 Jun Qin
Jun Qin Zhihui Ma
Zhihui Ma Shiyu Shu
Shiyu Shu
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Neurol. , 07 March 2023
Sec. Dementia and Neurodegenerative Diseases
Volume 14 - 2023 | https://doi.org/10.3389/fneur.2023.1103416
Microglia are the principal resident immune cells in the central nervous system (CNS) and play important roles in the development of CNS disorders. In recent years, there have been significant developments in our understanding of microglia, and we now have greater insight into the temporal and spatial patterns of microglia activation in a variety of CNS disorders, as well as the interactions between microglia and neurons. A variety of signaling pathways have been implicated. However, to date, all published clinical trials have failed to demonstrate efficacy over placebo. This review summarizes the results of recent important studies and attempts to provide a mechanistic view of microglia activation, inflammation, tissue repair, and CNS disorders.
In 1856, the German pathologist RUDOLF (1) coined the term “glia” to describe a distinct population of cells, other than neurons, in the brain. In the following decades, research in this field progressed slowly. It was not until 1919 that microglia were accepted as an independent cell type (2). Microglia account for 5–20% of all glial cells in the central nervous system (CNS) and are resident immune cells in the CNS (3). Under normal physiological conditions, microglia are in a resting state, with a branched cellular arrangement; they monitor their surroundings and play an important role in the regulation and monitoring of neurological homeostasis, proliferation and regeneration of neurons, and nourishment of nerves (4). Upon infection or injury, microglia are activated and move toward the site of infection/injury, releasing a variety of cytotoxic substances to attack the infecting agents and clear the damaged cells. When microglia are activated abnormally, the released cytotoxic substances can damage the surrounding normal tissues (5). Inhibition of aberrant microglial activation could limit the degree of neuroinflammation, and thus, represents a novel strategy for the treatment of a variety of CNS disorders (6). In humans, microglia emerge at 4.5 weeks gestation in the meninges, ventricular rim, and plexus, and migrate from these locations to the telencephalon and mesencephalon. During this process, microglia gradually develop into a branched form, appearing in this form from as early as 12 weeks gestation (7). Microglia differentiation is regulated by the transcription factor spleen focus forming virus proviral integration oncogene (SPI1, formerly known as PU.1) and interferon regulatory factor 8 (8). Microglia self-renewal in the CNS depends on the continuous activation of colony-stimulating factor 1 receptor (CSF1R) (9, 10).
Microglia produce opposing actions (i.e., neurotrophic vs. neurotoxic) depending on their activation status. Based on the classification of macrophages, microglia are usually divided into two groups: classically activated microglia (M1 type) and alternatively activated microglia (M2 type) (11). The inflammatory M1 type is usually caused by Toll-like receptors and γ-interferon signaling pathways. M1 microglia produce pro-inflammatory cytokines such as tumor necrosis factor-α (TNF-α), interleukin-6 (IL-6), IL-1, IL-1β, and chemokines, and express nicotinamide adenine dinucleotide phosphate (NADPH) oxidase and matrix metalloproteinases (MMP)-12. The anti-inflammatory M2 type plays a neuroprotective role. M2 microglia upregulate arginase 1 expression, secrete growth factors, and promote the release of anti-inflammatory cytokines such as IL-10 and transforming growth factor β (TGF-β) (12). Accumulating evidence suggests that microglia polarization is multidimensional, with extensive overlap in gene expression, rather than occurring on a simple linear spectrum (13). Single-cell RNA sequencing studies have shown that microglia can express both M1-type and M2-type activation marker genes (14). Transcriptomic studies have shown that microglia activation exhibits a broader transcriptional profile than M1 and M2 types (15). The in vivo phenotype of microglia is even more complex. A recent single-cell RNA sequencing study demonstrated at least nine subpopulations of microglia in the mouse brain (16). Similarly, nine microglia subpopulations have been identified in the cerebrocortex of the human brain (17). In an analysis based on the three-dimensional (3D) morphology of microglia, 10 distinct microglia subpopulations were proposed in the mouse and human brain (18). Spatial and temporal heterogeneity have also been observed at the single-cell level in mouse and human microglia (7). Moreover, distinct microglia subpopulations have been observed in specific CNS disorders, including amyotrophic lateral sclerosis (19) and Alzheimer's disease (AD) (20).
Microglia maintain CNS homeostasis in both the developing and mature brain. In the developing brain, microglia are implicated in neurogenesis, programmed cell death, synapse elimination, and neural circuit formation (21). During early development, microglia secrete neurotrophic factors to promote neurogenesis and maintain the survival and differentiation of specific neuronal lineages (22, 23). Immature neurons undergoing programmed cell death are cleared by microglia without triggering inflammatory processes (24). Microglia also remodel the CNS by selectively pruning redundant neuronal protrusions and defective synapses (25). In the adult brain, alterations in the microenvironment cause rapid and profound changes in microglia morphology and function as well as gene expression. Microglia also actively interact with neurons to influence neuronal regeneration, proliferation, and migration, and secrete neurotrophic factors such as insulin-like growth factor-1 (IGF-1) to promote the survival of neighboring neurons (26). Microglia are an important part of the innate immune sentinel network and serve as the first line of defense in the CNS. Cellular debris or dead cells can be cleared by microglia via phagocytosis, without triggering an inflammatory response (27). Upon invasion by pathogens, microglia respond by producing pro- and/or anti-inflammatory cytokines, chemokines, and complement proteins. These responses promote pathogen clearance but may, on occasion, elicit a strong collateral inflammatory response and exacerbate CNS pathology.
The activation of microglia is a complex process that involves multiple stimuli, including lipopolysaccharide (LPS), β-amyloid (Aβ), IL-1β, TNF-α, interferon (IFN)-γ, adenosine triphosphate (ATP), peptidoglycan (PG), and a variety of membrane receptors (Figure 1).
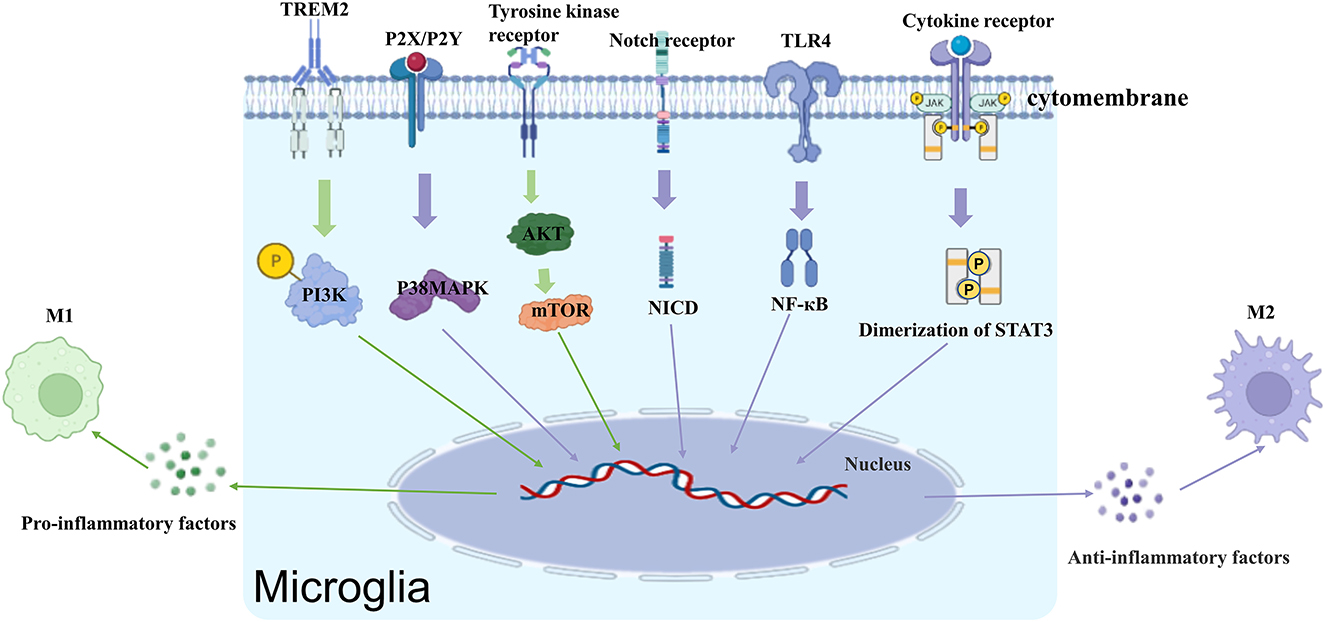
Figure 1. Receptors and signaling pathways that regulate microglia activation.
TLRs play an important role in inflammation, signal transduction, and apoptosis. The TLRs that are associated with microglia activation include TLR2, 3, 4, and 9. TLR4 is the primary receptor that mediates the activation of microglia by LPS (28). Kacimi et al. found that LPS bound to TLR4 on the cell membrane of microglia to activate its downstream signaling pathways, including the nuclear factor (NF)-κB, Janus kinase-signaling and transcriptional activating protein (JAK-STAT), C-Jun amino-terminal kinase (JNK), and p38/Mitogen-activated protein kinase (MAPK) pathways (29). In a study by Capiralla et al., LPS amplified TLR4/NF-κB/STAT signaling in microglia to increase the release of TNF-α and IL-6 (30).
Microglia express both TNFR1 and TNFR2. Exposure to IFN-γ or LPS increases TNFR1 expression in microglia (31). In the normal rat cortex, a portion of TNFR1 is located in lipid raft microdomains, Moderate TBI resulted in rapid recruitment of TNFR1, but not TNFR2 or Fas, to lipid rafts and induced alterations in the composition of signaling intermediates. Activation of TNFR1 after TBI induced the expression of caspase-8, thus initiating apoptosis and transient activation of NF-κB (32). Novel TNF-R1 inhibitors was found to significantly reduce cortical inflammatory cytokines 3 h after traumatic brain injury (TBI) and improved functional recovery following diffuse TBI in mice (33).
Microglia express both P2Y receptors coupled to G-proteins, as well as, P2X receptors coupled to ligand gated cation channels (34). They play an important role in microglia activation, motility and paracrine signaling. P2Y12R, a microglia-specific receptor that mediates microglial chemotaxis toward site of injury (35). Activation of P2Y12R results in the opening of K+ channel and increased cAMP levels that is implicated in microglia surveillance and chemotaxis toward injury site (36). Not only that P2Y12R on microglia in the spinal cord promotes the production of pro-inflammatory cytokines through the p38/MAPK pathway, and as such, has been implicated in the maintenance of neuropathic pain (37). Research has also shown that blockade of P2X7 receptors significantly reduces nociception in animal models of chronic neuropathic and inflammatory pain (38). The P2X7R is identified as a key player in the process of microgliosis, where by driving microglial activation, it can potentially lead to a deleterious cycle of neuroinflammation and neurodegeneration (39).
Microglia are the major source of IL-1 in the CNS. Upregulation of IL-1R in activated microglia plays an important role in microglia activation, neuronal damage, expression of pro-inflammatory cytokines, and degenerative CNS diseases (40). In newborn rats, LPS causes excessive microglia activation and nociceptive hypersensitivity that can persist into adulthood (41). In contrast, IL-1R antagonists can attenuate microglia activation and prevent nociceptive hypersensitivity. Silencing IL-1R can also attenuate microglia activation and reduce the expression of cyclooxyganese (COX)-2 and IL-6 (42).
Similar to astrocytes, both microglia and oligodendrocytes express IFN-γR (43). IFN-γ can produce seemingly opposing actions depending upon the specific circumstances. IFN-γ can exert protective effects by increasing phagocytosis and the clearance of neuronal debris and Aβ and inhibiting the expression of TNF-α and COX-2 (44). IFN-γ can also damage the CNS through microglia activation. In addition to activating resting microglia, stimulating the IFN-γR signaling pathway also upregulates the tyrosine kinase Lyn and purinergic P2X4 receptors of microglia, leading to abnormal neuronal excitation in dorsal horn neurons and neuropathic pain (45).
