 Mengyue Yao
Mengyue Yao Yulei Hao1†
Yulei Hao1† Tian Wang
Tian Wang Jiachun Feng
Jiachun Feng
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Neurol., 21 March 2023
Sec. Stroke
Volume 14 - 2023 | https://doi.org/10.3389/fneur.2023.1149671
This article is part of the Research TopicPost-Stroke Complications: Mechanisms, Diagnosis, and TherapiesView all 20 articles
The transient elevation of blood glucose produced following acute ischaemic stroke (AIS) has been described as stress-induced hyperglycaemia (SIH). SIH is common even in patients with AIS who have no previous diagnosis of diabetes mellitus. Elevated blood glucose levels during admission and hospitalization are strongly associated with enlarged infarct size and adverse prognosis in AIS patients. However, insulin-intensive glucose control therapy defined by admission blood glucose for SIH has not achieved the desired results, and new treatment ideas are urgently required. First, we explore the various definitions of SIH in the context of AIS and their predictive value in adverse outcomes. Then, we briefly discuss the mechanisms by which SIH arises, describing the dual effects of elevated glucose levels on the central nervous system. Finally, although preclinical studies support lowering blood glucose levels using insulin, the clinical outcomes of intensive glucose control are not promising. We discuss the reasons for this phenomenon.
Acute ischaemic stroke (AIS) is an acute brain injury that often occurs when an artery is suddenly blocked. It is one of the most common causes of severe disability and mortality worldwide (1). One description of stress-induced hyperglycaemia (SIH) is increased blood glucose due to a sudden clinical event that returns to baseline following the acute phase (2). SIH is commonly seen in AIS patients, even those with no history of diabetes mellitus (DM) diagnosis (3). Hyperglycaemia has been demonstrated to be independently linked to an adverse prognosis in AIS patients (4). Patients with previously undiagnosed DM are more likely to have increased short- and long-term mortality due to elevated blood glucose on admission to the hospital than patients with previously identified DM (3, 5). Hyperglycaemia has a dual effect on the central nervous system, with blood glucose above a specific range accelerating thrombosis, increasing stress and inflammatory responses, exacerbating reperfusion injury, and leading to lactate accumulation and mitochondrial dysfunction. This ultimately contributes to the transformation of the ischaemic penumbra into an infarcted region (6, 7). Therefore, it appears to be essential to maintain blood glucose levels in AIS. Although there are many published studies on SIH, there is no agreed-upon definition of SIH with AIS, such as admission blood glucose (ABG), stress hyperglycaemia ratio (SHR), or glucose variability (GV). The current large prospective controlled studies of SIH in patients with AIS treated with intravenous insulin have been designed using ABG as a definition and have not yielded desirable results (8–13).
Thus, in this review, we will discuss which optimal glycaemic definition of SIH is most associated with poor prognosis following AIS and whether the best definition cut-off value makes a difference in the presence and absence of recognized DM. Finally, we discuss whether insulin-intensive glucose control therapy can aid AIS patients with an improved prognosis.
In the context of AIS, SIH is measured in different ways. However, it is still being determined which are the optimal glucose metrics for measuring SIH, as well as the definition and cut-off value of SIH that lead to adverse outcomes in AIS patients in the presence and absence of recognized DM (Table 1).
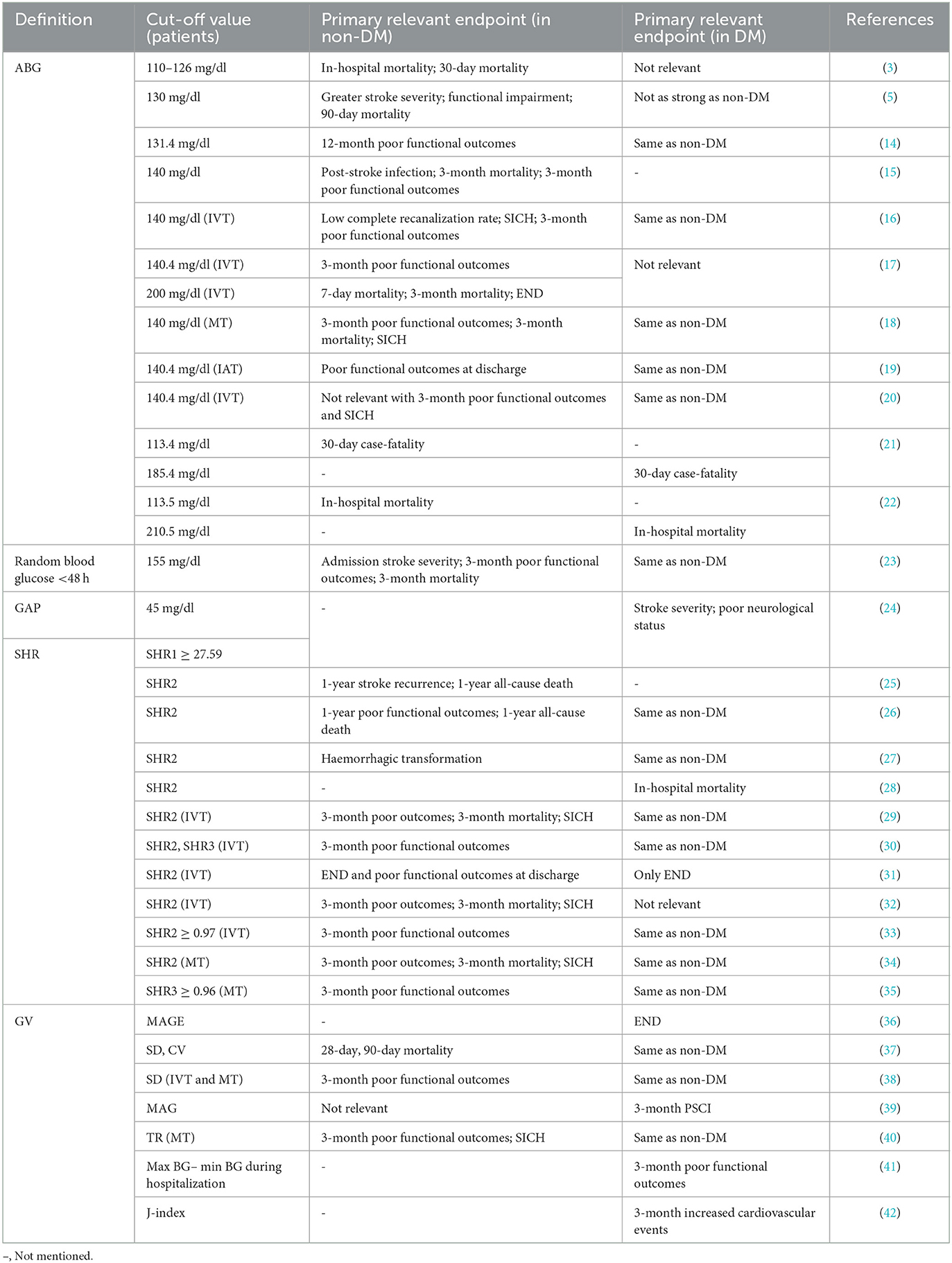
Table 1. The definition and a cut-off value of SIH related to adverse prognosis in ischaemic stroke patients with or without a previous DM diagnosis.
Several earlier clinical studies defined hyperglycaemia in terms of the blood glucose value, or ABG, within 24 h of admission to the hospital for AIS patients. The cut-off values for ABG to define hyperglycaemia in AIS patients vary between studies.
Whether elevated ABG has different effects on the prognosis of AIS patients with and without a DM diagnosis has been widely discussed. In a meta-analysis of 32 cohort studies, Capes and colleagues (3) observed that non-DM patients with ABG above 126 mg/dl had a threefold increased admission and 30-day mortality compared to patients with normal ABG. In addition, the researchers found that ABG was unrelated to an increase in short-term mortality in DM patients. A prospective study (5) including 447 AIS patients suggests that, in non-DM patients, 90-day mortality was 3.4 times greater in patients with ABG above 130 mg/ dl than in those with normoglycaemia. In comparison, for DM patients, the hazard ratio was 1.6. Another observational study (15) of 2,550 AIS patients found that ABG ≥140 mg/dl was independently correlated with post-stroke infection in non-DM patients. In contrast, the same result was not found in DM patients. However, a series of studies suggested that ABG ≥140 mg/dl is strongly correlated with the risk of symptomatic intracranial hemorrhage (SICH) after intravenous thrombolysis (IVT), mechanical thrombectomy (MT) or intra-arterial treatment (IAT) and a poor 3-month clinical prognosis in AIS patients. These studies did not specifically distinguish between non-DM and DM patient populations (16, 18–20). A later study (17) carefully differentiated between these two groups and showed that ABG ≥140 mg/dl was related to an adverse prognosis after IVT in non-DM AIS patients. In conclusion, absolute increases in hyperglycaemia were more strongly related to short- or long-term mortality, post-stroke infection, and adverse prognosis in AIS patients with non-DM than in patients with previously diagnosed DM.
Furthermore, Snarska et al. (22) discovered that the cut-off values for ABG, which predicted the risk of in-hospital death, were distinct in AIS patients without and with DM (113.5 mg/dl and 210.5 mg/dl). This difference is consistent with that reported by Farrokhnia et al. (21) and has been found similarly in patients with myocardial infarction (43). The reason for this discrepancy may be that the baseline glucose metabolism levels are usually significantly higher in DM patients than in non-DM patients. The ABG values reflect both acute stress and chronic glucose metabolism levels.
In addition, several studies (44) have focused on fasting blood glucose (FBG), an alternative metric for ABG. Elevated FBG on admission following AIS was reported to be appreciably correlated with poor function only in pre-DM patients, with no correlation in DM patients. It is inconclusive to define SIH in terms of ABG or FBG without considering the prior glucose metabolic status. As a result, new indexes such as glycaemic gap (GAP) and SHR were introduced, eliminating the interference of chronic glycaemic levels.
GAP is calculated as the difference between ABG and long-term mean glucose values determined from HbA1c. In a study aimed at comparing the ability of SHR, GAP, and ABG to assess poor prognosis in AIS patients, Yang et al. (24) demonstrated that a GAP of 45 mg/dl showed a superior ability to differentiate between AIS patients' severity and prognosis compared to ABG, as did SHR.
SHR is measured as ABG or FBG divisible by long-term mean glucose values determined from HbA1c and is also measured as FBG divisible directly by HbA1c in some articles. An analysis (26) of 8,622 AIS patients reported an association between high SHR and severe neurological deficits and all-cause mortality at 1 year in patients with and without DM. Interestingly, the mortality outcome was more significant in patients without DM, as found in a nationwide prospective registry study among AIS patients with non-DM in China (25). A recent retrospective study (28) assessed the impact of SHR, FBG, and HbA1c on in-hospital mortality in AIS patients with DM. SHR appeared to have a better predictive value than other absolute measures. Ngiam and colleagues (33) discovered that high SHR, particularly at SHR ≥ 0.97, was strongly related to 3-mouth adverse outcomes in AIS patients after IVT with or without DM. Other studies have also found this correlation (29, 31). The same has been confirmed in AIS patients after MT (34, 35). In addition, some studies have shown that in the above calculation, SIH obtained by direct division of FBG by HbA1c correlates more with poor functional outcomes after IVT (30, 45).
ABG and SHR cannot truly reflect the fluctuations in blood glucose under the influence of certain diseases due to the limitations of their numerical sources (obtained from HbA1c). Hyperglycaemic fluctuations caused by acute disease tend to exacerbate oxidative stress and damage endothelial cells, which results in a poor prognosis (46, 47). GV is the extent to which blood glucose levels fluctuate through time (48). An elevated GV is regarded as a sign of hypoglycaemia (49). There are two common types of GV, long-term with continuous blood glucose monitoring in years and short-term with days and months, and the most studied concerning stroke prognosis is short-term GV (50). Short-term GV is calculated in several ways, such as the mean amplitude of glycaemic excursions (MAGE), standard deviation, and coefficient of variation (50). In a 7.5-year follow-up of 28,354 patients with type 2 DM, the study observed that long-term high GV significantly enhanced stroke risk in DM patients (51). In a series of explorations of short-term GV, Hui et al. (36) found that altered GV in the first 3 days of hospitalization in AIS with DM patients was strongly related to early neurological deterioration (END) and that GV may be a more appropriate indicator than HbA1c. Yoon and colleagues (42) demonstrated that initial GV significantly enhanced the cardiovascular mortality risk at 3 months. In addition, elevated acute GV was related to poor functional outcomes (38, 41), impaired cognitive function (39), and a higher risk of haemorrhagic transformation (40), although GV was defined differently in these studies. GV has not been specifically studied in AIS populations with non-DM, possibly because diabetic populations are more likely to have measurable blood glucose changes.
In summary, we explored glycaemic indicators that could describe the full range of SIH following AIS. SHR has a similar predictor value for short- and long-term adverse outcomes in patients with and without DM and is independent of background glucose. Thus, SHR is expected to be a biomarker for SIH. Nevertheless, the current relevant studies do not provide a good head-to-head comparison of the different glucose markers. In the future, more prospective trials are needed to compare the clinical applicability of glycaemic indicators such as ABG, SHR, and GV or to attempt to apply them in combination.
The combined synergistic effects of SIH in patients with AIS can be induced by the glucose regulatory center, the hypothalamic–pituitary–adrenal (HPA) axis, the sympathetic adrenomedullary system, and humoral factors (Figure 1).
