 Ping Jin
Ping Jin Yu Wang
Yu Wang Na Nian1
Na Nian1 Xiao-Ming Fu
Xiao-Ming Fu- 1Department of Neurology, The Affiliated Hospital of Institute of Neurology, Anhui University of Chinese Medicine, Hefei, China
- 2Institute of Neurology, Anhui University of Chinese, Hefei, China
Hereditary spastic paraplegias (HSP) are inherited neurodegenerative disorders characterized by progressive paraplegia and spasticity in the lower limbs. SPG48 represents a rare genotype characterized by mutations in AP5Z1, a gene playing a role in intracellular membrane trafficking. This study describes a case of a 53-year-old male patient with SPG48 presenting spastic paraplegia, infertility, hearing impairment, cognitive abnormalities and peripheral neuropathy. The Sanger sequencing revealed a homozygous deletion in the chr 7:4785904-4786677 region causing a premature stop codon in exon 10. The patient's brother was heterozygous for the mutation. The brain magnetic resonance imaging found a mild brain atrophy and white matter lesions. In the analysis of the auditory thresholds, we found a significant hearing decrease in both ears.
Introduction
Hereditary spastic paraplegia (HSP) represents a rare group of neurodegenerative disorders characterized by progressive lower limb spasticity and weakness (1). HSP can be inherited in an autosomal dominant, autosomal recessive, X-linked recessive or mitochondrial manner (2). Several genotypes (from SPG1 to SPG82) and pathogenic genes have been identified, with the most common causative genes being SPAST, ATL1 and REEP1 (3). Damaging variants in AP5Z1 are associated with SPG48, a rare autosomal recessive condition with a variegated clinical phenotype (4). To date, we know of 14 case reports related to this gene. In this study, we described the clinical features and imaging findings of a 43-year-old Chinese male patient with a large deletion in the AP5Z1 gene. The patient reported spastic paraplegia, hearing impairment and infertility.
Case presentation
During April 2020, a 53-year-old Chinese male patient was admitted to our hospital due to progressive walking difficulties and distal weakness at lower limbs. He got low grades during primary school, and then did not complete it. He married at the age of 28 years. When he was 35 years old, he received a diagnosis of infertility. In 2010, the patient experienced instability and mild shaking during walking. Walking instability worsened in 2015, when the patient reported difficulties with standing up from a squatting position. In 2019, the patient started using crutches to walk. Since 2017, hearing capacity has gradually decreased in both ears. Since 2019, the patient only recognizes sounds spoken aloud, without pain, infection and/or tinnitus in the ears. The patient's parents were first cousins, the family history was unremarkable for similar neurologic and auditory findings.
On examination, we observed bilateral spastic gait, tendon hyperreflexia, ankle clonuses (+) and bilateral pyramidal signs. The Romberg's test was positive. A mild spastic dysarthria was also evident. Sensory examination revealed loss of vibratory sensations at the great toes and reduced pinprick sensations in to the lower limbs. The spastic paraplegia rating scale (SPRS) scored 19 points. The orientation in space and time was normal but attention and calculation wreere impaired. Hearing loss affected both ears, with positive left (+) and right (±) Rinne tests. The Weber test was biased toward the right ear. Both appearance and size of the testis were normal. Blood laboratory tests and cerebrospinal fluid evaluation were normal, including systemic hormones. Brain magnetic resonance imaging (MRI) showed a mild brain atrophy with periventricular white matter hyperintensities on fluid-attenuated inversion recovery (FLAIR) sequences (Figure 1). Cervical and lumbar MRI described degenerative changes in the lumbar spine and mild distension of multiple intervertebral disks, with no significant compression or other abnormal signals of the spinal cord. The electromyography investigation showed decreased amplitudes of compound muscle action potentials (CMAPs) in the right common peroneal nerve and left tibial nerve. A prolonged latency of sensory nerve action potentials (SNAPs) was observed in bilateral sural nerves, with an increased latency of the F wave in median and tibial nerves of both limbs. Bilateral tibial H-reflex investigation showed a prolonged latency, with a neurogenic damage in the tibial anterior muscles. The audiometry evaluation found a sensorineural deafness on the left side and a mixed deafness on the right side. The evoked potentials suggested an increased binaural auditory threshold. On the brain stem auditory evoked potentials, we found bilateral prolonged waves I and III. A prolonged left P40 latency was also described on somatosensory evoked cortical potentials. The results obtained on cognitive tests were 27 points on the Mini Mental State Examination (MMSE) and 18 points on the Montreal Cognitive Assessment (MoCA).