PPARs are a family of ligand-activated nuclear transcription factors in the cytoplasm. PPARs are composed of three different isomers (i.e., α, β/δ, and γ) (46). PPARγ forms a heterodimer with retinol-like X receptors and binds to promoter regions to regulate their transcription (47). In a mouse study by Pan et al., the resveratrol malibatol A produced neuroprotective effects by activating PPARγ, which, in turn, switched microglia from the pro-inflammatory M1 type to the anti-inflammatory M2 type (48). PPARα can also interact directly with pro-inflammatory transcription factors such as NF-κB and P38MAPK to negatively regulate their targets and produce anti-inflammatory effects through a non-DNA-dependent mechanism (49).
β-Adrenergic receptor antagonists have been shown to block CD11b upregulation, increase the production of a variety of pro-inflammatory cytokines by microglia, and attenuate anxiety-like behavior in mice subjected to repeated social defeat (50). Studies have also found that CXCL10, a chemokine released by neurons after brain injury, activates microglia to promote the dissipation of innervated dendrites from the injury site; these effects were obliterated in CXCR3 knockout mice. Other chemokines that have been implicated in microglia activation include C-C chemokine receptor 2 (CCR2), CXC Chemokine Receptor 3 (CXCR3), and chemokine (C-X3-C motif) receptor 1 (CX3CR1) (51).
The Notch pathway is highly conserved in a variety of organisms and is involved in the development of almost all organ systems. Upon binding with ligands, activated Notch receptors on the cell surface of microglia increase protein hydrolase cleavage and the release of intracellular segments of Notch into the nucleus, where they bind to the transcriptional repressor RBP-Jκ to activate the transcription of target genes and regulate cell proliferation, differentiation, and apoptosis (52). The Notch signaling pathway has been shown to mediate the release of TNF-α, IL-1β, IL-6, and other pro-inflammatory factors in activated microglia, promoting cell proliferation and migration. The Notch signaling pathway also promotes the activation of various key pathways such as NF-κB, p38 MAPK, and JNK in microglia by regulating the expression of the deubiquitinating enzyme CYLD, thereby facilitating microglial activation (53).
The MAPK, ERK, and NF-κB pathways have been shown to play key roles in the transcriptional and post-transcriptional regulation of iNOS and TNF-α gene expression in LPS-activated microglia (54). MAPK is an important molecule that carries signals from the cell surface to the nucleus. Previous studies have shown that the activation of p38MAPK signal pathway is closely related to neuroinflammation caused by activation of microglia in ischemic stroke (55). At the molecular level, MicroRNA22, Apelin-12 can inhibit p38MAPK signaling pathway to inhibit the microglia activation to improve ischemic brain injury (56, 57). After rat primary microglia were treated with thrombin, microglia were activated, iNOS expression increased, and ERK phosphorylation was induced (58). After treatment of lipopolysaccharide-activated BV-2 microglial cells with ERK inhibitor SCH772984, the release of nitric oxide was inhibited, and the phosphorylated ERK decreased (59). The role of NF-κB, a key factor in transcriptional regulation, is crucial in the regulation of inflammatory response and immune stress (60). It has been shown that microglia activation is closely related to NF-κB signaling pathway (61). Stepharine improves the outcome of middle cerebral artery ischemia rats by inhibiting TLR4/NF-κB signaling pathway, attenuating neuronal damage and suppressing microglia hyperactivation (62). Quercetin reduces microglia activation by inhibiting NF-κB signaling pathway and plays a neuroprotective role against brain injury caused by ischemia and hypoxia (63).
Src family kinases (SFKs), a class of non-receptor tyrosine kinases, play an important role in regulating cell proliferation, differentiation, metabolism, the immune response, and signal transduction. Evidence indicates that epidemic B encephalitis virus (JEV)-stimulated Src activation promotes TNF-α and IL-1β expression by initiating Src/Ras/Raf/ERK/NF-κB and Src/Raf/ERK/NF-κB in microglia (64).
Abundant research shows that a central role for the PI3K-AKT signaling pathway in health and disease (65, 66). In addition, activated AKT is vital for initiating immune responses, as evidenced by the functional defects observed in various leukocyte subsets when PI3K and AKT are ablated in experimental rodent models (67). For example, in vivo systemic challenge with LPS in rodents leads to an increase in phosphorylation of AKT via PI3K signaling in the CNS, which induces activation of microglial populations in the cortex, hippocampus, and thalamus, and is associated with exacerbated release of the pro-inflammatory factors TNF-α, IL-1β, iNOS, and IL-6 (68, 69).
Apelin-13 inhibits LPS-induced activation of M1-type microglia by downregulating the STAT3 signaling pathway (70). Binding of LPS to TLR4 receptors on the microglial membrane surface recruits downstream signaling molecules by activating the MyD88/IRAK/TRAF6 signaling cascade, which, in turn, causes IKKα/β phosphorylation, IκB-α phosphorylation, IκB-α degradation, NF-κB nuclear translocation, NF-κB p65 phosphorylation, and other signaling cascades, ultimately enhancing NF-κB transcriptional activity (71–73). Mammalian target of rapamycin (mTOR) is a member of a conserved serine/threonine kinase family that is implicated in many fundamental biological processes, including gene transcription, protein translation, and ribosome synthesis (74). mTOR interacts with the IKK complex and increases IKK activity, which, in turn, promotes NF-κB transcriptional activity (75, 76). Endogenous α-KG inhibits microglia activation by suppressing the mTOR/NF-κB pathway. TNF-α-sensitive pericytes release IL-6 to activate microglia through the synergistic action of the IκB-NF-κB and JAK-STAT3 pathways (77).
Ischemic stroke, also known as cerebral infarction, is the sudden interruption of cerebral blood flow in local brain tissue due to various reasons; it accounts for 85% of all strokes. Ischemic stroke leads to necrosis of local brain tissue and corresponding neurological deficits. Microglial activation has traditionally been considered to be detrimental in ischemic stroke (Table 1), and thus, has been extensively studied. However, a growing body of evidence suggests another side to the coin: microglial activation may be critical for neurogenesis, angiogenesis, and synaptic remodeling, thereby facilitating functional recovery after cerebral ischemia. When ischemic stroke occurs, the lack of blood sugar and oxygen in brain tissue leads to the death of neurons, the release of reactive oxygen species, the activation of complement, the up-regulation of endothelial cell adhesion factors, and the dead cells release danger signals to the brain microenvironment, including HMGB1 and ATP, to activate microglia (96). Overall, microglia activation consists of two phases: the M2-like anti-inflammatory phenotype, which is predominant from day 0.5 to 7 after cerebral ischemia, and the M1-like pro-inflammatory phenotype, which is predominant from day 7 to 14. M1-type microglia produce pro-inflammatory mediators, including TNF-α, IL-1β, interferon-c (IFN-c), IL-6, iNOS, MMP-9, and MMP-3 (78). On the one hand, these pro-inflammatory factors and cytotoxic substances secreted by M1-type microglia may directly or indirectly damage neuronal survival, prevent neurogenesis, and aggravate long-term neurological deficits after brain injury (97). On the other hand, they promote the formation of inflammatory microenvironment, which is conducive to the transformation of unpolarized microglia into M1 type, forming pro-inflammatory feedback and promoting the pathological process of cerebral ischemia, which is also unfavorable for neurons after ischemic stroke. In addition, M1-type microglia can prevent axon regeneration (98) and exacerbate oligodendrocyte death. This is not conducive to central nervous system recovery. In contrast, M2-type (alternative) microglia are characterized by the production of pro-angiogenic and anti-inflammatory molecules, including IL-10, TGF-β, insulin-like growth factor, and vascular endothelial growth factor (VEGF) (78). Anti-inflammatory factors secreted by M2-type microglia cells can reduce inflammation and improve ischemic brain injury. The neurotrophic factors secreted by it can promote the proliferation of neural precursor cells, accelerate the migration of neuroblasts (99), promote the maturation of new neurons, and promote the repair of the central nervous system. In addition, after ischemic stroke, M2-type microglia and macrophages, compared with M1 phenotypes, have a stronger phagocytosis capacity for necrotic neurons to remove damaged neurons, prevent secondary inflammatory reactions, and provide survival space for new neurons to maintain the homeostasis of the central nervous system under pathological conditions.
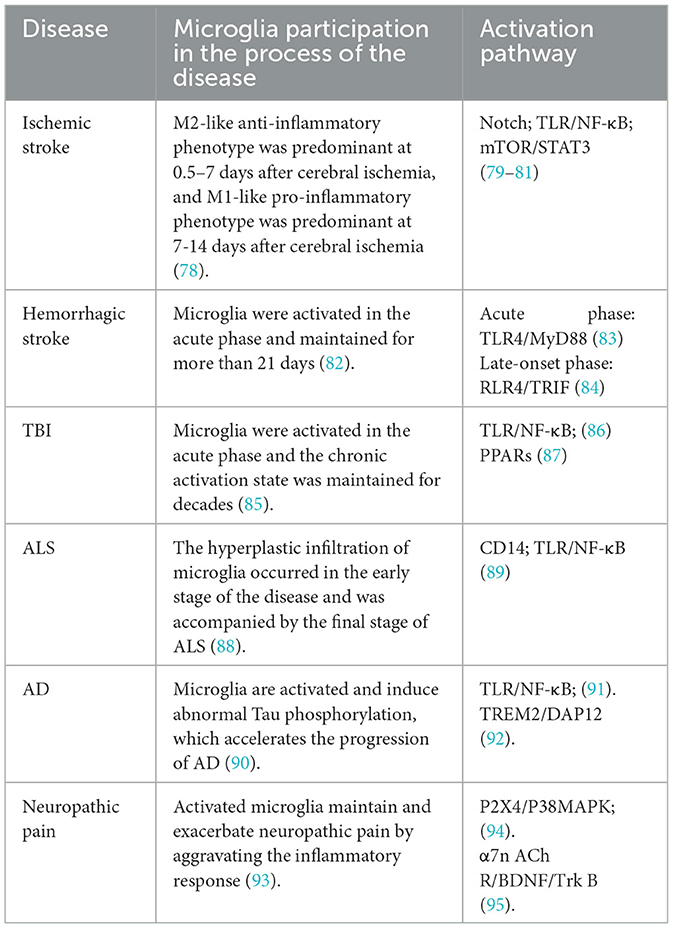
Table 1. Involvement of microglia in CNS disorders.
Hemorrhagic stroke is characterized by a sudden-onset cerebral aneurysm rupture or vascular leakage, leading to cerebral blood circulation disorders. Patients with cerebrovascular diseases may suffer from cerebral arterial stenosis, occlusion, or rupture due to various causes, thus resulting in acute cerebrovascular hemorrhage. Patients exhibit symptoms and signs reflective of temporary or permanent cerebral dysfunction. Hemorrhagic stroke accounts for 15% of all strokes. After intracerebral hemorrhage, a large amount of blood enters the brain tissue to form hematoma, resulting in space occupying lesions and release cell components, thrombin, chemokines and other bioactive substances, activating microglia cells through different ways. M1 microglia are largely pro-inflammatory and release high levels of pro-inflammatory factors upon activation, including IFN-γ, TNF-α, and IL-1β. This excessive inflammatory response is an important source of secondary injury in hemorrhagic stroke. M1 microglia also secrete large amounts of reactive oxygen species (ROS) and protein hydrolases, including heme oxygenase-1 (HO-1), MMP, and iNOS. ROS accumulation increases the expression of apoptotic genes and inflammatory mediators, which, in turn, induces apoptosis and disrupts the blood-brain barrier (BBB) to promote secondary injury (100). Iron accumulation caused by HO-1 overexpression also aggravates secondary injury by increasing lipid peroxidation and free radical formation. Overproduction of NO due to iNOS activation increases peroxynitrite to disrupt the BBB (101). Overexpression of MMP-2 and MMP-9 can also disrupt the BBB by injuring the tight junctions between capillary endothelial cells and the basement membrane (102). Activated M2-type microglia secrete anti-inflammatory factor IL-10 and up-regulate its phenotype markers to play an anti-inflammatory role (103), and can produce specific trophic factors to promote the proliferation of neuroprecursor cells and the migration of neuroblasts, and facilitate the functional integration of new neurons into the existing neural circuits to assist the recovery of nerve function after injury. Moreover, increased secretion of protective molecules such as IL-10 can promote axon regeneration and recovery (104).
