 Mackenzie M. Aychman
Mackenzie M. Aychman David L. Goldman
David L. Goldman Joshua S. Kaplan
Joshua S. Kaplan- Behavioral Neuroscience Program, Department of Psychology, Western Washington University, Bellingham, WA, United States
Cannabidiol (CBD) has numerous pharmacological targets that initiate anti-inflammatory, antioxidative, and antiepileptic properties. These neuroprotective benefits have generated interest in CBD's therapeutic potential against the secondary injury cascade from traumatic brain injury (TBI). There are currently no effective broad treatment strategies for combating the damaging mechanisms that follow the primary injury and lead to lasting neurological consequences or death. However, CBD's effects on different neurotransmitter systems, the blood brain barrier, oxidative stress mechanisms, and the inflammatory response provides mechanistic support for CBD's clinical utility in TBI. This review describes the cascades of damage caused by TBI and CBD's neuroprotective mechanisms to counter them. We also present challenges in the clinical treatment of TBI and discuss important future clinical research directions for integrating CBD in treatment protocols. The mechanistic evidence provided by pre-clinical research shows great potential for CBD as a much-needed improvement in the clinical treatment of TBI. Upcoming clinical trials sponsored by major professional sport leagues are the first attempts to test the efficacy of CBD in head injury treatment protocols and highlight the need for further clinical research.
Introduction
Traumatic Brain Injury (TBI) is a global public health epidemic that causes death or hospitalization in an estimated 27–69 million people annually (1, 2). Yet, TBI has been called the “silent epidemic” because of its range in acute symptoms and severity that lead to underdiagnosis and underreporting by patients or treatment facilities (3–6). In addition to acute symptomology that includes amnesia, disorientation, and changes to mental processing speed, even mild TBIs can have long-term mental health impacts including depression and changes in impulsivity, judgement, and memory. The severity of the impact (i.e., the direct trauma to the brain) often determines the severity of the TBI symptoms (7) and involve brain changes that underlie persistent neurological deficits and seizures. These post-concussion symptoms contribute to high hospitalization rates among TBI sufferers in which 43% require additional hospitalization during the first year post-injury (5). Patients with TBIs have financial hardships caused by their cognitive and physical disabilities that can require expensive medical treatments and limit work activities. There is also the societal economic burden that in the United States, alone, was $76.5 billion in 2010 dollars (5). Because of inconsistent diagnoses and subsequent underreporting of TBIs, the true cost and financial impact is expected to be much higher than this estimate.
The complexity of cellular, molecular, physiological, and neurometabolic mechanisms associated with different stages post-TBI makes it particularly difficult to treat. There is currently no single pharmacological approach that has been effective in treating TBIs (8). Yet, shared mechanisms of damage exist across TBI severity levels suggesting that a single strategy may be generally efficacious (9). Research into Cannabidiol (CBD), a non-intoxicating phytocannabinoid abundantly produced by some chemovars of Cannabis sativa L or synthetically produced from several biological systems (10), has revealed promising protective properties to counter the damaging effects of TBI that warrant concentrated investigation (11–13). CBD's unique pharmacodynamic profile (14) and high tolerability in adults (15–17) affords unique capabilities not shared by currently available treatment strategies. Here, we discuss CBD's proposed protective mechanisms against TBI-induced neuroinflammation and degeneration, which may be a plausible intervention for treating and reducing physiological damage and the associated symptoms that arise from TBI.
Mechanisms of injury
The clinical presentation of TBI symptoms results from a sequence of physiological, molecular, and chemical changes that occur immediately following the initial impact or following a delay. These two waves of disturbances are described as the primary and secondary injury, respectively.
Primary injury
The primary injury of TBI is the direct structural damage to neural tissues and blood vessels that derive from the impact event itself (18). During impact, there is a shockwave of brain compression and expansion that creates substantial mechanical forces within the skull. This shockwave causes immediate contusions to the area of impact, damages glial cells, induces localized hemorrhaging, shears axons and blood vessels, and disrupts cytoskeletal elements (19–23). Currently, helmets and mouthguards are the only known protective strategies against primary TBI injury resulting from non-vehicular sport or combat activities. These protective devices are effective at reducing the severity of TBIs but cannot prevent TBI when there is sufficiently forceful impact. Thus, a variety of TBIs occur both with (e.g., military combat) and without this protection (e.g., motor vehicle crashes). Due to the nature of the immediate physiological damage, pharmacological approaches are intended to reduce secondary injury rather than prevent the occurrence of primary injury.
