 Stefanie Perrier1,2
Stefanie Perrier1,2 Kether Guerrero1,2Luan T. Tran1,2
Kether Guerrero1,2Luan T. Tran1,2 Mackenzie A. Michell-Robinson1,2Geneviève Legault1,2,3
Mackenzie A. Michell-Robinson1,2Geneviève Legault1,2,3 Bernard Brais1,4,5Michel Sylvain6James Dorman7,8
Bernard Brais1,4,5Michel Sylvain6James Dorman7,8 Michelle Demos9
Michelle Demos9 Wolfgang Köhler10
Wolfgang Köhler10 Tomi Pastinen11,12
Tomi Pastinen11,12 Isabelle Thiffault11,12,13*†
Isabelle Thiffault11,12,13*† Geneviève Bernard1,2,3,5,14*†
Geneviève Bernard1,2,3,5,14*†- 1Department of Neurology and Neurosurgery, McGill University, Montreal, QC, Canada
- 2Child Health and Human Development Program, Research Institute of the McGill University Health Centre, Montreal, QC, Canada
- 3Department of Pediatrics, McGill University, Montreal, QC, Canada
- 4Montreal Neurological Institute, McGill University, Montreal, QC, Canada
- 5Department of Human Genetics, McGill University, Montreal, QC, Canada
- 6Division of Pediatric Neurology, Centre Mère-Enfant Soleil du CHU de Québec - Université Laval, Québec City, QC, Canada
- 7John H. Stroger Jr. Hospital of Cook County, Chicago, IL, United States
- 8Department of Neurological Sciences, Rush Medical College, Chicago, IL, United States
- 9Division of Neurology, Department of Pediatrics, University of British Columbia, BC Children's Hospital, Vancouver, BC, Canada
- 10Leukodystrophy Center, University of Leipzig Medical Center, Leipzig, Germany
- 11Genomic Medicine Center, Children's Mercy Hospital, Kansas City, MO, United States
- 12University of Missouri Kansas City School of Medicine, Kansas City, MO, United States
- 13Department of Pathology and Laboratory Medicine, Children's Mercy Hospital, Kansas City, MO, United States
- 14Department of Specialized Medicine, Division of Medical Genetics, McGill University Health Center, Montreal, QC, Canada
Introduction: Rare neurodevelopmental disorders, including inherited white matter disorders or leukodystrophies, often present a diagnostic challenge on a genetic level given the large number of causal genes associated with a range of disease subtypes. This study aims to demonstrate the challenges and lessons learned in the genetic investigations of leukodystrophies through presentation of a series of cases solved using exome or genome sequencing.
Methods: Each of the six patients had a leukodystrophy associated with hypomyelination or delayed myelination on MRI, and inconclusive clinical diagnostic genetic testing results. We performed next generation sequencing (case-based exome or genome sequencing) to further investigate the genetic cause of disease.
Results: Following different lines of investigation, molecular diagnoses were obtained for each case, with patients harboring pathogenic variants in a range of genes including TMEM106B, GJA1, AGA, POLR3A, and TUBB4A. We describe the lessons learned in reaching the genetic diagnosis, including the importance of (a) utilizing proper multi-gene panels in clinical testing, (b) assessing the reliability of biochemical assays in supporting diagnoses, and (c) understanding the limitations of exome sequencing methods in regard to CNV detection and region coverage in GC-rich areas.
Discussion: This study illustrates the importance of applying a collaborative diagnostic approach by combining detailed phenotyping data and metabolic results from the clinical environment with advanced next generation sequencing analysis techniques from the research environment to increase the diagnostic yield in patients with genetically unresolved leukodystrophies.
1. Introduction
Although genetic sequencing technologies have drastically evolved in recent years, identification and interpretation of rare variants associated with phenotypically similar but genetically heterogeneous diseases remains a challenge. Rare inherited white matter disorders, or leukodystrophies, can be especially difficult to genetically diagnose, given the growing number of causal genes associated with different disease subtypes (1, 2). Clinical presentation can be similar between patients, involving typically progressive neurological manifestations such as cerebellar, pyramidal, and extrapyramidal features, with or without cognitive involvement (3, 4). Upon MRI investigations to identify white matter abnormalities, leukodystrophies can be categorized based on imaging characteristics as hypomyelinating or as other pathologies, such as demyelinating leukodystrophies (5–7). In young children, it is important to differentiate hypomyelination from delayed myelination by repeating brain MRI as both have distinctive lists of differential diagnoses (6). Neuroimaging patterns and recognition of disease-specific MRI features can further aid in narrowing the underlying genetic cause of disease (5). On a clinical basis, diagnostic procedures typically combine brain MRI, genetic sequencing (e.g., multi-gene panels or exome sequencing) and metabolic investigations (e.g., enzyme deficiencies or CSF metabolite levels) to confirm or exclude diagnoses (3).
Since the rise of next generation sequencing (NGS) in the research environment, leukodystrophy diagnostic rates have seen significant increases, both in report of variants within known disease-associated genes, and in the discovery of novel disease-causing genes (8–12). Despite the increase in diagnostic rates, there still remain patients who are genetically unresolved following clinical and/or research investigations, which may result from limitations within the technology itself, challenges in variant identification, or evaluation of variant pathogenicity. Given that leukodystrophies can be associated with multi-systemic features, challenges may be faced when navigating differential genetic diagnoses, especially when presented with variants of unknown significance in multiple genes. Furthermore, when resolving the genetic basis of a leukodystrophy, it is essential to consider the patient's entire picture, including clinical presentation, disease progression, neuroimaging features, and metabolic test results.
This study presents an overview of several lessons learned in the diagnosis of patients with genetically unresolved white matter disorders. Each patient presented with hypomyelination or delayed myelination on MRI and was investigated genetically on a research basis by our laboratory after clinical testing remained inconclusive. We utilized NGS to determine the genetic cause of disease, noting the below challenges that were faced. The clinical and MRI features are described below with the corresponding genetic investigations, along with a discussion of the lessons learned on the path to resolving genetic diagnoses.
2. Methods
2.1. Protocol approvals, registrations, and patient consents
This research was approved by the Montreal Children's Hospital and McGill University Health Center Research Ethics Boards (11-105-PED; 2019-4972) and the Children's Mercy Institutional Review Board (study # 11120514). Informed consent was obtained from all participants and/or their parents/legal guardians.
2.2. Genetic sequencing and data interpretation
NGS data were either obtained from a clinical laboratory for further analysis or completed on a research basis using genomic DNA extracted from whole blood, fibroblasts, or saliva according to standard protocols. Exome sequencing (ES) and genome sequencing (GS) were performed on a case-by-case basis as described previously (13, 14). Potential disease-causing variants were identified and evaluated based on the American College of Medical Genetics Standards and Guidelines for the Interpretation of Sequence Variants (15). All variants were validated by Sanger sequencing or confirmed by clinical genetic testing. Medical records and brain MRIs were reviewed for each patient.
2.3. Data availability
Data supporting this study's findings are available upon reasonable request. Raw data from participants (i.e., raw genetic data and MRI data) are not publicly available to protect patient privacy. Pathogenic variants have been deposited in the National Center for Biotechnology Information (NCBI) ClinVar repository (ncbi.nlm.nih.gov/clinvar/) under the accession numbers: SCV002820973 - SCV002820979.