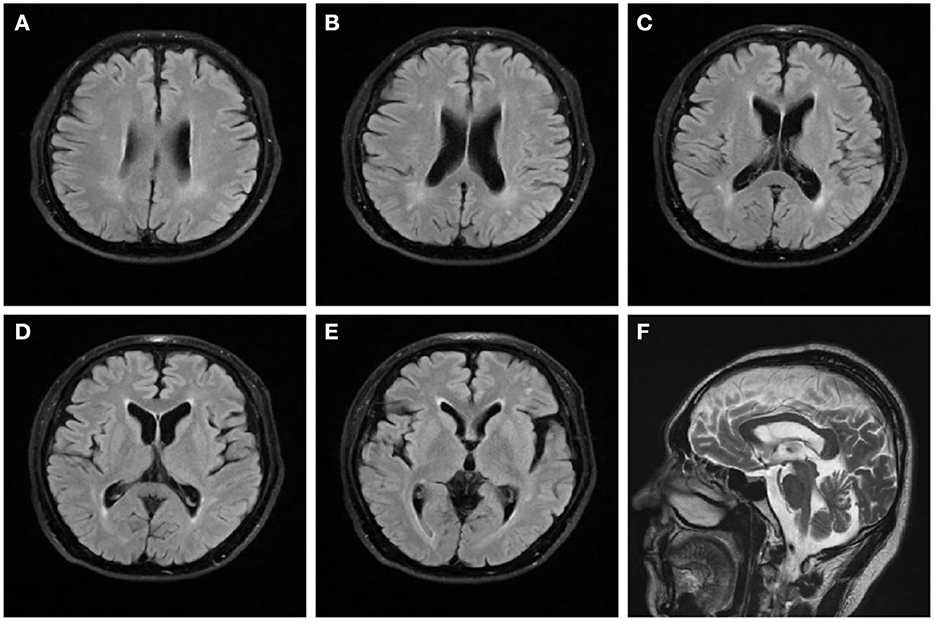
Figure 1. Brain magnetic resonance imaging of the patient. FLAIR sequences (A–E) reveal high signal changes in the periventricular white matter and mild brain atrophy. T2-weighted (F) sequence shows that the corpus callosum is normal.
We confirmed the location of gene deletion by Sanger sequencing, identifying homozygous deletions in the chr7:4785904-4786677 region involving exon 10 of the AP5Z1 gene, NM_014855.3, c.1133-345_1311 + 249del, p.G378Vfs*93X (Figure 2). Using the software Mutalyzer 2.0.35, we found that the deletion causes early termination of amino acid synthesis. The patient's brothers were asymptomatic, with heterozygous mutations identified by fluorescent quantitative polymerase chain reaction (PCR). The primers were as follows: AP5Z1-10 (forward: AACCAGTCACAGAAGCACGG, reverse: GAACAGGTGGAGGTTGTCCC), and the sequences of the PCR products were determined using the ABI 9700 Genetic analyzer (ABI, Foster City, CA). Reaction system: total volume 10ul, including 1ul of genomic DNA template, 5ul of SYBR Green Mix, 0.5ul of upstream and downstream primers, 3ul of ddH2O. Reaction conditions: 95°C, 30s, (95°C, 15s; 60°C, 15s; 72°C, 15s) × 46 cycles. The final data was analyzed by 2−ΔCt. The results showed that relative to two copies of healthy people, the patient had 0 copies, his brothers had a single copy (Figure 3). This deletion mutation is considered a pathogenic variant according to American College of Medical Genetics and Genomics standards and guidelines. Based on clinical features, imaging findings and genetic abnormalities, we diagnosed the patient with SPG48. Oral administration of baclofen and tizanidine improved his symptoms.