Secondary injury
The secondary injury is the additional systems damage that results from mechanisms induced by the original structural injury (i.e., the primary injury). It is characterized by a widespread cascade of cellular, molecular, and biochemical changes that include, but is not limited to, unregulated ion and neurotransmitter release, dysregulation of glial cells, neuronal hyperexcitability, excitotoxicity, increased blood brain barrier (BBB) permeability, and widespread neuroinflammation (24–27). Diffuse mechanoporation results in ion leakage and ultimately the unregulated release of glutamate that triggers apoptotic events (24, 28, 29), increases the presence of reactive oxygen species (ROS), and induces oxidative stress (30). Together, these outcomes further trigger a proinflammatory response involving microglial and macrophage activation and pro-inflammatory cytokine release that boost and sustains posttraumatic inflammation for an indeterminant amount of time (31). This protracted inflammatory response contributes to additional tissue damage and neurodegeneration (32, 33). Activated microglia in their M1 state are especially neurotoxic because of their ability to enhance pro-inflammatory and neurotoxic mediators [e.g., IL-1β, tumor necrosis factor (TNF)-α, superoxide radicals, nitric oxide] and decrease phagocytic activity (34). The resulting immune response and concurrent neuroinflammation create a toxic cellular environment that inhibits neuronal healing and perpetuates neuron loss (35) and atrophy (36), and is associated with white matter degradation (37). Therefore, this inflammatory cascade is a main target for pharmaceutical intervention (26).
Current clinical treatment practices and the role for CBD
Physicians managing patients with TBI are seeking safe and effective treatment options that would ameliorate the symptoms of TBI, reduce recovery time, and prevent or decrease chronic neurologic dysfunction from residual brain injuries (Figure 1). Similar underlying mechanisms of secondary injury are observed across TBI severities, but differences in their magnitude impact clinical treatment strategies and patient outcomes (38). To clinically verify that pharmacotherapy such as CBD would improve outcomes for patients with mild, moderate, and severe TBI, there must be a reliable way to determine the severity categories.
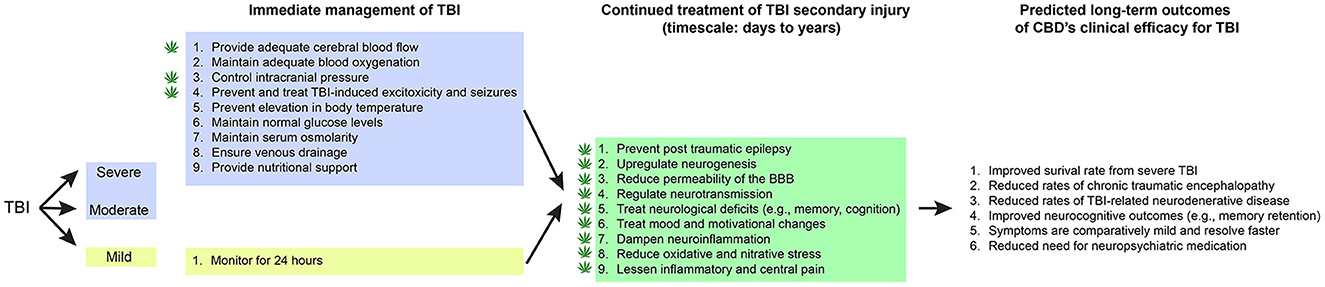
Figure 1. CBD's proposed role in immediate and continued treatment of TBI symptoms. TBI severity determines the scope of immediate clinical interventions. Preclinical evidence supports CBD's potential utility in some of these immediate treatment procedures (indicated by a cannabis leaf). However, CBD has broader potential to support TBI recovery by dampening the secondary injury cascade. If CBD is effective at improving some of these symptoms, there would be long-term predicted benefits across survival, neurocognitive, neurodegenerative, and neuropsychiatric measures.
There is ongoing debate in the medical community about the reliability of current clinical tools, such as the Glasgow Coma Scale (GCS), to assess and categorize TBI severity (39). The GCS measures the level of consciousness and has been commonly used to classify TBI severity as mild (GCS 13–15), moderate (GCS 9–12), and severe (GCS 3–8). Numerous influential medical organizations, including the World Health Organization, classify TBI using this GCS scale (40–42). However, one-third of trauma patients with GCS score of 13 were found to have intracranial lesions (43) raising concerns that these patients should not be categorized as mild TBI and instead categorized as experiencing moderate severity. Subsequently, recommendations were made to redefine mild TBI (GCS 14–15) and moderate TBI (GCS 9–13) (44). There are also ongoing discussions of ways to improve current TBI assessment and develop new tools that facilitate both clinical research and treatment (39).
