 Siqi Dong1,2†
Siqi Dong1,2† Yuanfeng Zhang
Yuanfeng Zhang Xiaoni Liu
Xiaoni Liu Ping Huang
Ping Huang Xiangjun Chen
Xiangjun Chen- 1Department of Neurology, Huashan Hospital and Institute of Neurology, Fudan University, Shanghai, China
- 2National Center for Neurological Disorders, Shanghai, China
- 3Department of Biochemistry and Physiology, Shanghai University of Medicine and Health Sciences, Shanghai, China
- 4Department of Neurology, Shanghai Children's Hospital, Shanghai Jiao Tong University, Shanghai, China
- 5Department of Forensic Pathology, Academy of Forensic Science, Shanghai, China
- 6Human Phenome Institute, Fudan University, Shanghai, China
The PANK2 gene, which encodes mitochondrial pantothenate kinase 2 protein, is the disease-causing gene for pantothenate kinase-associated neurodegeneration (PKAN). We report a case of atypical PKAN with autism-like symptoms presenting with speech difficulties, psychiatric symptoms, and mild developmental retardation. Magnetic resonance imaging (MRI) of the brain showed the typical “eye-of-the-tiger” sign. Whole-exon sequencing revealed PANK2 p.Ile501Asn/p.Thr498Ser compound heterozygous variants. Our study highlights the phenotypic heterogeneity of PKAN, which can be confused with autism spectrum disorder (ASD) and attention-deficit hyperactivity disorder (ADHD) and requires careful clinical identification.
Introduction
Mitochondrial pantothenate kinase 2 (PANK2) is a key regulatory enzyme in the biosynthesis of coenzyme A, which catalyzes the phosphorylation of pantothenate to generate 4′-phosphopantothenate. PANK2 is the only one out of four PANK genes encoding an isoform that localizes to mitochondria. Homozygous or compound heterozygous variants of the PANK2 gene can lead to pantothenate kinase-associated neurodegeneration (PKAN), the most prevalent type of neurodegeneration with brain iron accumulation (NBIA) disorders. PKAN patients present with a progressive movement disorder, dysarthria, cognitive impairment, and retinitis pigmentosa. In magnetic resonance imaging, PKAN patients exhibit the characteristic of the “eye of the tiger” sign in the globus pallidus, which corresponds to iron accumulation and gliosis as shown in neuropathological examinations (1).
Based on the age at onset and rate of disease progression, PKAN is classified into two major groups: (I) the classic form of PKAN characterized by early onset (<10 years old when first symptoms start), rapid progression, dysarthria, dystonia, rigidity, loss of ambulation ~15 years after the first symptoms, and pigmentary retinopathy and (II) atypical PKAN, which presents in the second decade of life, with slower progression, and heterogeneous phenotypes, such as extrapyramidal signs, neurobehavioral signs, and speech difficulties (2). Despite a series of reports, the phenotypic heterogeneity of PKAN is still not fully understood. Here we report a compound heterozygote for PANK2 variants in a Chinese patient with early-onset atypical PKAN, which is easily confused with ASD.
Case description
The patient, a 9-year-old boy, was recruited from a non-consanguineous family with no history of genetic diseases. His language and motor development were normal until he was 3 years old when his parents found that he rarely communicated or made eye contact with others. He did not catch up to their peers in language abilities. Psychiatric symptoms were also observed, including restricted or repetitive behaviors, emotional lability, attention deficit, and hyperactivity. At the age of 4, the patient gradually developed unstable walking, easy to fall, and often walked on tiptoes. He was taken to a local pediatric hospital and evaluated on a series of scales, including the Gesell test, Autism Behavior Checklist, psycho-educational profile, and Achenbach Child Behavior Checklist. The Gesell test suggested mild developmental retardation. The Autism Behavior Checklist scored 39. Psycho-educational profile suggested mild retardation in communication and moderate retardation in motor and maladaptive behaviors. Achenbach Child Behavior Checklist suggested significant social withdrawal. He was diagnosed with ASD and ADHD according to DSM-V criteria (persistent deficits in each of three areas of social communication and interaction plus two types of restricted, repetitive behaviors; eight symptoms for inattention and seven symptoms for hyperactivity and impulsivity). The patient thus underwent long-term rehabilitation behavior therapy, and the symptoms of unstable walking improved. At the age of 9, the patient's symptoms worsened after fever, characterized by frequent falls, tremors of the upper limbs, involuntary nodding of the head, and body shaking.