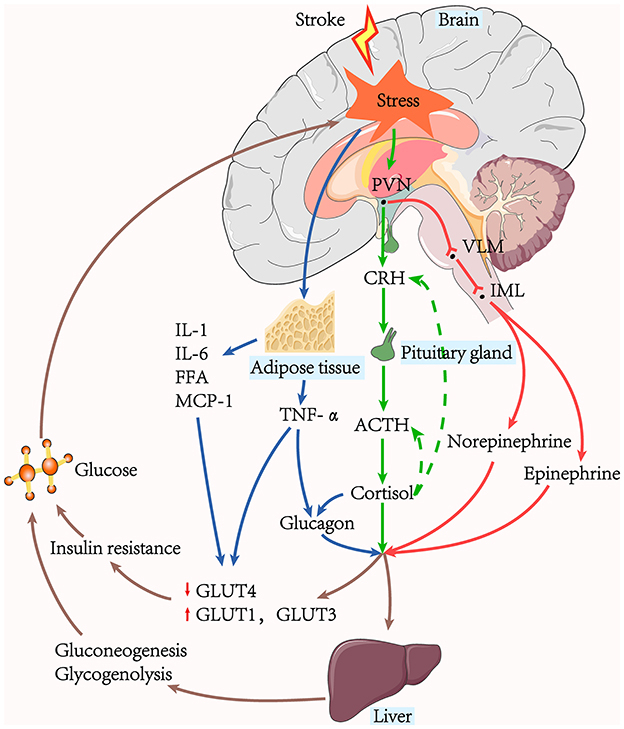
Figure 1. The pathway of stress-induced hyperglycaemia generation. Hepatic gluconeogenesis, glycogenolysis, and insulin resistance are the leading causes of hyperglycaemia. The sympathoadrenal system and the HPA axis are active in acute ischaemic stroke. The stressors stimulate the adrenal medulla to release catecholamine via the PVN-VLM-IML pathway, while the HPA axis stimulates the adrenal cortex to produce cortisol. Glucagon can be stimulated by cortisol, and TNF-α secreted from surrounding tissues. Glycaemic hormones such as catecholamine, cortisol, and glucagon, act on the liver to promote hepatic gluconeogenesis and glycogenolysis. Hyperglycaemia further exacerbates the stress response and contributes to the increased release of pro-inflammatory factors, creating a vicious cycle.
The insular cortex controls the output of the sympathetic and parasympathetic nervous systems, and several studies have shown it to be associated with elevated blood glucose following acute ischaemia (52–54). Many brain regions, including the hypothalamus and brainstem, have also been demonstrated to alter blood sugar levels and stress responses. The catecholamine neuronal system in the brainstem, including the locus coeruleus, nucleus tracts solitaries, and ventrolateral medulla (VLM), significantly contribute to stress responses. Catecholamine neurons in the VLM are the control centers of SIH and receive inputs directly from multiple stress-responsive brain regions, including the hyperglycaemic excitability of the hypothalamic paraventricular nucleus (PVN)-VLM pathway (55). In addition, the preganglionic neurons of the sympathetic medullary system are located in the intermediolateral nucleus (IML) (56, 57).
Gluconeogenesis, glycogenolysis, and excessive insulin resistance contribute significantly to the production and maintenance of hyperglycaemia during AIS (2, 58). Stressor stimulation causes excitation of the HPA axis and increases circulating cortisol. Cortisol has several metabolic effects to achieve elevated blood glucose levels, including activating vital hepatic gluconeogenesis enzymes and reducing glucose uptake in peripheral tissues (2). Stressor stimuli also converge on the brainstem catecholaminergic neurons and spinal cord efferent neurons in the medial column of preganglionic sympathetic neurons, activating the sympathetic adrenomedullary system and increasing blood levels of norepinephrine and epinephrine. Both epinephrine and norepinephrine stimulate the expression of essential genes that regulate glycogenolysis and gluconeogenesis (55). Norepinephrine additionally has the impact of increasing glycerol supply to the liver via lipolysis.
Excess glucagon is the main mediator of glucose metabolism and can be stimulated by cortisol (59). Studies have shown that glucagon, catecholamine, and cortisol act synergistically in changes in glucose metabolism to rapidly raise fasting plasma glucose. Insulin is a naturally significant hormone that lowers blood glucose levels. Its action mechanism is mainly through the mobilization of cells in the liver, skeletal muscle, and other peripheral tissues to synthesize and store glycogen, fats, and proteins and reduce their catabolism (60). Under physiological conditions, the increase of the plasma insulin level stimulates the onset of glucose storage activity mediated by the insulin-sensitive glucose transporter (GLUT)-4, occurring primarily in muscle and adipose tissue, where GLUT-4 translocates from intracellular storage to the membrane, thus enhancing glucose uptake (61). However, following AIS, decreased insulin-mediated glucose uptake was paralleled with over-expression of the insulin-insensitive GLUT-1 and GLUT-3 in other tissues throughout the body, blocking GLUT-4-mediated glucose storage, further exacerbating the increase in peripheral blood glucose (2).
Acute insulin resistance manifests as insulin-mediated glucose uptake reduction, mainly due to defective post-receptor insulin signaling and down-regulation of GLUT-4. Tumor necrosis factor-α (TNF-α), interleukin (IL)-1, and IL-6 can inhibit post-receptor insulin signaling, and TNF-α can also directly down-regulate the expression of the messenger RNA of GLUT-4 (62). An activated sympathetic nervous system induces adipocyte decomposition and increases free fatty acids (63). Excess circulating free fatty acids inhibit post-receptor insulin signaling and glycogen synthase to reduce glucose uptake (64, 65). In addition, fatty tissue secretes large amounts of pro-inflammatory cytokines, such as monocyte chemotactic protein-1 (MCP-1), an essential participant in insulin resistance (66, 67). TNF-α may also contribute to elevated blood glucose levels via the promotion of glucagon production (68).
Glucose is a major energy supplier to brain tissue and provides a valuable metabolic substrate during the acute disruption of cerebral blood flow. In animal models of recirculation after ischaemia, ATP (indicating the recovery of energy metabolism) tended to increase in hyperglycaemia groups more than in hypo- and normoglycaemia groups (69). A study of rabbit models of focal cerebral ischaemia observed a smaller area of cortical brain infarction in the glucose-perfused group compared to the saline group (70). Moreover, an animal model of haemorrhagic shock demonstrated that rapidly induced hyperglycaemia resulted in a significant elevation in blood pressure, cardiac output, and viability (71). However, saline or mannitol at similar osmolar doses did not achieve the above effects. These studies suggest that elevated glucose rescues damaged brain tissue to some extent and improves survival.
Glucose diffuses along a concentration gradient from the bloodstream into the cells in the ischaemic area. Appropriate hyperglycaemia maximizes the guarantee of cellular metabolism. Uytenboogaart et al. (72) reported the concentration-effect phenomenon in lacunar stroke, where glucose values above 144 mg/dl were linked to a good functional prognosis. It is worth noting that hyperglycaemia worsens the clinical prognosis in non-lacunar stroke compared with lacunar stroke (73). This is possibly due to non-lacunar infarction of an area known as the ischaemic penumbra, the site of reduced blood flow around the ischaemic core. Hyperglycaemia causes cellular acidosis by increasing its intracellular lactate content, leading to a poor prognosis (74). In lacunar infarcts, this is absent, and elevated blood glucose can supply energy to the surrounding tissues, leading to a better prognosis. However, its beneficial effects are diminished when severe hyperglycaemia exceeds 216 mg/dl (72).
The physiological protective mechanisms of short-term increases in hyperglycaemia have been extensively explored in cardiac ischaemia (75, 76). Acute hyperglycaemia reduces post-ischaemic cell death by producing cell survival proteins, releasing cellular survival factors, and favoring angiogenesis. In a porcine coronary ischaemia-reperfusion model, Chu et al. (77) demonstrated that hyperglycaemia during acute ischaemia increased the production of cell survival proteins, such as phosphorylated endothelial nitric oxide synthase (eNOS) and heat shock protein 27 and thus reduced infarct size. Malfitano et al. (78) showed that hyperglycaemia decreased pro-inflammatory cytokines, increased cell survival factors (hypoxia-inducible factor-1α, vascular endothelial growth factor), and reduced apoptosis in the rat myocardial infarction model, thereby improving systolic myocardial function and reducing the size of the myocardial infarction. In addition, hyperglycaemia contributes to an increase in capillaries and a reduction in fibrosis. Subsequently, the protective effect of hyperglycaemia on ischaemic injury in myocardial infarction was also shown to increase antioxidant enzyme activity, improve glutathione redox balance, and reduce sympathetic activity after infarction (79).
The presence of acute hyperglycaemia leading to adverse prognosis has been repeatedly validated in patients with AIS, irrespective of a prior diagnosis of DM, suggesting a possible causal relationship. Various studies have extensively explored these potential mechanisms (7), which are discussed below (Figure 2).
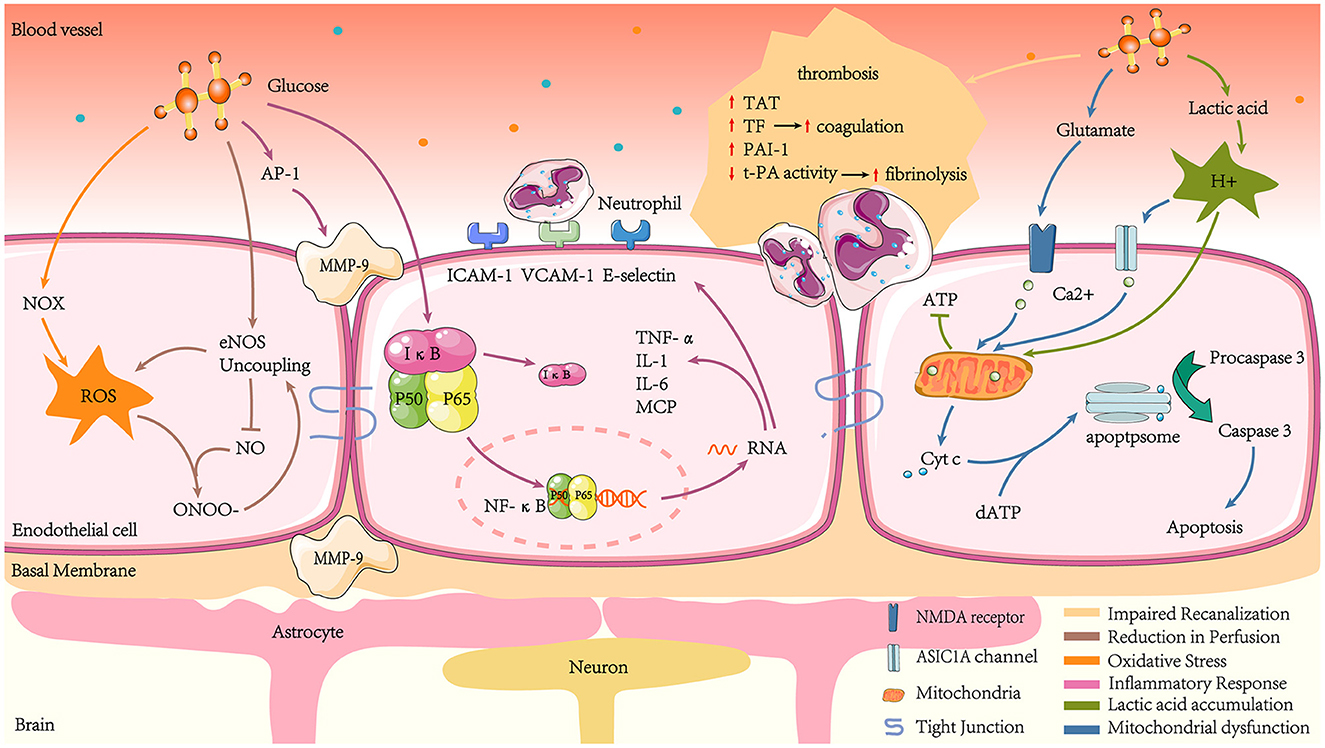
Figure 2. Mechanisms of hyperglycaemia-mediated damage in ischaemic stroke. Acute hyperglycaemia increases thrombosis by promoting coagulation and inhibiting hyperfibrinolysis, such as increased TAT, TF, and elevated PAI-1 activity, while t-PA activity decreases. Hyperglycaemia amplifies ROS production, which, together with NO, produces ONOO- and reduces NO bioavailability. At the same time, hyperglycaemia reduces NO production by promoting eNOS uncoupling, leading to endothelial cell diastolic dysfunction and reduced cerebral blood flow. Hyperglycaemia also increases NF-κB translocation to the nucleus by regulating IκB proteasomal degradation. NF-κB combines with specific κB sites on DNA sequence promoters to transcribe inflammatory factors such as TNF-α, IL-1, and IL-6. The NF-κB pathway also transcribes the chemokine MCP and adhesion factors such as ICAM-1, VCAM-1, and E-selectin, which induce leukocyte adhesion. Hyperglycaemia stimulates MMP-9 production, which can damage tight junctions, basement membrane proteins, and astrocyte peduncles, leading to BBB leakage. Hyperglycaemia exacerbates lactate accumulation in the ischaemic zone, leading to mitochondrial dysfunction and reduced ATP production, which fails to maintain intra- and extracellular osmotic and ionic gradients and mediates cytotoxic cell death. In addition, the overproduction of excitatory glutamate in response to hyperglycaemic stimulation activates NMDA receptors. At the same time, the lactate aggregation environment leads to increased H+ and promotes the opening of ASIC1 channels. Together, this results in increased intra-mitochondrial Ca2+. The imbalance of intra-mitochondrial Ca2+ exacerbates the leakage of Cyt c into the cytosol. Cyt c then interacts with dATP to activate downstream apoptogenic proteins, eventually triggering apoptosis.
Impaired recanalization is associated with enhanced coagulation and reduced fibrinolytic activity (80). In human studies, hyperglycaemia elevates thrombin-antithrombin (TAT) complexes and tissue factor (TF) to produce procoagulant effects (81). Animal and cytological studies revealed that acute hyperglycaemia resulted in elevated plasminogen activator inhibitor type 1 (PAI-1) and decreased plasminogen activator (t-PA) activity levels, leading to a hypercoagulable state by affecting fibrinolytic homeostasis, which was confirmed in human studies (82, 83). Ribo et al. (84) found that during tissue-type t-PA-induced recanalization, acute hyperglycaemia was related to lower recanalization rates compared to chronic hyperglycaemia, suggesting that hyperglycaemia affects reperfusion in the ischaemic penumbra by impairing the fibrinolytic system.
A considerable number of studies have reported the phenomenon of cerebral blood flow reduction in hyperglycaemic animals (85–87). In an animal model experiment, acute hyperglycaemia-induced by intraperitoneal injection of glucose was linked to a 24% decrease in cerebral blood flow in rats. Following the intraperitoneal injection of mannitol, the plasma osmolarity increased as much as that observed following glucose injection. In contrast, cerebral blood flow decreased by only 10% (88). This suggests hyperglycaemia may deteriorate brain function by inhibiting compensatory blood circulation after ischaemic injury. The possible mechanism is acute hyperglycaemia promoting the formation of eNOS uncoupling phenomenon in the microvascular system surrounding the ischaemic region, leading to increased reactive oxygen species (ROS) production and decreased NO production (89). The reaction between ROS and NO can rapidly form ONOO–, which exacerbates eNOS uncoupling and produces a vicious cycle of oxidative stress. Reduced NO production and bioavailability leads to local vasodilation and impaired reperfusion (90).