One approach to improve the assessment of TBI severity is to incorporate more clinical information, exemplified by the work of the American Congress of Rehabilitation Medicine (ACRM). In 1993, ACRM defined mild TBI as an event causing one or more of the following conditions: (1) loss of consciousness (LOC), (2) post traumatic amnesia (PTA), (3) mental status changes, and (4) focal neurologic deficits. The severity of this TBI was assessed by how long the patient was unconscious, how long the PTA lasted, and the GCS score [see Brasure et al. (45) for specific criteria]. Notably, the ACRM's original definition only pertained to mild TBI but the same measures of severity have been widely used to categorize mild, moderate, and severe TBI (41). In 2021, the ACRM announced plans to update their work on measures of TBI severity to more reliably distinguish a mild TBI, from moderate and severe injuries (46). Being able to match CBD's outcomes to reliably assessed TBI severities will lead to better predictive validity and more effective long-term care plans.
Clinical management of different TBI severities
It is estimated that mild TBI represents 70–90 percent of all traumatic brain injuries (47, 48). Even mild TBI can have symptoms that are severe and debilitating such as posttraumatic headaches, nausea/emesis, dizziness or impaired balance, light or noise sensitivity, blurry or double vision, fatigue, memory impairment, poor concentration, increased anxiety, irritability, emotional lability, worsened mood, and sleep disturbance (49). Patients suspected of having a mild TBI should be medically evaluated by a licensed health professional to provide clinical management, head injury instructions, and determine whether they will need referral to an emergency department for a head CT to evaluate for more dangerous brain injuries that require hospitalization (50).
Current management for mild TBI recommends observation for 24 h at a hospital or home to watch for neurologic deterioration (50). These patients are also instructed to refrain from strenuous activities and contact sports until the symptoms resolve which usually occurs around 4 weeks (51) with notable variability (52). Clinical research is needed to determine whether CBD can stop or reduce the severity of any of the symptoms caused by mild TBI and whether treatment with CBD reduces the recovery time. Another clinical issue is whether treatment with CBD lowers the risk of permanent injuries from one or multiple episodes of mild TBI and lowers the risk of CTE associated with multiple head injuries.
Patients with moderate and severe TBI can have primary injuries that require surgical intervention including: depressed skull fracture, subdural or epidural hematoma, intracerebral hemorrhage, and penetrating injury (44, 53). Hospitalized patients with moderate and severe TBI also require medical management, usually in the intensive care unit, with the goal of controlling secondary injuries (44, 53).
Current treatment of intermediate and severe TBI in a critical care unit (ICU) includes medical and surgical management known to mitigate the effects of secondary brain injury: (1) maintaining blood pressure to provide adequate cerebral blood flow, (2) maintaining adequate oxygenation and normal pCO2, (3) controlling intracranial pressure (ICP), (4) preventing and treating seizures, (5) preventing elevated body temperature, (6) maintaining normal glucose levels, (7) management of IV fluids to maintain normal serum osmolarity, (8) elevating and positioning the head to promote venous drainage, and (9) provide nutritional support (44, 53). The patient is at considerable risk of deteriorating to brain death if there is a progression of cerebral edema and elevated ICP that cannot be controlled. Bifrontal craniectomies with dural incisions to decompress the brain is a surgical option for patients not responding to medical management of elevated ICP. This is a lifesaving procedure that effectively reduces ICP but usually leaves survivors with severe disabilities (54). Despite the slow recovery, and prolonged rehabilitation, some of these patients regain enough activities of daily living to provide a degree of functional independence (53).
A clinical role for CBD
The clinical issue is whether CBD, a treatment that can decrease BBB permeability and reduce neuroinflammation (55), can be an effective tool to reduce cerebral edema and lower ICP, and reduce the number of patients who develop this life-threatening intracranial hypertension. CBD differs from current clinical management options because it directly repairs the underlying cause of vasogenic edema which include increased permeability of the BBB (56) and neuroinflammation (57). As presented later in this review, pre-clinical research has demonstrated CBD's efficacy against these TBI consequences as well as improve cerebral blood flow (58) and treatment of genetic and pharmacologically-induced seizures (59). However, clinical research is needed to verify that CBD can be an effective pharmaceutical agent for reducing cerebral edema in patients with moderate and severe TBI. This clinical research also needs to evaluate whether CBD can reduce the incidence of post traumatic epilepsy (PTE), and if improved management of CBF contributes to a better clinical outcome. There is also preclinical evidence that CBD promotes neurogenesis (60), and the clinical issue is whether the neurogenesis that normally occurs in TBI patients can be enhanced with CBD and if this will reduce head injury symptoms, speed the recovery, and provide a better restoration of neurological function. Following in-patient treatment and stabilization period, patients may be released with substantial impairments in brain function that results from their TBI. This damage to neurotransmitter signaling results in long-lasting changes in personality traits, mood, and neurological function. Below, we describe these changes and the relevant mechanisms through which CBD may target them.