On examination, the patient showed involuntary nodding of the head, unstable walking, and was unable to complete the hoop. There was obstructed upward movement of both eyes and increased muscle tone, which is more prominent in the right limbs. Muscle strength and deep tendon reflexes were normal. Romberg signs and cerebellar signs were absent. Wechsler Intelligence Scale test was 70, which was at the borderline. Ophthalmologic examinations were normal except for slightly thinner retinal epithelial cells. Electroencephalogram showed no abnormalities. Laboratory tests, including routine blood and urine tests, biochemistry, liver and kidney functions, electrolytes, autoantibodies, and trace element tests were all normal. T2-weighted brain magnetic resonance imaging (MRI) showed a characteristic of the “eye-of-the-tiger” sign (Figure 1).

Figure 1. T2-weighted brain MRI showing the “eye-of-the-tiger” sign.
Whole-exon sequencing revealed two compound heterozygous variants (p.Ile501Asn and p.Thr498Ser) involving exons 5 in PANK2 of the patient. The father was a heterozygote for the Ile501Asn variant, and the mother was a heterozygote for the Thr498Ser variant (Figure 2A). These two variants were absent in 1,000 g, ExAC, and gnomAD databases. In silico predictors, including SIFT, Polyphen2, and Variant Taster, predict Ile501Asn as “Damaging (0),” “Probably damaging,” and “Disease causing”; and Thr498Ser as “Damaging (0.004),” “Probably damaging,” and “Disease causing.” Amino acid sequence alignment shows that Ile501 and Thr498 are highly conserved both across species and in PANK family proteins (Figure 2B). Ile501Asn has been reported in several previous studies (2–4), while Thr498Ser was novel. According to the ACMG standard, PANK2 p.Ile501Asn was classified as likely pathogenic (PS1, PM1, PM2, and PP3), while PANK2 p.Thr498Ser was classified as a variant of uncertain significance (PM1, PM2, and PP3).
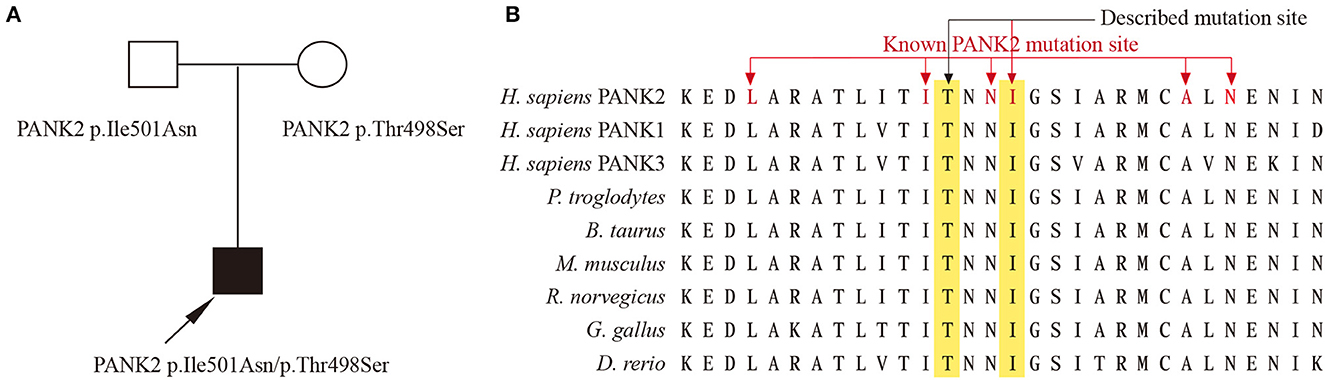
Figure 2. (A) Family with compound heterozygous PANK2 gene variants. Proband PKAN is indicated with an arrow. (B) Amino acid sequence alignment between species and PANK family proteins, the red font indicates amino acid changes previously reported in PKAN.
Discussion
In this study, we report a case of early-onset PKAN in a patient presenting with atypical symptoms, including speech difficulties, psychiatric symptoms, and mild developmental retardation, mimicking ASD and ADHD. Whole-exon sequencing revealed PANK2 p.Ile501Asn/p.Thr498Ser compound heterozygous variants, of which Thr498Ser has not been reported earlier.