Oxidative stress is a condition where the body is out of balance concerning oxidation and antioxidation, with a tendency to oxidize and produce large amounts of free radicals. ROS are mainly generated by the mitochondria and nicotinamide adenine dinucleotide phosphate oxidase (NOX) in biological systems (91). Notably, NOX is a major origin of ROS in neuronal cells after transient cerebral ischaemia-reperfusion (92). In a model in which endothelial cells were co-cultured with astrocytes to mimic the blood-brain barrier (BBB) environment, high glucose caused a marked increase in BBB permeability through enhanced oxidative stress. The mechanism was shown to be that high glucose enhanced NOX activity and expression levels (93). In addition, a neuronal cell culture experiment revealed that glucose provided the essential electron donor for ROS production in neurons after reperfusion (92). Excess reactive oxygen species lead to increased BBB permeability, brain oedema, and ultimately increased infarct size by peroxidising lipids, proteins, and nucleic acids (94–96). Furthermore, reactive oxygen species consume NO and generate a range of oxygen-free radicals and nitro compounds. The decrease in circulating NO levels leads to vascular endothelial relaxation dysfunction.
Another effect following ischaemia-reperfusion that is significantly enhanced by hyperglycaemia is inflammation. A study in rat stroke models demonstrated that hyperglycaemia triggers a thrombo-inflammatory cascade response and amplifies and exacerbates middle cerebral artery occlusion (MCAO)-induced downstream microvascular thrombosis. The thrombo-inflammatory cascade begins immediately after MCAO in hyperglycaemic rats and continues throughout the reperfusion phase, accompanied by increased matrix metalloproteinase-9 (MMP-9), serotonin, and TAT complex (97). The increase in MMP-9 may be explained by hyperglycaemia promoting the expression of the pro-inflammatory transcription factor nuclear factor-activated protein-1 (AP-1) (98, 99). The increased MMP-9 is implicated in BBB destruction, resulting in plasma protein and inflammatory cell leakage, brain oedema formation, and a worse neurological prognosis (100, 101). Further investigations of hyperglycaemia-induced BBB leakage during ischaemia/reperfusion may be due to tight junction injury, basement membrane proteins, and astrocytic peduncles (97).
In addition, glucose causes an increase in transcription factors in the nucleus: nuclear factor-kappaB (NF-κB) (102). NF-κB is stably coupled to the inhibitory protein IκB in the cytoplasm. After phosphorylation and hydrolysis of IκB due to inflammatory stimuli, the heterodimers of p50 and p65 (the active form of NF-κB) undergo nuclear translocation. They are transcribed and translated into inflammatory proteins, such as TNF- α, IL-1, IL-6, and MCP-1, amplifying and maintaining the inflammatory response (103). Hyperglycaemia induces the promoter of the NF-κB subunit p65, causing an increase in p65 gene expression, which accelerates inflammatory factor production (104). Intercellular adhesion molecule-1 (ICAM-1), vascular cell adhesion molecule-1 (VCAM-1), and E-selectin are also induced by NF-κB (105, 106). These cytokines, chemokines, and adhesion molecules attract leukocytes to areas of ischaemia (107–109). Excessive leukocyte infiltration destroys the BBB, which causes irreversible infarction in the ischaemic penumbra.
Lactic acid is a source of energy that is metabolized in the brain in the absence of energy (110, 111). Lactic acid accumulation is another risk factor caused by hyperglycaemia following a stroke. Compared with normoglycaemic animals, hyperglycaemia significantly increases lactate concentrations in ischaemic regions of a cat's brain, decreases high-energy phosphate and pH, and converts the ischaemic penumbra into infarct regions (112, 113). A human study found a similar phenomenon that hyperglycaemia in perfusion-diffusion mismatched patients led to enhanced brain lactate production, reducing rescue rates post-infarct penumbra (74). The cause of this phenomenon may be that the accumulation of lactic acidosis causes acidosis, damaging mitochondria and affecting ATP production, leading to depolarisation of cell membranes and the inability to maintain osmotic and ionic gradients inside and outside cells, ultimately leading to cytotoxic cell death (114).
The brain's energy supply is disrupted during AIS, and hypoxia depolarisation at presynaptic terminals results in the liberation of excitatory neurotransmitters such as glutamate (115). A study suggested that hyperglycaemia enhances extracellular glutamate build-up during cortical ischaemia (116). N-methyl-D-aspartate (NMDA) receptors are typically ionotropic glutamate receptors and are essential mediators of neuronal death during cerebral ischaemia (117). Excitatory glutamate activates NMDA receptors, allowing Ca2+ to effectively enter the cell and mitochondria. Hyperglycaemia induces cell apoptosis following a stroke by increasing Ca2+ in the mitochondria, a vital link being the entry of cytochrome c (Cyt c) across the mitochondrial membrane into the cytosol (118). Upon entry into the cytosol, Cyt c interacts with procaspase-9, apoptotic protease-activating factor-1, and dATP to form an activation complex (119). This complex then drives caspase-3 activation, which ultimately induces apoptosis (120).
The glutamate non-dependent Ca2+ loading pathway also contributes to Ca2+ toxicity in ischaemia. Acid-sensitive ion channels (ASICs) are proton-gated ion channels activated by protons and widely exist in the central and surrounding system (121, 122). In the center, ASICs are primarily expressed in neurons and act as pH sensors to cause neuronal excitation. In physiological conditions, pH is relatively stable in brain tissue. However, massive anaerobic glycolysis of glucose during hypoxic-ischaemia leads to higher lactate accumulation in ischaemic tissue, and the pH drops rapidly (112). ASIC1A, a subtype of ASIC, can be activated by acidosis and conduct Ca2+. An animal experiment has confirmed that ASIC1A knock-out mice resist ischaemia and acid damage (123). Acidosis induces Ca2+ into cells via ASIC1, independent of the glutamate pattern, leading to increased intracellular Ca2+ concentration, which activation can trigger/regulate multiple cellular processes (124). In addition, during the early stage of ischaemia-reperfusion, hyperglycaemia hinders Ca2+ recovery during reperfusion, prolonging the existence of intracellular Ca2+ (125).
Several laboratory studies have extensively explored the effects on infarct volume of pre- and post-infarction insulin administration in global or focal ischaemic stroke. All the results have shown that insulin has a potent neuroprotective effect, reducing infarct size in crucial brain areas such as the hippocampus, striatum, and cerebral cortex (12, 126). In a transient forebrain ischaemia model, direct injection of insulin or insulin-like growth factor-1 (IGF-1) into the ventricles reduced ischaemic neuronal damage, suggesting that insulin can achieve neuroprotective results through direct interaction action with brain tissue, possibly in part through the IGF-1 receptor (127). In a transient focal ischaemic stroke model, the combination of insulin and glucose nullified most of the apparent protective effects (128), suggesting that insulin's significant neuroprotective effect could be achieved by lowering blood glucose, in agreement with the results of another experiment (129). Additional animal experiments have confirmed that possible mechanisms for the protective effects of insulin also include the regulation of neurotransmitters, the promotion of glycogen synthesis, and the prevention of neuronal necrosis and apoptosis (Table 2).
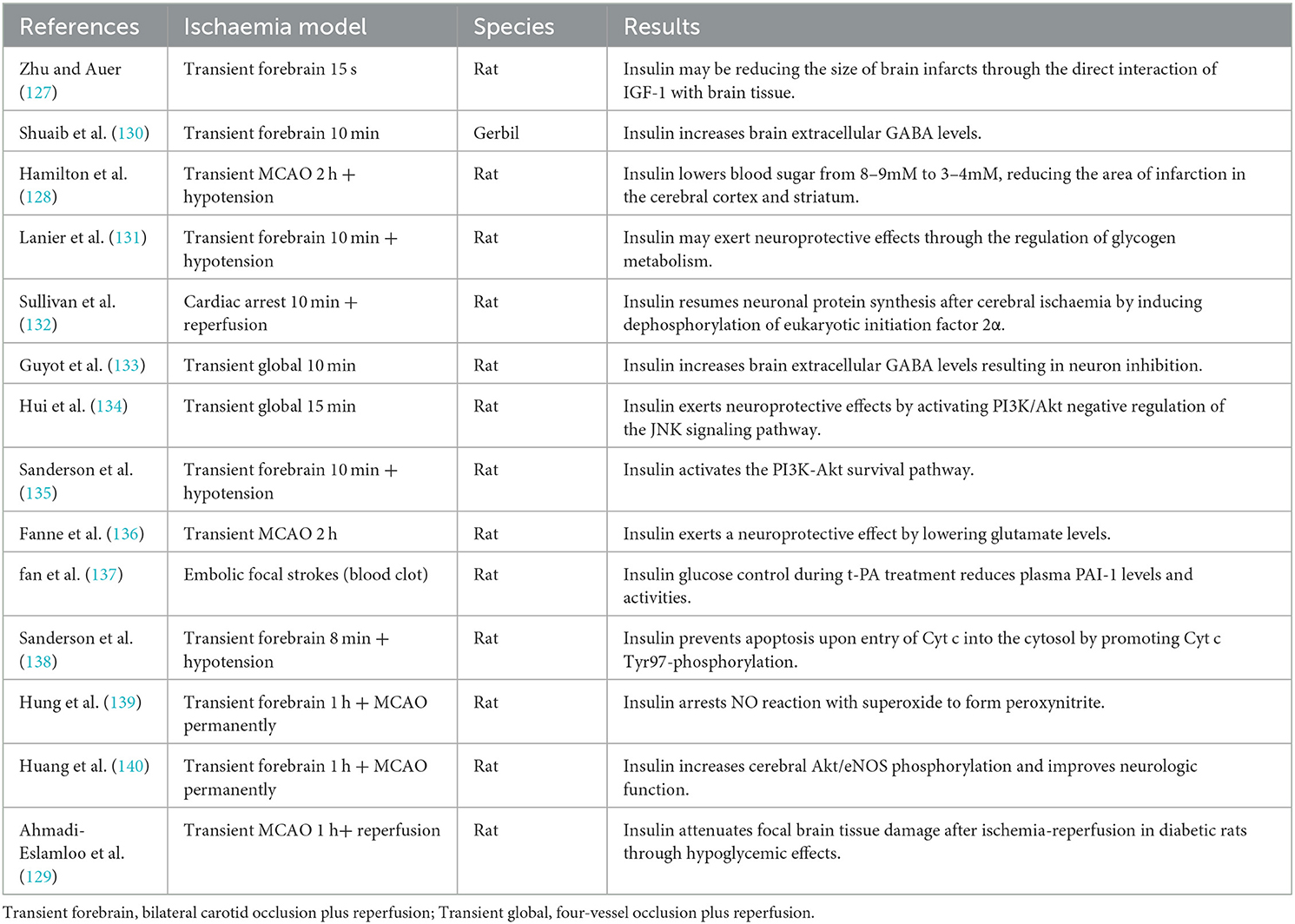
Table 2. Mechanism of the neuroprotective effect of insulin in acute ischaemic stroke animals during hyperglycaemia.
Intensive glucose control with intravenous insulin effectively reduces in-hospital mortality in critically ill non-stroke patients (141, 142). Thus, in recent years, some clinical trials have explored the optimal glycaemic targets for intensive insulin treatment in AIS patients with elevated ABG (Table 3).
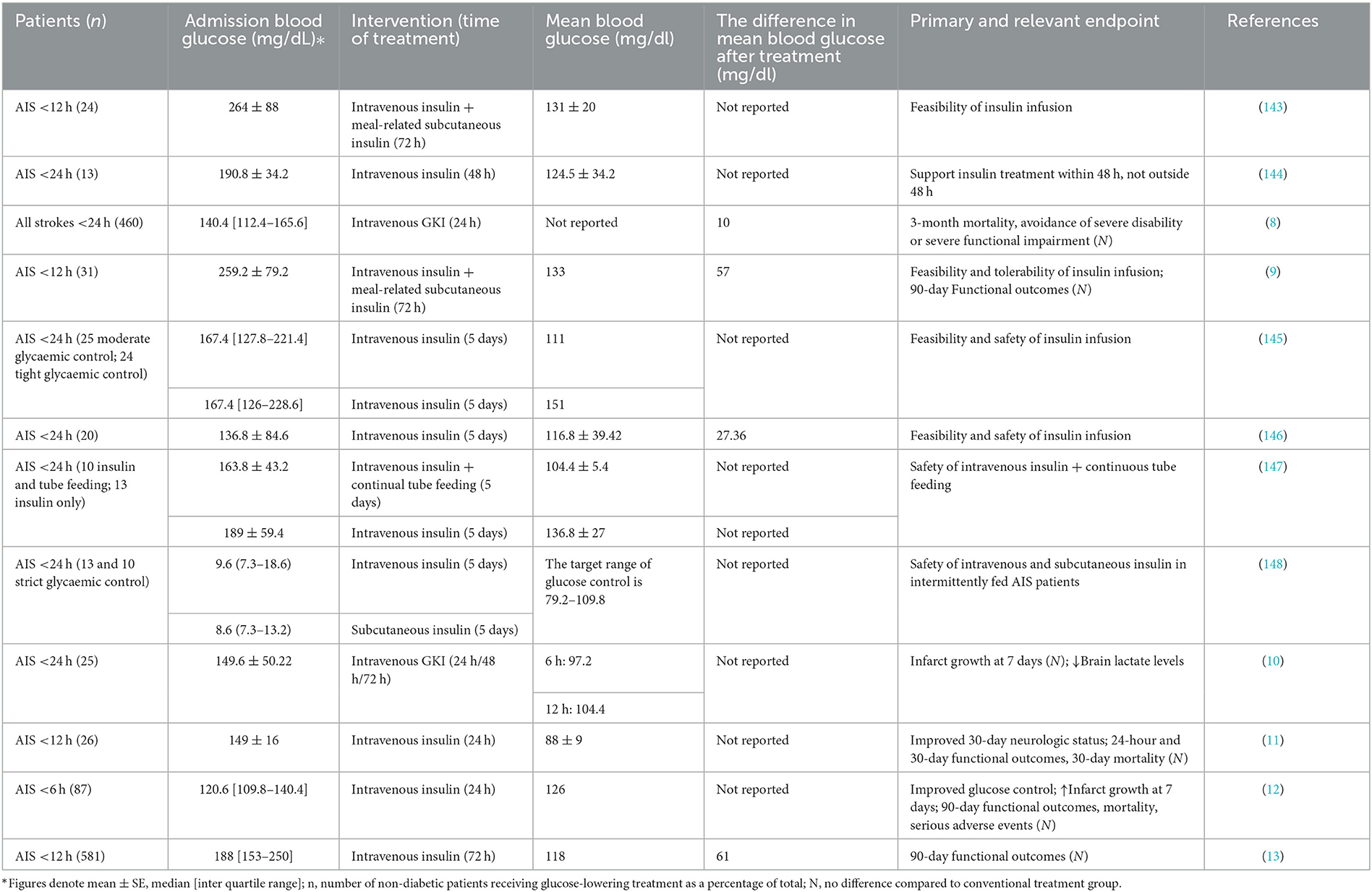
Table 3. Randomized trials aimed at comparing the effectiveness of intensive glycaemic treatment with standard treatment in acute ischaemic stroke patients with enhanced admission blood glucose.