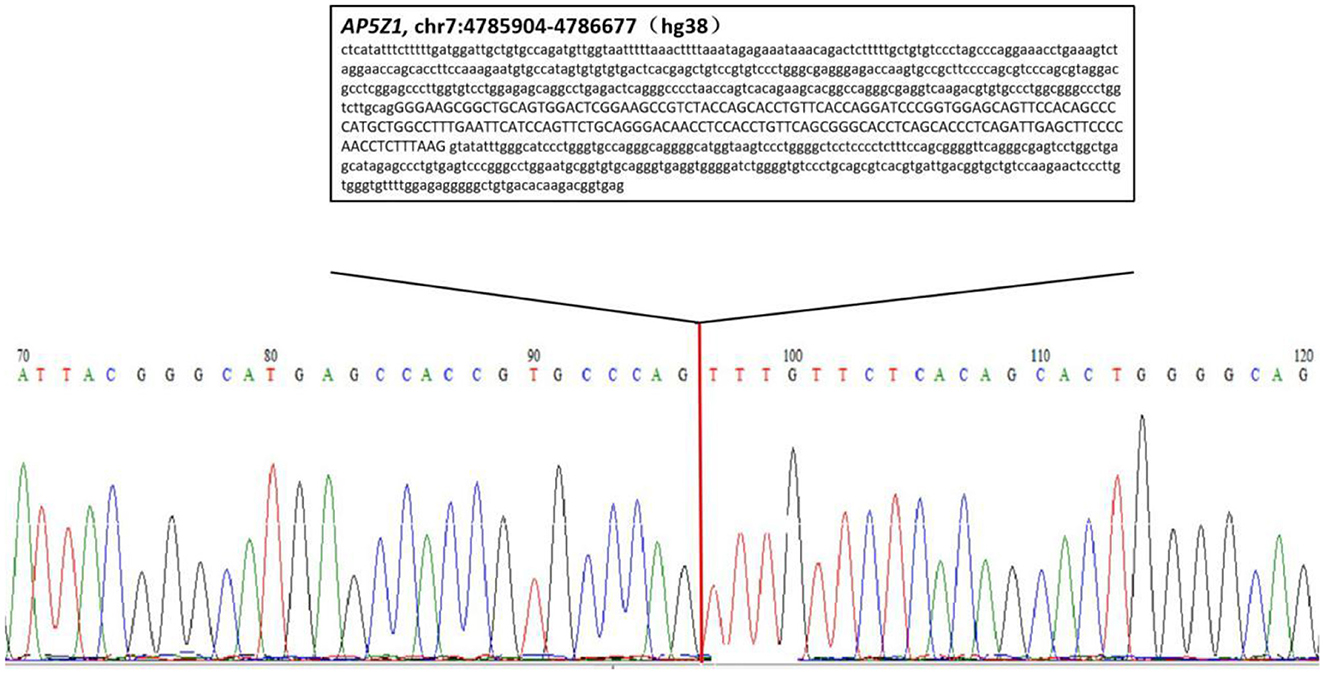
Figure 2. Sequencing chromatogram of the patient. The lower case letters represent the nucleotide sequence of intron regions while capital letters represent the nucleotide sequence of exon 10.
