
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
BRIEF RESEARCH REPORT article
Front. Neurol. , 09 February 2023
Sec. Neurogenetics
Volume 14 - 2023 | https://doi.org/10.3389/fneur.2023.1094234
 Teodor Angelov1*
Teodor Angelov1* Teodora Chamova1
Teodora Chamova1 Slavena Atemin2Tihomir Todorov2
Slavena Atemin2Tihomir Todorov2 Slavko Ormandzhiev2Ivan Tourtourikov2,3Albena Todorova2,3David Devos4Ivailo Tournev1,5
Slavko Ormandzhiev2Ivan Tourtourikov2,3Albena Todorova2,3David Devos4Ivailo Tournev1,5Objectives: Amyotrophic lateral sclerosis (ALS) is a neurodegenerative disorder characterized by progressive deterioration of motor function, disability, and death. Variants in the PFN1 gene, encoding the Profilin-1 protein, are related to ALS18.
Methods: We present a pedigree consisting of 3 generations and 4 affected individuals, 3 of which carry a novel heterozygous variant: c.92T > G (p.Val31Gly) in the PFN1 gene. This variant was discovered through means of whole exome sequencing (WES) and targeted analysis of ALS-related genes.
Results: The mean age of onset in our pedigree was 59.75 (±10.11 SD) years with a significant difference between the first two generations (females) and the third (male) of 22.33 (±3.4 SD) years. For this ALS form, we observed a longer disease progression of 4 (±1.87 SD) years (three of four affected are still alive). Clinical manifestations displayed predominant impairment of the lower motor neuron (LMN) in one limb, with gradual involvement of other limbs. A novel heterozygous missense variant c.92T > G, p. Val31Gly (NM_005022.4) in exon 1 in the PFN1 gene was discovered through means of whole exome sequencing (WES). Segregation analysis in the family showed that the detected variant was inherited from the affected mother, and the affected aunt also turned out to be a variant carrier.
Conclusions: ALS18 is a very rare form of the disease. We report here a relatively large pedigree with a novel variant, leading to late onset (after 50 years), initial involvement of the lower limbs and relatively slow progression.
Amyotrophic lateral sclerosis (ALS) is a neurodegenerative disorder characterized by progressive deterioration of motor function, disability and death (1). The disease is genetically heterogeneous and can be caused by variants in certain genes (36 at the moment, according to latest data), encoding proteins involved in the development and metabolism of motoneurons (2). Clinical cases can be divided in two major groups—sporadic ALS (sALS) and familial ALS (fALS), of which 90–95 and 35–40% of cases respectively, are left without genetic verification.
The PFN1 gene encodes the protein Profilin-1, pathogenic variants in which were found to cause ALS18 (3). Profilin-1 regulates the polymerization of actin, in response to extracellular signals, and thus is actively involved in cytoskeletal dynamics and axonal transport of motoneurons (4). As a result of the variations, monomeric globular (G)-actin cannot polymerize to filamentous (F)-actin (5). The reduced G/F-actin ratio, as well as reduced number of growth cones, are considered as a biochemical marker, when observing motoneurons at the microscopic level.
The first description of ALS18 was made by Wu et al. (3), who reported 22 patients with ALS, carrying 4 missense variants [c.211T > G (p.Cys71Gly); c.341T > C (p.Met114Thr); c.350A > G (p.Glu117Gly) and c.353G > T (p.Gly118Val)] in the PFN1 gene. In this report the mean age of onset was 44.8 years, with initial weakness and atrophies in the limbs. In the following year, screening studies of the PFN1 gene were performed and a new variant was demonstrated by Ingre et al. (6) [c.326C > T (p.Thr109Met)] in a German patient. A new variant was found by Chen et al. (7) [c.406C > T (p.Arg136Trp)] in a Chinese patient, and a single variant in the PFN1 gene has been reported from the Middle East [c.58G > A (p.Ala20Thr)] by Smith et al. (8). Two studies also indicated that a variant in the PFN1 gene [c.318_319dup (p.Asp107fs)] causes an early onset, polyostotic Paget-like disorder (9, 10).
We present a family, consisting of 3 generations and 4 affected individuals, with a clinical diagnosis ALS (Figure 1A).