M1-type and M2-type microglia exhibit different temporal patterns in a controlled cortical injury (CCI) model (105). Microglia can remain chronically activated for years after the acute inflammatory process has subsided. In patients with TBI, microglia activation was found to persist for decades (85, 106). The activation pattern of microglia also varies spatially. Microglia, macrophages, and astrocytes are predominantly located around the lesion site in the acute phase but spread to distant brain regions by 1-year post-injury (107). These findings highlight the long-lasting impact of TBI and suggest the presence of persistent neuroinflammation at the site of the injury and beyond. Studies of the heterogeneity of brain inflammation after injury, especially those focusing on the microglia polarization phenotype, are needed to develop treatments based on the regulation of microglia polarization.
The characteristic pathological features of AD include senile plaques, neurogenic fiber tangles, neuroinflammation, and neuronal loss (Figure 2). Deposition of Aβ secreted from neurons and the formation of extracellular senile plaques mark the first wave of damage to neurons. This is followed by the formation of neurogenic fiber tangles (hyperphosphorylated Tau aggregates) within nerves, which eventually leads to neuronal death. The interval between the two waves of damage may be years or even decades. Aβ has been found to induce the aggregation of microglia and infiltration around amyloid plaques. In vitro experiments have demonstrated that Aβ1-42 activates microglia via CD36 and TLR2-TLR6 heterodimers (108), followed by excessive expression of the proinflammatory factors IL-1β, TNF-α, molecularly imprinted polymer-1α, and monocyte chemotactic protein-1 (109). Aβ can also directly interact in a concentration-dependent manner with the amyloid precursor protein (APP), resulting in the activation of microglia and secretion of the inflammatory factor TNF-α (110). In addition, NO production by activated microglia can lead to nitration of Aβ peptide 10Y, a modification that promotes the formation of age spots (111). Activated microglia produce pro-inflammatory cytokines (e.g., TNF-α) and increase Aβ deposition by upregulating β-secretase, a rate-limiting enzyme for Aβ production (112). In another study, activated microglia and damaged neurons were found to release high mobility group protein B1 (HMBG1), which, in turn, was found to bind to Aβ to form Aβ oligomers, inhibit the clearance of Aβ by microglia, and increase the release of pro-inflammatory factors and oxygen radicals to aggravate neuroinflammation (113). Furthermore, Tau hyperphosphorylation and aggregation are driven by microglial activation (114, 115). Specifically, elevated IL-1 caused by CX3CR1 depletion in microglia leads to increased Tau hyperphosphorylation (116). Together, these studies strongly suggest that microglia activation may be an important link between the two waves of neuronal cell injury, i.e., Aβ deposition and hyperphosphorylated Tau protein aggregation, in AD.
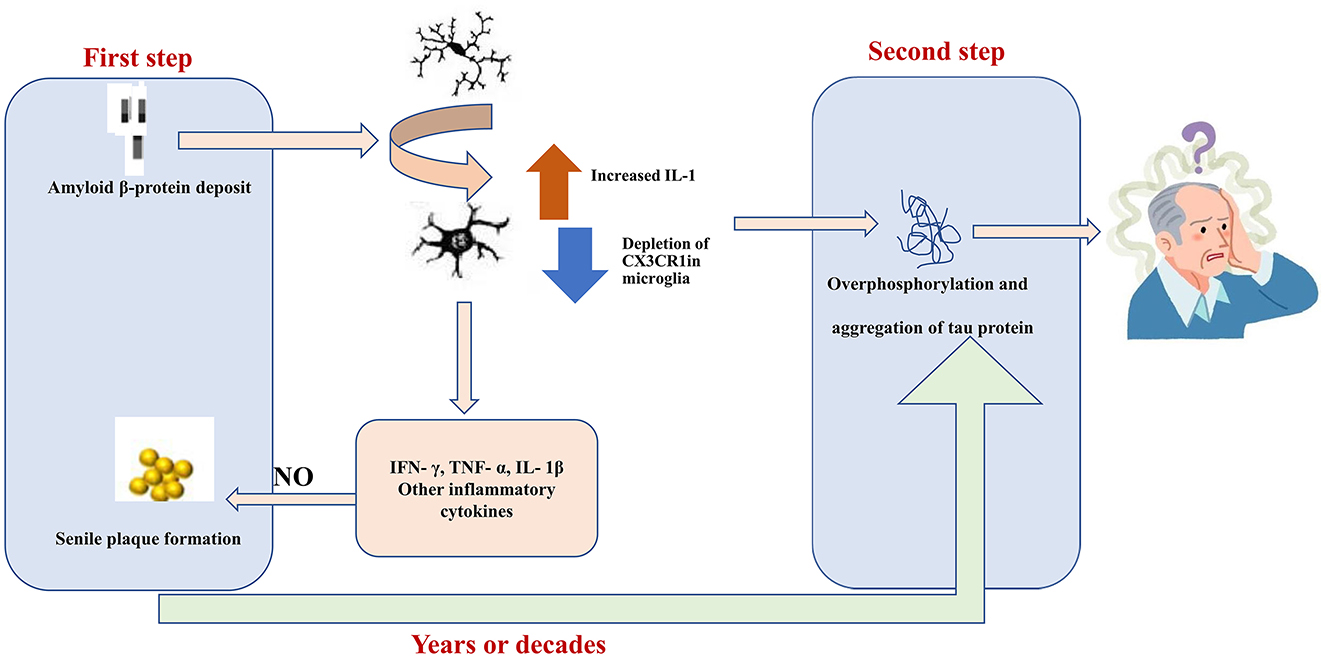
Figure 2. Aβ1-42 activates MG through CD36 and the toll-like receptor TLR2-TLR6 heterodimer. Subsequently, the expression levels of pro-inflammatory factors, interleukin (IL)-1β, and tumor necrosis factor (TNF)-α were significantly increased. In which nitric oxide produced by activated MG can lead to nitration of Aβ peptide 10Y, and this modification promotes the formation of senile plaques. Elevated IL-1 levels in MG caused by depletion of CX3C chemokine receptor 1 (CX3CR1) lead to hyperphosphorylation of tau. Hyperphosphorylated tau aggregates are formed in the nerves and eventually lead to neuronal cell death. The gap between these two waves of destruction could be years or even decades.
The main features of Parkinson's disease (PD) pathologies are selective degeneration and loss of dopamine (DA) neurons in the dense substantia nigra, deposition of α-synuclein (α-Syn), and the presence of Lewy bodies (LB) (117). Currently, there is no effective treatment for PD. Microglia-mediated neuroinflammation plays an important role in the pathogenesis of PD, controlling the survival and death of DA neurons. Activated MG was found in the midbrain substantia nigra dense zone (SNpc) of PD patients by immunofluorescence co-localization, and microglia-produced inflammatory factors cause the loss of DA neurons in the midbrain substantia nigra (118). When microglia are aberrantly activated, it produces inflammation and engulfs connections between brain cells, inducing neuronal death and resulting in brain damage triggering PD. Xia et al. found that microglia exosomes are involved in the α-Sny aggregation in the pathological process (119). Activated microglia releases exosomes that promote the infection of insoluble α-Sny with normal cells and propagate the aggregation of pathological α-Sny, which is accelerated by pro-inflammatory factors released in response to microglia inflammation, and the use of GW4869 (an inhibitor of sphingosine phosphodiesterase) significantly reduces the number of exosomes and decreases α -Sny proliferation and aggregation, leading to the possibility of targeting microglia exosomes for PD treatment (120). Also, age-related changes in the inflammatory response, as well as genetic and environmental factors, may contribute to the prevalence of pro-inflammatory microglia and affect the susceptibility to microglia activation, which may further influence the pathogenesis and lead to progressive neuronal loss.
Gerbino et al. (121) found large numbers of activated microglia and astrocytes in the brainstems and spinal cords of transgenic SOD1 ALS model mice as well as in human ALS patients. Positron emission tomography (PET) imaging also revealed activated microglia in the motor cortex, thalamus, and pons of ALS patients. Activated microglia produce large quantities of pro-inflammatory factors, such as TNF-α, monocyte chemotactic protein-1 (MCP-1), and MMP-9, as well as glutamate. This, in turn, activates glutamate receptors to produce excitotoxic damage to motor neurons. Activated microglia also release large amounts of chemokines and pro-inflammatory factors to activate surrounding astrocytes. The reciprocal interaction between activated microglia and reactive astrocytes forms a positive feedback loop to aggravate neuronal damage (122). Microglia can communicate with other cells by releasing microRNAs (miRNAs) which play a role in the post-transcriptional regulation of gene expression (123). Dysregulation of miRNAs has been observed in ALS patients, accompanied by inflammation and an altered microglial cell phenotype. Christoforidou et al. (124) found that microglia are the most likely source of miRNA dysregulation in ALS patients and correction of miRNA dysregulation through modulation of microglia function may be a new strategy for the treatment of ALS.
Multiple sclerosis is an inflammatory demyelinating disease mainly involving the CNS white matter of the central nervous system, and its pathological features include CNS inflammation, gliosis, demyelination and neurodegeneration (125). The experimental autoimmune encephalomyelitis (EAE) is the ideal animal model for the study of MS, which also exhibited similar pathological characteristics as MS. In EAE mouse model microglia release proteases, pro-inflammatory cytokines, ROS and reactive nitrogen species (RNS), and recruit reactive T lymphocytes, thus producing toxic effects on neurons and oligodendrocyte precursors. Targeted knockout of TGF-β-activated kinase 1 alleviates central nervous system inflammation and axonal and myelin damage (126). Other studies have shown that microglia cells are closely related to active demyelinating lesions (127). In fact, microglial activation can be detected before MS lesions damage the myelin sheath. Both white matter demyelination and diffuse axon injury have been shown to be associated with microglial activation. In addition, activation of microglia can be detected in the white matter prior to leukocyte infiltration, blood-brain barrier disruption, or demyelination (128). Activated microglia cause myelin loss and axonal damage. In addition, microglia may also affect gray matter. In MS patients, significant microglia activation is consistent with cortical damage in patients (129).
Upon neuropathic pain, nociceptive neurons are excited and release a variety of neurotransmitters (e.g., glutamate, ATP, and substance P) to increase the release of various cellular inflammatory factors such as MMPs, MCP-1/CCL-2, CX3CLI, and Toll receptor agonists (123). These pro-inflammatory factors activate microglia, which, in turn, release a variety of neuroexcitatory and pain-sensitizing substances. The expression of P2X4 receptors on the surface of microglia is regulated at the post-translational level by the CC chemokine receptor chemotactic cytokine receptor 2 (130). In addition, spinal microglia respond to extracellular stimuli through intracellular signaling pathways such as the mitogen-activated protein kinase, p38, and extracellular signal-regulated protein kinases pathways. After long-term repeated action of injurious stimulus signals on microglia, microglia cells will continue to be activated, increasing the degree of M1 polarization, and then evolves into chronic pain. This is one of the reasons why chronic neuralgia is so difficult to cure. Neuroinflammation mediated by microglia M1 polarization and its subsequent production of pro-inflammatory factors plays an important role in mediating neuropathic pain. At the same time, with the regression of inflammation in the later stage of this model, activated microglia began to transform from M1 type to M2 type, promoting the production of anti-inflammatory factors such as IL-4 and IL-10, helping to restore normal nerve function and transform the pain state. Thus, the polarization state of microglia cells is associated with pain. The status can appear dynamic changes in the occurrence and development of chronic neuropathic pain. Reasonable regulation of microglia polarization in the occurrence and development of chronic neuropathic pain is expected to be a potential strategy for the treatment of chronic neuropathic pain.