A total of 933 patients with acute stroke (ischaemic or haemorrhagic) were enrolled in the GIST-UK trial within 24 h (8), 80% of whom did not have a diagnosis of DM and had a median ABG of only 140.4 mg/dl. The experimental group maintained capillary glucose at 72–126 mg/dl by 24-h continuous intravenous infusion of glucose-potassium-insulin (GKI). Surprisingly, compared to the control group, the 90-day functional outcomes of the experimental group did not improve, and hypoglycaemic events occurred. By analyzing changes in blood glucose and blood pressure over 24 h, the benefits of lowering blood glucose may have been masked by lower blood pressure. The study was criticized for including a heterogeneous stroke type, with slow recruitment, late treatment initiation, and variability in glycaemic control of only 10 mg/dl (12, 13). Subsequent studies assessing the feasibility and safety of intensive glucose control with insulin after AIS have shown that such trials are feasible and necessary (9, 144–146).
In the SELESTIAL trial (10), 30% of AIS patients with a previous DM diagnosis were maintained on serum glucose between 72–126 mg/dl by GKI infusion. Brain lactate production level decreased at 6–12 h following insulin administration. However, insulin treatment was linked to a marked increase in cerebral infarct size in patients with complete intracranial vascular occlusion. In addition, 76% of patients in the experimental group developed asymptomatic hypoglycaemia. Similarly, a large study explored the effects of an intensive glycaemic regimen compared with usual care on glycaemic control and infarct size expansion. In the INSULINFARCT trial (12), 180 AIS patients were randomized within 6 h to receive 24 h of intravenous insulin therapy, with the vast majority of subjects having no history of DM. The study found that keeping blood glucose below 126 mg/dl did not prevent the transition from an ischaemic penumbra to an infarcted area.
Since all of the above studies failed to demonstrate a positive effect of intensive glucose therapy on stroke prognosis, Johnston and colleagues began to consider that variability in glycaemic control and duration of treatment may be the problem. In the SHINE trial (13), a large multicenter randomized controlled trial, 1,151 AIS patients underwent conventional (80–180 mg/dl) or strict glycaemic control (80–130 mg/dl) within 12 h. Of the enrolled patients, 68% had received reperfusion therapy, and 80% were diagnosed with DM. The change in glycaemic control compared to GIST-UK was significant (61 mg/dl). Intensive glucose control at up to 72 h did not significantly improve 90-day functional outcomes.
In reviewing the above clinical trials of intensive insulin glucose-lowering, only two studies distinguished between the two populations of AIS patients, with or without previously recognized DM, and discussed treatment prognosis. One study in AIS patients with DM revealed that maintaining blood glucose below 130 mg/dl with intensive insulin treatment was feasible but did not improve the prognosis at three months (9). In another clinical study of mild hyperglycaemia in non-DM patients after AIS, it was found that maintaining blood glucose levels at 81–126 mg/dl was relatively safe in the experimental group and improved neurological status after 30 days. However, there was no difference in functional outcomes compared to controls (11). The SHINE trial (13) did not yield the desired results. Torbey et al. (149) considered whether the different definitions of SIH determined the outcome. They performed a further subgroup analysis of SHINE, carefully selecting six glycaemic parameters: ABG, absence versus presence of diagnosed and undiagnosed DM, HbA1c, GAP, SHR (ABG/ average HbA1c-based daily blood glucose), and GV (SD). The results identified that patients with undiagnosed DM had the lowest likelihood of a good prognosis compared to those with true non-DM. However, no apparent benefit of SHINE treatment was observed in the small group of undiagnosed DM patients. After adjusting for baseline stroke severity and thrombolytic therapy, the researchers still found no benefit of intensive insulin therapy in any of the above subgroups. This study was a secondary analysis of the SHINE trial, and the results had unavoidable limitations. Because ABG cut-off values associated with poor prognosis are not the same in AIS patients with and without previously recognized DM, we suggest that future clinical studies should consider the different glycaemic backgrounds of the two populations, whose criteria for initiating glucose lowering, time of initiation and duration of glucose-lowering, and even glucose lowering goals may not be the same. Each characteristic needs to be determined by relevant basic and clinical trials. Next, we discuss the results based on the available experiments.
There is considerable heterogeneity in ABG treatment criteria in each study reporting functional outcomes, for example, 140.4 mg/dl (8), 259.2 mg/dl (9), 149 mg/dl (11), 120.6 mg/dl (12), and 188 mg/dl (13). Subsequent studies should include patients with glucose values above the cut-off value related to a worse prognosis, which is considered to be approximately 155 mg/dl (23). For patients treated with IVT or MT, or IAT, it is recommended to use ~140 mg/dl (16, 18, 19). Also, for non-DM patients with post-AIS hyperglycaemia, we recommend a glucose reduction criteria of ~126 mg/dl (3).
In studies exploring the effects of intensive glucose control, glucose lowering was initiated at 6 h (12), 12 h (9, 11, 13), and 24 h (8) after stroke. The duration of glucose lowering ranged from 24 to 72 h. Rosso and colleagues (12) found that in AIS patients who underwent intensive insulin treatment immediately within 6 h, baseline and day 7 MRI comparison revealed a more significant infarct growth in the experimental group. Therefore, intensive insulin treatment is not recommended in the hyperacute phase of cerebral infarction. The proper duration of glucose lowering after AIS is still being determined. Data obtained from continuous glucose monitoring indicate that continuous glycaemic control for 72 h after AIS is necessary (150–152).
Glucose-lowering targets are controversial. In a rodent model of focal ischaemia, insulin treatment after ischaemia resulted in minimal infarct size between 108–126 mg/dl glucose and increased infarct size between 36–54 mg/dl glucose (153). However, in two subsequent clinical trials measuring the controversial infarct size increase, serum glucose was kept below 126 mg/dl using insulin infusion. One of these trials did not find any marked variation in infarct size increase over 1 week between the intervention group and the control group (10). In the other trial, the intervention group's infarct size was more remarkable (12). Furthermore, in a study of patients with AIS, ABG values between 66.6–131.4 mg/dl were associated with 12-month positive functional outcomes (14). Gentile et al. (154) found a 4.6-fold lower in-hospital mortality rate in AIS patients who controlled their blood glucose below 130 mg/dl within 48 h of admission compared with those experiencing persistent hyperglycaemia. However, several clinical studies have shown similar mortality and neurological prognosis in intensive insulin therapy (118–133 mg/dl) and conventional insulin therapy groups (8, 9, 12, 13). There are no consistent recommendations for optimal glycaemic targets for SIH in treating AIS due to limited clinical trial results. Current American Heart Association/American Stroke Association (AHA/ASA) guidelines for AIS recommend treating hyperglycaemia to stabilize blood glucose levels at 140 to 180 mg/dl (155). However, most guidelines endorse that tight glycaemic control (blood glucose < 126 mg/dl) or hypoglycaemia is harmful and should be avoided (12, 156).
A recent meta-analysis on selecting an optimal glucose-lowering target for critical patients showed that glucose control levels < 110 and 110–144 mg/ dl had a greater hypoglycaemia incidence vs. 144–180 and >180 mg/dl (157). Hypoglycaemia exacerbates brain damage after AIS by increasing oxidative stress and inflammatory response. Studies on rodent models have shown that repeated hypoglycaemia increases post-ischaemic brain injury in diabetic rats by increasing mitochondrial ROS production and decreasing mitochondrial complex I activity after ischaemia (158, 159). In addition, hypoglycaemia induces increased IL-6 production, platelet aggregation, vascular adhesion molecule production, and inhibition of fibrinolytic mechanisms (160–162). These changes eventually lead to vascular injury and new thrombosis, increasing the risk of local cerebral ischaemia. Therefore, repeated hypoglycaemia has been indicated as an essential reason for the unsuccessful intensive glucose-lowering therapy. Future trials should be aimed at controlling blood glucose to “broad” levels rather than intensively lowering glucose to avoid harm from irreversible hypoglycaemic events.
Glucose-like peptide-1 (GLP-1) receptor agonists include exenatide, liraglutide, albiglutide, semaglutide, and dulaglutide considered to be the first alternative to insulin. GLP-1 is a peptide hormone secreted by the intestine following food stimulation and is implicated in regulating blood glucose homeostasis in the body. Natural GLP-1 is degraded by the endoprotease dipeptidyl-peptidase-4 (DPP-4) in vivo with an ~2-min half-life. GLP-1 receptor agonists can perform the same biological actions as natural GLP-1 but also avoid degradation and loss of activity, thus prolonging the duration of action. GLP-1 maintains glucose endocrine homeostasis through several mechanisms, including but not limited to promoting insulin secretion from pancreatic β-cells, inhibiting glucagon production, improving β cell quality and function, and decreasing appetite and gastric emptying. GLP-1 receptor agonists rarely cause hypoglycaemia, and their main drawback is mild to moderate gastrointestinal adverse effects. Findings from cardiovascular prognostic studies and meta-analyses suggest that GLP-1 receptor agonists reduce stroke incidence and have neuroprotective effects in DM patients (163). Some studies highlight that the neuroprotective effects of these drugs in AIS patients may be through the improvement of ischaemia-induced inflammation, support of BBB integrity by triggering the PI3K/AKT and mitogen-activated protein kinase (MAPK) pathway, as well as reducing ROS levels in ischaemic neurons (164, 165). The effectiveness and safeness of GLP-1 receptor agonists for treating hyperglycaemia after AIS has been demonstrated with exenatide at 9 h after AIS and continuing for 6 days to lower blood glucose (166). A recent multicenter randomized trial was designed to compare the difference in risk of hypoglycaemia and improvement in neurological prognosis with exenatide vs. standard of care. Exenatide 5 μg was administered subcutaneously twice daily in the treatment group, but the trial results have not yet been published (167).
Another recommended alternative therapy is DPP-4 inhibitors, which exert hypoglycaemic effects mainly by inhibiting the physiological degradation of GLP-1 in the blood, including alogliptin, linagliptin, saxagliptin, sitagliptin, and vildagliptin. Interestingly, DPP-4 inhibitors improve neurological prognosis after stroke in rodents, independent of GLP-1, possibly by increasing the bioavailability of other bioactive DPP-4 substrates, such as stromal cell-derived factor-1α (167). In addition, preclinical studies have found that these drugs can activate the Akt/mTOR pathway and anti-apoptotic and anti-inflammatory mechanisms to exert neuroprotective effects (168). However, two recent extensive randomized clinical studies have shown that DPP-4 inhibitors are ineffective in reducing ischaemic stroke risk following cardiovascular disease (169, 170). This is inconsistent with the neuroprotective conclusions drawn from preclinical studies and requires further experimental validation.
In addition to the two drugs mentioned above, the new glucose-lowering drug type, sodium glucose-linked cotransporter 2 (SGLT2) inhibitors, are recommended. Besides reducing blood glucose, SGLT2 inhibitors have antioxidant, anti-inflammatory, insulin resistance, and atherosclerotic plaque formation-reducing properties (171). In addition, SGLT2 inhibitors reversed hyperglycaemia-induced neuronal damage after acute cerebral ischaemia by inhibiting SGLT in a mouse model of temporary bilateral carotid stenosis (172). However, the current debate on whether SGLT2 inhibitors prevent strokes after cardiovascular disease is divided. In the EMPA-REG OUTCOM trial (173), there was a slight increase in stroke risk with empagliflozin. Some have attributed this trend to an elevated haematocrit in the empagliflozin group, which corresponds to an increased stroke risk due to increased blood viscosity. Fortunately, no such trend was found in the CANVAS trial (174). However, a meta-analysis concluded that SGLT2 inhibitors increased non-fatal stroke risk by 30% (175).
Metformin, a biguanide derivative, is the principal drug indicated for type 2 DM (176). The hypoglycaemic pharmacological effects of metformin are mainly through reducing hepatic glycogenolysis and enhancing peripheral tissue glucose uptake and utilization (177). Studies have shown that metformin can exert post-stroke neuroprotective effects, possibly inhibiting ischaemia-induced neuronal death, oxidative stress, and inflammatory responses (178, 179). The opening of the mitochondrial permeability transition pore (MPTP) is a significant cause of neuronal mortality due to ischaemia (180). Metformin may prevent neuronal death by inhibiting mitochondrial respiratory chain complex I, preventing MPTP opening, and activating AMPK (181, 182). Furthermore, metformin was demonstrated to regulate AIS-induced oxidative stress damage and reduce markers of brain inflammation via the lncRNA-H19/miR-148a-3p/Rock2 axis (183, 184).
The drawbacks of metformin compared to intravenous insulin application in critical care patients are apparent. First, compared with the immediate hypoglycaemic effect of insulin, the blood dose concentration peaked approximately 3 h following oral metformin (185). It fails to adjust rapidly to the dramatic changes in blood glucose concentration in patients. An exploratory safety study of metformin in patients with the transient ischemic attack or minor ischaemic attack found that 20% of patients in the experimental group permanently discontinued the drug due to gastrointestinal side effects (186) and its metabolic characteristics of being excreted mainly through the kidneys (185), which both limit its application. Finally, intravenous insulin can be quickly adjusted to each patient's diet, facilitating the maintenance of blood glucose stability during hospitalization. Other oral hypoglycaemic agents are available for similar reasons, such as sulfonylureas, glinides, α-glucosidase inhibitors, thiazolidinediones, and DPP-4 inhibitors. Therefore, intravenous insulin application seems to be a more appropriate treatment option than oral agents, such as metformin, in the case of elevated blood glucose after AIS.
In this review, we discuss the definition of SIH in the background of AIS, its formative mechanisms, and its dual impact on the central nervous system. Insulin is currently a widely accepted clinical treatment for post-AIS hyperglycaemia, but it does not achieve the desired effect in practice. In our analysis of intensive insulin treatment failure, the best definition of SIH should be independent of the existence of DM and strongly linked to short- and long-term efficacy. SHR, or the application of these definitions in combination, is a good option for future studies and needs further exploration. In addition, the treatment of SIH should consider the criteria for initiating glucose lowering after AIS, the timing of dosing and duration of treatment, the target range of glycaemic control, and treatment options other than insulin. GLP-1 receptor agonists are a potential new research target for treating SIH.