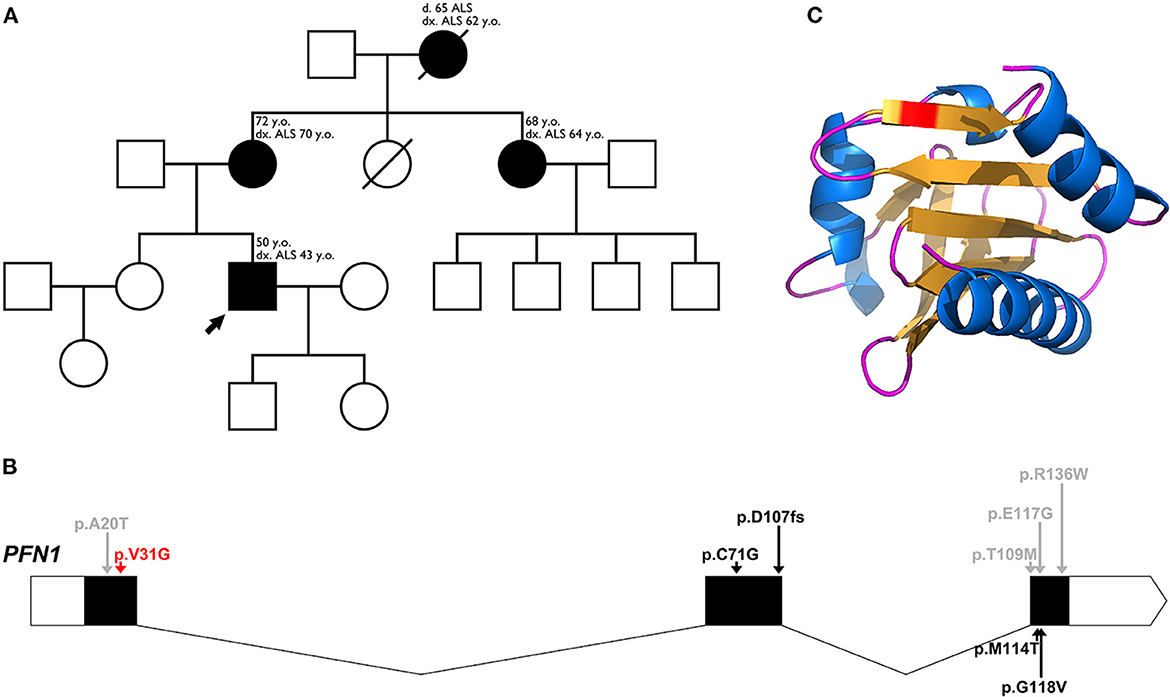
Figure 1. (A) Pedigree family tree for our family with 4 affected individuals. The proband of the family is marked with an arrow. For each affected individual, the age of onset and current age is indicated, as well as age of death for the deceased grandmother. (B) Schematic representation of the genomic structure of the PFN1 gene, with intron-exon boundaries. 5′-3′ UTRs are indicated in white, coding regions are indicated in black and noncoding regions are indicated as thin arrow lines. Arrows indicate major variants, identified in ALS, as well as one variant associated with a Paget-like disorder (red arrow for our novel variant, black arrows for pathogenic/likely pathogenic variants, and gray arrows for variants of uncertain significance). (C) Three-dimensional structure of the altered sequence of human PFN1 protein, modeled with AlphaFold2.
All patients underwent neurological examination, electromyography (EMG), electroneurography (ENG), magnetic resonance imaging (MRI) of the brain and spine, evaluation by an otolaryngologist to assess bulbar dysfunction and neuropsychological assessment. Patients were assessed using the most common scale to determine the degree of UMN and LMN damage—ALS Functional Rating Scale-Revised (ALSFRS-R; Table 1).