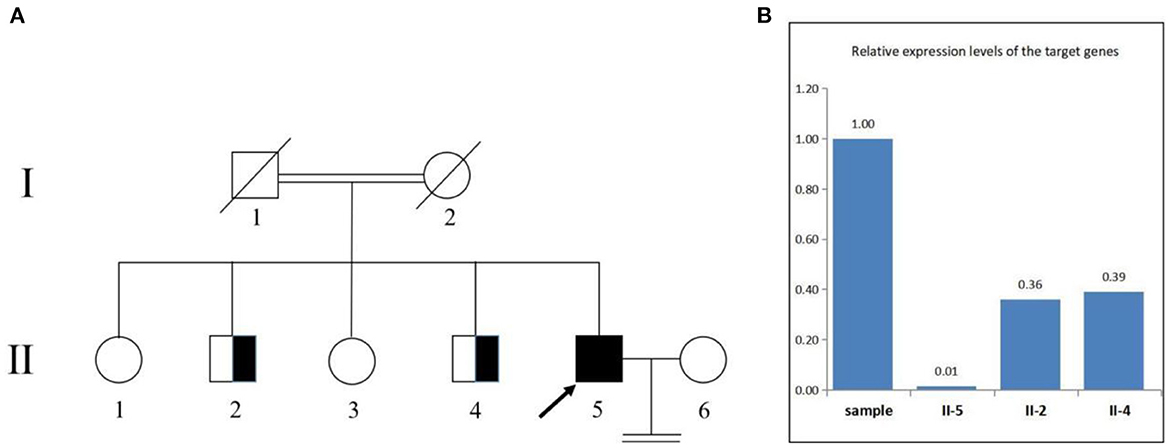
Figure 3. Pedigree of the family (A). The deletion involving exon 10 of the AP5Z1 gene was verified by fluorescence quantitative polymerase chain reaction (B). The squares indicate males, the circles indicate females. The pedigree chart shows that the patient's father (I-1) and mother (I-2) were cousins and deceased. The proband (II-5) is indicated by a black arrow. The patient's first brother (II-2) and second brother (II-4) were characterized by heterozygous mutations. The patient's sister refused to be tested.
Discussion
In 2010, Slabicki et al. (5) identified SPG48 as a novel genotype associated with HSP (5). SPG48 can be defined as a secondary lysosomal storage disease involving adaptor proteins (APs), ubiquitously expressed protein complexes ranging from AP1 to AP5 (6). AP5 is a cation-independent mannose 6-phosphate receptor located in endosomes and lysosomes that transports Golgi membrane protein 1 to the Golgi apparatus (7). AP5 is expressed in non-neuronal and neuronal tissues, such as cerebral cortex, hippocampus and cerebellum. Neuronal cells are abnormally sensitive to dysfunction of lysosomes and protein accumulation (8). The subtype SPG48 is associated with mutations in the zeta subunit of AP5, impairing the complex formation of AP5. This, in turn, results in defects in lysosomal structure and function (9). A mouse model of SPG48 showed that loss-of-function AP5 variants blocked autophagy, leading to an aberrant accumulation of autophagic vacuoles and axon degeneration (10). Postmortem findings in patients with bi-allelic AP5Z1 mutations demonstrated an extensive degeneration of cortical and subcortical regions, including basal ganglia, brainstem and spinal cord (8). Such alterations are common in neurodegenerative disorders, including HSP, amyotrophic lateral sclerosis and spinocerebellar ataxia.
As of today, 14 cases of SPG48 have been reported worldwide (Table 1). Most of them were characterized by point mutations, whereas our patient reported a segmental deletion in the AP5Z1 gene. Overall, the age of onset of SPG48 spans many decades and clinical manifestations are highly heterogeneous, including spastic paraplegia, urinary incontinence, ataxia, intellectual disability, sensorimotor neuropathy, Parkinson's syndrome, dystonia and eye disturbances [including pigmentary retinopathy, optic atrophy, cataract, glaucoma and ophthalmoplegia (11)], the atypical SPG48 did not even show spastic paraplegia (8). In most patients, brain MRI shows white matter lesions around corona radiata, semioval center and lateral ventricles. The “ears of the lynx” imaging sign suggests the presence of a genetic mutation, likely characteristic of HSP (12). The narrowing of corpus callosum is another relevant imaging characteristic in these patients. A few patients showed a normal brain MRI, whereas individual cases reported a diffuse brain atrophy and/or abnormal spinal cord signals.