3. Results and discussion
For each of the patients described below, a genetic diagnosis was resolved following ES or GS completed on a research basis. We noted several challenges and lessons learned throughout our investigations, which are presented below and summarized in Table 1. Summaries of clinical and MRI features are described, along with genetic investigations and discussion of each lesson with insight on applications to future studies. Additionally, Table 2 describes the advantages and disadvantages of each sequencing modality utilized in this study (16–18).
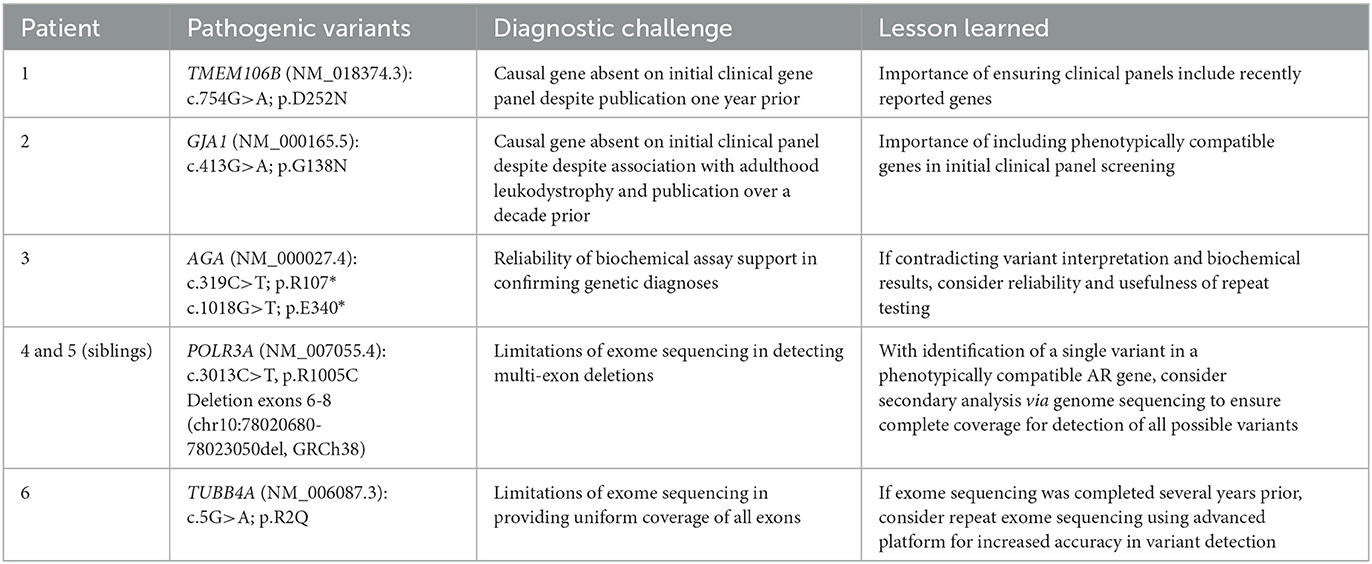
Table 1. Summary of solved cases and lessons learned on the path to genetic diagnosis.
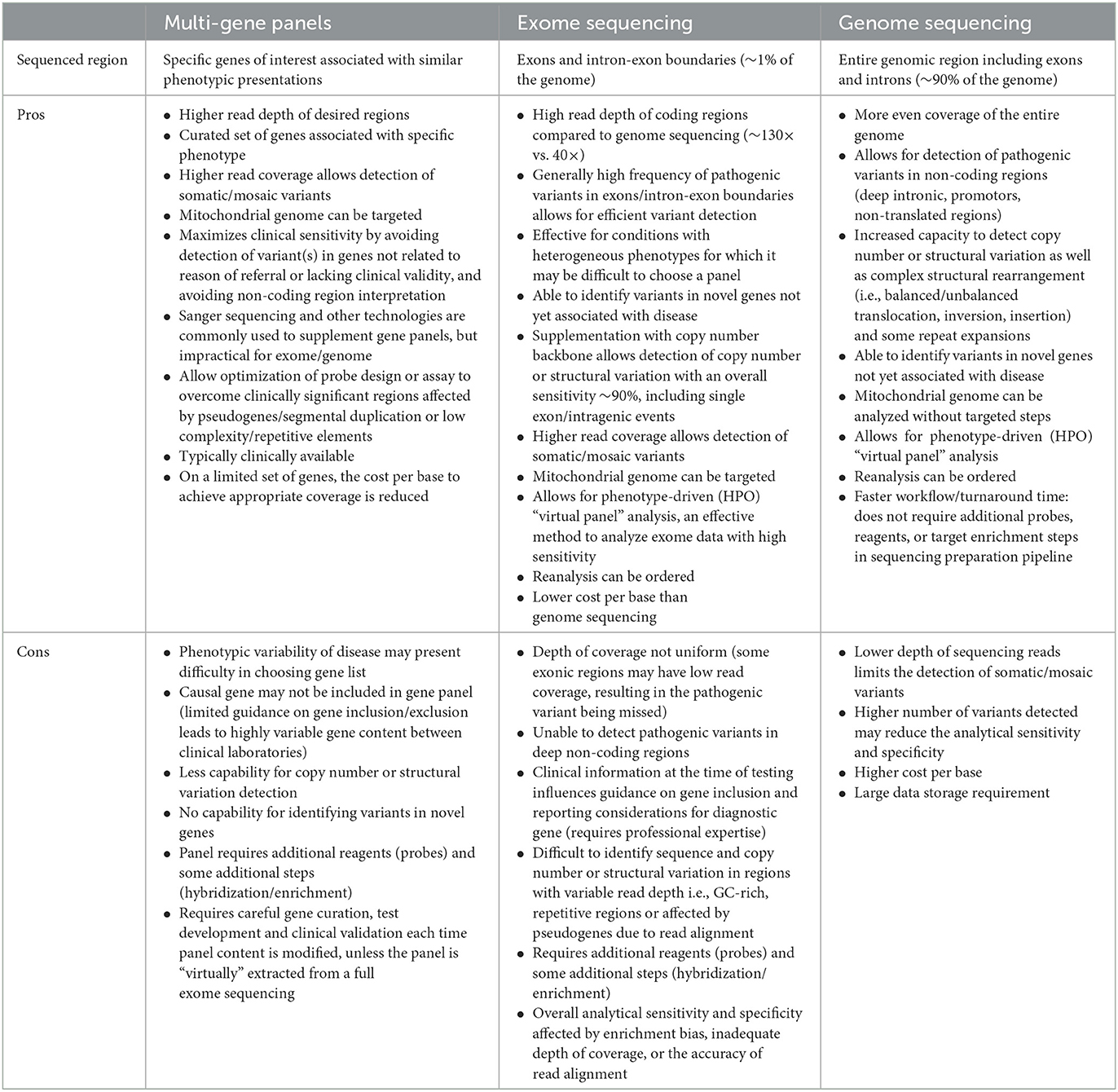
Table 2. Description of the advantages and disadvantages of using the three main discussed sequencing techniques: multi-gene panels, exome sequencing, and genome sequencing (16–18).
3.1. Lesson I: Inclusion of phenotypically compatible genes in clinical gene-panel testing
The following two patients demonstrate the importance of utilizing updated and broad leukodystrophy-associated targeted gene panels in the context of clinical genetic testing. Furthermore, this applies to both patients with early-onset (Patient 1) and adult-onset (Patient 2) diseases.