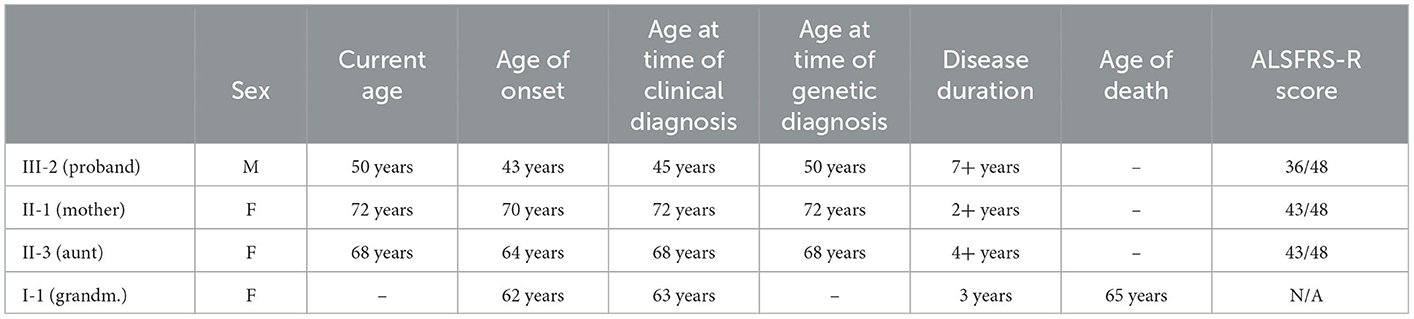
Table 1. Epidemiological data and ALSFRS-R score for the affected patients.
Venous blood was taken for genetic testing (whole exome sequencing (WES) and targeted analysis of ALS-related genes).
Next generation sequencing—whole exome sequencing (NGS-WES, Illumina) was applied for evaluation of ALS related genes, associated with clinical symptoms of the target patient. Data interpretation was performed with the Software Gensearch NGS (Phenosystems). The detected variant in the PFN1 gene was confirmed and studied for segregation in the family by Sanger sequencing.
Sanger sequencing of the targeted gene was performed with the Big Dye® Terminator cycle sequencing kit v.3.1 (Applied Biosystems). Sequencing profiles were interpreted with the Sequencing Analysis v.5.1.1 software.
The study complies with the ethical guidelines of the institutions involved.
A novel heterozygous variant: c.92T > G (p.Val31Gly), in the PFN1 gene, was found in 3 of the affected individuals (one of the affected individuals passed away before the conducted genetic tests) (Figure 1B). The novel variant was found in the proband (onset of disease at the age of 43, currently 50 years old) and segregation analysis was conducted for the remaining affected individuals—his mother (onset of disease at the age of 70, currently 72 years old) and his aunt (onset of disease at the age of 64, currently 68 years old). The grandmother of the proband had an onset of disease at the age of 62, and died at the age of 65 before the genetic tests were conducted. All of the affected members are of Bulgarian ethnicity and no healthy family members were available for genetic testing. Occupational risk factors were not observed, as patients were not professionally engaged in heavy physical labor or in an environment with toxins and industrial hazards.
The epidemiological and clinical features of the affected patients are presented in Tables 1, 2.
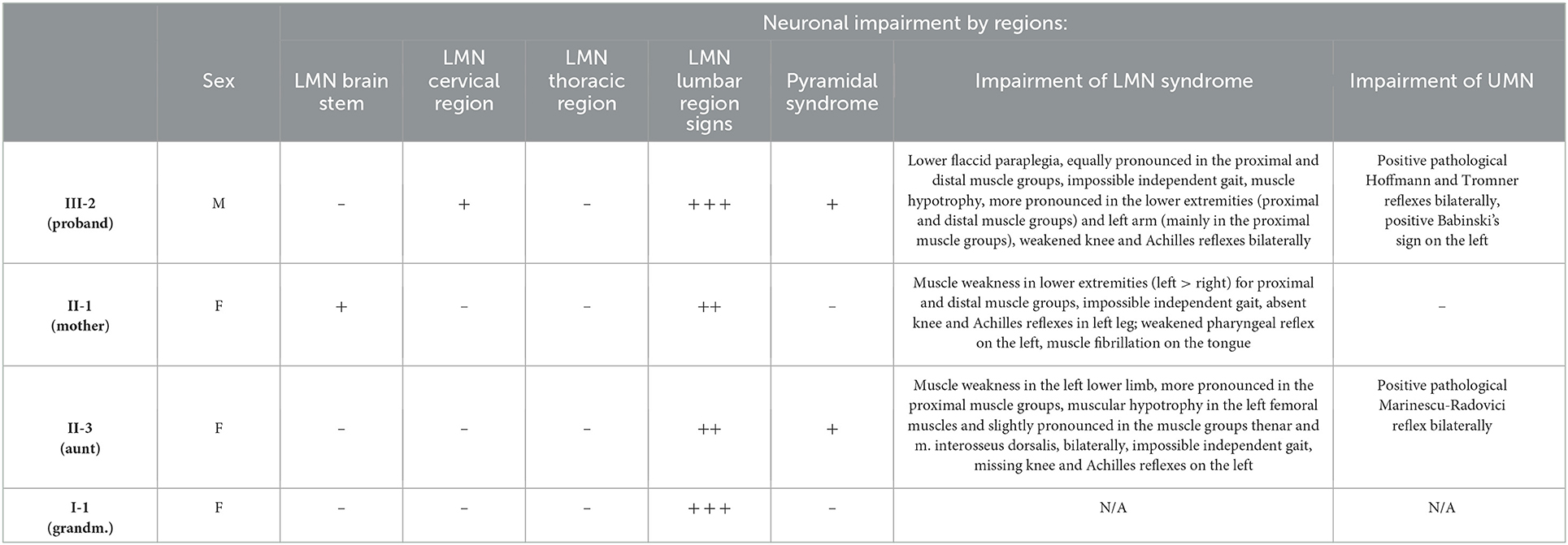
Table 2. Impairment of the UMN and LMN by regions.
The mean age of onset in our family is 59.75 (±10.11 SD)−62 years for generation I, 67 years for generation II and 43 years for generation III, with a significant difference between the first two generations (females) and the third (male) of 22.33 (±3.4 SD) years. The mean disease duration is 4 (±1.87 SD) years, as of making of this article (three of four affected individuals are still alive). Neurological assessment was consistent, with LMN involvement predominantly in the lower limbs, and milder involvement in the upper limbs for the proband. His mother had bulbar involvement as well, consisting of weakened pharyngeal reflex on the left and muscle fibrillation on the tongue.