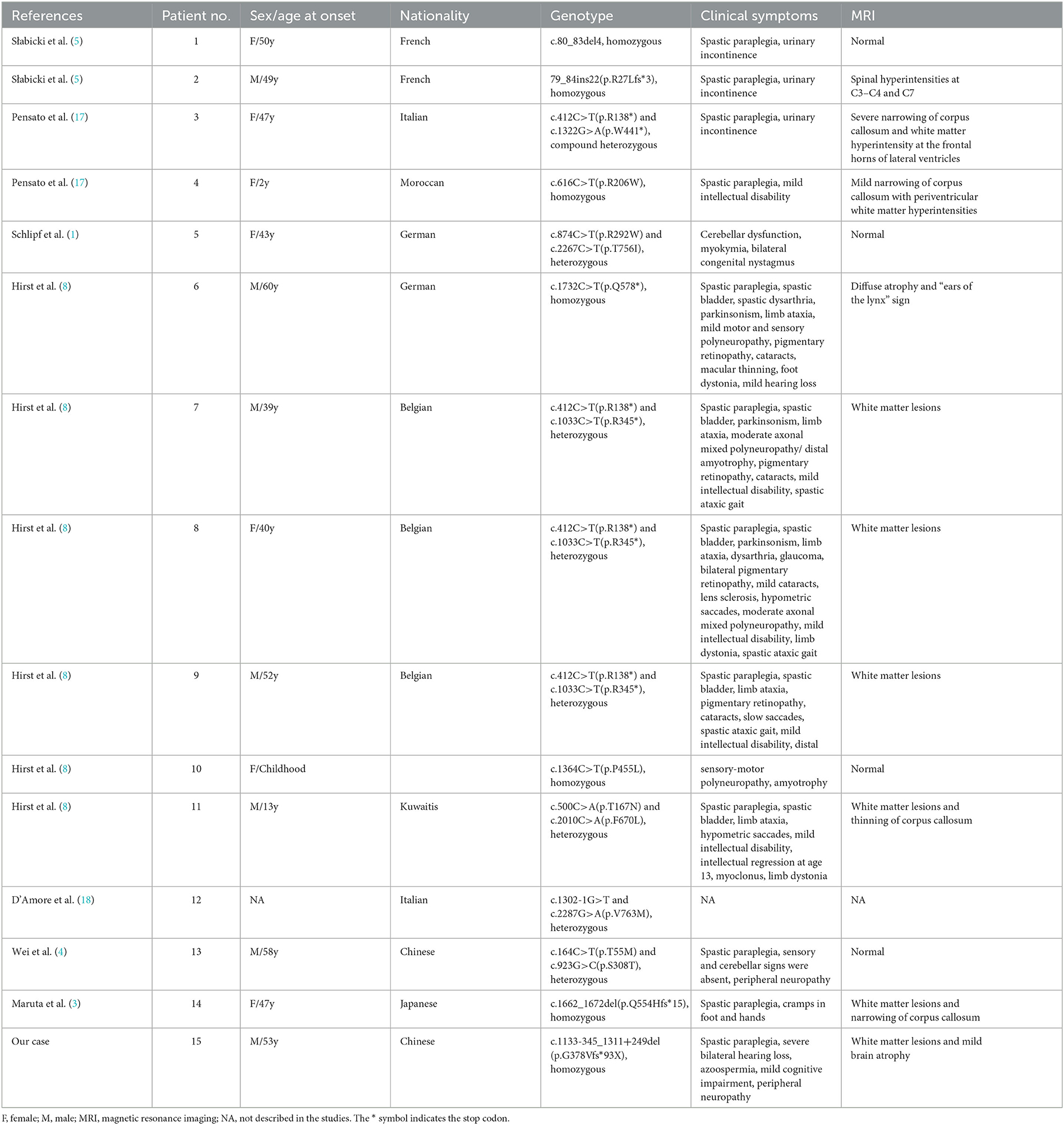
Table 1. Clinical, radiological and genetic features of SPG48/AP5Z1 patients.