Molecular targets of classical NSAIDs (i.e., aspirin, ibuprofen, naproxen, diclofenac, and sulforaphane) include COX-1 and COX-2. COX-1 is constitutively expressed in most tissues, including microglia, whereas COX-2 is inducible by inflammatory processes. The selective COX-2 inhibitor celecoxib attenuates LPS-induced systemic inflammation and brain white matter injury in neonatal rats (Table 2); this action is associated with a reduction in the number of activated microglia and astrocytes, a decrease in the IL-1β and TNF-α secretion levels, and inhibition of phosphorylated p38MAPK (139). In the Tg2576 transgenic AD mouse model, long-term administration of ibuprofen attenuates the activation of microglia, the release of IL-1β, axonal dystrophy, and amyloid plaques (140). Although observational epidemiological data show that NSAIDs have protective effects, and there is evidence that anti-inflammatory therapy has biological paradoxical effects, the first large primary prevention study of naproxen and celecoxib [Alzheimer's Disease Anti-inflammatory Prevention Test (ADAPT)] does not support the hypothesis that celecoxib or naproxen prevent adult AD with family history of dementia (141). A 2-year primary prevention trial [effect of naproxen treatment on pre-symptomatic Alzheimer's disease (INTREPAD)] showed that naproxen had no benefit in slowing the progression of pre-symptomatic AD compared with placebo (142). In addition to the gastrointestinal and cardiovascular safety issues most often mentioned by COX-2 selective inhibitors, other drug side effects caused by its inhibition of COX-2 but not COX-1 have also gradually attracted clinical attention. This reduces the therapeutic advantage of COX-2 selective inhibitors.
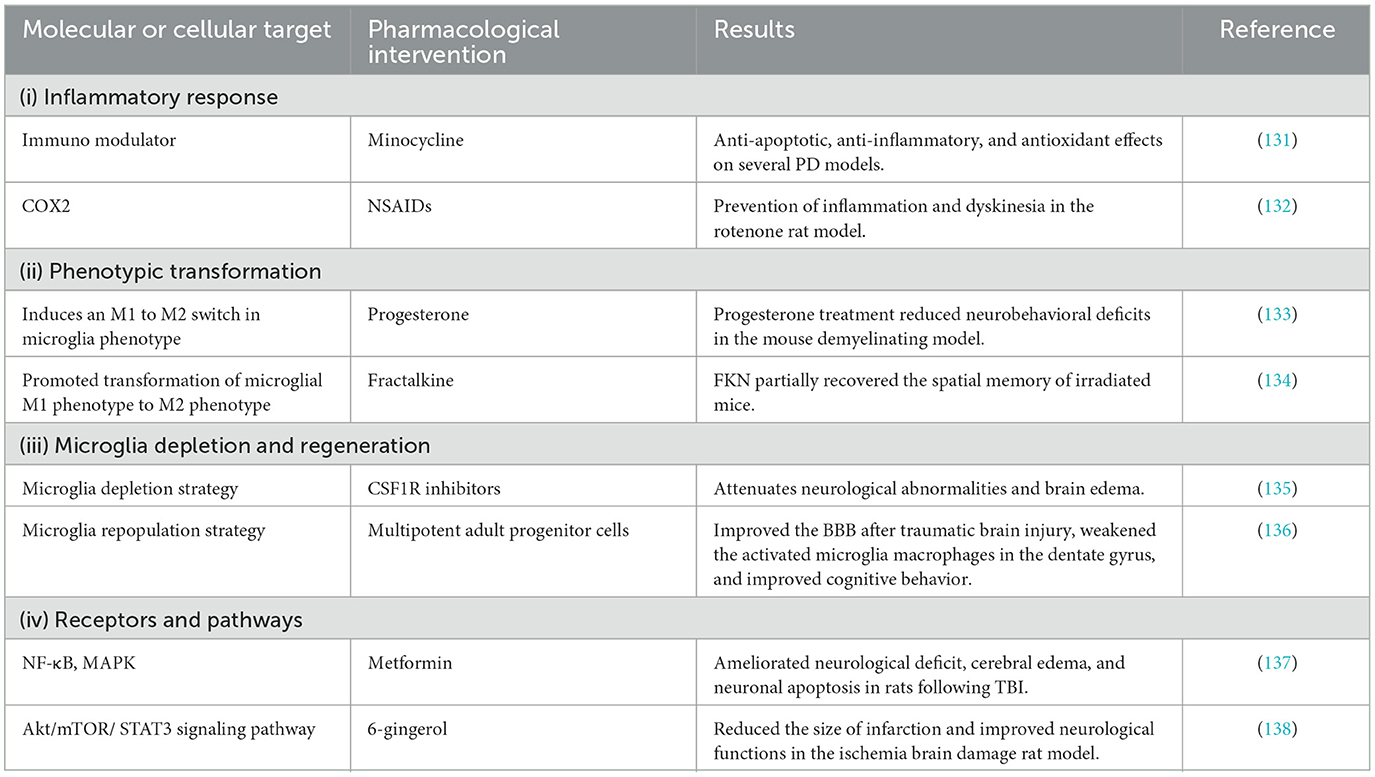
Table 2. Therapeutic strategy targeting microglia for CNS disorders.
Minocycline and related tetracyclines possess anti-inflammatory activity and neuroprotective effects in several models of CNS disorders. In a transgenic McGill-Thy1-APP mice for Alzheimer's disease, early treatment with minocycline was found to reduce iNOS and COX-2 expression (143), protect hippocampal neurogenesis (144), and improve cognition (145). However, inhibition of microglial activation failed to improve cognition if minocycline treatment was initiated after the onset of Aβ deposition (146). The neuroprotective effect of minocycline on animals has initiated the study of its clinical efficacy in AD and PD patients, but the results are uncertain. In addition, the safety data of long-term use of doxycycline and minocycline in these patients are insufficient. Safety issues should be considered from two levels. The fact that AD/PD patients (especially the changes of intestinal flora caused by antibiotics and their consequences), as well as AD and PD are universal incurable diseases that require long-term antibiotic treatment every day, constitutes a worldwide threat of bacterial resistance to these antibiotics (147).
Plant-derived natural compounds are a major source of novel neuroprotective agents. Such compounds include flavonoids, glycosides, phenolics, terpenoids, quinones, alkaloids, lignans, coumarins, chalcone, stilbene, and others (biphenyl, phenylpropanoid, oxy carotenoid). These compounds can reduce the expression of neurotoxic mediators (NO, PGE2, iNOS, and COX-2) and pro-inflammatory cytokines (IL-6, TNF-α, and IL-1β), down-regulate inflammatory markers, and prevent neural damage (148). Hyperoside, a natural flavonoid glycoside, has anti-neuroinflammatory properties. In MPTP-induced PD mice, hyperoside inhibited the activation of glia and reduced the secretion of inflammatory factors, protecting DA neurons and reversing the motor dysfunction caused by MPTP (149). Morin is a natural flavonol extracted from plants from the Moraceae family. It can reduce the expression of TNF-α, IL-1β, IL-6, and increased the expression of brain-derived neurotrophic factor in the hippocampus of Aβ1-42-injected rats, attenuated memory deficits in a rat model of Alzheimer's disease (150). Although various preclinical studies have been conducted on the role of natural compounds in neurodegenerative diseases, little is known about their exact effects in humans, and further clinical trials are needed to confirm the neuroprotective efficacy of this natural compound and to assess its safety (151). At the same time, natural compounds have some limitations, such as their pharmacokinetic properties and stability, which hinder their clinical development and use as drugs.
In many studies, anti-inflammatory drugs (e.g., aspirin, celecoxib, and naproxen) have failed to prevent or treat neurodegenerative diseases (152–154), suggesting that inhibition of M1 microglia alone is not sufficient; concomitant M2 microglia activation may be needed. In a mouse model of demyelination induced by a neurotoxic copper reagent (CPZ), progesterone reduced the level of demyelination and had an anti-inflammatory role in CNS demyelination by inducing M2 microglia polarization and suppressing the M1 phenotype through the inhibition of NF-κB and NLRP3 inflammasome (155). The latest studies in animal stroke models demonstrate that curcumin can exerts their anti-inflammatory effects via attenuation of brain proinflammatory microglia M1 polarization while promoting anti-inflammatory microglial M2 polarization. As a result, stroked animals treated with curcumin have significantly fewer brain active M1 microglia, smaller brain infarct volume, better functional recovery, and better survival rate (156). Semaglutide, a long-acting glucagon-like peptide-1 (GLP-1) receptor agonist, was found to inhibit the neuroinflammatory response and promote neural repair by promoting microglia conversion to the M2-like phenotype and inhibiting M1-like phenotype conversion (possibly mediated by the NF-κB pathway p65) in rats with cerebral ischemia-reperfusion (157). Although these targeted microglia therapies can improve the disease symptoms to a certain extent, they do not achieve precise treatment effects due to the heterogeneity of microglia. Thus, techniques and methods to target functionally-specific microglia to achieve precise disease treatment must be explored in the future.
Accumulating evidence suggests that microglia depletion may represent a therapeutic approach for the prevention and treatment of CNS disorders that involve neuroinflammation. The CSF1R inhibitor PLX3397 can deplete microglia and attenuate brain injury and neurological deficits in an animal model of hemorrhagic stroke (158). Interestingly, microglia regeneration has been shown to promote hippocampal neural regeneration and improve spatial learning through the IL-6 signaling pathway in the early stages of TBI (69). In the CaM/Tet-DTA mouse model of induced neuronal loss, microglia depletion and then regeneration promotes functional recovery (159). Thus, in preclinical studies, depletion of microglia by removing the trophic factors required to maintain them, and subsequent repopulation with a more beneficial microglia phenotype, may produce therapeutic effects. The key problem with this approach is the unknown risk of microglial depletion and the ethical concerns of repopulating microglia in the CNS. In addition, it has been found that depletion and regeneration of microglia can induce the increase of gray matter microglia, neuronal death in somatosensory cortex and atax-like behavior.
Activation of microglia and astrocytes exacerbates glyoxylate deprivation in rats with ischemia-reperfusion injury to the brain; P2X7 receptor antagonists can attenuate memory deficits and inflammatory factor production in the hippocampus and improve animal survival (160). TLR4 gene mutation blocks the activation and release of pro-inflammatory factors by microglia (161). Metformin inhibits the translocation of NF-κB p65 from the cytoplasm into the nucleus and the phosphorylation of ERK1/2 and p38 MAPK inhibits microglia activation and reduces the production of pro-inflammatory cytokines to improve neurobehavioral function in an animal model of TBI (137). The 20-hydroxyeicosatetraenoic acid (20-HETE) inhibitor HET0016 inhibits microglia activation and reduces the release of pro-inflammatory factors through the NF-?B signaling pathway, demonstrating promising protective effects against TBI in the developing brain (162). It is generally believed that the anti-inflammatory effect of α7 nAChR is mediated by NF-κB and JAK2/STAT3 (163). Nicotine can significantly decrease the levels of TNF-α and IL-1β in the hippocampal CA1 region of ischemic stroke rats by up-regulating α7 nAChR and inhibiting microglia inflammation (164). The signal pathways and molecular mechanisms involved in the activation and polarization of microglia are complex and diverse. The activation of microglia can be regulated by targeting different pathways and molecules. However, the exact mechanism of microglial activation pathway, especially the molecular basis of signal transduction in the process of activation, is not completely clear. At the same time, different signal pathways are also linked and can be regulated. These results in that the treatment strategy of treating related diseases by interfering with microglia activated receptors and signal pathways cannot be fully effective.
Despite the accumulating preclinical evidence suggesting that the targeting of microglia could potentially be applied in the treatment of CNS disorders, numerous obstacles remain before this approach can be translated into clinical use. First, the phenotypic switch of microglia exhibits complex and varying spatiotemporal patterns during the course of CNS disorders. Second, the mechanisms underlying the regulation of microglia activation and phenotypic switch are implicated in many other biological processes and functions. Third, the tools used in preclinical studies to selectively manipulate microglia activation are often based on genetic approaches (e.g., knockout and small interfering RNAs), and thus, lack specificity. Fourth, the impacts of age and sex differences on microglia function have been largely overlooked in preclinical studies. Fifth, preclinical animal models often do not have solid construct validity and do not capture the heterogeneity of human disease. Finally, alteration of the microglia phenotype, loss of neuroprotective function, and acquisition of neurotoxic function are complex and may vary with the stage and severity of CNS disorders.
In recent years, there has been increasing interest in studying the activation of microglia cells because of their potential role in the development of neurodegenerative diseases and related impairment disorders in these patients. This paper reviews the role of microglia in neurological diseases and injuries. As the main innate immune cells of the central nervous system, microglia cells are important immune cells in the CNS, playing a variety of functions such as proinflammatory, anti-inflammatory, chemotactic, phagocytotic and neuroprotective in nervous system diseases and injuries.