The ALSFRS-R scale for all affected individuals showed evidence of predominant LMN impairment at the lower limb region. The mother has also discrete involvement in the brainstem, which does not affect her daily functions. There was no significant reduction in daily functions due to impairment of the UMN. These data explain the relatively high points score on this scale (40.66/48), and if we take into account the lower sum of the proband (36/48), the mother's and aunt's functions are largely preserved.
A novel heterozygous missense variant: c.92T > G (p.Val31Gly) (NM_005022.4) in exon 1 of the PFN1 gene was found by WES. Based on the standards and guidelines of ACMG/AMP for interpretation of sequence variants (11), we currently classified it as likely pathogenic (categories: PP1, PP3, PP4, and PM2). The variant is not found in gnomAD genomes and exomes. The CADD score for the variant is 32, with REVEL scoring at 0.895. In total, 11 in silico predictors classify the variant as pathogenic and 7 classify it as likely pathogenic. The variant was confirmed by Sanger sequencing, and segregation analysis in the family showed that the detected variant is inherited from the affected mother, and the affected aunt also turned out to be a variant carrier.
The variant is located within the critical Profilin-1 domain, between amino acids 2-106. Simulating the altered sequence, using AlphaFold2 (12), did not show any structural alterations (Figure 1C). While the actin interaction site of PFN1 lies within amino acids 60-130, there have been reports of likely pathogenic/pathogenic variants outside of this sequence. The valine residue at position 31 is highly conserved among several species, including yeast, rat and nematode. The neighboring positions 30 and 32 are highly conserved as well, and act as poly-proline binding sites.
ALS18 is a very rare form of ALS. The pedigree reported by us carries a novel, and so far undescribed variant, c.92T > G (p.Val31Gly), which is characterized by specific clinical features, consistent with the ALS18 form. In the same exon 1 of the PFN1 gene, apart from the detected in our study variant, two non-pathogenic (synonymous) variants have also been detected [c.93C > G (p.Val31Val) and c.93C > T (p.Val31Val), gnomAD browser].