Our patient reported spastic paraplegia, peripheral neuropathy and mild cognitive impairment, and for the first time findings such as azoospermia and severe bilateral hearing loss. Previous studies found a significant reduction in mitochondrial length and density in HSP neurons, suggesting that an abnormal mitochondrial morphology may contribute to axonal defects (13). A recent study found that an impaired mitochondrial dynamics contributed to axonal degeneration in SPG11 and SPG48 neurons, resulting in less efficient and shortened axons (14). Mitochondrial function is also related to human sperm motility and morphology (15). In addition, loss of mitochondrial function has been associated with deafness (16). Therefore, we might speculate that AP5Z1 gene mutations may affect both spermatogenesis and hearing.
SPG48 greatly impacts the central nervous system, with complex and varying clinical and radiographic characteristics. Clinicians should be aware of non-neurological findings in the presence of suspected SGP48 cases, as early recognition of the disease will minimize unnecessary evaluations and treatments. At present, there is no effective treatment for SPG48, symptomatic and supportive treatment including baclofen and tizanidine can moderately improve the symptoms of spastic paraplegia and urinary incontinence. Future therapies might restore mitochondrial function.
Data availability statement
The original contributions presented in the study are included in the article/supplementary material, further inquiries can be directed to the corresponding author.
Ethics statement
Written informed consent was obtained from the individual(s) and/or minor(s)' legal guardian/next of kin for the publication of any potentially identifiable images or data included in this article.
Author contributions
PJ wrote the manuscript with input from all other authors. All authors contributed to the article and approved the submitted version.
Acknowledgments
We thank the patient and his family for placing their trust in us. We also acknowledge TopEdit LLC for the linguistic editing and proofreading during the preparation of this manuscript.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher's note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Schlipf NA, Schüle R, Klimpe S, Karle KN, Synofzik M, Wolf J, et al. AP5Z1/SPG48 frequency in autosomal recessive and sporadic spastic paraplegia. Mol Genet Genomic Med. (2014) 2:379–82. doi: 10.1002/mgg3.87
2. Klebe S, Stevanin G, Depienne C. Clinical and genetic heterogeneity in hereditary spastic paraplegias: from SPG1 to SPG72 and still counting. Rev Neurol. (2015) 171:505–30. doi: 10.1016/j.neurol.2015.02.017
3. Maruta K, Ando M, Otomo T, Takashima H. A case of spastic paraplegia 48 with a novel mutation in the AP5Z1 gene. Rinsho Shinkeigaku. (2020) 60:543–8. doi: 10.5692/clinicalneurol.60.cn-001419
4. Wei Q, Dong HL, Pan LY, Chen CX, Yan YT, Wang RM, et al. Clinical features and genetic spectrum in Chinese patients with recessive hereditary spastic paraplegia. Transl Neurodegener. (2019) 8:19. doi: 10.1186/s40035-019-0157-9
5. Słabicki M, Theis M, Krastev DB, Samsonov S, Mundwiller E, Junqueira M, et al. A genome-scale DNA repair RNAi screen identifies SPG48 as a novel gene associated with hereditary spastic paraplegia. PLoS Biol. (2010) 8:e1000408. doi: 10.1371/journal.pbio.1000408