MY and YH reviewed the literature and wrote the draft manuscript. TW helped assess the literature. MX, HL, and JF drew the pictures. LF designed and revised the work. DM supervised the work. All authors contributed to the article and approved the submitted version.
This study was funded by the Natural Science Foundation of Jilin Province (212558JC010685134 and 202512JC010376367).
We would like to thank Editage (www.editage.cn) for English language editing. Figures 1, 2 were modified from Servier Medical Art (http://smart.servier.com/), licensed under a Creative Common Attribution 3.0 Generic License (https://creativecommons.org/licenses/by/3.0/).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
ABG, admission blood glucose; ACTH, adrenocorticotrophic hormone; AIS, acute ischaemic stroke; AP-1, activated protein-1; ASICs, acid-sensitive ion channels; BBB, blood-brain barrier; BG, blood glucose; CRH, corticotrophin-releasing hormone; CV, coefficient of variance; Cyt c, cytochrome c; DM, diabetes mellitus; DPP-4, dipeptidyl-peptidase-4; END, early neurological deterioration; eNOS, endothelial nitric oxide synthase; FBG, fasting blood glucose; FFA, free fatty acids; GABA, gamma-aminobutyric acid; GAP, glycaemic gap = ABG (mg/dl) - [28.7 × HbA1c (%) - 46.7]; GKI, glucose-potassium-insulin; GLP-1, Glucose-like peptide-1; GLUT, glucose transporter; GV, glucose variability; HPA axis, hypothalamic–pituitary–adrenal axis; IAT, intra-arterial treatment; ICAM-1, intercellular adhesion molecule-1; IGF-1, insulin-like growth factor-1; IL-1, interleukin-1; IL-6, interleukin-6; IML, intermediolateral nucleus; IVT, intravenous thrombolysis; JNK, c-Jun N-terminal kinase; MAG, mean absolute glucose; MAGE, the mean amplitude of glycaemic excursions; MCAO, middle cerebral artery occlusion; MCP-1, monocyte chemotactic protein-1; MMP-9, matrix metalloproteinase-9; MPTP, mitochondrial permeability transition pore; MT, mechanical thrombectomy; NMDA, N-methyl-D-aspartate; ROS, reactive oxygen species; SHR, stress hyperglycaemia ratio; SHR1, ABG (mg/dl) / HbA1c (%); SHR2, FBG (mg/dl) / HbA1c (%); SHR3, FBG (mg/dl) / HbA1c (%) (derived from the glycosylated hemoglobin level); SIH, stress-induced hyperglycaemia; PAI-1, plasminogen activator inhibitor type 1; PSCI, post-stroke cognitive impairment; PVN, paraventricular nucleus; SD, standard deviation; SICH, symptomatic intracranial hemorrhage; TAT, thrombin-antithrombin; TF, tissue factor; TNF-α, tumor necrosis factor-α; t-PA, tissue-type plasminogen activator; TR, time rate; VCAM-1, vascular cell adhesion molecule-1; VLM, ventrolateral medulla.
1. Donkor ES. Stroke in the 21(st) century: A snapshot of the burden, epidemiology, and quality of life. Stroke Res Treat. (2018) 2018:3238165. doi: 10.1155/2018/3238165
2. Dungan KM, Braithwaite SS, Preiser JC. Stress hyperglycaemia. Lancet. (2009) 373:1798–807. doi: 10.1016/S0140-6736(09)60553-5
3. Capes SE, Hunt D, Malmberg K, Pathak P, Gerstein HC. Stress hyperglycemia and prognosis of stroke in nondiabetic and diabetic patients: a systematic overview. Stroke. (2001) 32:2426–32. doi: 10.1161/hs1001.096194
4. Kim JT, Jahan R, Saver JL, Investigators S. Impact of glucose on outcomes in patients treated with mechanical thrombectomy: a post hoc analysis of the solitaire flow restoration with the intention for thrombectomy study. Stroke. (2016) 47:120–7. doi: 10.1161/STROKEAHA.115.010753
5. Stead LG, Gilmore RM, Bellolio MF, Mishra S, Bhagra A, Vaidyanathan L, et al. Hyperglycemia as an independent predictor of worse outcome in non-diabetic patients presenting with acute ischemic stroke. Neurocrit Care. (2009) 10:181–6. doi: 10.1007/s12028-008-9080-0
6. Marik PE, Bellomo R. Stress hyperglycemia: an essential survival response! Crit Care. (2013) 17:305. doi: 10.1186/cc12514
7. Kruyt ND, Biessels GJ, Devries JH, Roos YB. Hyperglycemia in acute ischemic stroke: pathophysiology and clinical management. Nat Rev Neurol. (2010) 6:145–55. doi: 10.1038/nrneurol.2009.231
8. Gray CS, Hildreth AJ, Sandercock PA, O'Connell JE, Johnston DE, Cartlidge NE, et al. Glucose-potassium-insulin infusions in the management of post-stroke hyperglycaemia: the uk glucose insulin in stroke trial (gist-uk). Lancet Neurol. (2007) 6:397–406. doi: 10.1016/S1474-4422(07)70080-7
9. Bruno A, Kent TA, Coull BM, Shankar RR, Saha C, Becker KJ, et al. Treatment of hyperglycemia in ischemic stroke (this): a randomized pilot trial. Stroke. (2008) 39:384–9. doi: 10.1161/STROKEAHA.107.493544
10. McCormick M, Hadley D, McLean JR, Macfarlane JA, Condon B, Muir KW. Randomized, controlled trial of insulin for acute poststroke hyperglycemia. Ann Neurol. (2010) 67:570–8. doi: 10.1002/ana.21983
11. Staszewski J, Brodacki B, Kotowicz J, Stepien A. Intravenous insulin therapy in the maintenance of strict glycemic control in nondiabetic acute stroke patients with mild hyperglycemia. J Stroke Cerebrovasc Dis. (2011) 20:150–4. doi: 10.1016/j.jstrokecerebrovasdis.2009.11.013
12. Rosso C, Corvol JC, Pires C, Crozier S, Attal Y, Jacqueminet S, et al. Intensive versus subcutaneous insulin in patients with hyperacute stroke: results from the randomized insulinfarct trial. Stroke. (2012) 43:2343–9. doi: 10.1161/STROKEAHA.112.657122
13. Johnston KC, Bruno A, Pauls Q, Hall CE, Barrett KM, Barsan W, et al. Intensive vs standard treatment of hyperglycemia and functional outcome in patients with acute ischemic stroke: The shine randomized clinical trial. JAMA. (2019) 322:326–35. doi: 10.1001/jama.2019.9346
14. Ntaios G, Egli M, Faouzi M, Michel P. J-shaped association between serum glucose and functional outcome in acute ischemic stroke. Stroke. (2010) 41:2366–70. doi: 10.1161/STROKEAHA.110.592170
15. Zonneveld TP, Nederkoorn PJ, Westendorp WF, Brouwer MC, van de Beek D, Kruyt ND. Hyperglycemia predicts poststroke infections in acute ischemic stroke. Neurology. (2017) 88:1415–21. doi: 10.1212/WNL.0000000000003811
16. Saqqur M, Shuaib A, Alexandrov AV, Sebastian J, Khan K, Uchino K. The correlation between admission blood glucose and intravenous rt-PA-induced arterial recanalization in acute ischemic stroke: a multi-centre TCD study. Int J Stroke. (2015) 10:1087–92. doi: 10.1111/ijs.12517
17. Fang HJ, Pan YS, Wang YJ, Wang CX, Wang YL, Zhong LY. Prognostic value of admission hyperglycemia on outcomes of thrombolysis in ischemic stroke patients with or without diabetes. Chin Med J. (2020) 133:2244–6. doi: 10.1097/CM9.0000000000001005
18. Goyal N, Tsivgoulis G, Pandhi A, Dillard K, Katsanos AH, Magoufis G, et al. Admission hyperglycemia and outcomes in large vessel occlusion strokes treated with mechanical thrombectomy. J Neurointerv Surg. (2018) 10:112–7. doi: 10.1136/neurintsurg-2017-012993
19. Osei E, Den Hertog HM, Berkhemer OA, Fransen PS, Roos YB, Beumer D, et al. Increased admission and fasting glucose are associated with unfavorable short-term outcome after intra-arterial treatment of ischemic stroke in the mr clean pretrial cohort. J Neurol Sci. (2016) 371:1–5. doi: 10.1016/j.jns.2016.10.003
20. Osei E, Den Hertog HM, Berkhemer OA, Fransen PSS, Roos Y, Beumer D, et al. Admission glucose and effect of intra-arterial treatment in patients with acute ischemic stroke. Stroke. (2017) 48:1299–305. doi: 10.1161/STROKEAHA.116.016071
21. Farrokhnia N, Björk E, Lindbäck J, Terent A. Blood glucose in acute stroke, different therapeutic targets for diabetic and non-diabetic patients? Acta Neurol Scand. (2005) 112:81–7. doi: 10.1111/j.1600-0404.2005.00440.x
22. Snarska KK, Bachórzewska-Gajewska H, Kapica-Topczewska K, Drozdowski W, Chorazy M, Kułakowska A, et al. Hyperglycemia and diabetes have different impacts on outcome of ischemic and hemorrhagic stroke. Arch Med Sci. (2017) 13:100–8. doi: 10.5114/aoms.2016.61009
23. Fuentes B, Castillo J, San José B, Leira R, Serena J, Vivancos J, et al. The prognostic value of capillary glucose levels in acute stroke: The glycemia in acute stroke (glias) study. Stroke. (2009) 40:562–8. doi: 10.1161/STROKEAHA.108.519926
24. Yang CJ, Liao WI, Wang JC, Tsai CL, Lee JT, Peng GS, et al. Usefulness of glycated hemoglobin a1c-based adjusted glycemic variables in diabetic patients presenting with acute ischemic stroke. Am J Emerg Med. (2017) 35:1240–6. doi: 10.1016/j.ajem.2017.03.049
25. Zhu B, Pan Y, Jing J, Meng X, Zhao X, Liu L, et al. Stress hyperglycemia and outcome of non-diabetic patients after acute ischemic stroke. Front Neurol. (2019) 10:1003. doi: 10.3389/fneur.2019.01003
26. Li J, Quan K, Wang Y, Zhao X, Li Z, Pan Y, et al. Effect of stress hyperglycemia on neurological deficit and mortality in the acute ischemic stroke people with and without diabetes. Front Neurol. (2020) 11:576895. doi: 10.3389/fneur.2020.576895
27. Yuan C, Chen S, Ruan Y, Liu Y, Cheng H, Zeng Y, et al. The stress hyperglycemia ratio is associated with hemorrhagic transformation in patients with acute ischemic stroke. Clin Interv Aging. (2021) 16:431–42. doi: 10.2147/CIA.S280808
28. Mi D, Li Z, Gu H, Jiang Y, Zhao X, Wang Y, et al. Stress hyperglycemia is associated with in-hospital mortality in patients with diabetes and acute ischemic stroke. CNS Neurosci Ther. (2022) 28:372–81. doi: 10.1111/cns.13764
29. Merlino G, Smeralda C, Gigli GL, Lorenzut S, Pez S, Surcinelli A, et al. Stress hyperglycemia is predictive of worse outcome in patients with acute ischemic stroke undergoing intravenous thrombolysis. J Thromb Thrombolysis. (2021) 51:789–97. doi: 10.1007/s11239-020-02252-y
30. Shen CL, Xia NG, Wang H, Zhang WL. Association of stress hyperglycemia ratio with acute ischemic stroke outcomes post-thrombolysis. Front Neurol. (2021) 12:785428. doi: 10.3389/fneur.2021.785428
31. Wang L, Cheng Q, Hu T, Wang N, Wei X, Wu T, et al. Impact of stress hyperglycemia on early neurological deterioration in acute ischemic stroke patients treated with intravenous thrombolysis. Front Neurol. (2022) 13:870872. doi: 10.3389/fneur.2022.870872
32. Merlino G, Pez S, Tereshko Y, Gigli GL, Lorenzut S, Surcinelli A, et al. Stress hyperglycemia does not affect clinical outcome of diabetic patients receiving intravenous thrombolysis for acute ischemic stroke. Front Neurol. (2022) 13:903987. doi: 10.3389/fneur.2022.903987
33. Ngiam JN, Cheong CWS, Leow AST, Wei YT, Thet JKX, Lee IYS, et al. Stress hyperglycaemia is associated with poor functional outcomes in patients with acute ischaemic stroke after intravenous thrombolysis. QJM. (2022) 115:7–11. doi: 10.1093/qjmed/hcaa253
34. Merlino G, Pez S, Gigli GL, Sponza M, Lorenzut S, Surcinelli A, et al. Stress hyperglycemia in patients with acute ischemic stroke due to large vessel occlusion undergoing mechanical thrombectomy. Front Neurol. (2021) 12:725002. doi: 10.3389/fneur.2021.725002
35. Chen X, Liu Z, Miao J, Zheng W, Yang Q, Ye X, et al. High stress hyperglycemia ratio predicts poor outcome after mechanical thrombectomy for ischemic stroke. J Stroke Cerebrovasc Dis. (2019) 28:1668–73. doi: 10.1016/j.jstrokecerebrovasdis.2019.02.022
36. Hui J, Zhang J, Mao X, Li Z, Li X, Wang F, et al. The initial glycemic variability is associated with early neurological deterioration in diabetic patients with acute ischemic stroke. Neurol Sci. (2018) 39:1571–7. doi: 10.1007/s10072-018-3463-6
37. Lin J, Cai C, Xie Y, Yi L. Acute glycemic variability and mortality of patients with acute stroke: a meta-analysis. Diabetol Metab Syndr. (2022) 14:69. doi: 10.1186/s13098-022-00826-9
38. Baudu J, Gerbaud E, Catargi B, Montaudon M, Beauvieux MC, Sagnier S, et al. High glycemic variability: an underestimated determinant of stroke functional outcome following large vessel occlusion. Rev Neurol. (2022) 178:732–40. doi: 10.1016/j.neurol.2021.12.010
39. Lim JS, Kim C, Oh MS, Lee JH, Jung S, Jang MU, et al. Effects of glycemic variability and hyperglycemia in acute ischemic stroke on post-stroke cognitive impairments. J Diabetes Complications. (2018) 32:682–7. doi: 10.1016/j.jdiacomp.2018.02.006