3.1.1. Patient 1: Clinical & MRI summary
Patient 1, a female, was born at term following a normal pregnancy, and presented shortly after birth with nystagmus, episodes of rapid tremor of the hands, and tremor of the mandibula while feeding. She had mildly reduced axial tone, and increased tone in all four limbs. Through infancy to early childhood, she continued to have mild to moderate axial hypotonia and spastic quadriparesis, with brisk deep tendon reflexes. She also had dysmetria, bilateral sensorineural hearing loss, and dysphagia. At age 3 years, she began to demonstrate additional neurological features, including dysarthria, mild sialorrhea, dysdiadochokinesis, and a slightly unstable gait. She had ophthalmic abnormalities involving strabismus and abnormal pursuits and saccades (hyper/hypometric). Despite showing an initial neurological deterioration, she later stabilized and then started improving, with developmental progress, resolution of sensorineural hearing loss, and amelioration of her dysphagia.
MRI at age 3 weeks revealed abnormal myelination, with myelin deposition being insufficient for age. At the time, due to her young age, it was not possible to determine if she had hypomyelination or myelination delay. There was also involvement of the corticospinal tracts in the c-spine, pyramids, and pons, as well as T2-hyperintensity of the cerebellar white matter. MRI at age 7 months revealed progression of the white matter signal changes, concerning both the deep and subcortical white matter and corpus callosum, and involvement of the middle cerebellar peduncles. An abnormal lactate peak was detected in the white matter on MR spectroscopy. Additionally, available sequences of the orbits demonstrated normal size but abnormal signal intensity of the intraorbital segments of the optic nerves characterized by T2 weighted imaging hyperintensity and increased signal on DWI, not confirmed in the ADC map.
3.1.2. Patient 1: Genetic investigations
Following the initial abnormal MRI findings, genetic investigations for Patient 1 began with multi-gene panel sequencing on a clinical basis. After screening a panel of 163 known leukodystrophy and leukoencephalopathy genes, no conclusive variants were identified. On a research basis, ES was performed, leading to the identification of a de novo pathogenic variant in the gene TMEM106B (NM_018374.3): c.754G>A (p.D252N). This variant has been reported in six unrelated individuals with a hypomyelinating leukodystrophy as a single-nucleotide hotspot associated with this disease (19–21). In retrospect, this gene was not included in the clinical panel, and therefore the pathogenic variant was not detected during the first genetic investigation. The length of time between the first publication of the gene as disease-causing and the completion of the clinical multi-gene panel sequencing was approximately one year.
3.1.3. Patient 2: Clinical & MRI summary
Patient 2, whose MRI was previously published in a Neuroimage case report (22), presented in adulthood with slowly progressive gait disturbances, falls, and issues with memory, starting at age 35. As a child, he was known to have stomach malrotation and syndactyly, the latter of which was surgically corrected. Despite minor motor problems since childhood, he reported normal activities of daily life, and remained fully ambulatory. He had slight dysmetria and mild gait ataxia. He was also noted to have mild facial dysmorphisms, dental abnormalities, and severe myopia.
MRI in adulthood revealed a pattern of diffuse hypomyelination, with involvement of the posterior limb of the internal capsule, pons, and cerebellar peduncles. T2-weighted hypointensities were also noted in both the Rolandic cortex and the dentate nucleus. The corpus callosum was thin and there was mild vermian atrophy (22).
Regarding significant family history, his mother also experienced slowly progressive neurodegeneration in adulthood, involving pyramidal signs and gait ataxia, worsening over several years. She experienced dysphagia, speech and cognitive difficulties, and epileptic seizures. She died in her early 80s after experiencing recurrent aspiration pneumonias.
Brain MRI of the patient's mother at age 80 showed diffuse white matter abnormalities, with involvement of the posterior limbs of the internal capsule and the pons (corticospinal tracts and medial lemniscus), along with significant atrophy of the posterior white matter. She also had a thin corpus callosum, cerebellar vermis atrophy, and ventriculomegaly (22).
3.1.4. Patient 2: Genetic investigations
Following identification of white matter abnormalities on MRI of Patient 2, a gene panel including 122 leukodystrophy or leukoencephalopathy genes was completed on a clinical basis. Results were inconclusive, and the patient was referred to our research study for further investigations. ES was completed on DNA from the proband, revealing a heterozygous pathogenic missense variant in GJA1 (NM_000165.5): c.413G>A; p.G138D. This variant was previously reported as pathogenic in association with autosomal dominant Oculodentodigital dysplasia (ODDD) syndrome (23). Parental DNA was unavailable for segregation analysis, but given the similar presentation of the mother, autosomal dominant inheritance was presumed. Notably, on the initial clinical gene panel, the GJA1 gene (published over a decade prior) was not included, and therefore not identified on the first investigation.
3.1.5. Inclusion of novel and compatible genes on clinical panels
Gene panels which target a specific set of genes known to be associated with disease phenotypes are often used as a first-line diagnostic tool in the clinical setting. While the use of gene panels may provide an effective means for identifying pathogenic variants in known genes, patients with pathogenic variants outside of those remain genetically undiagnosed, often leading to a long diagnostic odyssey. Advantages of using multi-gene panels compared to ES or GS have been studied in the past, and there remain benefits and limitations for each of these diagnostic techniques (Table 2) (24–28). While multi-gene panel sequencing can be a powerful molecular diagnostic tool, without inclusion of up-to-date causal genes, they remain limited in effectiveness for investigations of rare diseases. Regarding Patient 1, the TMEM106B gene was published as associated with hypomyelination nearly a year prior to the initial clinical screening (19). Contrarily, Patient 2 had a pathogenic variant in GJA1, published as associated with ODDD syndrome in the early 2000s (29). As neurological features are only seen in a portion of patients with ODDD, it is likely to be an often-unrecognized form of adulthood leukodystrophy. Thus, it remains possible it was excluded from the large leukodystrophy-associated gene panel due to the leukodystrophy features in ODDD remaining overlooked, as the phenotypic bias in diagnosis may lean toward the other associated features present on clinical evaluation.
Both above cases had to transition from clinical to research-based studies to be resolved, thus increasing the time to diagnosis, which could have been minimized had the initial panel screening contained the causal genes. Furthermore, these cases demonstrate the importance of maintaining clinical gene panels to be both updated with newly published genes and expansive enough to include likely causative genes. In the field of medical genetics, discovery of novel disease-associated genes advances at a rapid pace, with unique genetic causes of leukodystrophies continuously reported, which in parallel should be included in gene panels. It is also imperative that clinicians ordering clinical gene panels are knowledgeable regarding whether the proper genes are included when there is a high index of suspicion for a phenotypically distinct condition.
3.2. Lesson II: Reliability of biochemical assay support in confirming genetic diagnoses
Often, variants identified on ES may be classified as being of unknown significance if they have not been previously linked to a disease, or if functional evidence is lacking. For some implicated genes, clinical biochemical assay results may be used as biomarkers to support variant pathogenicity. The following case presents a scenario in which likely pathogenic variants were identified in a causal gene; however, this finding was initially unsupported by biochemical testing results. On repeat biochemical testing, results were verified and the genetic diagnosis was confirmed. Thus, this case demonstrates the importance of considering the validity of biochemical testing results when evaluating the likelihood of variant pathogenicity, especially when contradicting evidence is provided in the case of likely pathogenic variants.