At present, With the development of neuroimaging, three-dimensional modeling, genomics, transcriptomics, proteomics and other technologies, combined with the study of temporal dynamics, spatial microenvironments and complex signal networks, we will have a more comprehensive and in-depth understanding of the role of microglia. This provides insight into the different distribution of microglia activation phenotypes at the injury site and the time course by which inflammation spreads to other areas of the brain, such as the thalamus.
The dual role of microglia cells often brings negative effects and is not conducive to the repair of the nervous system after injury. Recent studies have shown that simple inhibition of microglia activation has only a limited beneficial effect, and inhibition of M1-like responses can be harmful (165). Furthermore, as seen in clinical trials of AD, pharmacological treatments of anti-inflammatory agents tested in several models may not have similar effects when administered in patients with TBI, thus limiting the potential therapeutic effect (166). Therefore, it will become the focus of future research to continue to explore the rules and mechanisms of microglia activation and explore new ways to control the changes in microglia cell function. At the same time, the current studies are based on animal models, and it is unclear whether the characteristics of rodent cells apply to humans. Moreover, the controversy about the classification of microglia cells is fierce at the present stage, so strengthening the understanding of microglia cells and rational typing of them will also become the focus and hot spot of future research. Therefore, future studies must consider microglial cell polarization as a therapeutic strategy, evaluating several markers in target cells within a sufficient time window to show long-term positive results.
JQ wrote the main manuscript text. XC and ZM prepared table and figures. SS did language modification. All authors reviewed the manuscript.
This work was supported by National Natural Science Foundation of China (No. 82071377), Chongqing Science and Technology Commission grants of China (No. cstc2015jcyjA10095), and Chongqing medical scientific research project (Joint project of Chongqing Health Commission and Science and Technology Bureau) (No. 2022GDRCO02). The study was partly supported by The sixth batch of Young and Middle-aged High-level Medical Talents Training Project of Chongqing Municipal Health Commission and Kuanren Talents Program of the Second Affiliated Hospital of Chongqing Medical University, Chongqing, China.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
1. Almad AA, Maragakis NJ. Glia: an emerging target for neurological disease therapy. Stem Cell Res Ther. (2012) 3:37. doi: 10.1186/scrt128
2. Pérez-Cerdá F, Sánchez-Gómez MV, Matute C. Pío del Río Hortega and the discovery of the oligodendrocytes. Front Neuroanat. (2015) 9:92. doi: 10.3389/fnana.2015.00092
3. Arcuri C, Mecca C, Bianchi R, Giambanco I, Donato R. The Pathophysiological Role of Microglia in Dynamic Surveillance, Phagocytosis and Structural Remodeling of the Developing CNS. Front Mol Neurosci. (2017) 10:191. doi: 10.3389/fnmol.2017.00191
4. Cornell J, Salinas S, Huang HY, Zhou M. Microglia regulation of synaptic plasticity and learning and memory. Neural Regen Res. (2022) 17:705–16. doi: 10.4103/1673-5374.322423
5. Fleiss B, Van Steenwinckel J, Bokobza CI KS, Ross-Munro E, Gressens P. Microglia-mediated neurodegeneration in perinatal brain injuries. Biomolecules. (2021) 11:11. doi: 10.3390/biom11010099
6. Song GJ, Suk K. Pharmacological Modulation of Functional Phenotypes of Microglia in Neurodegenerative Diseases. Front Aging Neurosci. (2017) 9:139. doi: 10.3389/fnagi.2017.00139
7. Masuda T, Sankowski R, Staszewski O, Böttcher C, Amann L, Sagar C, et al. Kunz Spatial and temporal heterogeneity of mouse and human microglia at single-cell resolution. Nature. (2019) 566:388–92. doi: 10.1038/s41586-019-0924-x
8. Kierdorf K, Erny D, Goldmann T, Sander V, Schulz C, Perdiguero EG, et al. Microglia emerge from erythromyeloid precursors via Pu.1- and Irf8-dependent pathways. Nat Neurosci. (2013) 16:273–80. doi: 10.1038/nn.3318
9. Ginhoux F, Greter M, Leboeuf M, Nandi S, See P, Gokhan S, et al. Fate mapping analysis reveals that adult microglia derive from primitive macrophages. Science. (2010) 330:841–5. doi: 10.1126/science.1194637
10. Li Q, Barres BA. Microglia and macrophages in brain homeostasis and disease. Nat Rev Immunol. (2018) 18:225–42. doi: 10.1038/nri.2017.125
11. Xiong XY, Liu L, Yang QW. Functions and mechanisms of microglia/macrophages in neuroinflammation and neurogenesis after stroke. Prog Neurobiol. (2016) 142:23–44. doi: 10.1016/j.pneurobio.2016.05.001
12. Colonna M, Butovsky O. Microglia Function in the Central Nervous System During Health and Neurodegeneration. Annu Rev Immunol. (2017) 35:441–68. doi: 10.1146/annurev-immunol-051116-052358
13. Ransohoff RM. A polarizing question: do M1 and M2 microglia exist? Nat Neurosci. (2016) 19:987–91. doi: 10.1038/nn.4338
14. Sousa C, Golebiewska A, Poovathingal SK, Kaoma T, Pires-Afonso Y, Martina S, et al. Single-cell transcriptomics reveals distinct inflammation-induced microglia signatures. EMBO Rep. (2018) 19:e46171. doi: 10.15252/embr.201846171
15. Xue J, Schmidt SV, Sander J, Draffehn A, Krebs W, Quester I, et al. Transcriptome-based network analysis reveals a spectrum model of human macrophage activation. Immunity. (2014) 40:274–88. doi: 10.1016/j.immuni.2014.01.006
16. Hammond TR, Dufort C, Dissing-Olesen L, Giera S, Young A, Wysoker A, et al. Single-cell RNA sequencing of microglia throughout the mouse lifespan and in the injured brain reveals complex cell-state changes. Immunity. (2019) 50:253–271.e6. doi: 10.1016/j.immuni.2018.11.004
17. Olah M, Menon V, Habib N, Taga MF, Ma Y, Yung CJ, et al. Single cell RNA sequencing of human microglia uncovers a subset associated with Alzheimer's disease. Nat Commun. (2020) 11:6129. doi: 10.1038/s41467-020-19737-2
18. Salamanca L, Mechawar N, Murai KK, Balling R, Bouvier DS, Skupin A, et al. MIC-MAC: An automated pipeline for high-throughput characterization and classification of three-dimensional microglia morphologies in mouse and human postmortem brain samples. Glia. (2019) 67:1496–509. doi: 10.1002/glia.23623
19. Chiu IM, Morimoto ET, Goodarzi H, Liao JT, O'Keeffe S, Phatnani HP, et al. A neurodegeneration-specific gene-expression signature of acutely isolated microglia from an amyotrophic lateral sclerosis mouse model. Cell Rep. (2013) 4:385–401. doi: 10.1016/j.celrep.2013.06.018
20. Keren-Shaul H, Spinrad A, Weiner A, Matcovitch-Natan O, Dvir-Szternfeld R, Ulland TK, et al. A unique microglia type associated with restricting development of Alzheimer's disease. Cell. (2017) 169:1276–1290.e17. doi: 10.1016/j.cell.2017.05.018
21. Schafer DP, Stevens B. Microglia function in central nervous system development and plasticity. Cold Spring Harb Perspect Biol. (2015) 7:a020545. doi: 10.1101/cshperspect.a020545
22. Trang T, Beggs S, Salter MW. Brain-derived neurotrophic factor from microglia: a molecular substrate for neuropathic pain. Neuron Glia Biol. (2011) 7:99–108. doi: 10.1017/S1740925X12000087
23. Ueno M, Fujita Y, Tanaka T, Nakamura Y, Kikuta J, Ishii M, et al. Layer V cortical neurons require microglial support for survival during postnatal development. Nat Neurosci. (2013) 16:543–51. doi: 10.1038/nn.3358
24. Prinz M, Priller J. Microglia and brain macrophages in the molecular age: from origin to neuropsychiatric disease. Nat Rev Neurosci. (2014) 15:300–12. doi: 10.1038/nrn3722
25. Paolicelli RC, Bolasco G, Pagani F, Maggi L, Scianni M, Panzanelli P, et al. Synaptic pruning by microglia is necessary for normal brain development. Science. (2011) 333:1456–8. doi: 10.1126/science.1202529
26. Bilbo S, Stevens B. Microglia: The Brain's First Responders. In: Cerebrum: the Dana forum on brain science. Dana Foundation. (2017).
27. Tremblay M, Stevens B, Sierra A, Wake H, Bessis A, Nimmerjahn A, et al. The role of microglia in the healthy brain. J Neurosci. (2011) 31:16064–9. doi: 10.1523/JNEUROSCI.4158-11.2011
28. Shabab T, Khanabdali R, Moghadamtousi SZ, Kadir HA, Mohan G. Neuroinflammation pathways: a general review. Int J Neurosci. (2017) 127:624–33. doi: 10.1080/00207454.2016.1212854
29. Kacimi R, Giffard RG, Yenari MA. Endotoxin-activated microglia injure brain derived endothelial cells via NF-κB, JAK-STAT and JNK stress kinase pathways. J Inflamm (Lond). (2011) 8:7. doi: 10.1186/1476-9255-8-7
30. Capiralla H, Vingtdeux V, Zhao H, Sankowski R, Al-Abed Y, Davies P, et al. Resveratrol mitigates lipopolysaccharide- and Aβ-mediated microglial inflammation by inhibiting the TLR4/NF-κB/STAT signaling cascade. J Neurochem. (2012) 120:461–72. doi: 10.1111/j.1471-4159.2011.07594.x
31. Veroni C, Gabriele L, Canini I, Castiello L, Coccia E, Remoli ME, et al. Activation of TNF receptor 2 in microglia promotes induction of anti-inflammatory pathways. Mol Cell Neurosci. (2010) 45:234–44. doi: 10.1016/j.mcn.2010.06.014
32. Lotocki G, Alonso OF, Dietrich WD, Keane RW. Tumor necrosis factor receptor 1 and its signaling intermediates are recruited to lipid rafts in the traumatized brain. J Neurosci. (2004) 24:11010–6. doi: 10.1523/JNEUROSCI.3823-04.2004
33. Rowe RK, Harrison JL, Zhang H, Bachstetter AD, Hesson DP, O'Hara BF, et al. Novel TNF receptor-1 inhibitors identified as potential therapeutic candidates for traumatic brain injury. J Neuroinflammation. (2018) 15:154. doi: 10.1186/s12974-018-1200-y
34. Domercq M, Zabala A, Matute C. Purinergic receptors in multiple sclerosis pathogenesis. Brain Res Bull. (2019) 151:38–45. doi: 10.1016/j.brainresbull.2018.11.018
35. Sasaki Y, Hoshi M, Akazawa C, Nakamura Y, Tsuzuki H, Inoue K, et al. Selective expression of Gi/o-coupled ATP receptor P2Y12 in microglia in rat brain. Glia. (2003) 44:242–50. doi: 10.1002/glia.10293