40. Kim TJ, Lee JS, Park SH, Ko SB. Short-term glycemic variability and hemorrhagic transformation after successful endovascular thrombectomy. Transl Stroke Res. (2021) 12:968–75. doi: 10.1007/s12975-021-00895-4
41. Kim YS, Kim C, Jung KH, Kwon HM, Heo SH, Kim BJ, et al. Range of glucose as a glycemic variability and 3-month outcome in diabetic patients with acute ischemic stroke. PLoS ONE. (2017) 12:e0183894. doi: 10.1371/journal.pone.0183894
42. Yoon JE, Sunwoo JS, Kim JS, Roh H, Ahn MY, Woo HY, et al. Poststroke glycemic variability increased recurrent cardiovascular events in diabetic patients. J Diabetes Complications. (2017) 31:390–4. doi: 10.1016/j.jdiacomp.2016.11.014
43. Capes SE, Hunt D, Malmberg K, Gerstein HC. Stress hyperglycaemia and increased risk of death after myocardial infarction in patients with and without diabetes: a systematic overview. Lancet. (2000) 355:773–8. doi: 10.1016/S0140-6736(99)08415-9
44. Yao M, Ni J, Zhou LX, Peng B, Zhu YC, Cui LY, et al. Elevated fasting blood glucose is predictive of poor outcome in non-diabetic stroke patients: a sub-group analysis of smart. PLoS ONE. (2016) 11:e0160674. doi: 10.1371/journal.pone.0160674
45. Chen G, Ren J, Huang H, Shen J, Yang C, Hu J, et al. Admission random blood glucose, fasting blood glucose, stress hyperglycemia ratio, and functional outcomes in patients with acute ischemic stroke treated with intravenous thrombolysis. Front Aging Neurosci. (2022) 14:782282. doi: 10.3389/fnagi.2022.782282
46. Ceriello A, Esposito K, Piconi L, Ihnat MA, Thorpe JE, Testa R, et al. Oscillating glucose is more deleterious to endothelial function and oxidative stress than mean glucose in normal and type 2 diabetic patients. Diabetes. (2008) 57:1349–54. doi: 10.2337/db08-0063
47. Allen CL, Bayraktutan U. Oxidative stress and its role in the pathogenesis of ischaemic stroke. Int J Stroke. (2009) 4:461–70. doi: 10.1111/j.1747-4949.2009.00387.x
48. Monnier L, Colette C, Owens DR. Glycemic variability: The third component of the dysglycemia in diabetes. Is it important? How to measure it? J Diabetes Sci Technol. (2008) 2:1094–100. doi: 10.1177/193229680800200618
49. Rama Chandran S, Tay WL, Lye WK, Lim LL, Ratnasingam J, Tan ATB, et al. Beyond hba1c: comparing glycemic variability and glycemic indices in predicting hypoglycemia in type 1 and type 2 diabetes. Diabetes Technol Ther. (2018) 20:353–62. doi: 10.1089/dia.2017.0388
50. Zhou Z, Sun B, Huang S, Zhu C, Bian M. Glycemic variability: adverse clinical outcomes and how to improve it? Cardiovasc Diabetol. (2020) 19:102. doi: 10.1186/s12933-020-01085-6
51. Lin CC, Yang CP Li CI, Liu CS, Chen CC, Lin WY, et al. Visit-to-visit variability of fasting plasma glucose as predictor of ischemic stroke: Competing risk analysis in a national cohort of taiwan diabetes study. BMC Med. (2014) 12:165. doi: 10.1186/s12916-014-0165-7
52. Allport LE, Butcher KS, Baird TA, MacGregor L, Desmond PM, Tress BM, et al. Insular cortical ischemia is independently associated with acute stress hyperglycemia. Stroke. (2004) 35:1886–91. doi: 10.1161/01.STR.0000133687.33868.71
53. Moreton FC, McCormick M, Muir KW. Insular cortex hypoperfusion and acute phase blood glucose after stroke: a ct perfusion study. Stroke. (2007) 38:407–10. doi: 10.1161/01.STR.0000254487.73282.4d
54. Pettersen JA, Pexman JH, Barber PA, Demchuk AM, Buchan AM, Hill MD. Insular cortical ischaemia does not independently predict acute hypertension or hyperglycaemia within 3 h of onset. J Neurol Neurosurg Psychiatry. (2006) 77:885–7. doi: 10.1136/jnnp.2005.087494
55. Zhao Z, Wang L, Gao W, Hu F, Zhang J, Ren Y, et al. A central catecholaminergic circuit controls blood glucose levels during stress. Neuron. (2017) 95:138–52e5. doi: 10.1016/j.neuron.2017.05.031
56. Sawchenko PE, Swanson LW. The organization of noradrenergic pathways from the brainstem to the paraventricular and supraoptic nuclei in the rat. Brain Res. (1982) 257:275–325. doi: 10.1016/0165-0173(82)90010-8
57. Ulrich-Lai YM, Herman JP. Neural regulation of endocrine and autonomic stress responses. Nat Rev Neurosci. (2009) 10:397–409. doi: 10.1038/nrn2647
58. Vanhorebeek I, Van den Berghe G. Diabetes of injury: novel insights. Endocrinol Metab Clin North Am. (2006) 35:859–72. doi: 10.1016/j.ecl.2006.09.002
59. Wise JK, Hendler R, Felig P. Influence of glucocorticoids on glucagon secretion and plasma amino acid concentrations in man. J Clin Invest. (1973) 52:2774–82. doi: 10.1172/JCI107473
60. Kahn CR, White MF. The insulin receptor and the molecular mechanism of insulin action. J Clin Invest. (1988) 82:1151–6. doi: 10.1172/JCI113711
61. Saltiel AR, Pessin JE. Insulin signaling pathways in time and space. Trends Cell Biol. (2002) 12:65–71. doi: 10.1016/S0962-8924(01)02207-3
62. Stephens JM, Pekala PH. Transcriptional repression of the glut4 and c/ebp genes in 3t3-l1 adipocytes by tumor necrosis factor-alpha. J Biol Chem. (1991) 266:21839–45. doi: 10.1016/S0021-9258(18)54714-1
63. Raje V, Ahern KW, Martinez BA, Howell NL, Oenarto V, Granade ME, et al. Adipocyte lipolysis drives acute stress-induced insulin resistance. Sci Rep. (2020) 10:18166. doi: 10.1038/s41598-020-75321-0
64. Dresner A, Laurent D, Marcucci M, Griffin ME, Dufour S, Cline GW, et al. Effects of free fatty acids on glucose transport and irs-1-associated phosphatidylinositol 3-kinase activity. J Clin Invest. (1999) 103:253–9. doi: 10.1172/JCI5001
65. Kelley DE, Mokan M, Simoneau JA, Mandarino LJ. Interaction between glucose and free fatty acid metabolism in human skeletal muscle. J Clin Invest. (1993) 92:91–8. doi: 10.1172/JCI116603
66. Wang YY, Lin SY, Chuang YH, Chen CJ, Tung KC, Sheu WH. Adipose proinflammatory cytokine expression through sympathetic system is associated with hyperglycemia and insulin resistance in a rat ischemic stroke model. Am J Physiol Endocrinol Metab. (2011) 300:E155–63. doi: 10.1152/ajpendo.00301.2010
67. Fantuzzi G. Adipose tissue, adipokines, and inflammation. J Allergy Clin Immunol. (2005) 115:911–9; 20. doi: 10.1016/j.jaci.2005.02.023
68. Blumberg D, Hochwald S, Burt M, Donner D, Brennan MF. Tumor necrosis factor alpha stimulates gluconeogenesis from alanine in vivo. J Surg Oncol. (1995) 59:220–4; 4–5. doi: 10.1002/jso.2930590404
69. Ibayashi S, Fujishima M, Sadoshima S, Yoshida F, Shiokawa O, Ogata J, et al. Cerebral blood flow and tissue metabolism in experimental cerebral ischemia of spontaneously hypertensive rats with hyper-, normo-, and hypoglycemia. Stroke. (1986) 17:261–6. doi: 10.1161/01.STR.17.2.261
70. Kraft SA, Larson CP Jr, Shuer LM, Steinberg GK, Benson GV, Pearl RG. Effect of hyperglycemia on neuronal changes in a rabbit model of focal cerebral ischemia. Stroke. (1990) 21:447–50. doi: 10.1161/01.STR.21.3.447
71. McNamara JJ, Mills D, Aaby GV. Effect of hypertonic glucose on hemorrhagic shock in rabbits. Ann Thorac Surg. (1970) 9:116–21. doi: 10.1016/S0003-4975(10)65784-0
72. Uyttenboogaart M, Koch MW, Stewart RE, Vroomen PC, Luijckx GJ, De Keyser J. Moderate hyperglycaemia is associated with favourable outcome in acute lacunar stroke. Brain. (2007) 130:1626–30. doi: 10.1093/brain/awm087
73. Bruno A, Biller J, Adams HP Jr, Clarke WR, Woolson RF, Williams LS, et al. Acute blood glucose level and outcome from ischemic stroke. Trial of org 10172 in acute stroke treatment (toast) investigators. Neurology. (1999) 52:280–4. doi: 10.1212/WNL.52.2.280
74. Parsons MW, Barber PA, Desmond PM, Baird TA, Darby DG, Byrnes G, et al. Acute hyperglycemia adversely affects stroke outcome: a magnetic resonance imaging and spectroscopy study. Ann Neurol. (2002) 52:20–8. doi: 10.1002/ana.10241
75. Xu G, Takashi E, Kudo M, Ishiwata T, Naito Z. Contradictory effects of short- and long-term hyperglycemias on ischemic injury of myocardium via intracellular signaling pathway. Exp Mol Pathol. (2004) 76:57–65. doi: 10.1016/j.yexmp.2003.08.003
76. Ma G, Al-Shabrawey M, Johnson JA, Datar R, Tawfik HE, Guo D, et al. Protection against myocardial ischemia/reperfusion injury by short-term diabetes: Enhancement of vegf formation, capillary density, and activation of cell survival signaling. Naunyn Schmiedebergs Arch Pharmacol. (2006) 373:415–27. doi: 10.1007/s00210-006-0102-1
77. Chu LM, Osipov RM, Robich MP, Feng J, Oyamada S, Bianchi C, et al. Is hyperglycemia bad for the heart during acute ischemia? J Thorac Cardiovasc Surg. (2010) 140:1345–52. doi: 10.1016/j.jtcvs.2010.05.009
78. Malfitano C, Alba Loureiro TC, Rodrigues B, Sirvente R, Salemi VM, Rabechi NB, et al. Hyperglycaemia protects the heart after myocardial infarction: Aspects of programmed cell survival and cell death. Eur J Heart Fail. (2010) 12:659–67. doi: 10.1093/eurjhf/hfq053
79. Malfitano C, Barboza CA, Mostarda C, da Palma RK, dos Santos CP, Rodrigues B, et al. Diabetic hyperglycemia attenuates sympathetic dysfunction and oxidative stress after myocardial infarction in rats. Cardiovasc Diabetol. (2014) 13:131. doi: 10.1186/s12933-014-0131-x
80. Lemkes BA, Hermanides J, Devries JH, Holleman F, Meijers JC, Hoekstra JB. Hyperglycemia: a prothrombotic factor? J Thromb Haemost. (2010) 8:1663–9. doi: 10.1111/j.1538-7836.2010.03910.x
81. Stegenga ME, Van der Crabben SN, Levi M, de Vos AF, Tanck MW, Sauerwein HP, et al. Hyperglycemia stimulates coagulation, whereas hyperinsulinemia impairs fibrinolysis in healthy humans. Diabetes. (2006) 55:1807–12. doi: 10.2337/db05-1543
82. Pandolfi A, Giaccari A, Cilli C, Alberta MM, Morviducci L, De Filippis EA, et al. Acute hyperglycemia and acute hyperinsulinemia decrease plasma fibrinolytic activity and increase plasminogen activator inhibitor type 1 in the rat. Acta Diabetol. (2001) 38:71–6. doi: 10.1007/s005920170016
83. Pandolfi A, Iacoviello L, Capani F, Vitacolonna E, Donati MB, Consoli A. Glucose and insulin independently reduce the fibrinolytic potential of human vascular smooth muscle cells in culture. Diabetologia. (1996) 39:1425–31. doi: 10.1007/s001250050594
84. Ribo M, Molina C, Montaner J, Rubiera M, Delgado-Mederos R, Arenillas JF, et al. Acute hyperglycemia state is associated with lower tpa-induced recanalization rates in stroke patients. Stroke. (2005) 36:1705–9. doi: 10.1161/01.STR.0000173161.05453.90.9f
85. Nakai H, Yamamoto YL, Diksic M, Worsley KJ, Takara E. Triple-tracer autoradiography demonstrates effects of hyperglycemia on cerebral blood flow, ph, and glucose utilization in cerebral ischemia of rats. Stroke. (1988) 19:764–72. doi: 10.1161/01.STR.19.6.764
86. Duckrow RB, Beard DC, Brennan RW. Regional cerebral blood flow decreases during chronic and acute hyperglycemia. Stroke. (1987) 18:52–8. doi: 10.1161/01.STR.18.1.52
87. Ginsberg MD, Welsh FA, Budd WW. Deleterious effect of glucose pretreatment on recovery from diffuse cerebral ischemia in the cat. I. Local cerebral blood flow and glucose utilization. Stroke. (1980) 11:347–54. doi: 10.1161/01.STR.11.4.347
88. Duckrow RB, Beard DC, Brennan RW. Regional cerebral blood flow decreases during hyperglycemia. Ann Neurol. (1985) 17:267–72. doi: 10.1002/ana.410170308
89. Fabian RH, Kent TA. Hyperglycemia accentuates persistent “functional uncoupling” of cerebral microvascular nitric oxide and superoxide following focal ischemia/reperfusion in rats. Transl Stroke Res. (2012) 3:482–90. doi: 10.1007/s12975-012-0210-9
90. Meza CA, La Favor JD, Kim DH, Hickner RC. Endothelial dysfunction: Is there a hyperglycemia-induced imbalance of nox and nos? Int J Mol Sci. (2019) 20:3775. doi: 10.3390/ijms20153775
91. Valko M, Leibfritz D, Moncol J, Cronin MT, Mazur M, Telser J. Free radicals and antioxidants in normal physiological functions and human disease. Int J Biochem Cell Biol. (2007) 39:44–84. doi: 10.1016/j.biocel.2006.07.001
92. Suh SW, Shin BS, Ma H, Van Hoecke M, Brennan AM, Yenari MA, et al. Glucose and nadph oxidase drive neuronal superoxide formation in stroke. Ann Neurol. (2008) 64:654–63. doi: 10.1002/ana.21511