3.2.1. Patient 3: Clinical & MRI summary
Patient 3, a female, presented to the clinic with a wobbly gait at age 4 years and was found to have diffuse abnormal myelination on MRI, associated with a pattern of hypomyelination. MRI at age 5 was significant for diffuse hypomyelination/delayed myelination, where slight progression of myelination of the peripheral subcortical white matter was seen on T1-weighted imaging, with the degree of myelination on T2-weighted imaging remaining stable. Upon review of the latest MRI at age 8, there was also mild thinning of the corpus callosum, mild cerebellar vermis atrophy, and a mild increase in VR spaces, with T2-hypointensity of the pulvinar.
Clinically, she had a history of global developmental delay from age 6 months, and throughout childhood she continued to have mild developmental delay, without focal abnormalities. She was known for ophthalmic issues, including right eye esotropia and left optic nerve pallor. She had mild difficulties with tandem gait. Nerve conduction studies were normal. Through to age 8, she did not experience signs of regression, and only continued to have mild developmental challenges. She also had persistent mildly low platelets levels, which appeared to normalize at age 7. Initial urine oligosaccharides testing revealed a faint abnormal band, which was further investigated clinically via HPLC analysis, and reported to be unremarkable for the known lysosomal storage disorders (i.e., alpha mannosidosis, alpha fucidosis, sialidosis, galactosialidosis, GM1 gangliosidosis, GM2 gangliosidosis type Sandhoff, GSD11 Pompe disease infantile). On biochemical testing of CSF, amino acids and neurotransmitter metabolites were unremarkable.
3.2.2. Patient 3: Genetic investigations
ES was performed using patient genomic DNA, and upon initial data analysis, two compound heterozygous nonsense variants were identified in the gene AGA (NM_000027.4): c.319C>T; p.R107*, and c.1018G>T; p.E340*. AGA encodes for the enzyme aspartylglucosaminidase, and pathogenic variants which impair its function are known to cause aspartylglucosaminuria (OMIM: 208400), an autosomal recessive neurodegenerative lysosomal storage disease. The first AGA nonsense variant (p.R107*) results in a truncated protein product lacking 240 amino acids (aa) from the 347-aa length wildtype protein. Although this specific variant has not been reported in published cases, similar truncating variants are known to be associated with AGA loss-of-function and cause disease (30, 31). The second nonsense variant (p.E340*) results in a truncated protein lacking only 7-aa compared to the wildtype protein. While this variant has also not been reported previously, the nearby p.E334* variant has been studied functionally to cause a reduction in the production of an active enzyme (30). Based on ACMG criteria (15), both variants were predicted to be likely pathogenic. Each variant was validated using Sanger sequencing, however, parental DNA was unavailable to confirm segregation.
Regarding confirmatory biochemical testing, diagnosis of aspartylglucosaminuria is supported by screening urine oligosaccharides, as aspartylglucosamine accumulates in the urine of affected individuals (32). In this patient, initial urine oligosaccharides analysis revealed an abnormal band, however, further HPLC analysis did not detect a pattern associated with disease. This result contradicted with the predicted pathogenic variant interpretation as both variants were thought to be associated with impaired enzyme function. Therefore, it was decided to repeat clinical urine oligosaccharides screening to verify the results, and on the second screen, an abnormal pattern associated with aspartylglucosaminuria was detected, providing support for the diagnosis. Additional tests were performed, including a sialic acid assay which showed mild elevation of total sialic acid, thought to result from abnormal accumulation of an oligosaccharide species associated with aspartylglucosaminuria. Finally, enzymatic testing demonstrated low aspartylglucosaminidase activity, further confirming the diagnosis of aspartylglucosaminuria.
3.2.3. Importance of biochemical assay results in supporting genetic diagnoses
Biochemical and functional assays are an important diagnostic component for many genetic disorders as metabolic results often provide clues to a diagnosis, such as in lysosomal storage disorders. In the presented case, initial urine oligosaccharides screening results did not support the most likely candidate gene, and therefore difficulties were met in resolving the diagnosis. Variants were predicted to be pathogenic, however, we were unable to confirm their inheritance on trans alleles. In addition, biochemical testing provided conflicting evidence, thus causing uncertainty in either the genetic or biochemical results. The diagnosis was only confirmed after repeated biochemical screening for urine oligosaccharides, which also resulted in a prolonged diagnostic period. Therefore, this case demonstrates the importance of maintaining a degree of trust in variant interpretation guidelines when evaluating pathogenicity, and that repeat biochemical screening may be necessary should results contradict the genetic diagnosis. This case also stresses the importance of remaining cautious when presented with conflicting diagnostic evidence and approaching the reassessment of either genetic or biochemical results with a high level of clinical reasoning. Contrary to what was seen in our case, in which molecular genetic investigations allowed for the diagnosis of a metabolic disease where the biochemical genetic tests were initially normal, it is possible that genetic variants initially thought to be likely pathogenic can be reclassified as benign based on biochemical investigations. For example, in considering pathogenic variants in the ABCD1 gene which cause adrenoleukodystrophy, the detection of true pathogenic variants is complicated by the fact that several non-functional pseudogenes exist on different chromosomes, which may result in false-positive variant detection (33). In this case, it is crucial to confirm the pathogenicity of the variants with very long chain fatty acids. In conclusion, special attention should be given to cases with conflicting genetic and biochemical evidence, and next steps for confirming or rejecting diagnoses should be considered only after critical evaluation of all provided evidence.
3.3. Lesson III: Understanding limitations of exome sequencing in variant detection
While the use of ES has demonstrated high success rates in resolving genetic causes of many rare diseases, it is well-known that this technology is associated with several limitations in its capacity to detect all disease-causing variants (34). The following three cases illustrate the shortcomings of ES in variant detection, including the limited ability to detect CNVs (Patients 4 and 5) and variants in GC-rich regions (Patient 6).
3.3.1. Patients 4 and 5: Clinical & MRI summary
Patients 4 and 5 were siblings who each experienced increasing cognitive and behavioral concerns in early adulthood and were found to have hypomyelination on MRI. Patient 4, a female, had behavioral problems since childhood, with intellectual and learning disabilities. She had severe myopia, as well as growth and pubertal abnormalities, experiencing menarche at age 14 but requiring hormonal treatment for irregular menstruations. In early adulthood, she was diagnosed with bipolar disorder, and further MRI investigations revealed white matter abnormalities, leading to a diagnosis of leukodystrophy. In adulthood, she experienced further neurological deterioration, exhibiting dysarthria, intention tremor, gait ataxia, as well as dystonia. She also had saccadic pursuits, with limitations in upgaze. She further experienced dysphagia and required a gastrostomy. Delayed dentition was noted in childhood, and in adulthood it was reported her teeth were becoming loose. MRI at age 30 years was significant for hypomyelination with a pattern compatible with POLR3-related leukodystrophy. There was also moderate diffuse cerebral and cerebellar atrophy. Interestingly, bone abnormalities were noted in the skull, described as diffuse thickening of the calvarium.