36. Madry C, Kyrargyri V, Arancibia-Cárcamo IL, Jolivet R, Kohsaka S, Bryan RM, et al. Microglial ramification, surveillance, and interleukin-1β release are regulated by the two-pore domain K(+) channel THIK-1. Neuron. (2018) 97:299–312.e6. doi: 10.1016/j.neuron.2017.12.002
37. Liu M, Yao M, Wang H, Xu L, Zheng Y, Huang B, et al. P2Y(12) receptor-mediated activation of spinal microglia and p38MAPK pathway contribute to cancer-induced bone pain. J Pain Res. (2017) 10:417–26. doi: 10.2147/JPR.S124326
38. Ren WJ, Illes P. Involvement of P2X7 receptors in chronic pain disorders. Purinergic Signal. (2022) 18:83–92. doi: 10.1007/s11302-021-09796-5
39. Monif M, Burnstock G, Williams DA. Microglia: proliferation and activation driven by the P2X7 receptor. Int J Biochem Cell Biol. (2010) 42:1753–6. doi: 10.1016/j.biocel.2010.06.021
40. Girard S, Sébire G, Kadhim H. Proinflammatory orientation of the interleukin 1 system and downstream induction of matrix metalloproteinase 9 in the pathophysiology of human perinatal white matter damage. J Neuropathol Exp Neurol. (2010) 69:1116–29. doi: 10.1097/NEN.0b013e3181f971e4
41. Wang KC, Wang SJ, Fan LW, Cai Z, Rhodes PG, Tien LT, et al. Interleukin-1 receptor antagonist ameliorates neonatal lipopolysaccharide-induced long-lasting hyperalgesia in the adult rats. Toxicology. (2011) 279:123–9. doi: 10.1016/j.tox.2010.10.002
42. Boraschi D, Italiani P, Weil S, Martin MU. The family of the interleukin-1 receptors. Immunol Rev. (2018) 281:197–232. doi: 10.1111/imr.12606
43. Hashioka S, Klegeris A, Schwab C, Yu S, McGeer PL. Differential expression of interferon-gamma receptor on human glial cells in vivo and in vitro. J Neuroimmunol. (2010) 225:91–9. doi: 10.1016/j.jneuroim.2010.04.023
44. Zhang J, He H, Qiao Y, Zhou T, He H, Yi S, et al. Priming of microglia with IFN-γ impairs adult hippocampal neurogenesis and leads to depression-like behaviors and cognitive defects. Glia. (2020) 68:2674–92. doi: 10.1002/glia.23878
45. Fiore NT, Yin Z, Guneykaya D, Gauthier CD, Hayes JP, D'Hary A, et al. Sex-specific transcriptome of spinal microglia in neuropathic pain due to peripheral nerve injury. Glia. (2022) 70:675–96. doi: 10.1002/glia.24133
46. Honda A, Kamata S, Akahane M, Machida Y, Uchii K, Shiiyama Y, et al. Functional and structural insights into human PPARα/δ/γ subtype selectivity of bezafibrate. Fenofibric Acid, and Pemafibrate. Int J Mol Sci. (2022) 23:4726. doi: 10.3390/ijms23094726
47. Zhao XR, Gonzales N, Aronowski J. Pleiotropic role of PPARγ in intracerebral hemorrhage: an intricate system involving Nrf2. RXR, and NF-κB, CNS. Neurosci Ther. (2015) 21:357–66. doi: 10.1111/cns.12350
48. Pan J, Jin JL, Ge HM, Yin KL, Chen X, Han LJ, et al. Malibatol A regulates microglia M1/M2 polarization in experimental stroke in a PPARγ-dependent manner. J Neuroinflammation. (2015) 12:51. doi: 10.1186/s12974-015-0270-3
49. Bougarne N, Weyers B, Desmet SJ, Deckers J, Ray DW, Staels B, et al. Molecular Actions of PPARα in lipid metabolism and inflammation. Endocr Rev. (2018) 39:760–802. doi: 10.1210/er.2018-00064
50. Wohleb ES, Hanke ML, Corona AW, Powell ND, Stiner LM, Bailey MT, et al. β-Adrenergic receptor antagonism prevents anxiety-like behavior and microglial reactivity induced by repeated social defeat. J Neurosci. (2011) 31:6277–88. doi: 10.1523/JNEUROSCI.0450-11.2011
51. Umeda K, Goto Y, Watanabe K, Ushio N, Fereig RM, Ihara F, et al. Transcriptomic Analysis of the Effects of Chemokine Receptor CXCR3 Deficiency on Immune Responses in the Mouse Brain during Toxoplasma gondii Infection. Microorganisms. (2021) 9:2340. doi: 10.3390/microorganisms9112340
52. Pagie S, Gérard N, Charreau B. Notch signaling triggered via the ligand DLL4 impedes M2 macrophage differentiation and promotes their apoptosis. Cell Commun Signal. (2018) 16:4. doi: 10.1186/s12964-017-0214-x
53. Yao L, Cao Q, Wu C, Kaur C, Hao A, Ling EA, et al. Notch signaling in the central nervous system with special reference to its expression in microglia. CNS Neurol Disord Drug Targets. (2013) 12:807–14. doi: 10.2174/18715273113126660172
54. Park J, Jang KM, Park KK. Apamin Suppresses LPS-Induced Neuroinflammatory Responses by Regulating SK Channels and TLR4-Mediated Signaling Pathways. Int J Mol Sci. (2020) 21:4319. doi: 10.3390/ijms21124319
55. Zhang BY, Wang GR, Ning WH, Liu J, Yang S, Shen Y, et al. Tolerance to Cerebral Ischemia/Reperfusion through Inhibition of the GluN2B/m-Calpain/p38 MAPK Proapoptotic Pathway. Neural Plast. (2020) 2020:8840675. doi: 10.1155/2020/8840675
56. Dong H, Cui B, Hao X. MicroRNA-22 alleviates inflammation in ischemic stroke via p38 MAPK pathways. Mol Med Rep. (2019) 20:735–44. doi: 10.3892/mmr.2019.10269
57. Liu DR, Hu W, Chen GZ. Apelin-12 exerts neuroprotective effect against ischemia-reperfusion injury by inhibiting JNK and P38MAPK signaling pathway in mouse. Eur Rev Med Pharmacol Sci. (2018) 22:3888–95. doi: 10.26355/eurrev_201806_15273
58. Akaishi T, Yamamoto S, Abe K. The Synthetic Curcumin Derivative CNB-001 Attenuates Thrombin-Stimulated Microglial Inflammation by Inhibiting the ERK and p38 MAPK Pathways. Biol Pharm Bull. (2020) 43:138–44. doi: 10.1248/bpb.b19-00699
59. Liu M, Jiang L, Wen M, Ke Y, Tong X, Huang W, et al. Microglia depletion exacerbates acute seizures and hippocampal neuronal degeneration in mouse models of epilepsy. Am J Physiol Cell Physiol. (2020) 319:C605–c610. doi: 10.1152/ajpcell.00205.2020
60. Van der Heiden K, Cuhlmann S, Luong le A, Zakkar M, Evans PC. Role of nuclear factor kappaB in cardiovascular health and disease. Clin Sci. (2010) 118:593–605. doi: 10.1042/CS20090557
61. Zhuang P, Wan Y, Geng S, He Y, Feng B, Ye Z, et al. Salvianolic Acids for Injection (SAFI) suppresses inflammatory responses in activated microglia to attenuate brain damage in focal cerebral ischemia. J Ethnopharmacol. (2017) 198:194–204. doi: 10.1016/j.jep.2016.11.052
62. Hao T, Yang Y, Li N, Mi Y, Zhang G, Song J, et al. Inflammatory mechanism of cerebral ischemia-reperfusion injury with treatment of stepharine in rats. Phytomedicine. (2020) 79:153353. doi: 10.1016/j.phymed.2020.153353
63. Wu M, Liu F, Guo Q. Quercetin attenuates hypoxia-ischemia induced brain injury in neonatal rats by inhibiting TLR4/NF-κB signaling pathway. Int Immunopharmacol. (2019) 74:105704. doi: 10.1016/j.intimp.2019.105704
64. Chen CJ, Ou YC, Chang CY, Pan HC, Lin SY, Liao SL, et al. Src signaling involvement in Japanese encephalitis virus-induced cytokine production in microglia. Neurochem Int. (2011) 58:924–33. doi: 10.1016/j.neuint.2011.02.022
65. Choi Y, Park NJ, Le TM, Lee E, Lee D, Nguyen HDT, et al. Immune Pathway and Gene Database (IMPAGT) Revealed the Immune Dysregulation Dynamics and Overactivation of the PI3K/Akt Pathway in Tumor Buddings of Cervical Cancer. Curr Issues Mol Biol. (2022) 44:5139–52. doi: 10.3390/cimb44110350
66. Chu E, Mychasiuk R, Hibbs ML, Semple BD. Dysregulated phosphoinositide 3-kinase signaling in microglia: shaping chronic neuroinflammation. J Neuroinflammation. (2021) 18:276. doi: 10.1186/s12974-021-02325-6
67. Xie S, Chen M, Yan B, He X, Chen X, Li D. Identification of a role for the PI3K/AKT/mTOR signaling pathway in innate immune cells. PLoS ONE. (2014) 9:e94496. doi: 10.1371/journal.pone.0094496
68. Hoogland ICM, Westhoff D, Engelen-Lee JY, Melief J, Valls Serón M, Houben-Weerts J, et al. Microglial Activation After Systemic Stimulation With Lipopolysaccharide and Escherichia coli. Front Cell Neurosci. (2018) 12:110. doi: 10.3389/fncel.2018.00110
69. Willis EF, MacDonald KPA, Nguyen QH, Garrido AL, Gillespie ER, Harley SBR, et al. Repopulating Microglia Promote Brain Repair in an IL-6-Dependent Manner. Cell. (2020) 180:833–846.e16. doi: 10.1016/j.cell.2020.02.013
70. Zhou S, Guo X, Chen S, Xu Z, Duan W, Zeng B, et al. Apelin-13 regulates LPS-induced N9 microglia polarization involving STAT3 signaling pathway. Neuropeptides. (2019) 76:101938. doi: 10.1016/j.npep.2019.101938
71. Chen H, Wang Z, Gao X, Lv J, Hu Y, Jung YS, et al. ASFV pD345L protein negatively regulates NF-κB signalling by inhibiting IKK kinase activity. Vet Res. (2022) 53:32. doi: 10.1186/s13567-022-01050-z
72. Sun SC. The non-canonical NF-κB pathway in immunity and inflammation. Nat Rev Immunol. (2017) 17:545–58. doi: 10.1038/nri.2017.52
73. Zhang X, Kracht L, Lerario AM, Dubbelaar ML, Brouwer N, Wesseling EM, et al. Epigenetic regulation of innate immune memory in microglia. J Neuroinflammation. (2022) 19:111. doi: 10.1186/s12974-022-02463-5
74. Chen QY, Costa M. PI3K/Akt/mTOR Signaling Pathway and the Biphasic Effect of Arsenic in Carcinogenesis. Mol Pharmacol. (2018) 94:784–92. doi: 10.1124/mol.118.112268
75. Gao Y, Gartenhaus RB, Lapidus RG, Hussain A, Zhang Y, Wang X, et al. Differential IKK/NF-κB activity is mediated by TSC2 through mTORC1 in PTEN-Null prostate cancer and tuberous sclerosis complex tumor cells. Mol Cancer Res. (2015) 13:1602–14. doi: 10.1158/1541-7786.MCR-15-0213
76. Hou YC, Chiu WC, Yeh CL, Yeh SL. Glutamine modulates lipopolysaccharide-induced activation of NF-κB via the Akt/mTOR pathway in lung epithelial cells. Am J Physiol Lung Cell Mol Physiol. (2012) 302:L174–83. doi: 10.1152/ajplung.00066.2011
77. Matsumoto J, Dohgu S, Takata F, Machida T, Bölükbaşi Hatip FF, Hatip-Al-Khatib I, et al. TNF-α-sensitive brain pericytes activate microglia by releasing IL-6 through cooperation between IκB-NFκB and JAK-STAT3 pathways. Brain Res. (2018) 1692:34–44. doi: 10.1016/j.brainres.2018.04.023
78. Yenari MA, Kauppinen TM, Swanson RA. Microglial activation in stroke: therapeutic targets. Neurotherapeutics. (2010) 7:378–91. doi: 10.1016/j.nurt.2010.07.005
79. Li QQ, Ding DH, Wang XY, Sun YY, Wu J. Lipoxin A4 regulates microglial M1/M2 polarization after cerebral ischemia-reperfusion injury via the Notch signaling pathway. Exp Neurol. (2021) 339:113645. doi: 10.1016/j.expneurol.2021.113645
80. Li S, Hua X, Zheng M, Wu J, Ma Z, Xing X, et al. PLXNA2 knockdown promotes M2 microglia polarization through mTOR/STAT3 signaling to improve functional recovery in rats after cerebral ischemia/reperfusion injury. Exp Neurol. (2021) 346:113854. doi: 10.1016/j.expneurol.2021.113854