93. Allen CL, Bayraktutan U. Antioxidants attenuate hyperglycaemia-mediated brain endothelial cell dysfunction and blood-brain barrier hyperpermeability. Diabetes Obes Metab. (2009) 11:480–90. doi: 10.1111/j.1463-1326.2008.00987.x
94. Zhang Z, Yan J, Shi H. Role of hypoxia inducible factor 1 in hyperglycemia-exacerbated blood-brain barrier disruption in ischemic stroke. Neurobiol Dis. (2016) 95:82–92. doi: 10.1016/j.nbd.2016.07.012
95. Venkat P, Chopp M, Chen J. Blood-brain barrier disruption, vascular impairment, and ischemia/reperfusion damage in diabetic stroke. J Am Heart Assoc. (2017) 6:e005819. doi: 10.1161/JAHA.117.005819
96. Bémeur C, Ste-Marie L, Montgomery J. Increased oxidative stress during hyperglycemic cerebral ischemia. Neurochem Int. (2007) 50:890–904. doi: 10.1016/j.neuint.2007.03.002
97. Guo Y, Dong L, Gong A, Zhang J, Jing L, Ding T, et al. Damage to the blood-brain barrier and activation of neuroinflammation by focal cerebral ischemia under hyperglycemic condition. Int J Mol Med. (2021) 48:142. doi: 10.3892/ijmm.2021.4975
98. Aljada A, Ghanim H, Mohanty P, Syed T, Bandyopadhyay A, Dandona P. Glucose intake induces an increase in activator protein 1 and early growth response 1 binding activities, in the expression of tissue factor and matrix metalloproteinase in mononuclear cells, and in plasma tissue factor and matrix metalloproteinase concentrations. Am J Clin Nutr. (2004) 80:51–7. doi: 10.1093/ajcn/80.1.51
99. Ahn JD, Morishita R, Kaneda Y, Lee KU, Park JY, Jeon YJ, et al. Transcription factor decoy for activator protein-1 (ap-1) inhibits high glucose- and angiotensin ii-induced type 1 plasminogen activator inhibitor (pai-1) gene expression in cultured human vascular smooth muscle cells. Diabetologia. (2001) 44:713–20. doi: 10.1007/s001250051680
100. Hawkins BT, Lundeen TF, Norwood KM, Brooks HL, Egleton RD. Increased blood-brain barrier permeability and altered tight junctions in experimental diabetes in the rat: contribution of hyperglycaemia and matrix metalloproteinases. Diabetologia. (2007) 50:202–11. doi: 10.1007/s00125-006-0485-z
101. Kamada H, Yu F, Nito C, Chan PH. Influence of hyperglycemia on oxidative stress and matrix metalloproteinase-9 activation after focal cerebral ischemia/reperfusion in rats: relation to blood-brain barrier dysfunction. Stroke. (2007) 38:1044–9. doi: 10.1161/01.STR.0000258041.75739.cb
102. Dhindsa S, Tripathy D, Mohanty P, Ghanim H, Syed T, Aljada A, et al. Differential effects of glucose and alcohol on reactive oxygen species generation and intranuclear nuclear factor-kappab in mononuclear cells. Metabolism. (2004) 53:330–4. doi: 10.1016/j.metabol.2003.10.013
103. Barnes PJ, Karin M. Nuclear factor-kappab: a pivotal transcription factor in chronic inflammatory diseases. N Engl J Med. (1997) 336:1066–71. doi: 10.1056/NEJM199704103361506
104. El-Osta A, Brasacchio D, Yao D, Pocai A, Jones PL, Roeder RG, et al. Transient high glucose causes persistent epigenetic changes and altered gene expression during subsequent normoglycemia. J Exp Med. (2008) 205:2409–17. doi: 10.1084/jem.20081188
105. Jing L, Wang JG, Zhang JZ, Cao CX, Chang Y, Dong JD, et al. Upregulation of icam-1 in diabetic rats after transient forebrain ischemia and reperfusion injury. J Inflamm. (2014) 11:35. doi: 10.1186/s12950-014-0035-2
106. Morigi M, Angioletti S, Imberti B, Donadelli R, Micheletti G, Figliuzzi M, et al. Leukocyte-endothelial interaction is augmented by high glucose concentrations and hyperglycemia in a nf-kb-dependent fashion. J Clin Invest. (1998) 101:1905–15. doi: 10.1172/JCI656
107. Okada Y, Copeland BR, Mori E, Tung MM, Thomas WS, del Zoppo GJ. P-selectin and intercellular adhesion molecule-1 expression after focal brain ischemia and reperfusion. Stroke. (1994) 25:202–11. doi: 10.1161/01.STR.25.1.202
108. Clark WM, Lauten JD, Lessov N, Woodward W, Coull BM. Time course of icam-1 expression and leukocyte subset infiltration in rat forebrain ischemia. Mol Chem Neuropathol. (1995) 26:213–30. doi: 10.1007/BF02815139
109. Del Zoppo GJ, Mabuchi T. Cerebral microvessel responses to focal ischemia. J Cereb Blood Flow Metab. (2003) 23:879–94. doi: 10.1097/01.WCB.0000078322.96027.78
110. Gallagher CN, Carpenter KL, Grice P, Howe DJ, Mason A, Timofeev I, et al. The human brain utilizes lactate via the tricarboxylic acid cycle: A 13c-labelled microdialysis and high-resolution nuclear magnetic resonance study. Brain. (2009) 132:2839–49. doi: 10.1093/brain/awp202
111. Sotelo-Hitschfeld T, Fernández-Moncada I, Barros LF. Acute feedback control of astrocytic glycolysis by lactate. Glia. (2012) 60:674–80. doi: 10.1002/glia.22304
112. Wagner KR, Kleinholz M, de Courten-Myers GM, Myers RE. Hyperglycemic versus normoglycemic stroke: Topography of brain metabolites, intracellular ph, and infarct size. J Cereb Blood Flow Metab. (1992) 12:213–22. doi: 10.1038/jcbfm.1992.31
113. Chew W, Kucharczyk J, Moseley M, Derugin N, Norman D. Hyperglycemia augments ischemic brain injury: in vivo mr imaging/spectroscopic study with nicardipine in cats with occluded middle cerebral arteries. AJNR Am J Neuroradiol. (1991) 12:603–9.
114. Lipton P. Ischemic cell death in brain neurons. Physiol Rev. (1999) 79:1431–568. doi: 10.1152/physrev.1999.79.4.1431
115. Obrenovitch TP, Urenjak J, Richards DA, Ueda Y, Curzon G, Symon L. Extracellular neuroactive amino acids in the rat striatum during ischaemia: Comparison between penumbral conditions and ischaemia with sustained anoxic depolarisation. J Neurochem. (1993) 61:178–86. doi: 10.1111/j.1471-4159.1993.tb03553.x
116. Li PA, Shuaib A, Miyashita H, He QP, Siesjö BK, Warner DS. Hyperglycemia enhances extracellular glutamate accumulation in rats subjected to forebrain ischemia. Stroke. (2000) 31:183–92. doi: 10.1161/01.STR.31.1.183
117. Martel MA, Ryan TJ, Bell KF, Fowler JH, McMahon A, Al-Mubarak B, et al. The subtype of glun2 c-terminal domain determines the response to excitotoxic insults. Neuron. (2012) 74:543–56. doi: 10.1016/j.neuron.2012.03.021
118. Luetjens CM, Bui NT, Sengpiel B, Münstermann G, Poppe M, Krohn AJ, et al. Delayed mitochondrial dysfunction in excitotoxic neuron death: cytochrome c release and a secondary increase in superoxide production. J Neurosci. (2000) 20:5715–23. doi: 10.1523/JNEUROSCI.20-15-05715.2000
119. Jiang X, Wang X. Cytochrome c promotes caspase-9 activation by inducing nucleotide binding to apaf-1. J Biol Chem. (2000) 275:31199–203. doi: 10.1074/jbc.C000405200
120. Porter AG, Jänicke RU. Emerging roles of caspase-3 in apoptosis. Cell Death Differ. (1999) 6:99–104. doi: 10.1038/sj.cdd.4400476
121. Cheng YR, Jiang BY, Chen CC. Acid-sensing ion channels: dual function proteins for chemo-sensing and mechano-sensing. J Biomed Sci. (2018) 25:46. doi: 10.1186/s12929-018-0448-y
122. Storozhuk M, Cherninskyi A, Maximyuk O, Isaev D, Krishtal O. Acid-sensing ion channels: focus on physiological and some pathological roles in the brain. Curr Neuropharmacol. (2021) 19:1570–89. doi: 10.2174/1570159X19666210125151824
123. Xiong ZG, Zhu XM, Chu XP, Minami M, Hey J, Wei WL, et al. Neuroprotection in ischemia: blocking calcium-permeable acid-sensing ion channels. Cell. (2004) 118:687–98. doi: 10.1016/j.cell.2004.08.026
124. Li M, Inoue K, Branigan D, Kratzer E, Hansen JC, Chen JW, et al. Acid-sensing ion channels in acidosis-induced injury of human brain neurons. J Cereb Blood Flow Metab. (2010) 30:1247–60. doi: 10.1038/jcbfm.2010.30
125. Araki N, Greenberg JH, Sladky JT, Uematsu D, Karp A, Reivich M. The effect of hyperglycemia on intracellular calcium in stroke. J Cereb Blood Flow Metab. (1992) 12:469–76. doi: 10.1038/jcbfm.1992.64
126. Voll CL, Auer RN. The effect of postischemic blood glucose levels on ischemic brain damage in the rat. Ann Neurol. (1988) 24:638–46. doi: 10.1002/ana.410240508
127. Zhu CZ, Auer RN. Intraventricular administration of insulin and igf-1 in transient forebrain ischemia. J Cereb Blood Flow Metab. (1994) 14:237–42. doi: 10.1038/jcbfm.1994.30
128. Hamilton MG, Tranmer BI, Auer RN. Insulin reduction of cerebral infarction due to transient focal ischemia. J Neurosurg. (1995) 82:262–8. doi: 10.3171/jns.1995.82.2.0262
129. Ahmadi-Eslamloo H, Dehghani GA, Moosavi SMS. Long-term treatment of diabetic rats with vanadyl sulfate or insulin attenuate acute focal cerebral ischemia/reperfusion injury via their antiglycemic effect. Metab Brain Dis. (2018) 33:225–35. doi: 10.1007/s11011-017-0153-7
130. Shuaib A, Ijaz MS, Waqar T, Voll C, Kanthan R, Miyashita H, et al. Insulin elevates hippocampal gaba levels during ischemia. This is independent of its hypoglycemic effect. Neuroscience. (1995) 67:809–14. doi: 10.1016/0306-4522(95)00093-X
131. Lanier WL, Hofer RE, Gallagher WJ. Metabolism of glucose, glycogen, and high-energy phosphates during transient forebrain ischemia in diabetic rats: effect of insulin treatment. Anesthesiology. (1996) 84:917–25. doi: 10.1097/00000542-199604000-00020
132. Sullivan JM, Alousi SS, Hikade KR, Bahu NJ, Rafols JA, Krause GS, et al. Insulin induces dephosphorylation of eukaryotic initiation factor 2alpha and restores protein synthesis in vulnerable hippocampal neurons after transient brain ischemia. J Cereb Blood Flow Metab. (1999) 19:1010–9. doi: 10.1097/00004647-199909000-00009
133. Guyot LL, Diaz FG, O'Regan MH, Song D, Phillis JW. The effect of topical insulin on the release of excitotoxic and other amino acids from the rat cerebral cortex during streptozotocin-induced hyperglycemic ischemia. Brain Res. (2000) 872:29–36. doi: 10.1016/S0006-8993(00)02426-4
134. Hui L, Pei DS, Zhang QG, Guan QH, Zhang GY. The neuroprotection of insulin on ischemic brain injury in rat hippocampus through negative regulation of jnk signaling pathway by pi3k/akt activation. Brain Res. (2005) 1052:1–9. doi: 10.1016/j.brainres.2005.05.043
135. Sanderson TH, Kumar R, Murariu-Dobrin AC, Page AB, Krause GS, Sullivan JM. Insulin activates the pi3k-akt survival pathway in vulnerable neurons following global brain ischemia. Neurol Res. (2009) 31:947–58. doi: 10.1179/174313209X382449
136. Fanne RA, Nassar T, Heyman SN, Hijazi N, Higazi AA. Insulin and glucagon share the same mechanism of neuroprotection in diabetic rats: role of glutamate. Am J Physiol Regul Integr Comp Physiol. (2011) 301:R668–73. doi: 10.1152/ajpregu.00058.2011
137. Fan X, Ning M, Lo EH, Wang X. Early insulin glycemic control combined with tpa thrombolysis reduces acute brain tissue damages in a focal embolic stroke model of diabetic rats. Stroke. (2013) 44:255–9. doi: 10.1161/STROKEAHA.112.663476
138. Sanderson TH, Mahapatra G, Pecina P, Ji Q, Yu K, Sinkler C, et al. Cytochrome c is tyrosine 97 phosphorylated by neuroprotective insulin treatment. PLoS ONE. (2013) 8:e78627. doi: 10.1371/journal.pone.0078627
139. Hung LM, Huang JP, Liao JM, Yang MH Li DE, Day YJ, et al. Insulin renders diabetic rats resistant to acute ischemic stroke by arresting nitric oxide reaction with superoxide to form peroxynitrite. J Biomed Sci. (2014) 21:92. doi: 10.1186/s12929-014-0092-0
140. Huang SS, Lu YJ, Huang JP, Wu YT, Day YJ, Hung LM. The essential role of endothelial nitric oxide synthase activation in insulin-mediated neuroprotection against ischemic stroke in diabetes. J Vasc Surg. (2014) 59:483–91. doi: 10.1016/j.jvs.2013.03.023
141. Malmberg K, Rydén L, Efendic S, Herlitz J, Nicol P, Waldenström A, et al. Randomized trial of insulin-glucose infusion followed by subcutaneous insulin treatment in diabetic patients with acute myocardial infarction (digami study): Effects on mortality at 1 year. J Am Coll Cardiol. (1995) 26:57–65. doi: 10.1016/0735-1097(95)00126-K
142. Van den Berghe G, Wouters P, Weekers F, Verwaest C, Bruyninckx F, Schetz M, et al. Intensive insulin therapy in critically ill patients. N Engl J Med. (2001) 345:1359–67. doi: 10.1056/NEJMoa011300