Patient 5, a male, had onset of behavioral issues and personality changes in his late 20s. His development was reported as normal, having graduated high school after attending special education classes. He was reported to have puberty and growth abnormalities, receiving growth hormone treatment in early adolescence. He had severe myopia but reported no dental abnormalities through childhood. In early adulthood, he began to experience cognitive decline, with severely impaired intellectual function, while behavioral difficulties, specifically irritability, persisted. Family noted episodes of pseudobulbar affect. He was reported to have a high-pitched voice, but no dysarthria. He had hypotonia and mild gait ataxia. Brain MRI in adulthood revealed hypomyelination with a pattern compatible with POLR3-related leukodystrophy. Similar to his sister, he also had thickening of the calvarium.
3.3.2. Patients 4 and 5: Genetic investigations
ES was first completed on both siblings, and one pathogenic missense variant was discovered in POLR3A (NM_007055.4): c.3013C>T; p.R1005C. This variant was previously reported in a compound heterozygous state in patients with POLR3-related leukodystrophy (35, 36). As POLR3-related leukodystrophy is an autosomal recessive condition and only one variant was identified, data reanalysis was performed to attempt to identify an alternate genetic cause, however, no strong candidates were found. GS was then completed, identifying a maternally inherited POLR3A deletion of approximately 3 kb on 10q22.3, including exons 6-8 [(chr10:78020680-78023050)x1, GRCh38], thereby resolving the genetic diagnosis as POLR3-related leukodystrophy. Notably, a similar deletion of POLR3A exons 6-8 (NG_029648.1; NC_000010.11: g.78020702_78023071del; c.646–687_c.1185+844del; p.E216_K395del) has been associated with spastic ataxia when in compound heterozygous state with the POLR3A c.1909+22G>A variant (37). Additionally, large deletions in the RNA polymerase III subunit gene POLR3B, which forms the catalytic core of the enzyme along with POLR3A, are associated with a similar phenotype of POLR3-related leukodystrophy (38). This diagnosis aligns with the clinical features seen in both patients including the typical MRI pattern, severe myopia, and growth and endocrine abnormalities, as well as the delayed dentition in Patient 4.
3.3.3. Advantages of genome sequencing in CNV detection
ES is known for several limitations in variant detection, inherent to its technological design. Moreover, this includes the inability to detect certain types of variants, including CNVs like the large deletion described in the above cases. This limitation is a direct result of the lack of sequencing depth uniformity in ES, as the enriched exonic regions interspaced by non-coding intronic regions are not evenly sequenced. Therefore, deletions are difficult to detect through conventional ES methods, and GS offers a more effective means to resolve the deleted region. While ES generally captures a higher sequencing depth of the targeted exonic regions, GS offers a more uniform coverage of the genome (39). Indeed, as it provides more even and unbiased coverage of all exonic regions, GS delivers more accurate variant detection, especially in the recognition of large deletions (40). The described cases offer a practical example of the limitations of ES in detecting CNVs, demonstrating the importance of pursuing additional investigations when a single pathogenic variant is identified in a gene associated with a phenotypically compatible autosomal recessive disease. The POLR3A gene could have been investigated in more detail on a research basis through long-range PCR, primer walking, or quantitative PCR to resolve the genetic diagnosis. Considering the limited ability to perform such experiments in the clinical setting, GS is a better approach, and in this case, the benefits of GS were clearly demonstrated as it provided an efficient means to detect the deletion. In conclusion, these cases demonstrate a lesson in harnessing knowledge of the limitations associated with variant detection via ES when investigating genetic diseases.
3.3.4. Patient 6: Clinical & MRI summary
Patient 6, a male, was born at 40 weeks and 6 days following an induced vaginal delivery, and was treated for hyperbilirubinemia with phototherapy for 2 days. Shortly after birth, he presented with nystagmus, and further experienced severe developmental delay and failure to thrive. He demonstrated neurological features including severe axial and appendicular hypotonia, moderate spasticity, and generalized dystonia. He was severely delayed in motor development, unable to gain normal head control or the ability to sit independently or walk. He experienced sialorrhea and dysphagia, requiring a gastrostomy at age 3. He had epilepsy from early childhood, which was well-controlled by age 11. He also experienced asymptomatic subluxation of both hips. Additionally, he had bilateral myopia and strabismus, with bilateral optic atrophy and restriction of extraocular movements. At age 13, his condition was relatively stable, although he had slight motor deterioration, losing the ability to pick up objects with his hands.
Brain MRIs demonstrated diffuse hypomyelination with relative preservation of the cerebellum and brainstem. On follow-up MRIs, cerebral and cerebellar atrophy were noted.
3.3.5. Patient 6: Genetic investigations
Patient 6 was first investigated using ES on a research basis, however, no strong candidates were identified despite data reanalysis. Five years after the initial sequencing, we opted to repeat ES using an updated platform in an attempt to identify variants which may have been previously undetected. This resulted in identification of a de novo pathogenic variant in TUBB4A (NM_006087.3): c.5G>A; p.R2Q, resolving the diagnosis. This variant was reported in two individuals having hypomyelination with atrophy of the basal ganglia and cerebellum (H-ABC) (41, 42). Although TUBB4A variants are mainly associated with H-ABC, isolated hypomyelination has also been described (42–45), aligning with Patient 6's MRI pattern, which did not display basal ganglia involvement.
Upon review of the initial ES data, the genomic region containing the TUBB4A variant was only covered by five sequencing reads, only one of which carried the variant. Therefore, due to suboptimal coverage and read depth, the variant was not detected on the first investigation. Indeed, this variant was located near the beginning of the gene's first exon, a region generally known to contain an increased GC content (46) and consequently associated with reduced coverage on ES.
3.3.6. Coverage bias in exome sequencing and impact on variant detection
The case of Patient 6 is a direct example of the limitations of ES in detecting variants in areas commonly subject to coverage bias. It is well-known that ES is associated with non-uniform coverage in specific regions, including those that are GC-rich. Uneven coverage of these regions may result from technological challenges in either library or cluster amplification, the sequencing step itself, or the alignment of sequencing data (47–49). Updates to sequencing platforms aim to mitigate this through advanced methods, which have led to more consistent coverage in recent years. Therefore, when evaluating the best course of action for persistently unsolved cases sequenced several years prior, it is important to consider the benefits of re-sequencing using updated platforms. Through this case, the value of re-sequencing was certainly demonstrated as a diagnosis was easily resolved on the second round of sequencing, with the causal variant being located in an area which was not initially covered on first round sequencing, and thereby not detected. Furthermore, it is important to remain knowledgeable of the efficacy and utility of sequencing technologies used in genetic analyses, and whether updated technologies for repeat sequencing could be valuable for subsequent investigations.