Despite having been diagnosed with ASD, a diagnosis of PKAN was more suitable in our patient since there was an “eye-of-the-tiger” sign in the T2-weighted brain MRI and the PANK2 compound heterozygous variant. The “Eye-of-the-tiger” sign is almost never excluded in PKAN patients, whether classic or atypical ones (2). Our patient was classified as atypical PKAN, as extrapyramidal signs, including dystonia, dysarthria, rigidity, and choreoathetosis, which is prominent in classic PKAN patients, were not severe and progressed slowly. While our patients present prominent speech difficulties, psychiatric symptoms, and mild developmental retardation, which were more common in atypical PKAN patients (2). Ile501Asn homozygous or complex heterozygous variants have previously been reported to cause atypical PKAN (2–4), consistent with our findings.
Interestingly, atypical PKAN is often present in the second decade of life, while our patient developed the disease at age 3, contrary to previous reports, making it easy to be confused with some neurodevelopmental diseases, such as ASD. By the revision of previous reports of PKAN in China (Table 1), we find that there are 42.9% of early-onset patients who showed slow progression, suggesting that some early-onset patients do not necessarily exhibit typical PKAN, consistent with our findings. Dystonia and dysarthria are the most common symptoms of PKAN, presenting in 77.4% of patients. Psychiatric symptoms, which are prominent in our patient, are rare, accounting for 6.5% by revision, and reported to be 9.1% in the previous literature (2). Therefore, PKAN patients with prominent psychiatric symptoms are rare, which requires careful identification. For children suspected of ASD and ADHD with poor sensory integration, MRI and genetic examination are needed to diagnose the disease at an early stage and give reasonable treatment.
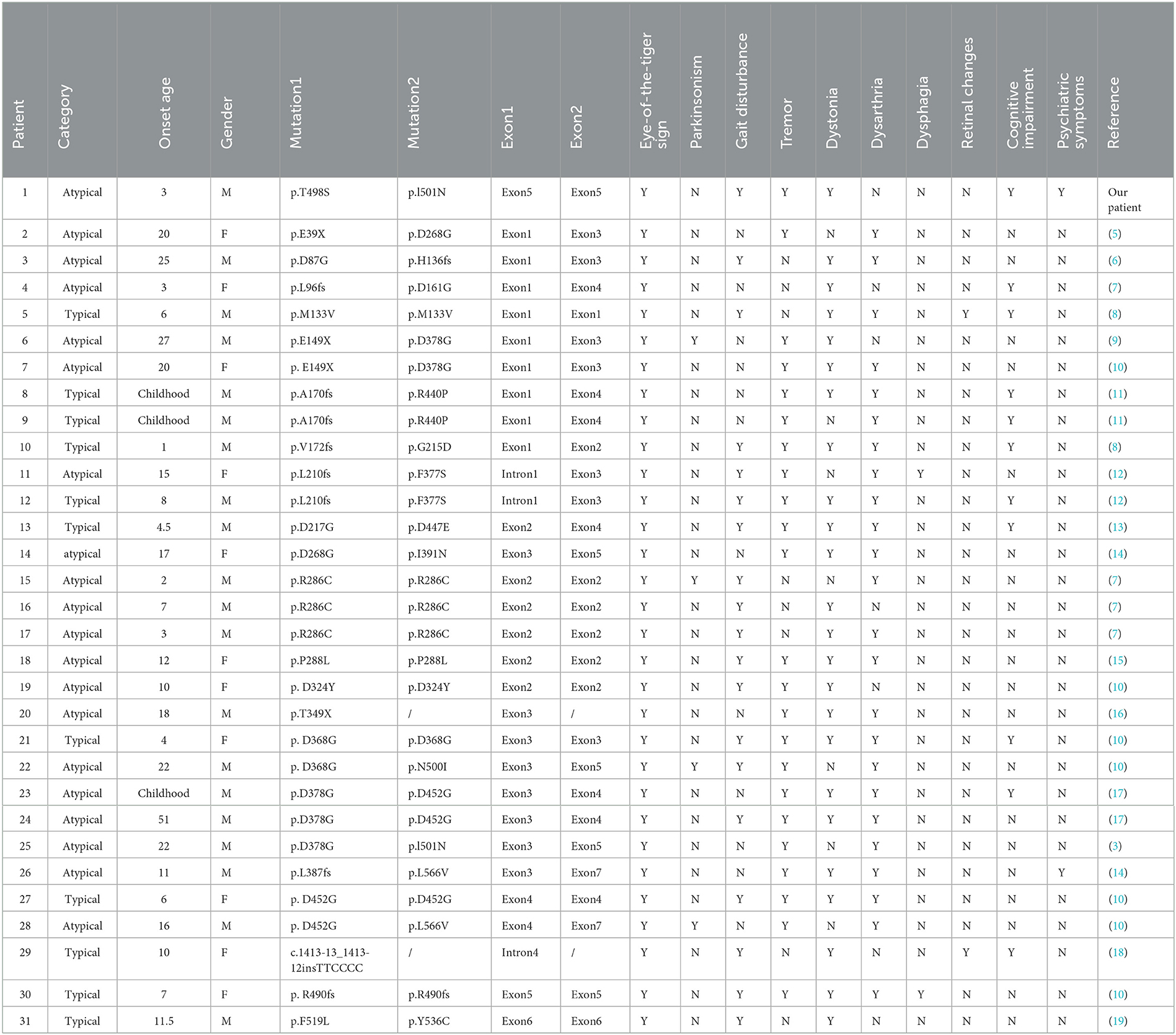
Table 1. Clinical, radiological, and genetic characteristics in Chinese patients with PANK.