81. Ma H, Su D, Wang Q, Chong Z, Zhu Q, He W, et al. Phoenixin 14 inhibits ischemia/reperfusion-induced cytotoxicity in microglia. Arch Biochem Biophys. (2020) 689:108411. doi: 10.1016/j.abb.2020.108411
82. Kooijman E, Nijboer CH, van Velthoven CT, Mol W, Dijkhuizen RM, Kesecioglu J, et al. Long-term functional consequences and ongoing cerebral inflammation after subarachnoid hemorrhage in the rat. PLoS ONE. (2014) 9:e90584. doi: 10.1371/journal.pone.0090584
83. Sun XG, Zhang MM, Liu SY, Chu XH, Xue GQ, Zhang BC, et al. of TREM-1 in the development of early brain injury after subarachnoid hemorrhage. Exp Neurol. (2021) 341:113692. doi: 10.1016/j.expneurol.2021.113692
84. Schneider UC, Davids AM, Brandenburg S, Müller A, Elke A, Magrini S, et al. Microglia inflict delayed brain injury after subarachnoid hemorrhage. Acta Neuropathol. (2015) 130:215–31. doi: 10.1007/s00401-015-1440-1
85. Johnson VE, Stewart JE, Begbie FD, Trojanowski JQ, Smith DH, Stewart W. Inflammation and white matter degeneration persist for years after a single traumatic brain injury. Brain. (2013) 136:28–42. doi: 10.1093/brain/aws322
86. Jassam YN, Izzy S, Whalen M, McGavern DB, El Khoury J. Neuroimmunology of Traumatic Brain Injury: Time for a Paradigm Shift. Neuron. (2017) 95:1246–65. doi: 10.1016/j.neuron.2017.07.010
87. Sauerbeck A, Gao J, Readnower R, Liu M, Pauly JR, Bing G, et al. Pioglitazone attenuates mitochondrial dysfunction, cognitive impairment, cortical tissue loss, and inflammation following traumatic brain injury. Exp Neurol. (2011) 227:128–35. doi: 10.1016/j.expneurol.2010.10.003
88. Philips T, Robberecht W. Neuroinflammation in amyotrophic lateral sclerosis: role of glial activation in motor neuron disease. Lancet Neurol. (2011) 10:253–63. doi: 10.1016/S1474-4422(11)70015-1
89. Liu J, Wang F. Role of neuroinflammation in amyotrophic lateral sclerosis: cellular mechanisms and therapeutic implications. Front Immunol. (2017) 8:1005. doi: 10.3389/fimmu.2017.01005
90. Guo S, Wang H, Yin Y. Microglia polarization from M1 to M2 in neurodegenerative diseases. Front Aging Neurosci. (2022) 14:815347. doi: 10.3389/fnagi.2022.815347
91. Yang J, Wise L, Fukuchi KI. TLR4 Cross-Talk With NLRP3 inflammasome and complement signaling pathways in Alzheimer's disease. Front Immunol. (2020) 11:724. doi: 10.3389/fimmu.2020.00724
92. Zhou Y, Song WM, Andhey PS, Swain A, Levy T, Miller KR, et al. Human and mouse single-nucleus transcriptomics reveal TREM2-dependent and TREM2-independent cellular responses in Alzheimer's disease. Nat Med. (2020) 26:131–42. doi: 10.1038/s41591-019-0695-9
93. Tsuda M, Masuda T, Tozaki-Saitoh H, Inoue K. Microglial regulation of neuropathic pain. J Pharmacol Sci. (2013) 121:89–94. doi: 10.1254/jphs.12R14CP
94. Tsuda M, Masuda T, Tozaki-Saitoh H, Inoue K. P2X4 receptors and neuropathic pain. Front Cell Neurosci. (2013) 7:191. doi: 10.3389/fncel.2013.00191
95. Gu W, Zhang W, Lei Y, Cui Y, Chu S, Gu X, et al. Activation of spinal alpha-7 nicotinic acetylcholine receptor shortens the duration of remifentanil-induced postoperative hyperalgesia by upregulating KCC2 in the spinal dorsal horn in rats. Mol Pain. (2017) 13:1744806917704769. doi: 10.1177/1744806917704769
96. Iadecola C, Anrather J. The immunology of stroke: from mechanisms to translation. Nat Med. (2011) 17:796–808. doi: 10.1038/nm.2399
97. Roughton K, Andreasson U, Blomgren K, Kalm M. Lipopolysaccharide-induced inflammation aggravates irradiation-induced injury to the young mouse brain. Dev Neurosci. (2013) 35:406–15. doi: 10.1159/000353820
98. Kitayama M, Ueno M, Itakura T, Yamashita T. Activated microglia inhibit axonal growth through RGMa. PLoS ONE. (2011) 6:e25234. doi: 10.1371/journal.pone.0025234
99. Mijailović NR, Vesic K, Arsenijevic D, Milojević-Rakić M, Borovcanin MM. Galectin-3 involvement in cognitive processes for new therapeutic considerations. Front Cell Neurosci. (2022) 16:923811. doi: 10.3389/fncel.2022.923811
100. Shao Z, Tu S, Shao A. Pathophysiological mechanisms and potential therapeutic targets in intracerebral hemorrhage. Front Pharmacol. (2019) 10:1079. doi: 10.3389/fphar.2019.01079
101. Feng J, Chen X, Guan B, Li C, Qiu J, Shen J, et al. Inhibition of peroxynitrite-induced mitophagy activation attenuates cerebral ischemia-reperfusion injury. Mol Neurobiol. (2018) 55:6369–86. doi: 10.1007/s12035-017-0859-x
102. Song Y, Yang Y, Cui Y, Gao J, Wang K, Cui J, et al. Lipoxin A4 Methyl Ester Reduces Early Brain injury by inhibition of the nuclear factor kappa B (NF-κB)-dependent matrix metallopeptidase 9 (MMP-9) pathway in a rat model of intracerebral hemorrhage. Med Sci Monit. (2019) 25:1838–47. doi: 10.12659/MSM.915119
103. Hu X, Leak RK, Shi Y, Suenaga J, Gao Y, Zheng P, et al. Microglial and macrophage polarization—new prospects for brain repair. Nat Rev Neurol. (2015) 11:56–64. doi: 10.1038/nrneurol.2014.207
104. Butovsky O, Ziv Y, Schwartz A, Landa G, Talpalar AE, Pluchino S, et al. Microglia activated by IL-4 or IFN-gamma differentially induce neurogenesis and oligodendrogenesis from adult stem/progenitor cells. Mol Cell Neurosci. (2006) 31:149–60. doi: 10.1016/j.mcn.2005.10.006
105. Morganti JM, Riparip LK, Rosi S. Call Off the Dog(ma): M1/M2 Polarization Is Concurrent following Traumatic Brain Injury. PLoS ONE. (2016) 11:e0148001. doi: 10.1371/journal.pone.0148001
106. Ramlackhansingh AF, Brooks DJ, Greenwood RJ, Bose SK, Turkheimer FE, Kinnunen KM, et al. Inflammation after trauma: microglial activation and traumatic brain injury. Ann Neurol. (2011) 70:374–83. doi: 10.1002/ana.22455
107. Hosomi S, Ohnishi M, Ogura H, Shimazu T. Traumatic brain injury-related inflammatory projection: beyond local inflammatory responses. Acute Med Surg. (2020) 7:e520. doi: 10.1002/ams2.520
108. Stewart CR, Stuart LM, Wilkinson K, van Gils JM, Deng J, Halle A, et al. CD36 ligands promote sterile inflammation through assembly of a Toll-like receptor 4 and 6 heterodimer. Nat Immunol. (2010) 11:155–61. doi: 10.1038/ni.1836
109. Yoo HJ, Kwon MS. Aged Microglia in Neurodegenerative Diseases: Microglia Lifespan and Culture Methods. Front Aging Neurosci. (2021) 13:766267. doi: 10.3389/fnagi.2021.766267
110. Manocha GD, Floden AM, Rausch K, Kulas JA, McGregor BA, Rojanathammanee L, et al. APP Regulates Microglial Phenotype in a Mouse Model of Alzheimer's Disease. J Neurosci. (2016) 36:8471–86. doi: 10.1523/JNEUROSCI.4654-15.2016
111. Kummer MP, Hermes M, Delekarte A, Hammerschmidt T, Kumar S, Terwel D, et al. Nitration of tyrosine 10 critically enhances amyloid β aggregation and plaque formation. Neuron. (2011) 71:833–44. doi: 10.1016/j.neuron.2011.07.001
112. Cheng F, Fransson L, Mani K. Proinflammatory cytokines induce accumulation of glypican-1-derived heparan sulfate and the C-terminal fragment of β-cleaved APP in autophagosomes of dividing neuronal cells. Glycobiology. (2020) 30:539–49. doi: 10.1093/glycob/cwaa011
113. He W, Yuan K, Ji B, Han Y, Li J. Protective effects of curcumin against neuroin?ammation induced by Aβ25-35 in primary rat microglia: modulation of high-mobility group box 1, toll-like receptor 4 and receptor for advanced glycation end products expression. Ann Transl Med. (2020) 8:88. doi: 10.21037/atm.2019.12.147
114. Lee CY, Landreth GE. The role of microglia in amyloid clearance from the AD brain. J Neural Transm (Vienna). (2010) 117:949–60. doi: 10.1007/s00702-010-0433-4
115. McFarland KN, Chakrabarty P. Microglia in Alzheimer's Disease: a Key Player in the Transition Between Homeostasis and Pathogenesis. Neurotherapeutics. (2022) 19:186–208. doi: 10.1007/s13311-021-01179-3
116. Bhaskar K, Konerth M, Kokiko-Cochran ON, Cardona A, Ransohoff RM, Lamb BT, et al. Regulation of tau pathology by the microglial fractalkine receptor. Neuron. (2010) 68:19–31. doi: 10.1016/j.neuron.2010.08.023
117. Hayes MT. Parkinson's disease and parkinsonism. Am J Med. (2019) 132:802–7. doi: 10.1016/j.amjmed.2019.03.001
118. Zhang YN, Fan JK, Gu L, Yang HM, Zhan SQ, Zhang H, et al. Metabotropic glutamate receptor 5 inhibits α-synuclein-induced microglia inflammation to protect from neurotoxicity in Parkinson's disease. J Neuroinflammation. (2021) 18:23. doi: 10.1186/s12974-021-02079-1
119. Xia Y, Zhang G, Han C, Ma K, Guo X, Wan F, et al. Microglia as modulators of exosomal alpha-synuclein transmission. Cell Death Dis. (2019) 10:174. doi: 10.1038/s41419-019-1404-9
120. Jiang T, Xu C, Gao S, Zhang J, Zheng J, Wu X, et al. Cathepsin L-containing exosomes from α-synuclein-activated microglia induce neurotoxicity through the P2X7 receptor. NPJ Parkinsons Dis. (2022) 8:127. doi: 10.1038/s41531-022-00394-9
121. Gerbino V, Kaunga E, Ye J, Canzio D, O'Keeffe S, Rudnick ND, et al. The Loss of TBK1 kinase activity in motor neurons or in all cell types differentially impacts ALS disease progression in SOD1 Mice. Neuron. (2020) 106:789–805.e5. doi: 10.1016/j.neuron.2020.03.005
122. Pajarillo E, Rizor A, Lee J, Aschner M, Lee E. The role of astrocytic glutamate transporters GLT-1 and GLAST in neurological disorders: Potential targets for neurotherapeutics. Neuropharmacology. (2019) 161:107559. doi: 10.1016/j.neuropharm.2019.03.002
123. Lu Y, Cao DL, Ma LJ, Gao YJ. TRAF6 Contributes to CFA-Induced Spinal Microglial Activation and Chronic Inflammatory Pain in Mice. Cell Mol Neurobiol. (2022) 42:1543–55. doi: 10.1007/s10571-021-01045-y
124. Christoforidou E, Joilin G, Hafezparast M. Potential of activated microglia as a source of dysregulated extracellular microRNAs contributing to neurodegeneration in amyotrophic lateral sclerosis. J Neuroinflammation. (2020) 17:135. doi: 10.1186/s12974-020-01822-4
125. Reich DS, Lucchinetti CF, Calabresi PA. Multiple Sclerosis. N Engl J Med. (2018) 378:169–80. doi: 10.1056/NEJMra1401483
126. Ransohoff RM, El Khoury J. Microglia in health and disease. Cold Spring Harb Perspect Biol. (2015) 8:a020560. doi: 10.1101/cshperspect.a020560