4. Conclusion
In this study, we illustrate several lessons learned when investigating genetic diagnoses in a subset of patients with previously genetically unresolved leukodystrophies following negative clinical investigations. The encountered challenges represent common lessons and pitfalls that clinicians and researchers may face when navigating pathogenic variant identification and interpretation in genetic diagnostics. The first two cases highlight the importance of utilizing appropriate gene panels in first line clinical investigations, ensuring that they contain both recently published and phenotypically compatible genes. The next case demonstrates the importance of using clinical reasoning when evaluating biochemical results that conflict with probable genetic diagnoses. Finally, the last three cases illustrate the technical limitations associated with variant detection via ES, which were resolved using repeat sequencing to identify previously undetected variants. When investigating genetically unresolved cases, remaining knowledgeable of the challenges associated with variant identification can provide insight on the most beneficial and effective course of action. However, it is important to note that although technological limitations may impede detection of pathogenic variants, cases could remain genetically unresolved due to other factors, such as challenges in accurate variant pathogenicity interpretation due to gaps in knowledge base, or simply because the causal gene has not yet been associated with a disease. It is imperative that unsolved cases are regularly re-evaluated as the field advances in both sequencing technologies, analysis techniques, and reports of novel genes. Notably, protocols for the genetic diagnosis of both pediatric and adult-onset leukodystrophies can aid clinicians in evaluating the most optimal approach to sequencing. Several molecular diagnostic workflows and step-by-step approaches based on large cohorts and expert consensus recommendations have been described in the literature, which can provide guidance in the clinical setting (3, 50–54). Furthermore, in a clinical context, resolving genetic diagnoses of rare inherited diseases is especially important for understanding of the disease course and prognosis, as well as tailoring supportive care, and further advising through genetic counseling.
Data availability statement
The data presented in the study are deposited in the National Center for Biotechnology Information (NCBI) ClinVar repository (ncbi.nlm.nih.gov/clinvar/) under the accession numbers: SCV002820973 - SCV002820979.
Ethics statement
The studies involving human participants were reviewed and approved by the Montreal Children's Hospital and McGill University Health Center Research Ethics Boards (11-105-PED; 2019-4972) and the Children's Mercy Institutional Review Board (study # 11120514). Written informed consent to participate in this study was provided by the participants and/or their legal guardians/next of kin.
Author contributions
SP, IT, and GB designed the study, analyzed and interpreted the data, and drafted the manuscript. KG, LT, MM-R, GL, BB, MS, JD, MD, WK, and TP contributed to the acquisition and interpretation of the clinical and genetic data and revised the manuscript for intellectual content. GB and IT oversaw the entire study. GB, IT, and TP secured funding for the study. All authors approved the final version of the manuscript.
Funding
This research study was supported by project grants from the Canadian Institutes of Health Research (377869 and 426534). The work from IT and TP were made possible by the generous gifts to Children's Mercy Research Institute and Genomic Answers for Kids Program at Children's Mercy Kansas City. GB has received the Clinical Research Scholar Junior 1 Award from the Fonds de Recherche du Quebec-Santé (FRQS) (2012–2016), the New Investigator Salary Award from the Canadian Institutes of Health Research (2017–2022), and the Clinical Research Scholar Senior award from the FRQS (2022–2025). SP has been supported by the FRQS Doctoral Scholarship, the Fondation du Grand défi Pierre Lavoie Doctoral Scholarship, the McGill Faculty of Medicine F.S.B. Miller Fellowship, and the Research Institute of the McGill University Health Center Desjardins Studentship in Child Health Research. MM-R was supported by the Vanier Canada Graduate Scholarship and the McGill University Faculty of Medicine. TP holds the Dee Lyons/Missouri Endowed Chair in Pediatric Genomic Medicine.
Acknowledgments
The authors acknowledge the McGill University and Genome Quebec Innovation Center. This research was enabled in part by support provided by Compute Canada. We also would like to thank the GA4K team at Children's Mercy Kansas City and Laura Puckett, Adam Walters, and Margaret Gibson for their work in library preparation and sequencing.
Conflict of interest
GB is a consultant for Passage Bio Inc. (2020-2022) and Ionis (2019). She is a site investigator for the Alexander's disease trial of Ionis Pharmaceuticals (2021-present), Metachromatic leukodystrophy of Shire/Takeda (2020-2021), Krabbe and GM1 gangliosidosis gene therapy trials of Passage Bio (2021-present), GM1 gangliosidosis natural history study sponsored by the University of Pennsylvania with funding from Passage Bio (2021-present) and Adrenoleukodystrophy/Hematopoietic stem cell transplantation natural history study of Bluebird Bio (2019), a site sub-investigator for the MPS II gene therapy trial of Regenxbio (2021-present) and the MPS II clinical trial of Denali (2022-present). She has received unrestricted educational grants from Takeda (2021-2022). She serves on the scientific advisory board of the Pelizaeus-Merzbacher Foundation, the Yaya Foundation Scientific and Clinical Advisory Council and is the Chair of the Medical and Scientific Advisory Board of the United Leukodystrophy Foundation. She is a member of the Vanishing White Matter Consortium, the H-ABC Clinical Advisory Board and the Chair of the POLR3-related (4H) Leukodystrophy Consortium. She is on the editorial boards of Neurology Genetics, Frontiers in Neurology – Neurogenetics, and Journal of Medical Genetics. LT currently manages sponsored clinical trials at the site level for Ionis Pharmaceuticals (Alexander disease clinical trial, 2021-present), Passage Bio (Krabbe disease and GM1 gangliosidosis clinical trials, 2021-present), and Teva Pharmaceuticals (chronic and episodic migraine clinical trials, 2022-present). He also manages a GM1 gangliosidosis natural history study sponsored by the University of Pennsylvania with funding from Passage Bio. MS has received honorarium from Takeda for speaking presentations. WK receives research support from Alexion, Bluebird Bio and MedDay, and is consultant and co-investigator for AMN disease trial of Minoryx. He also serves on scientific advisory boards of the United Leukodystrophy Foundation, the European Leukodystrophy Association and the Myelin Project Germany.