Damage to neurotransmitter systems
Both primary and secondary injury cascades impact neurotransmitter systems that result in sustained imbalance of cortical excitation and inhibition (24). Rodent models of TBI have implicated shifts in glutamate, GABA, serotonin, and catecholamine signaling in the persistent disruptions and damage to neural circuits (25).
Compression of tissue during TBI causes morphological damage and leads to a rapid increase in intracellular Ca2+ (61–63). This elevation in intracellular calcium contributes to the excessive glutamate release and region-specific changes in excitatory amino acid transporter (EAAT) expression that's observed following TBI (64–66). The outcome is a reduction in glutamate uptake resulting in excitotoxicity and apoptosis (67, 68). In response to elevated synaptic glutamate concentrations, AMPA receptors undergo shifts in subunit expression and become calcium permeable (69) which may enhance plasticity following injury, but an ultimate reduction in NMDA signaling—following a robust increase post-injury—reduces excitatory output of affected neurons (70, 71). Together, the initial wave of acute posttraumatic glutamate release results in excitotoxicity, apoptosis, and dysfunction of surviving neurons, while the following depression of signaling is responsible for some motor and cognitive deficits associated with TBI (24).
In parallel, the loss of GABAergic neurons elevates the disparity in the excitation/inhibition ratio and augments apoptotic processes and cellular injury. Apoptotic events resulting from the primary injury and an increase in extracellular glutamate promote the loss of GABAergic neurons. Furthermore, TBI induced dysfunction to GABAA receptor subunits (decreased α1, α4, γ2, and δ) leads to abnormal patterns of phasic and tonic inhibition, with a higher reduction in phasic inhibition (72). Additionally, activation of the JaK/STAT and Egr3 pathways results in decreased GABAA receptor signaling and hyperexcitability (72–75). This GABAergic neuron loss and reduced inhibitory tone is associated with both cognitive and motor deficits in addition to TBI-induced seizures (74). The remaining GABA neurons show elevated levels of GABAB receptor signaling (76), which serve as autoreceptors and further decrease GABAA receptor-mediated transmission. Changes to the expression of genes regulating glutamate and GABA result in long-lasting changes in homeostatic control and hinder endogenous mechanisms to restore the excitatory/inhibitory balance (25).
TBI negatively impacts attention, memory, and mood through impairments to cholinergic (77) and serotonergic systems (78). Biphasic changes in acetylcholine signaling begin with initial unregulated acetylcholine release and are followed by chronic cholinergic hypofunction (77, 79). This is eventually reflected in decreased receptor binding (80) and reduced choline acetyltransferase activity (81). Ultimately, this results in impaired attention and disrupted memory consolidation (82). Mood changes following TBI are thought to stem from sustained decreases in both serotonin receptors and transporters, contributing to increased rates of anxiety and depression in TBI sufferers (83).
TBI also triggers an increase in tyrosine hydroxylase and enhances the synthesis of dopamine and norepinephrine (84–86). However, persistent neuroinflammation contributes to a downregulation of receptors and decreased transmitter release (87, 88). Further, direct axonal damage from the primary injury negatively impacts signaling (89, 90). Sustained dopamine impairment can have feed-forward effects that further increase inflammation (91), disrupt metabolism (92), and reduce levels of brain-derived neurotropic factor (BDNF) (93), which normally has a role in stimulating neurogenesis, promoting neuronal survival, facilitating regeneration, and protecting tissue from oxidative stress and apoptosis following TBI (94). Lower baseline levels of BDNF among older adults hinders recovery and is one factor that may lead to lower rates of survival from TBI (95).
The consequence of TBI on several neurotransmitters systems causes many of the clinical and pathological hallmark symptoms of injury and underlie changes to cognitive and motor processes. Furthermore, structural damage to the lesioned areas contributes to mood and cognitive disturbances, such as damage to the forebrain cholinergic (96, 97) and catecholaminergic afferents (86, 98). An effective pharmacological approach will protect against any perturbation to these systems to prevent further neurological damage from the secondary injury response.
