387–92. doi: 10.3174/ajnr.A3856
22. Zou J, Duan X, Zheng G, Zhao Z, Chen S, Dai P, et al. A novel PIK3CD C896T mutation detected in bilateral sudden sensorineural hearing loss using next generation sequencing: An indication of primary immunodeficiency. J Otol. (2016) 11:78–83. doi: 10.1016/j.joto.2016.06.001
23. Ioannidis NM, Rothstein JH, Pejaver V, Middha S, McDonnell SK, Baheti S, et al. REVEL: An ensemble method for predicting the pathogenicity of rare missense variants. Am J Hum Genet. (2016) 99:877–85. doi: 10.1016/j.ajhg.2016.08.016
24. Richards S, Aziz N, Bale S, Bick D, Das S, Gastier-Foster J, et al. Standards and guidelines for the interpretation of sequence variants: A joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet Med. (2015) 17:405–24. doi: 10.1038/gim.2015.30
25. Kim SH, Park HY, Choi HS, Chung HP, Choi JY. Functional and molecular expression of epithelial sodium channels in cultured human endolymphatic sac epithelial cells. Otol Neurotol. (2009) 30:529–34. doi: 10.1097/MAO.0b013e31819a8e0e
26. Szekanecz Z, McInnes IB, Schett G, Szamosi S, Benko S, Szucs G. Autoinflammation and autoimmunity across rheumatic and musculoskeletal diseases. Nat Rev Rheumatol. (2021) 17:585–95. doi: 10.1038/s41584-021-00652-9
27. Ou-Yang LJ, Tang KT. A case of adult onset Still's disease with mutations of the MEFV gene who is partially responsive to colchicine. Medicine. (2018) 97:e0333. doi: 10.1097/MD.0000000000010333
28. Yang N, Ma Y, Yao H, Chang Q, Zhang V, Liang Z, et al. Novel biallelic variants in COL7A1 cause recessive dystrophic epidermolysis bullosa. Mol Genet Genomic Med. (2020) 8:e1347. doi: 10.1002/mgg3.1347
29. Weinel S, Lucky AW, Uitto J, Pfendner EG, Choo D. Dystrophic epidermolysis bullosa with one dominant and one recessive mutation of the COL7A1 gene in a child with deafness. Pediatr Dermatol. (2008) 25:210–4. doi: 10.1111/j.1525-1470.2008.00636.x
30. Tanaka C, Hara T, Suzaki I, Maegaki Y, Takeshita K. Sensorineural deafness in siblings with adenosine deaminase deficiency. Brain Dev. (1996) 18:304–6. doi: 10.1016/0387-7604(96)00014-9
31. Xu X, Negandhi J, Min W, Tsui M, Post M, Harrison RV, et al. Early enzyme replacement therapy improves hearing and immune defects in adenosine deaminase deficient-mice. Front Immunol. (2019) 10:416. doi: 10.3389/fimmu.2019.00416
32. Venkatraman Girija U, Furze CM, Gingras AR, Yoshizaki T, Ohtani K, Marshall JE, et al. Molecular basis of sugar recognition by collectin-K1 and the effects of mutations associated with 3MC syndrome. BMC Biol. (2015) 13:27. doi: 10.1186/s12915-015-0136-2
33. Chen K, Wu W, Mathew D, Zhang Y, Browne SK, Rosen LB, et al. Autoimmunity due to RAG deficiency and estimated disease incidence in RAG1/2 mutations. J Allergy Clin Immunol. (2014) 133:880-2e10. doi: 10.1016/j.jaci.2013.11.038
34. Montenegro MM, Quaio CR, Palmeira P, Gasparini Y, Rangel-Santos A, Damasceno J, et al. Gene expression profile suggesting immunological dysregulation in two Brazilian Bloom's syndrome cases. Mol Genet Genomic Med. (2020) 8:e1133. doi: 10.1002/mgg3.1133
35. Boisson B, Laplantine E, Prando C, Giliani S, Israelsson E, Xu Z, et al. Immunodeficiency, autoinflammation and amylopectinosis in humans with inherited HOIL-1 and LUBAC deficiency. Nat Immunol. (2012) 13:1178–86. doi: 10.1038/ni.2457
36. Nakamura A, Tanaka R, Morishita K, Yoshida H, Higuchi Y, Takashima H, et al. Neuron-specific knockdown of the Drosophila fat induces reduction of life span, deficient locomotive ability, shortening of motoneuron terminal branches and defects in axonal targeting. Genes Cells. (2017) 22:662–9. doi: 10.1111/gtc.12500
37. Wilk P, Uehlein M, Piwowarczyk R, Dobbek H, Mueller U, Weiss MS. Structural basis for prolidase deficiency disease mechanisms. FEBS J. (2018) 285:3422–41. doi: 10.1111/febs.14620