In conclusion, our study highlights the phenotypic heterogeneity of PKAN, which can be confused with ASD and requires careful clinical identification.
Data availability statement
The original contributions presented in the study are included in the article/supplementary material, further inquiries can be directed to the corresponding authors.
Ethics statement
The studies involving human participants were reviewed and approved by the Ethics Committee of Huashan Hospital, Fudan University. Written informed consent to participate in this study was provided by the participants' legal guardian/next of kin. Written informed consent was obtained from the minor(s)' legal guardian/next of kin for the publication of any potentially identifiable images or data included in this article.
Author contributions
Conception and study design: XC and PH. Data collection or acquisition: SD, YT, YZ, PH, and XC. Review of previous literatures: SD, ZQ, and XL. Drafting the manuscript work: SD, YT, PH, and XC. All authors contributed to the approval of final version to be published and agreement to be accountable for the integrity and accuracy of all aspects of the work.
Funding
This study was supported by 2020 Medical Service and Support Capacity Improvement Project: Construction of the Cohort-Based Multidisciplinary Accurate Diagnosis and Treatment Platform for Neurological Autoimmune and Infectious Diseases, Shanghai Fudan University Education Development Foundation and State Key Laboratory of Genetic Engineering, Human Phenome Institute, Zhangjiang Fudan International Innovation Center, and Fudan University.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher's note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Zhou B, Westaway SK, Levinson B, Johnson MA, Gitschier J, Hayflick SJ, et al. A novel pantothenate kinase gene (PANK2) is defective in Hallervorden-Spatz syndrome. Nat Genet. (2001) 28:345–9. doi: 10.1038/ng572
2. Hayflick SJ, Westaway SK, Levinson B, Zhou B, Johnson MA, Ching KHL, et al. Genetic, clinical, and radiographic delineation of Hallervorden–Spatz syndrome. New Engl J Med. (2003) 348:33–40. doi: 10.1056/NEJMoa020817
3. Pan S, Zhu C. Atypical pantothenate kinase-associated neurodegeneration with PANK2 variants: clinical description and a review of the literature. Neurocase. (2020) 26:175–82. doi: 10.1080/13554794.2020.1752739
4. Zhang Y, Tang B, Zhao A, Xia K, Long Z, Guo J, et al. Novel compound heterozygous variants in the PANK2 gene in a Chinese patient with atypical pantothenate kinase-associated neurodegeneration. Movement Disord. (2005) 20:819–21. doi: 10.1002/mds.20408
5. Song XW, Wang YL, Shi YW, Deng WY, Chen SQ, Lin H, et al. Clinical manifestations and detection of pantothenate kinase 2 gene mutation in a patient with Hallervorden-Spatz syndrome. Zhonghua Yi Xue Za Zhi. (2009) 89:3320–3.
6. Cheng Y, Liu YT, Yang ZH, Yang J, Shi CH, Xu YM, et al. Novel compound heterozygous PANK2 gene mutations in a Chinese patient with atypical pantothenate kinase-associated neurodegeneration. Int J Neurosci. (2018) 128:1109–13. doi: 10.1080/00207454.2018.1483364
7. Zeng J, Xing W, Liao W, Wang X. Magnetic resonance imaging, susceptibility weighted imaging and quantitative susceptibility mapping findings of pantothenate kinase-associated neurodegeneration. J Clin Neurosci. (2019) 59:20–8. doi: 10.1016/j.jocn.2018.10.090
8. Du X, Guo H, He Y, Liang C. Pantothenate kinase-associated neurodegeneration: two cases report and literature review. J Clin Pediatr. (2020) 38:452–5.