The remaining authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher's note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Vanderver A, Prust M, Tonduti D, Mochel F, Hussey HM, Helman G, et al. Case definition and classification of leukodystrophies and leukoencephalopathies. Mol Genet Metab. (2015) 114:494–500. doi: 10.1016/j.ymgme.2015.01.006
2. Urbik VM, Schmiedel M, Soderholm H, Bonkowsky JL. Expanded phenotypic definition identifies hundreds of potential causative genes for leukodystrophies and leukoencephalopathies. Child Neurol Open. (2020) 7:2329048X20939003. doi: 10.1177/2329048X20939003
3. Parikh S, Bernard G, Leventer RJ, van der Knaap MS, van Hove J, Pizzino A, et al. A Clinical approach to the diagnosis of patients with leukodystrophies and genetic leukoencephelopathies. Mol Genet Metab. (2015) 114:501–15. doi: 10.1016/j.ymgme.2014.12.434
4. Adang LA, Sherbini O, Ball L, Bloom M, Darbari A, Amartino H, et al. Revised consensus statement on the preventive and symptomatic care of patients with leukodystrophies. Mol Genet Metab. (2017) 122:18–32. doi: 10.1016/j.ymgme.2017.08.006
5. Schiffmann R, van der Knaap MS. Invited article: an mri-based approach to the diagnosis of white matter disorders. Neurology. (2009) 72:750–9. doi: 10.1212/01.wnl.0000343049.00540.c8
6. Steenweg ME, Vanderver A, Blaser S, Bizzi A, de Koning TJ, Mancini GM, et al. Magnetic resonance imaging pattern recognition in hypomyelinating disorders. Brain. (2010) 133:2971–82. doi: 10.1093/brain/awq257
7. Barkovich AJ, Deon S. Hypomyelinating disorders: an mri approach. Neurobiol Dis. (2016) 87:50–8. doi: 10.1016/j.nbd.2015.10.015
8. Vanderver A, Simons C, Helman G, Crawford J, Wolf NI, Bernard G, et al. Whole exome sequencing in patients with white matter abnormalities. Ann Neurol. (2016) 79:1031–7. doi: 10.1002/ana.24650
9. Kaur P, do Rosario MC, Hebbar M, Sharma S, Kausthubham N, Nair K, et al. Clinical and genetic spectrum of 104 indian families with central nervous system white matter abnormalities. Clin Genet. (2021) 100:542–50. doi: 10.1111/cge.14037
10. Yan H, Ji H, Kubisiak T, Wu Y, Xiao J, Gu Q, et al. Genetic analysis of 20 patients with hypomyelinating leukodystrophy by trio-based whole-exome sequencing. J Hum Genet. (2021) 66:761–8. doi: 10.1038/s10038-020-00896-5
11. Kevelam SH, Steenweg ME, Srivastava S, Helman G, Naidu S, Schiffmann R, et al. Update on leukodystrophies: a historical perspective and adapted definition. Neuropediatrics. (2016) 47:349–54. doi: 10.1055/s-0036-1588020
12. Helman G, Lajoie BR, Crawford J, Takanohashi A, Walkiewicz M, Dolzhenko E, et al. Genome sequencing in persistently unsolved white matter disorders. Ann Clin Transl Neurol. (2020) 7:144–52. doi: 10.1002/acn3.50957
13. Cohen ASA, Farrow EG, Abdelmoity AT, Alaimo JT, Amudhavalli SM, Anderson JT, et al. Genomic answers for children: dynamic analyses of >1000 pediatric rare disease genomes. Genet Med. (2022) 24:1336–48. doi: 10.1016/j.gim.2022.02.007
14. Thiffault I, Farrow E, Zellmer L, Berrios C, Miller N, Gibson M, et al. Clinical genome sequencing in an unbiased pediatric cohort. Genet Med. (2019) 21:303–10. doi: 10.1038/s41436-018-0075-8
15. Richards S, Aziz N, Bale S, Bick D, Das S, Gastier-Foster J, et al. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the american college of medical genetics and genomics and the association for molecular pathology. Genet Med. (2015) 17:405–24. doi: 10.1038/gim.2015.30
16. Marchuk DS, Crooks K, Strande N, Kaiser-Rogers K, Milko LV, Brandt A, et al. Increasing the diagnostic yield of exome sequencing by copy number variant analysis. PLoS ONE. (2018) 13:e0209185. doi: 10.1371/journal.pone.0209185
17. Rehder C, Bean LJH, Bick D, Chao E, Chung W, Das S, et al. Next-generation sequencing for constitutional variants in the clinical laboratory, 2021 revision: a technical standard of the American College of Medical Genetics and Genomics (ACMG). Genet Med. (2021) 23:1399–415. doi: 10.1038/s41436-021-01139-4
18. Wang X, Shen X, Fang F, Ding CH, Zhang H, Cao ZH, et al. Phenotype-driven virtual panel is an effective method to analyze wes data of neurological disease. Front Pharmacol. (2018) 9:1529. doi: 10.3389/fphar.2018.01529
19. Simons C, Dyment D, Bent SJ, Crawford J, D'Hooghe M, Kohlschütter A, et al. A recurrent de novo mutation in TMEM106B causes hypomyelinating leukodystrophy. Brain. (2017) 140:3105–11. doi: 10.1093/brain/awx314
20. Yan H, Kubisiak T, Ji H, Xiao J, Wang J, Burmeister M. The recurrent mutation in TMEM106B also causes hypomyelinating leukodystrophy in China and is a Cpg hotspot. Brain. (2018) 141:e36. doi: 10.1093/brain/awy029
21. Ikemoto S, Hamano SI, Kikuchi K, Koichihara R, Hirata Y, Matsuura R, et al. A recurrent TMEM106B mutation in hypomyelinating leukodystrophy: a rapid diagnostic assay. Brain Dev. (2020) 42:603–6. doi: 10.1016/j.braindev.2020.06.002
22. Michell-Robinson MA, Perrier S, Lucia C, Tran LT, Thiffault I, Köhler W, et al. Oculodentodigital dysplasia: a cause of hypomyelinating leukodystrophy in adults. Neurology. (2022) 98:675–7. doi: 10.1212/WNL.0000000000200228
23. Fenwick A, Richardson RJ, Butterworth J, Barron MJ, Dixon MJ. Novel mutations in GJA1 cause oculodentodigital syndrome. J Dent Res. (2008) 87:1021–6. doi: 10.1177/154405910808701108
24. Saudi Mendeliome G. Comprehensive gene panels provide advantages over clinical exome sequencing for mendelian diseases. Genome Biol. (2015) 16:134. doi: 10.1186/s13059-015-0693-2
25. Molina-Ramírez LP, Kyle C, Ellingford JM, Wright R, Taylor A, Bhaskar SS, et al. Personalised virtual gene panels reduce interpretation workload and maintain diagnostic rates of proband-only clinical exome sequencing for rare disorders. J Med Genet. (2022) 59:393–8. doi: 10.1136/jmedgenet-2020-107303
26. Lionel AC, Costain G, Monfared N, Walker S, Reuter MS, Hosseini SM, et al. Improved diagnostic yield compared with targeted gene sequencing panels suggests a role for whole-genome sequencing as a first-tier genetic test. Genet Med. (2018) 20:435–43. doi: 10.1038/gim.2017.119
27. de Koning TJ, Jongbloed JDH, Sikkema-Raddatz B, Sinke RJ. Targeted next-generation sequencing panels for monogenetic disorders in clinical diagnostics: the opportunities and challenges. Expert Rev Mol Diagn. (2015) 15:61–70. doi: 10.1586/14737159.2015.976555
28. Xue Y, Ankala A, Wilcox WR, Hegde MR. Solving the molecular diagnostic testing conundrum for mendelian disorders in the era of next-generation sequencing: single-gene, gene panel, or exome/genome sequencing. Genet Med. (2015) 17:444–51. doi: 10.1038/gim.2014.122
29. Paznekas WA, Boyadjiev SA, Shapiro RE, Daniels O, Wollnik B, Keegan CE, et al. Connexin 43 (GJA1) mutations cause the pleiotropic phenotype of oculodentodigital dysplasia. Am J Hum Genet. (2003) 72:408–18. doi: 10.1086/346090