38. Pulvirenti F, Zuntini R, Milito C, Specchia F, Spadaro G, Danieli MG, et al. Clinical associations of biallelic and monoallelic TNFRSF13B variants in Italian primary antibody deficiency syndromes. J Immunol Res. (2016) 2016:8390356. doi: 10.1155/2016/8390356
39. Zorn M, Kuhnisch J, Bachmann S, Seifert W. Disease relevance of rare VPS13B missense variants for neurodevelopmental Cohen syndrome. Sci Rep. (2022) 12:9686. doi: 10.1038/s41598-022-13717-w
40. Karasawa T, Sato R, Imaizumi T, Hashimoto S, Fujita M, Aizawa T, et al. Glomerular endothelial expression of type I IFN-stimulated gene, DExD/H-Box helicase 60 via toll-like receptor 3 signaling: possible involvement in the pathogenesis of lupus nephritis. Ren Fail. (2022) 44:137–45. doi: 10.1080/0886022X.2022.2027249
41. Ostrowski M, Carmo NB, Krumeich S, Fanget I, Raposo G, Savina A, et al. Rab27a and Rab27b control different steps of the exosome secretion pathway. Nat Cell Biol. (2010) 12:19–30. doi: 10.1038/ncb2000
42. Oud MM, Tuijnenburg P, Hempel M, van Vlies N, Ren Z, Ferdinandusse S, et al. Mutations in EXTL3 cause neuro-immuno-skeletal dysplasia syndrome. Am J Hum Genet. (2017) 100:281–96. doi: 10.1016/j.ajhg.2017.01.013
43. Keller MD, Ganesh J, Heltzer M, Paessler M, Bergqvist AG, Baluarte HJ, et al. Severe combined immunodeficiency resulting from mutations in MTHFD1. Pediatrics. (2013) 131:e629–34. doi: 10.1542/peds.2012-0899
44. Sumpter R Jr, Sirasanagandla S, Fernandez AF, Wei Y, Dong X, Franco L, et al. Fanconi anemia proteins function in mitophagy and immunity. Cell. (2016) 165:867–81. doi: 10.1016/j.cell.2016.04.006
45. Castella M, Pujol R, Callen E, Trujillo JP, Casado JA, Gille H, et al. Origin, functional role, and clinical impact of Fanconi anemia FANCA mutations. Blood. (2011) 117:3759–69. doi: 10.1182/blood-2010-08-299917
46. Staufner C, Peters B, Wagner M, Alameer S, Baric I, Broue P, et al. Defining clinical subgroups and genotype-phenotype correlations in NBAS-associated disease across 110 patients. Genet Med. (2020) 22:610–21. doi: 10.1038/s41436-019-0698-4
47. Munthe-Fog L, Hummelshoj T, Honore C, Madsen HO, Permin H, Garred P. Immunodeficiency associated with FCN3 mutation and ficolin-3 deficiency. N Engl J Med. (2009) 360:2637–44. doi: 10.1056/NEJMoa0900381
48. Cabrera S, Sanchez E, Requena T, Martinez-Bueno M, Benitez J, Perez N, et al. Intronic variants in the NFKB1 gene may influence hearing forecast in patients with unilateral sensorineural hearing loss in Meniere's disease. PLoS ONE. (2014) 9:e112171. doi: 10.1371/journal.pone.0112171
49. Yamada T, Ogi K, Sakashita M, Kanno M, Kubo S, Ito Y, et al. Toll-like receptor ligands induce cytokine and chemokine production in human inner ear endolymphatic sac fibroblasts. Auris Nasus Larynx. (2017) 44:398–403. doi: 10.1016/j.anl.2016.10.007
50. Martin-Sierra C, Requena T, Frejo L, Price SD, Gallego-Martinez A, Batuecas-Caletrio A, et al. A novel missense variant in PRKCB segregates low-frequency hearing loss in an autosomal dominant family with Meniere's disease. Hum Mol Genet. (2016) 25:3407–15. doi: 10.1093/hmg/ddw183
51. Zou J, Zhao Z, Song X, Zhang G, Li H, Zhang Q, et al. Elevated G-CSF, IL8, and HGF in patients with definite Meniere's disease may indicate the role of NET formation in triggering autoimmunity and autoinflammation. Sci Rep. (2022) 22:8. doi: 10.1038/s41598-022-20774-8
52. Onuki J, Takahashi M, Odagiri K, Wada R, Sato R. Comparative study of the daily lifestyle of patients with Meniere's disease and controls. Ann Otol Rhinol Laryngol. (2005) 114:927–33. doi: 10.1177/000348940511401206
53. Kitahara T, Okayasu T, Ito T, Fujita H, Ueda K. Endolymphatic Sac drainage surgery and plasma stress hormone vasopressin levels in Meniere's disease. Front Neurol. (2021) 12:722217. doi: 10.3389/fneur.2021.722217
54. Pullens B, Verschuur HP, van Benthem PP. Surgery for Meniere's disease. Cochrane Database Syst Rev. (2013) 2013:CD005395. doi: 10.1002/14651858.CD005395.pub3