Furthermore, ALS18 is distinguished by a specific phenotype: an age of onset after 50 years, an initial region of involvement in the lower extremities (and very rarely in the upper extremities) and long-term disease course without severe disability. These hallmarks should be carefully considered in newly diagnosed patients, as they largely resemble those of sporadic ALS cases, especially ones without symptoms, characterized by late onset and a long clinical course, or in cases, which are often interpreted as secondary ALS-syndrome (due to disc pathology or other causes). From this point of view, ALS18 may not be as rare, if a large number of cases have been misdiagnosed. An additional factor in their potential underestimation is the fact that increasingly genetic data is accumulating for variants in non-coding regions of the genome (e.g., introns, 5′ and 3′ untranslated regions etc.), defects in which can also lead to disease development (13–15).
ALS18 should be taken into account in cases with main key features, such as late age of onset (after 50 years) and/or initial region of involvement in the lower extremities (with or without discrete migration in other regions), and/or long-term course of the disease without severe disability significantly impairing daily functions.
We observed discrete differences in the epidemiological and clinical data for our described family, and for previously described patients with ALS18 (Table 3). The novel variant, described in this study [c.92T > G (p.Val31Gly)], allows to further strengthen the association of pathogenic variants in the PFN1 gene (which have been reported to induce disturbed dynamics of microtubules and/or troubled coordination between actin and microtubule filaments in motoneurons) with development of ALS18 (16). Different variants in the PFN1 gene lead to different flexibility and stability of microtubule entities, reported in a recent study using proteomic analysis in mammalian cells (17). According to this study, the variants c.341T > C (p.Met114Thr) and c.353G > T (p.Gly118Val) are indicated as partially destabilizing the Profilin-1 protein, while the variant c.211T > G (p.Cys71Gly) leads to severe destabilization at the microtubule level, due to its significant instability and increased accumulation of insoluble fractions.
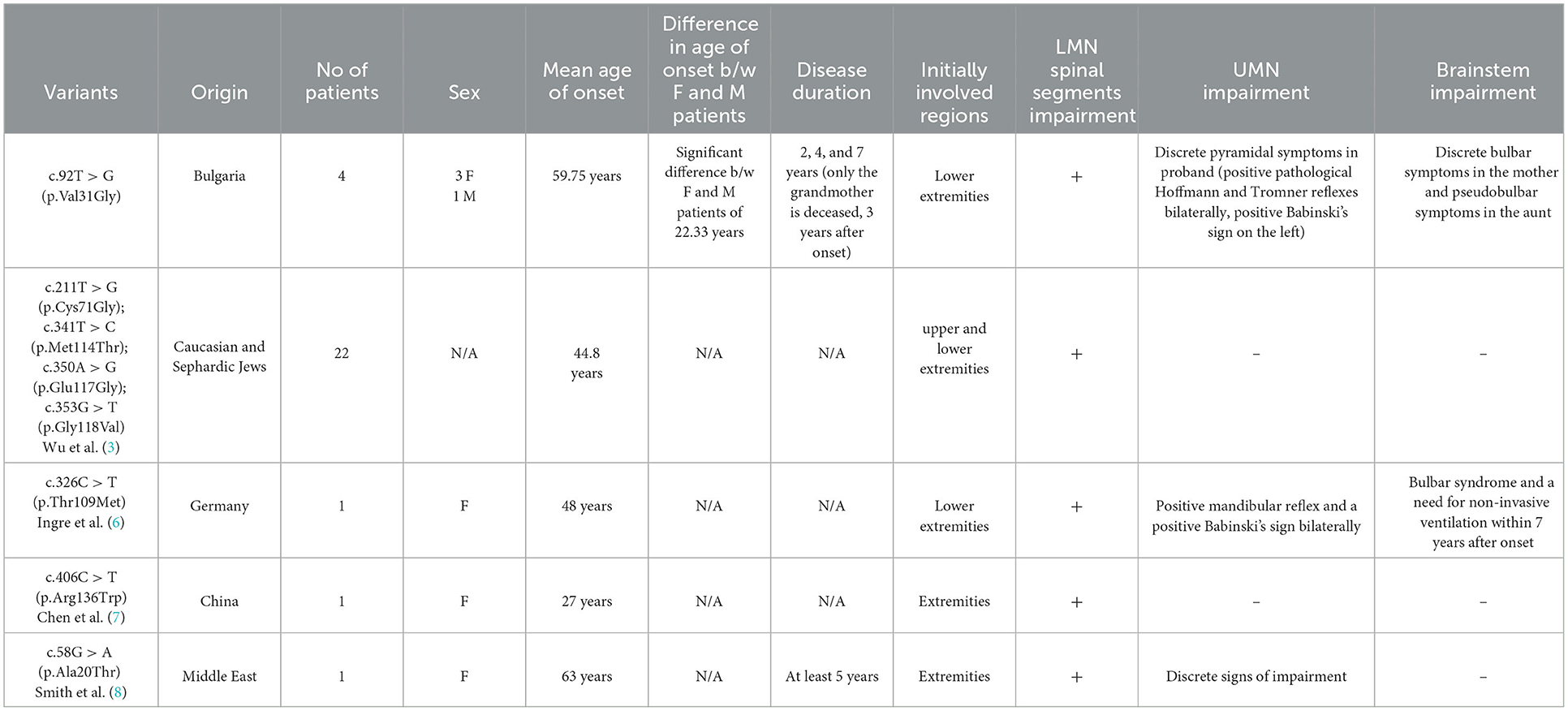
Table 3. Epidemiological and clinical characteristics, with comparison of our novel variant and others described in scientific literature.