143. Bruno A, Saha C, Williams LS, Shankar R. Iv insulin during acute cerebral infarction in diabetic patients. Neurology. (2004) 62:1441–2. doi: 10.1212/01.WNL.0000120754.73348.02
144. Walters MR, Weir CJ, Lees KR. A randomised, controlled pilot study to investigate the potential benefit of intervention with insulin in hyperglycaemic acute ischaemic stroke patients. Cerebrovasc Dis. (2006) 22:116–22. doi: 10.1159/000093239
145. Johnston KC, Hall CE, Kissela BM, Bleck TP, Conaway MR. Glucose regulation in acute stroke patients (grasp) trial: a randomized pilot trial. Stroke. (2009) 40:3804–9. doi: 10.1161/STROKEAHA.109.561498
146. Kreisel SH, Berschin UM, Hammes HP, Leweling H, Bertsch T, Hennerici MG, et al. Pragmatic management of hyperglycaemia in acute ischaemic stroke: Safety and feasibility of intensive intravenous insulin treatment. Cerebrovasc Dis. (2009) 27:167–75. doi: 10.1159/000185608
147. Kruyt ND, Biessels GJ, Vriesendorp TM, Devries JH, Hoekstra JB, Elbers PW, et al. Subjecting acute ischemic stroke patients to continuous tube feeding and an intensive computerized protocol establishes tight glycemic control. Neurocrit Care. (2010) 12:62–8. doi: 10.1007/s12028-009-9230-z
148. Vriesendorp TM, Roos YB, Kruyt ND, Biessels GJ, Kappelle LJ, Vermeulen M, et al. Efficacy and safety of two 5 day insulin dosing regimens to achieve strict glycaemic control in patients with acute ischaemic stroke. J Neurol Neurosurg Psychiatry. (2009) 80:1040–3. doi: 10.1136/jnnp.2008.144873
149. Torbey MT, Pauls Q, Gentile N, Falciglia M, Meurer W, Pettigrew CL, et al. Intensive versus standard treatment of hyperglycemia in acute ischemic stroke patient: a randomized clinical trial subgroups analysis. Stroke. (2022) 53:1510–5. doi: 10.1161/STROKEAHA.120.033048
150. Baird TA, Parsons MW, Phan T, Butcher KS, Desmond PM, Tress BM, et al. Persistent poststroke hyperglycemia is independently associated with infarct expansion and worse clinical outcome. Stroke. (2003) 34:2208–14. doi: 10.1161/01.STR.0000085087.41330.FF
151. Allport L, Baird T, Butcher K, Macgregor L, Prosser J, Colman P, et al. Frequency and temporal profile of poststroke hyperglycemia using continuous glucose monitoring. Diabetes Care. (2006) 29:1839–44. doi: 10.2337/dc06-0204
152. Wada S, Yoshimura S, Inoue M, Matsuki T, Arihiro S, Koga M, et al. Outcome prediction in acute stroke patients by continuous glucose monitoring. J Am Heart Assoc. (2018) 7:e008744. doi: 10.1161/JAHA.118.008744
153. Zhu CZ, Auer RN. Optimal blood glucose levels while using insulin to minimize the size of infarction in focal cerebral ischemia. J Neurosurg. (2004) 101:664–8. doi: 10.3171/jns.2004.101.4.0664
154. Gentile NT, Seftchick MW, Huynh T, Kruus LK, Gaughan J. Decreased mortality by normalizing blood glucose after acute ischemic stroke. Acad Emerg Med. (2006) 13:174–80. doi: 10.1197/j.aem.2005.08.009
155. Powers WJ, Rabinstein AA, Ackerson T, Adeoye OM, Bambakidis NC, Becker K, et al. Guidelines for the early management of patients with acute ischemic stroke: 2019 update to the 2018 guidelines for the early management of acute ischemic stroke: a guideline for healthcare professionals from the american heart association/american stroke association. Stroke. (2019) 50:e344–418. doi: 10.1161/STR.0000000000000211
156. Wijdicks EF, Sheth KN, Carter BS, Greer DM, Kasner SE, Kimberly WT, et al. Recommendations for the management of cerebral and cerebellar infarction with swelling: a statement for healthcare professionals from the american heart association/american stroke association. Stroke. (2014) 45:1222–38. doi: 10.1161/01.str.0000441965.15164.d6
157. Yatabe T, Inoue S, Sakaguchi M, Egi M. The optimal target for acute glycemic control in critically ill patients: a network meta-analysis. Intensive Care Med. (2017) 43:16–28. doi: 10.1007/s00134-016-4558-2
158. Dave KR, Tamariz J, Desai KM, Brand FJ, Liu A, Saul I, et al. Recurrent hypoglycemia exacerbates cerebral ischemic damage in streptozotocin-induced diabetic rats. Stroke. (2011) 42:1404–11. doi: 10.1161/STROKEAHA.110.594937
159. Shukla V, Fuchs P, Liu A, Cohan CH, Dong C, Wright CB, et al. Recurrent hypoglycemia exacerbates cerebral ischemic damage in diabetic rats via enhanced post-ischemic mitochondrial dysfunction. Transl Stroke Res. (2019) 10:78–90. doi: 10.1007/s12975-018-0622-2
160. Gogitidze Joy N, Hedrington MS, Briscoe VJ, Tate DB, Ertl AC, Davis SN. Effects of acute hypoglycemia on inflammatory and pro-atherothrombotic biomarkers in individuals with type 1 diabetes and healthy individuals. Diabetes Care. (2010) 33:1529–35. doi: 10.2337/dc09-0354
161. Wright RJ, Newby DE, Stirling D, Ludlam CA, Macdonald IA, Frier BM. Effects of acute insulin-induced hypoglycemia on indices of inflammation: putative mechanism for aggravating vascular disease in diabetes. Diabetes Care. (2010) 33:1591–7. doi: 10.2337/dc10-0013
162. Dantz D, Bewersdorf J, Fruehwald-Schultes B, Kern W, Jelkmann W, Born J, et al. Vascular endothelial growth factor: a novel endocrine defensive response to hypoglycemia. J Clin Endocrinol Metab. (2002) 87:835–40. doi: 10.1210/jcem.87.2.8215
163. Goldenberg RM, Cheng AYY, Fitzpatrick T, Gilbert JD, Verma S, Hopyan JJ. Benefits of glp-1 (glucagon-like peptide 1) receptor agonists for stroke reduction in type 2 diabetes: a call to action for neurologists. Stroke. (2022) 53:1813–22. doi: 10.1161/STROKEAHA.121.038151
164. Zhu H, Zhang Y, Shi Z, Lu D, Li T, Ding Y, et al. The neuroprotection of liraglutide against ischaemia-induced apoptosis through the activation of the pi3k/akt and mapk pathways. Sci Rep. (2016) 6:26859. doi: 10.1038/srep26859
165. Shan Y, Tan S, Lin Y, Liao S, Zhang B, Chen X, et al. The glucagon-like peptide-1 receptor agonist reduces inflammation and blood-brain barrier breakdown in an astrocyte-dependent manner in experimental stroke. J Neuroinflammation. (2019) 16:242. doi: 10.1186/s12974-019-1638-6
166. Daly SC, Chemmanam T, Loh PS, Gilligan A, Dear AE, Simpson RW, et al. Exenatide in acute ischemic stroke. Int J Stroke. (2013) 8:E44. doi: 10.1111/ijs.12073
167. Muller C, Cheung NW, Dewey H, Churilov L, Middleton S, Thijs V, et al. Treatment with exenatide in acute ischemic stroke trial protocol: A prospective, randomized, open label, blinded end-point study of exenatide vs. standard care in post stroke hyperglycemia. Int J Stroke. (2018) 13:857–62. doi: 10.1177/1747493018784436
168. Zhang G, Kim S, Gu X, Yu SP, Wei L. Dpp-4 inhibitor linagliptin is neuroprotective in hyperglycemic mice with stroke via the akt/mtor pathway and anti-apoptotic effects. Neurosci Bull. (2020) 36:407–18. doi: 10.1007/s12264-019-00446-w
169. Scirica BM, Bhatt DL, Braunwald E, Steg PG, Davidson J, Hirshberg B, et al. Saxagliptin and cardiovascular outcomes in patients with type 2 diabetes mellitus. N Engl J Med. (2013) 369:1317–26. doi: 10.1056/NEJMoa1307684
170. White WB, Cannon CP, Heller SR, Nissen SE, Bergenstal RM, Bakris GL, et al. Alogliptin after acute coronary syndrome in patients with type 2 diabetes. N Engl J Med. (2013) 369:1327–35. doi: 10.1056/NEJMoa1305889
171. Han JH, Oh TJ, Lee G, Maeng HJ, Lee DH, Kim KM, et al. The beneficial effects of empagliflozin, an sglt2 inhibitor, on atherosclerosis in apoe (-/-) mice fed a western diet. Diabetologia. (2017) 60:364–76. doi: 10.1007/s00125-016-4158-2
172. Harada S, Yamazaki Y, Nishioka H, Tokuyama S. Neuroprotective effect through the cerebral sodium-glucose transporter on the development of ischemic damage in global ischemia. Brain Res. (2013) 1541:61–8. doi: 10.1016/j.brainres.2013.09.041
173. Fitchett D, Inzucchi SE, Cannon CP, McGuire DK, Scirica BM, Johansen OE, et al. Empagliflozin reduced mortality and hospitalization for heart failure across the spectrum of cardiovascular risk in the empa-reg outcome trial. Circulation. (2019) 139:1384–95. doi: 10.1161/CIRCULATIONAHA.118.037778
174. Zhou Z, Lindley RI, Rådholm K, Jenkins B, Watson J, Perkovic V, et al. Canagliflozin and stroke in type 2 diabetes mellitus. Stroke. (2019) 50:396–404. doi: 10.1161/STROKEAHA.118.023009
175. Wu JH, Foote C, Blomster J, Toyama T, Perkovic V, Sundström J, et al. Effects of sodium-glucose cotransporter-2 inhibitors on cardiovascular events, death, and major safety outcomes in adults with type 2 diabetes: A systematic review and meta-analysis. Lancet Diabetes Endocrinol. (2016) 4:411–9. doi: 10.1016/S2213-8587(16)00052-8
176. Bailey CJ. Metformin: historical overview. Diabetologia. (2017) 60:1566–76. doi: 10.1007/s00125-017-4318-z
177. Hostalek U, Gwilt M, Hildemann S. Therapeutic use of metformin in prediabetes and diabetes prevention. Drugs. (2015) 75:1071–94. doi: 10.1007/s40265-015-0416-8
178. Cheng YY, Leu HB, Chen TJ, Chen CL, Kuo CH, Lee SD, et al. Metformin-inclusive therapy reduces the risk of stroke in patients with diabetes: a 4-year follow-up study. J Stroke Cerebrovasc Dis. (2014) 23:e99–105. doi: 10.1016/j.jstrokecerebrovasdis.2013.09.001
179. Mima Y, Kuwashiro T, Yasaka M, Tsurusaki Y, Nakamura A, Wakugawa Y, et al. Impact of metformin on the severity and outcomes of acute ischemic stroke in patients with type 2 diabetes mellitus. J Stroke Cerebrovasc Dis. (2016) 25:436–46. doi: 10.1016/j.jstrokecerebrovasdis.2015.10.016
180. Baines CP. The mitochondrial permeability transition pore and ischemia-reperfusion injury. Basic Res Cardiol. (2009) 104:181–8. doi: 10.1007/s00395-009-0004-8
181. Skemiene K, Rekuviene E, Jekabsone A, Cizas P, Morkuniene R, Borutaite V. Comparison of effects of metformin, phenformin, and inhibitors of mitochondrial complex i on mitochondrial permeability transition and ischemic brain injury. Biomolecules. (2020) 10:1400. doi: 10.3390/biom10101400
182. Li J, Benashski SE, Venna VR, McCullough LD. Effects of metformin in experimental stroke. Stroke. (2010) 41:2645–52. doi: 10.1161/STROKEAHA.110.589697
183. Zeng J, Zhu L, Liu J, Zhu T, Xie Z, Sun X, et al. Metformin protects against oxidative stress injury induced by ischemia/reperfusion via regulation of the incrna-h19/mir-148a-3p/rock2 axis. Oxid Med Cell Longev. (2019) 2019:8768327. doi: 10.1155/2019/8768327
184. Zemgulyte G, Tanaka S, Hide I, Sakai N, Pampuscenko K, Borutaite V, et al. Evaluation of the effectiveness of post-stroke metformin treatment using permanent middle cerebral artery occlusion in rats. Pharmaceuticals. (2021) 14:312. doi: 10.3390/ph14040312
185. Tucker GT, Casey C, Phillips PJ, Connor H, Ward JD, Woods HF. Metformin kinetics in healthy subjects and in patients with diabetes mellitus. Br J Clin Pharmacol. (1981) 12:235–46. doi: 10.1111/j.1365-2125.1981.tb01206.x
186. Den Hertog HM, Vermeer SE, Zandbergen AA, Achterberg S, Dippel DW, Algra A, et al. Safety and feasibility of metformin in patients with impaired glucose tolerance and a recent tia or minor ischemic stroke (limit) trial - a multicenter, randomized, open-label phase ii trial. Int J Stroke. (2015) 10:105–9. doi: 10.1111/ijs.12023
Keywords: stress-induced hyperglycaemia, acute ischaemic stroke, admission blood glucose, intensive glucose control, insulin
Citation: Yao M, Hao Y, Wang T, Xie M, Li H, Feng J, Feng L and Ma D (2023) A review of stress-induced hyperglycaemia in the context of acute ischaemic stroke: Definition, underlying mechanisms, and the status of insulin therapy. Front. Neurol. 14:1149671. doi: 10.3389/fneur.2023.1149671
Received: 22 January 2023; Accepted: 21 February 2023;
Published: 21 March 2023.
Edited by:
Cheong-Meng Chong, University of Macau, ChinaReviewed by:
Chen-Chih Chung, Taipei Medical University, TaiwanCopyright © 2023 Yao, Hao, Wang, Xie, Li, Feng, Feng and Ma. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Di Ma, bWFkaTIwMTdAamx1LmVkdS5jbg==; Liangshu Feng, ZmVuZ2xpYW5nc2h1QGpsdS5lZHUuY24=
†These authors have contributed equally to this work and share first authorship
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.