9. Mak CM, Sheng B, Lee HH, Lau KK, Chan WT, Lam CW, et al. Young-onset parkinsonism in a Hong Kong Chinese man with adult-onset Hallervorden-Spatz syndrome. Int J Neurosci. (2011) 121:224–7. doi: 10.3109/00207454.2010.542843
10. Li WB, Shen NX, Zhang C, Xie HC, Li ZY, Cao L, et al. Novel PANK2 mutations in patients with pantothenate kinase-associated neurodegeneration and the genotype-phenotype correlation. Front Aging Neurosci. (2022) 14:848919. doi: 10.3389/fnagi.2022.848919
11. Yang F, Wang J, Yang Z, Ren Z, Zeng F. PANK2 p.A170fs: a novel pathogenetic mutation, compound with PANK2 p.R440P, causing pantothenate kinase associated neurodegeneration in a Chinese family. Int J Neurosci. (2022) 132:582–8. doi: 10.1080/00207454.2020.1828883
12. Shan J, Wen B, Zhu J, Lin P, Zheng J, Yan C, et al. Novel PANK2 gene mutations in two Chinese siblings with atypical pantothenate kinase-associated neurodegeneration. Neurol Sci. (2013) 34:561–3. doi: 10.1007/s10072-012-1177-8
13. Shi X, Zheng F, Ye X, Li X, Zhao Q, Lin Z, Hu Y, Wang J. Basal ganglia calcification and novel compound heterozygous mutations in the PANK2 gene in a Chinese boy with classic Pantothenate kinase-associated neurodegeneration: a case report. Medicine (Baltimore). (2018) 97:e0316. doi: 10.1097/MD.0000000000010316
14. Zhang Y, Tang B, Guo J, Long Z, Xia K, Pan Q, et al. Studies on PANK2 gene mutations in Chinese patients with Hallervorden-Spatz syndrome. Zhonghua Yi Xue Yi Chuan Xue Za Zhi. (2005) 22:189–91.
15. Li YF, Li HF, Zhang YB, Wu JM. Novel homozygous PANK2 mutation identified in a consanguineous Chinese pedigree with pantothenate kinase-associated neurodegeneration. Biomed Rep. (2016) 5:217–20. doi: 10.3892/br.2016.715
16. Lee CH, Lu CS, Chuang WL, Yeh TH, Jung SM, Huang CL, et al. Phenotypes and genotypes of patients with pantothenate kinase-associated neurodegeneration in Asian and Caucasian populations: 2 cases and literature review. Sci World J. (2013) 2013:860539. doi: 10.1155/2013/860539
17. Wu YR, Chen CM, Chao CY, Lyu RK, Lee-Chen GJ. Pantothenate kinase-associated neurodegeneration in two Taiwanese siblings: identification of a novel PANK2 gene mutation. Mov Disord. (2009) 24:940–1. doi: 10.1002/mds.22458
18. Han J, Kim DW, Lee CH, Han SH. Optic atrophy in a patient with atypical pantothenate kinase-associated neurodegeneration. J Neuroophthalmol. (2016) 36:182–6. doi: 10.1097/WNO.0000000000000335
Keywords: PANK2 mutation, pantothenate kinase-associated neurodegeneration (PKAN), atypical PKAN, case report, autism spectral disorder (ASD)
Citation: Dong S, Tuo Y, Qi Z, Zhang Y, Liu X, Huang P and Chen X (2023) Case report: Novel compound heterozygous variants in the PANK2 gene in a Chinese patient diagnosed with ASD and ADHD. Front. Neurol. 14:1118076. doi: 10.3389/fneur.2023.1118076
Received: 07 December 2022; Accepted: 20 March 2023;
Published: 17 April 2023.
Edited by:
Kinya Ishikawa, Tokyo Medical and Dental University, JapanReviewed by:
Thea Giacomini, Giannina Gaslini Institute (IRCCS), ItalyAdriana Prato, University of Messina, Italy
Copyright © 2023 Dong, Tuo, Qi, Zhang, Liu, Huang and Chen. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Ping Huang, aHVhbmdwQHNzZmpkLmNu; Xiangjun Chen, eGlhbmdqY2hlbkBmdWRhbi5lZHUuY24=
†These authors have contributed equally to this work
‡ORCID: Xiangjun Chen orcid.org/0000-0002-8108-9013