In this publication we describe a novel variant [c.92T > G (p.Val31Gly)] in the PFN1 gene, responsible for development of ALS18. The family we report consists of 3 generations and 4 affected individuals, making it one of the largest with this form of ALS. The data reported by us help to broaden the clinical-genetic phenotype of ALS and to improve the differential-diagnostic notions, given its specific clinical phenotype.
The datasets presented in this article are not readily available because of ethical and privacy restrictions. Requests to access the datasets should be directed to the corresponding author.
The studies involving human participants were reviewed and approved by Ethics Committee at Medical University of Sofia. The patients/participants provided their written informed consent to participate in this study.
TA, TC, TT, AT, DD, and ITourn: formal analysis (equal), investigation (equal), methodology (equal), writing-original draft (equal), writing-review and editing (equal), and conceptualization (equal). SA and SO: formal analysis (equal), investigation (equal), methodology (equal), writing-original draft (equal), and writing-review and editing (equal). ITourt: formal analysis and conceptualization. All authors contributed to the article and approved the submitted version.
The Article Processing Fee for this manuscript is provided by the Medical University of Sofia.
The authors thank all of the patients and their caregivers who participated in the study. Part of this work was supported by project BG-RRP-2.004-0004-C01, a part of The Bulgarian National Recovery and Resilience Plan, financed by the National Science Fund of Bulgaria (BNSF).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
1. Ustyantseva E, Medvedev S, Zakian S. Studying ALS: current approaches, effect on potential treatment strategy. Adv Exp Med Biol. (2020) 1241:195–217. doi: 10.1007/978-3-030-41283-8_11
2. Suzuki N, Nishiyama A, Warita H, Aoki M. Genetics of amyotrophic lateral sclerosis: seeking therapeutic targets in the era of gene therapy. J Hum Genet. (2022) 13:1–22. doi: 10.1038/s10038-022-01055-8
3. Wu C-H, Fallini C, Ticozzi N, Keagle P, Sapp P, Piotrowska K, et al. Mutations in the profilin 1 gene cause familial amyotrophic lateral sclerosis. Nature. (2012) 488:499–503. doi: 10.1038/nature11280
4. Kwiatkowski D, Bruns G. Human profilin. Molecular cloning, sequence comparison, and chromosomal analysis. J BiolChem. (1988) 263:5910–5.