127. Zrzavy T, Hametner S, Wimmer I, Butovsky O, Weiner HL, Lassmann H, et al. Loss of 'homeostatic' microglia and patterns of their activation in active multiple sclerosis. Brain. (2017) 140:1900–13. doi: 10.1093/brain/awx113
128. Chiang GC, Pinto S, Comunale JP, Gauthier SA. Gadolinium-enhancing lesions lead to decreases in white matter tract fractional anisotropy in multiple sclerosis. J Neuroimaging. (2016) 26:289–95. doi: 10.1111/jon.12309
129. Herranz E, Giannì C, Louapre C, Treaba CA, Govindarajan ST, Ouellette R, et al. Neuroinflammatory component of gray matter pathology in multiple sclerosis. Ann Neurol. (2016) 80:776–90. doi: 10.1002/ana.24791
130. Inoue K. Purinergic signaling in microglia in the pathogenesis of neuropathic pain. Proc Jpn Acad Ser B Phys Biol Sci. (2017) 93:174–82. doi: 10.2183/pjab.93.011
131. Cankaya S, Cankaya B, Kilic U, Kilic E, Yulug B. The therapeutic role of minocycline in Parkinson's disease. Drugs Context. (2019) 8:212553. doi: 10.7573/dic.212553
132. Teema AM, Zaitone SA, Moustafa YM. Ibuprofen or piroxicam protects nigral neurons and delays the development of l-dopa induced dyskinesia in rats with experimental Parkinsonism: Influence on angiogenesis. Neuropharmacology. (2016) 107:432–50. doi: 10.1016/j.neuropharm.2016.03.034
133. Aryanpour R, Pasbakhsh P, Zibara K, Namjoo Z, Beigi Boroujeni F, Shahbeigi S, et al. Progesterone therapy induces an M1 to M2 switch in microglia phenotype and suppresses NLRP3 inflammasome in a cuprizone-induced demyelination mouse model. Int Immunopharmacol. (2017) 51:131–9. doi: 10.1016/j.intimp.2017.08.007
134. Wang J, Pan H, Lin Z, Xiong C, Wei C, Li H, et al. Neuroprotective effect of fractalkine on radiation-induced brain injury through promoting the M2 polarization of microglia. Mol Neurobiol. (2021) 58:1074–87. doi: 10.1007/s12035-020-02138-3
135. Henry RJ, Ritzel RM, Barrett JP, Doran SJ, Jiao Y, Leach JB, et al. Microglial Depletion with CSF1R inhibitor during chronic phase of experimental traumatic brain injury reduces neurodegeneration and neurological deficits. J Neurosci. (2020) 40:2960–74. doi: 10.1523/JNEUROSCI.2402-19.2020
136. Bao J, Zheng L, Zhang Q, Li X, Zhang X, Li Z, et al. Deacetylation of TFEB promotes fibrillar Aβ degradation by upregulating lysosomal biogenesis in microglia. Protein Cell. (2016) 7:417–33. doi: 10.1007/s13238-016-0269-2
137. Tao L, Li D, Liu H, Jiang F, Xu Y, Cao Y, et al. Neuroprotective effects of metformin on traumatic brain injury in rats associated with NF-κB and MAPK signaling pathway. Brain Res Bull. (2018) 140:154–61. doi: 10.1016/j.brainresbull.2018.04.008
138. Liu Y, Deng S, Zhang Z, Gu Y, Xia S, Bao X, et al. 6-Gingerol attenuates microglia-mediated neuroinflammation and ischemic brain injuries through Akt-mTOR-STAT3 signaling pathway. Eur J Pharmacol. (2020) 883:173294. doi: 10.1016/j.ejphar.2020.173294
139. Fan LW, Kaizaki A, Tien LT, Pang Y, Tanaka S, Numazawa S, et al. Celecoxib attenuates systemic lipopolysaccharide-induced brain inflammation and white matter injury in the neonatal rats. Neuroscience. (2013) 240:27–38. doi: 10.1016/j.neuroscience.2013.02.041
140. Chokshi R, Bennett O, Zhelay T, Kozak JA. NSAIDs Naproxen, Ibuprofen, Salicylate, and Aspirin Inhibit TRPM7 Channels by Cytosolic Acidification. Front Physiol. (2021) 12:727549. doi: 10.3389/fphys.2021.727549
141. Alzheimer's Disease Anti-inflammatory Prevention Trial Research Group. Results of a follow-up study to the randomized Alzheimer's Disease Anti-inflammatory Prevention Trial (ADAPT). Alzheimers Dement. (2013) 9:714–23. doi: 10.1016/j.jalz.2012.11.012
142. Meyer PF, Tremblay-Mercier J, Leoutsakos J, Madjar C, Lafaille-Maignan M, Savard M, et al. INTREPAD: A randomized trial of naproxen to slow progress of presymptomatic Alzheimer disease. Neurology. (2019) 92:e2070–80. doi: 10.1212/WNL.0000000000007232
143. Ferretti MT, Allard S, Partridge V, Ducatenzeiler A, Cuello AC. Minocycline corrects early, pre-plaque neuroinflammation and inhibits BACE-1 in a transgenic model of Alzheimer's disease-like amyloid pathology. J Neuroinflammation. (2012) 9:62. doi: 10.1186/1742-2094-9-62
144. Biscaro B, Lindvall O, Tesco G, Ekdahl CT, Nitsch RM. Inhibition of microglial activation protects hippocampal neurogenesis and improves cognitive deficits in a transgenic mouse model for Alzheimer's disease. Neurodegener Dis. (2012) 9:187–98. doi: 10.1159/000330363
145. Seabrook TJ, Jiang L, Maier M, Lemere CA. Minocycline affects microglia activation. Abeta deposition, and behavior in APP-tg mice. Glia. (2006) 53:776–82. doi: 10.1002/glia.20338
146. Adapt-Fs Research Group. Follow-up evaluation of cognitive function in the randomized Alzheimer's disease anti-inflammatory prevention trial and its follow-up study. Alzheimers Dement. (2015) 11:216–25.e1. doi: 10.1016/j.jalz.2014.03.009
147. Markulin I, Matasin M, Turk VE, Salković-Petrisic M. Challenges of repurposing tetracyclines for the treatment of Alzheimer's and Parkinson's disease. J Neural Transm (Vienna). (2022) 129:773–804. doi: 10.1007/s00702-021-02457-2
148. An J, Chen B, Kang X, Zhang R, Guo Y, Zhao J, et al. Neuroprotective effects of natural compounds on LPS-induced inflammatory responses in microglia. Am J Transl Res. (2020) 12:2353–78.
149. Wang K, Lu C, Wang T, Qiao C, Lu L, Wu D, et al. Hyperoside suppresses NLRP3 inflammasome in Parkinson's disease via pituitary adenylate cyclase-activating polypeptide. Neurochem Int. (2022) 152:105254. doi: 10.1016/j.neuint.2021.105254
150. Mohammadi N, Asle-Rousta M, Rahnema M, Amini R. Morin attenuates memory deficits in a rat model of Alzheimer's disease by ameliorating oxidative stress and neuroinflammation. Eur J Pharmacol. (2021) 910:174506. doi: 10.1016/j.ejphar.2021.174506
151. Hajialyani M, Hosein Farzaei M, Echeverría J, Nabavi SM, Uriarte E, Sobarzo-Sánchez E, et al. Hesperidin as a neuroprotective agent: a review of animal and clinical evidence. Molecules. (2019) 24:648. doi: 10.3390/molecules24030648
152. Jaturapatporn D, Isaac MG, McCleery J, Tabet N. Aspirin, steroidal and non-steroidal anti-inflammatory drugs for the treatment of Alzheimer's disease. Cochrane Database Syst Rev. (2012) 2012:Cd006378. doi: 10.1002/14651858.CD006378.pub2
153. Villarejo-Galende A, González-Sánchez M, Blanco-Palmero VA, Llamas-Velasco S, Benito-León J. Non-steroidal anti-inflammatory drugs as candidates for the prevention or treatment of alzheimer's disease: do they still have a role? Curr Alzheimer Res. (2020) 17:1013–22. doi: 10.2174/1567205017666201127163018
154. Wang M, Pan W, Xu Y, Zhang J, Wan J, Jiang H, et al. Microglia-mediated neuroinflammation: a potential target for the treatment of cardiovascular diseases. J Inflamm Res. (2022) 15:3083–94. doi: 10.2147/JIR.S350109
155. Mohammadi M, Abdi M, Alidadi M, Mohamed W, Zibara K, Ragerdi Kashani I, et al. Medroxyprogesterone acetate attenuates demyelination, modulating microglia activation, in a cuprizone neurotoxic demyelinating mouse model. Am J Neurodegener Dis. (2021) 10:57–68.
156. Subedi L, Gaire BP. Phytochemicals as regulators of microglia/macrophages activation in cerebral ischemia. Pharmacol Res. (2021) 165:105419. doi: 10.1016/j.phrs.2021.105419
157. Basalay MV, Davidson SM, Yellon DM. Neuroprotection in rats following ischaemia-reperfusion injury by GLP-1 analogues-liraglutide and semaglutide. Cardiovasc Drugs Ther. (2019) 33:661–7. doi: 10.1007/s10557-019-06915-8
158. Li M, Li Z, Ren H, Jin WN, Wood K, Liu Q, et al. Colony stimulating factor 1 receptor inhibition eliminates microglia and attenuates brain injury after intracerebral hemorrhage. J Cereb Blood Flow Metab. (2017) 37:2383–95. doi: 10.1177/0271678X16666551
159. Rice RA, Pham J, Lee RJ, Najafi AR, West BL, Green KN, et al. Microglial repopulation resolves inflammation and promotes brain recovery after injury. Glia. (2017) 65:931–44. doi: 10.1002/glia.23135
160. Chu K, Yin B, Wang J, Peng G, Liang H, Xu Z, et al. Inhibition of P2X7 receptor ameliorates transient global cerebral ischemia/reperfusion injury via modulating inflammatory responses in the rat hippocampus. J Neuroinflammation. (2012) 9:69. doi: 10.1186/1742-2094-9-69
161. Walter S, Letiembre M, Liu Y, Heine H, Penke B, Hao W, et al. Role of the toll-like receptor 4 in neuroinflammation in Alzheimer's disease. Cell Physiol Biochem. (2007) 20:947–56. doi: 10.1159/000110455
162. Shu S, Zhang Z, Spicer D, Kulikowicz E, Hu K, Babapoor-Farrokhran S, et al. Administration of a 20-hydroxyeicosatetraenoic acid synthesis inhibitor improves outcome in a rat model of pediatric traumatic brain injury. Dev Neurosci. (2019) 41:166–76. doi: 10.1159/000500895
163. Zhang W, Sun Q, Gao X, Jiang Y, Li R, Ye J, et al. Anti-inflammation of spirocyclopiperazinium salt compound LXM-10 targeting α7 nAChR and M4 mAChR and inhibiting JAK2/STAT3 pathway in rats. PLoS ONE. (2013) 8:e66895. doi: 10.1371/journal.pone.0066895
164. Guan YZ, Jin XD, Guan LX, Yan HC, Wang P, Gong Z, et al. Nicotine inhibits microglial proliferation and is neuroprotective in global ischemia rats. Mol Neurobiol. (2015) 51:1480–8. doi: 10.1007/s12035-014-8825-3
165. Aldrich A, Kielian T. Central nervous system fibrosis is associated with fibrocyte-like infiltrates. Am J Pathol. (2011) 179:2952–62. doi: 10.1016/j.ajpath.2011.08.036
Keywords: microglia, activation, membrane receptor, signaling pathway, inflammatory response
Citation: Qin J, Ma Z, Chen X and Shu S (2023) Microglia activation in central nervous system disorders: A review of recent mechanistic investigations and development efforts. Front. Neurol. 14:1103416. doi: 10.3389/fneur.2023.1103416
Received: 20 November 2022; Accepted: 13 February 2023;
Published: 07 March 2023.
Edited by:
Yijing Chen, Shenzhen Institutes of Advanced Technology (CAS), ChinaReviewed by:
Jing Qiu, University of Edinburgh, United KingdomCopyright © 2023 Qin, Ma, Chen and Shu. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Shiyu Shu, c2h1c2hpeXVAY3FtdS5lZHUuY24=
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.