30. Saarela J, Laine M, Oinonen C, von Schantz C, Jalanko A, Rouvinen J, et al. Molecular pathogenesis of a disease: structural consequences of aspartylglucosaminuria mutations. Hum Mol Genet. (2001) 10:983–95. doi: 10.1093/hmg/10.9.983
31. Isoniemi A, Hietala M, Aula P, Jalanko A, Peltonen L. Identification of a novel mutation causing aspartylglucosaminuria reveals a mutation hotspot region in the aspartylglucosaminidase gene. Hum Mutat. (1995) 5:318–26. doi: 10.1002/humu.1380050408
32. Goodspeed K, Feng C, Laine M, Lund TC. Aspartylglucosaminuria: clinical presentation and potential therapies. J Child Neurol. (2021) 36:403–14. doi: 10.1177/0883073820980904
33. Wiesinger C, Eichler FS, Berger J. The genetic landscape of X-linked adrenoleukodystrophy: inheritance, mutations, modifier genes, and diagnosis. Appl Clin Genet. (2015) 8:109–21. doi: 10.2147/TACG.S49590
34. Meienberg J, Zerjavic K, Keller I, Okoniewski M, Patrignani A, Ludin K, et al. New insights into the performance of human whole-exome capture platforms. Nucleic Acids Res. (2015) 43:e76. doi: 10.1093/nar/gkv216
35. Bernard G, Chouery E, Putorti ML, Tétreault M, Takanohashi A, Carosso G, et al. Mutations of POLR3A encoding a catalytic subunit of rna polymerase pol iii cause a recessive hypomyelinating leukodystrophy. Am J Hum Genet. (2011) 89:415–23. doi: 10.1016/j.ajhg.2011.07.014
36. Saitsu H, Osaka H, Sasaki M, Takanashi J, Hamada K, Yamashita A, et al. Mutations in POLR3A and POLR3B encoding rna polymerase iii subunits cause an autosomal-recessive hypomyelinating leukoencephalopathy. Am J Hum Genet. (2011) 89:644–51. doi: 10.1016/j.ajhg.2011.10.003
37. Infante J, Serrano-Cárdenas KM, Corral-Juan M, Farré X, Sánchez I, de Lucas EM, et al. POLR3A-related spastic ataxia: new mutations and a look into the phenotype. J Neurol. (2020) 267:324–30. doi: 10.1007/s00415-019-09574-9
38. Gutierrez M, Thiffault I, Guerrero K, Martos-Moreno G, Tran LT, Benko W, et al. Large exonic deletions in POLR3B gene cause POLR3-related leukodystrophy. Orphanet J Rare Dis. (2015) 10:69. doi: 10.1186/s13023-015-0279-9
39. Lelieveld SH, Spielmann M, Mundlos S, Veltman JA, Gilissen C. Comparison of exome and genome sequencing technologies for the complete capture of protein-coding regions. Hum Mutat. (2015) 36:815–22. doi: 10.1002/humu.22813
40. Belkadi A, Bolze A, Itan Y, Cobat A, Vincent QB, Antipenko A, et al. Whole-genome sequencing is more powerful than whole-exome sequencing for detecting exome variants. Proc Natl Acad Sci USA. (2015) 112:5473–8. doi: 10.1073/pnas.1418631112
41. Hamilton EM, Polder E, Vanderver A, Naidu S, Schiffmann R, Fisher K, et al. Hypomyelination with atrophy of the basal ganglia and cerebellum: further delineation of the phenotype and genotype-phenotype correlation. Brain. (2014) 137(Pt 7):1921–30. doi: 10.1093/brain/awu110
42. Miyatake S, Osaka H, Shiina M, Sasaki M, Takanashi J, Haginoya K, et al. Expanding the phenotypic spectrum of TUBB4A-associated hypomyelinating leukoencephalopathies. Neurology. (2014) 82:2230–7. doi: 10.1212/WNL.0000000000000535
43. Pizzino A, Pierson TM, Guo Y, Helman G, Fortini S, Guerrero K, et al. TUBB4A de novo mutations cause isolated hypomyelination. Neurology. (2014) 83:898–902. doi: 10.1212/WNL.0000000000000754
44. Shimojima K, Okumura A, Ikeno M, Nishimura A, Saito A, Saitsu H, et al. A de novo TUBB4A mutation in a patient with hypomyelination mimicking Pelizaeus-Merzbacher disease. Brain Dev. (2015) 37:281–5. doi: 10.1016/j.braindev.2014.05.004
45. Purnell SM, Bleyl SB, Bonkowsky JL. Clinical exome sequencing identifies a novel TUBB4A mutation in a child with static hypomyelinating leukodystrophy. Pediatr Neurol. (2014) 50:608–11. doi: 10.1016/j.pediatrneurol.2014.01.051
46. Kalari KR, Casavant M, Bair TB, Keen HL, Comeron JM, Casavant TL, et al. First exons and introns–a survey of GC content and gene structure in the human genome. In Silico Biol. (2006) 6:237–42.
47. Aird D, Ross MG, Chen W-S, Danielsson M, Fennell T, Russ C, et al. Analyzing and minimizing PCR amplification bias in illumina sequencing libraries. Genome Biol. (2011) 12:1–14. doi: 10.1186/gb-2011-12-2-r18
48. Chen Y-C, Liu T, Yu C-H, Chiang T-Y, Hwang C-C. Effects of GC bias in next-generation-sequencing data on de novo genome assembly. PLoS ONE. (2013) 8:e62856. doi: 10.1371/journal.pone.0062856
49. Ross MG, Russ C, Costello M, Hollinger A, Lennon NJ, Hegarty R, et al. Characterizing and measuring bias in sequence data. Genome Biol. (2013) 14:1–20. doi: 10.1186/gb-2013-14-5-r51
50. Resende LL, de Paiva ARB, Kok F, da Costa Leite C, Lucato LT. Adult leukodystrophies: a step-by-step diagnostic approach. Radiographics. (2019) 39:153–68. doi: 10.1148/rg.2019180081
51. Schlüter A, Rodríguez-Palmero A, Verdura E, Vélez-Santamaría V, Ruiz M, Fourcade S, et al. Diagnosis of genetic white matter disorders by singleton whole-exome and genome sequencing using interactome-driven prioritization. Neurology. (2022) 98:e912–23. doi: 10.1212/WNL.0000000000013278
52. Stutterd CA, Vanderver A, Lockhart PJ, Helman G, Pope K, Uebergang E, et al. Unclassified white matter disorders: a diagnostic journey requiring close collaboration between clinical and laboratory services. Eur J Med Genet. (2022) 65:104551. doi: 10.1016/j.ejmg.2022.104551
53. Lynch DS, Wade C, Paiva ARB, John N, Kinsella JA, Merwick Á, et al. Practical approach to the diagnosis of adult-onset leukodystrophies: an updated guide in the genomic era. J Neurol Neurosurg Psychiatry. (2019) 90:543–54. doi: 10.1136/jnnp-2018-319481
Keywords: leukodystrophy, hypomyelination, next generation sequencing (NGS), genetic diagnosis, medical genetics, pediatric neurology, white matter disorders, neurogenetics
Citation: Perrier S, Guerrero K, Tran LT, Michell-Robinson MA, Legault G, Brais B, Sylvain M, Dorman J, Demos M, Köhler W, Pastinen T, Thiffault I and Bernard G (2023) Solving inherited white matter disorder etiologies in the neurology clinic: Challenges and lessons learned using next-generation sequencing. Front. Neurol. 14:1148377. doi: 10.3389/fneur.2023.1148377
Received: 20 January 2023; Accepted: 23 February 2023;
Published: 03 April 2023.
Edited by:
Shanshan Mao, Zhejiang University School of Medicine, ChinaReviewed by:
Charles Marques Lourenco, Faculdade de Medicina de São José do Rio Preto, BrazilCesar Augusto Alves, Children's Hospital of Philadelphia, United States
Copyright © 2023 Perrier, Guerrero, Tran, Michell-Robinson, Legault, Brais, Sylvain, Dorman, Demos, Köhler, Pastinen, Thiffault and Bernard. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Geneviève Bernard, Z2VuZXZpZXZlLmJlcm5hcmRAbWNnaWxsLmNh; Isabelle Thiffault, aXRoaWZmYXVsdEBjbWguZWR1
†These authors have contributed equally to this work and share senior authorship