5. Mockrin S, Korn E. Acanthamoeba profilin interacts with G-actin to increase the rate of exchange of actin-bound adenosine 59-triphosphate. Biochemistry. (1980) 19:5359–62. doi: 10.1021/bi00564a033
6. Ingre C, Landers J, Rizik N, Volk AE, Akimoto C, Birve A, et al. A novel phosphorylation site mutation in profilin 1 revealed in a large screen of US, Nordic, and German amyotrophic lateral sclerosis/frontotemporal dementia cohorts. Neurobiol Aging. (2013) 34:e1–6. doi: 10.1016/j.neurobiolaging.2012.10.009
7. Chen Y, Zheng Z-Z, Huang R, Chen K, Song W, Zhao B, et al. PFN1 mutations are rare in Han Chinese populations with amyotrophic lateral sclerosis. Neurobiol Aging. (2013) 34:e1–5. doi: 10.1016/j.neurobiolaging.2013.01.013
8. Smith B, Vance C, Scotter E, Troakes C, Hao C. Novel mutations support a role for profilin 1 in the pathogenesis of ALS. Neurobiol Aging. (2015) 36:e17–27. doi: 10.1016/j.neurobiolaging.2014.10.032
9. Merlotti D, Materozzi M, Bianciardi S, Guarnieri V, Rendina D, Volterrani L, et al. Mutation of PFN1 gene in an early onset, polyostotic paget-like disease. J Clin Endocrinol Metab. (2020) 105:2553–65. doi: 10.1210/clinem/dgaa252
10. Scotto di Carlo F, Pazzaglia L, Esposito T, Gianfrancesco F. The loss of Profilin 1 causes early onset Paget's disease of bone. J Bone Miner Res. (2020) 35:1387–98. doi: 10.1002/jbmr.3964
11. Richards S, Aziz N, Bale S, Bick D, Das S, Gastier-Foster J, et al. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of medical genetics and genomics and the association for molecular pathology. Genet Med. (2015) 17:405–24. doi: 10.1038/gim.2015.30
12. Jumper J, Evans R, Pritzel A, Green T, Figurnov M, Ronneberger O, et al. Highly accurate protein structure prediction with AlphaFold. Nature. (2021) 596:583–9. doi: 10.1038/s41586-021-03819-2
13. Lu Y, Chen J, Lin H, Feng S, Che C, Liu CY, et al. Novel intronic mutations of TBK1 promote aberrant splicing modes in amyotrophic lateral sclerosis. Front Mol Neurosci. (2022) 15:691534. doi: 10.3389/fnmol.2022.691534
14. Morgan S, Shatunov A, Sproviero W, Jones A, Shoai M, Hughes D, et al. A comprehensive analysis of rare genetic variation in amyotrophic lateral sclerosis in the UK. Brain. (2017) 140:1611–8. doi: 10.1093/brain/awx082
15. Sabatelli M, Moncada A, Conte A, Lattante S, Marangi G, Luigetti M, et al. Mutations in the 3' untranslated region of FUS causing FUS overexpression are associated with amyotrophic lateral sclerosis. Hum Mol Genet. (2013) 22:4748–55. doi: 10.1093/hmg/ddt328
16. Henty-Ridilla J, Juanes M, Goode B. Profilin directly promotes microtubule growth through residues mutated in amyotrophic lateral sclerosis. Curr Biol. (2017) 27:3535–43.e4. doi: 10.1016/j.cub.2017.10.002
Keywords: ALS, genetics, phenotype, pedigree, PFN1
Citation: Angelov T, Chamova T, Atemin S, Todorov T, Ormandzhiev S, Tourtourikov I, Todorova A, Devos D and Tournev I (2023) Novel variant c.92T > G (p.Val31Gly) in the PFN1 gene (ALS18) responsible for a specific phenotype in a large Bulgarian amyotrophic lateral sclerosis pedigree. Front. Neurol. 14:1094234. doi: 10.3389/fneur.2023.1094234
Received: 09 November 2022; Accepted: 25 January 2023;
Published: 09 February 2023.
Edited by:
Georgios Koutsis, National and Kapodistrian University of Athens, GreeceReviewed by:
Ioannis Zaganas, University of Crete, GreeceCopyright © 2023 Angelov, Chamova, Atemin, Todorov, Ormandzhiev, Tourtourikov, Todorova, Devos and Tournev. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Teodor Angelov,  dGVvZG9yX2FAYWJ2LmJn
dGVvZG9yX2FAYWJ2LmJn
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.