6. Platt FM, Boland B, van der Spoel AC. The cell biology of disease: lysosomal storage disorders: the cellular impact of lysosomal dysfunction. J Cell Biol. (2012) 199:723–34. doi: 10.1083/jcb.201208152
7. Hirst J, Itzhak DN, Antrobus R, Borner GHH, Robinson MS. Role of the AP-5 adaptor protein complex in late endosome-to-Golgi retrieval. PLoS Biol. (2018) 16:e2004411. doi: 10.1371/journal.pbio.2004411
8. Hirst J, Madeo M, Smets K, Edgar JR, Schols L, Li J, et al. Complicated spastic paraplegia in patients with AP5Z1 mutations (SPG48). Neurol Genet. (2016) 2:e98. doi: 10.1212/NXG.0000000000000098
9. Hirst J, Edgar JR, Esteves T, Darios F, Madeo M, Chang J, et al. Loss of AP-5 results in accumulation of aberrant endolysosomes: defining a new type of lysosomal storage disease. Hum Mol Genet. (2015) 24:4984–96. doi: 10.1093/hmg/ddv220
10. Khundadze M, Ribaudo F, Hussain A, Rosentreter J, Nietzsche S, Thelen M, et al. A mouse model for SPG48 reveals a block of autophagic flux upon disruption of adaptor protein complex five. Neurobiol Dis. (2019) 127:419–31. doi: 10.1016/j.nbd.2019.03.026
11. de Freitas JL, Rezende Filho FM, Sallum JMF, França MC Jr, Pedroso JL, Barsottini OGP. Ophthalmological changes in hereditary spastic paraplegia and other genetic diseases with spastic paraplegia. J Neurol Sci. (2020) 409:116620. doi: 10.1016/j.jns.2019.116620
12. Breza M, Hirst J, Chelban V, Banneau G, Tissier L, Kol B. et al. Expanding the spectrum of AP5Z1-related hereditary spastic paraplegia (HSP-SPG48): a multicenter study on a rare disease. Mov Disord. (2021) 36:1034–8. doi: 10.1002/mds.28487
13. Denton K, Mou Y, Xu CC, Shah D, Chang J, Blackstone C, et al. Impaired mitochondrial dynamics underlie axonal defects in hereditary spastic paraplegias. Hum Mol Genet. (2018) 27:2517–30. doi: 10.1093/hmg/ddy156
14. Chen Z, Chai E, Mou Y, Roda RH, Blackstone C, Li XJ. Inhibiting mitochondrial fission rescues degeneration in hereditary spastic paraplegia neurons. Brain. (2022) 145:awab488. doi: 10.1093/brain/awab488
15. Boguenet M, Bouet PE, Spiers A, Reynier P, May-Panloup P. Mitochondria: their role in spermatozoa and in male infertility. Hum Reprod Update. (2021) 27:697–719. doi: 10.1093/humupd/dmab001
16. Xue L, Chen Y, Tang X, Yao J, Huang H, Wang M, et al. A deafness-associated mitochondrial DNA mutation altered the tRNASer(UCN) metabolism and mitochondrial function. Mitochondrion. (2019) 46:370–9. doi: 10.1016/j.mito.2018.10.001
17. Pensato V, Castellotti B, Gellera C, Pareyson D, Ciano C, Nanetti L, et al. Overlapping phenotypes in complex spastic paraplegias SPG11, SPG15, SPG35 and SPG48. Brain. (2014) 137:1907–20. doi: 10.1093/brain/awu121
Keywords: AP5Z1 gene mutation, SPG48, azoospermia, impaired hearing, brain MRI
Citation: Jin P, Wang Y, Nian N, Wang G-Q and Fu X-M (2023) Hereditary spastic paraplegia (SPG 48) with deafness and azoospermia: A case report. Front. Neurol. 14:1156100. doi: 10.3389/fneur.2023.1156100
Received: 01 February 2023; Accepted: 21 March 2023;
Published: 03 April 2023.
Edited by:
Huifang Shang, Sichuan University, ChinaReviewed by:
Gabriella Silvestri, Catholic University of the Sacred Heart, ItalyAlice Barbara Schindler, National Institute of Neurological Disorders and Stroke, United States
Copyright © 2023 Jin, Wang, Nian, Wang and Fu. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Xiao-Ming Fu, MTUzMTk0OTM1NkBxcS5jb20=
†ORCID: Ping Jin orcid.org/0000-0001-6528-8330
Xiao-Ming Fu orcid.org/0000-0002-9127-081X