55. Zou J, Pyykkö I. Outcomes of endolymphatic shunt surgery in Ménière's disease indicate potential contribution of shear stress instead of relieving the endolymphatic hydrops. Otolaryngology. (2014) 4:171. doi: 10.4172/2161-119X.1000171
56. Koelsch S, Boehlig A, Hohenadel M, Nitsche I, Bauer K, Sack U. The impact of acute stress on hormones and cytokines, and how their recovery is affected by music-evoked positive mood. Sci Rep. (2016) 6:23008. doi: 10.1038/srep23008
57. Requena T, Cabrera S, Martin-Sierra C, Price SD, Lysakowski A, Lopez-Escamez JA. Identification of two novel mutations in FAM136A and DTNA genes in autosomal-dominant familial Meniere's disease. Hum Mol Genet. (2015) 24:1119–26. doi: 10.1093/hmg/ddu524
58. Gallego-Martinez A, Requena T, Roman-Naranjo P, Lopez-Escamez JA. Excess of rare missense variants in hearing loss genes in sporadic Meniere disease. Front Genet. (2019) 10:76. doi: 10.3389/fgene.2019.00076
3M syndrome, Carnevale, Mingarelli, Malpuech, and Michels syndrome; ADA, adenosine deaminase; BCR, B-cell receptor; BLM, Bloom syndrome; COLEC11, collectin subfamily member 11; COL7A1, collagen type VII α1 chain; DTNA, dystrobrevin α; DPT, dermatopontin; EH, endolymphatic hydrops; ERCC6L2, ERCC excision repair 6 Like 2; ER, endoplasmic reticulum; ESCS, high-dose steroid delivery to the surface of the intact endolymphatic sac and incus; EXTL3:, exostosin like glycosyltransferase 3; FAM136A:, family with sequence similarity 136 member A; FANCA, Fanconi anemia, complementation group A; FAT4, FAT atypical cadherin 4; FCN3, Ficolin 3; G-CSF, granulocyte colony-stimulating factor; Gd-DTPA, gadolinium-diethylenetriamine pentaacetic acid; HGF, hepatocyte growth factor; hT2W-FLAIR, heavily T2-weighted 3-dimensional fluid-attenuated inversion recovery; hT2W-FLAIR-ZFI, hT2W-FLAIR reconstructed with magnitude and zero-filled interpolation; IL-1β, interleukin-1β; IL-1R, interleukin-1 receptor; IL2RG, interleukin-2 receptor-γ; IL6 and 8, interleukin-6 and 8; ITDex, intratympanic dexamethasone; LPIN2, lipin 2; MD, Meniere's disease; MEFV, Mediterranean fever innate immunity regulator; MIIRMR, hT2W-FLAIR with medium inversion time inversion recovery imaging and magnitude reconstruction; MOGS, mannosyl-oligosaccharide glucosidase; MRI, magnetic resonance imaging; MTHFD1, methylenetetrahydrofolate dehydrogenase, cyclohydrolase, and formyltetrahydrofolate synthetase 1; NBAS, neuroblastoma amplified sequence; PEPD, Peptidase D; POLE, DNA polymerase ε; PRF1, perforin 1; RAG2, recombination activating 2; RNF31, ring finger protein 31; SEMA3D, semaphorin 3D; SPACE, T2-sampling perfection with application-optimized contrasts using a flip angle evolution; TCR, T-cell receptor; TIRAP, Toll-interleukin 1 receptor domain containing adaptor protein; TLR3 and TLR4, toll-like receptor 3 and 4; TNF-α, tumor necrosis factor-α; TNFR1, tumor necrosis factor receptor 1; TNFRSF13B, tumor necrosis factor receptor superfamily member 13B; TNFSF12, tumor necrosis factor ligand superfamily member 12; UNC13D, unc-13 homolog D; VPS13B, vacuolar protein sorting 13 homolog B.
Keywords: Meniere's disease, immunology, genetics, mutation, diagnostics
Citation: Zou J, Zhang G, Li H, Zhao Z, Zhang Q, Pyykkö I and Mäkitie A (2023) Multiple genetic variants involved in both autoimmunity and autoinflammation detected in Chinese patients with sporadic Meniere's disease: a preliminary study. Front. Neurol. 14:1159658. doi: 10.3389/fneur.2023.1159658
Received: 06 February 2023; Accepted: 20 April 2023;
Published: 18 May 2023.
Edited by:
Craig Kinnear, Stellenbosch University, South AfricaReviewed by:
Paola Perin, University of Pavia, ItalyCopyright © 2023 Zou, Zhang, Li, Zhao, Zhang, Pyykkö and Mäkitie. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Jing Zou, amluZy56b3VAaGVsc2lua2kuZmk=
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.