The impact of ECS signaling on neuroinflammation during secondary injury
The endocannabinoid system (ECS) influences TBI outcomes as increasing ECS tone protects against synaptic hyperexcitability, reduces neuroinflammation, and improves blood brain barrier integrity (99–101). The ECS is comprised of cannabinoid type I and II receptors (CB1 and CB2) and lipid signaling messengers, anandamide, and 2-AG. Among its numerous functional roles, its powerful influence over the immune response and neuroinflammatory signaling confers neuroprotective qualities that are relevant in the TBI secondary injury cascade. Endocannabinoids are involved in regulating inflammation by acting on cannabinoid receptors as well as targets beyond the canonical endocannabinoid system including TRPV1 and PPARγ (102). Yet, their neuroprotective potential has been most thoroughly studied at the endocannabinoid receptors. CB1 receptors are expressed in a variety of cell types and tissues but are expressed in highest abundance in the central nervous system (CNS) (103). CB1 receptors are involved in regulating inflammation in both the CNS and PNS and play an important role in dampening proinflammatory chemokine secretion (104). CB1 activation inhibits adenylate cyclase activity and decreases levels of cAMP, in addition to activating inwardly rectifying K+ channel conductance, decreasing N-type and P/Q-type voltage-operated Ca2+ channel conductance, ultimately reducing intracellular Ca2+ influx (105). The consequence of CB1 activation is therefore a reduction in neurotransmitter release, however its net impact on glutamate vs. GABA release is regionally determined and based on endocannabinoid concentrations (106–108).
CB2 receptors are present in higher concentrations in the immune system and on microglia than on neurons (55, 103, 109, 110). They have notable immunomodulatory and neuroprotective roles via the MAPK pathway and regulation of ERK-1/2 phosphorylation, which aids in reducing inflammation (109). Additionally, activation of CB2Rs is important for decreasing M1 state macrophages and increasing bias toward anti-inflammatory M2 polarization (111). CB1 and CB2 are highly involved in TBI and a potential reduction in inflammation and Ca2+ influx, which can prevent excitotoxicity, inhibit inflammatory cytokine production, and may therefore be neuroprotective (105).
Previous studies in a mouse model of TBI have demonstrated that CB1 and CB2 antagonists prevent activation of neuroprotective mechanisms in response to brain edema, diffuse axonal injury, and microglial activation (101, 102, 112). Therefore, endogenous ECS signaling can be neuroprotective, but it can also be a target of therapeutic intervention. For instance, boosting 2-AG levels protects against neurodegeneration, normalizes ionotropic glutamate and GABAA receptor expression levels, prevents additional tau pathologies, and improves behavioral outcomes in a mouse model of repeated TBI (113, 114). Since CB1 and CB2 antagonists only partially blocked neuroprotective benefits of increasing 2-AG levels, it suggests that ECS neurotransmitters may confer neuroprotection against secondary injury through actions at both ECS and non-ECS receptors. One potential non-ECS receptor target could be δ-subunit containing GABAA receptors (115) that are found extrasynaptically and mediate a tonic-inhibitory current (116). Further mechanistic investigation is needed to determine the ECS and non-ECS receptor contributions to neuroprotection in TBI. Importantly, the neuroprotective potential of solely targeting the ECS receptors is limited by the sensitivity to desensitization following prolonged pharmacological activation (117, 118). Therefore, pharmacological interventions for TBIs may be more effective if they target additional mechanisms beyond solely CB1 and CB2 receptors.
CBD's neuroprotective potential in TBI
Cannabidiol (CBD) is a non-intoxicating, non-psychedelic phytocannabinoid that has over 65 known ECS and non-ECS targets in the brain and body that are influenced dose-dependently [for a detailed review of relevant molecular targets, see (14)]. Not all of these targets are relevant in the context of TBI therapy, but several of these targets directly mitigate inflammation which has prompted interest in the potential utility of CBD to dampen secondary injury in TBI. These relevant targets and therapeutic mechanisms are discussed below. However, there are currently no clear dosing guidelines for these neuroprotective benefits in TBI. Effective CBD dosing has proven challenging for other therapeutic purposes such as anxiety (119), deficits in prosocial behavior (120), and cocaine-induced reinstatement (121) that demonstrate inverted-U dose response curves. It's unclear if a similar inverted-U response will be observed for CBD's neuroprotection against the multitude of secondary injury cascade mechanisms or if treatment efficacy is retained with escalating dosing such is observed with CBD's anti-epileptic effects (120, 122).
Achieving the optimal therapeutic dose for combating secondary injury in TBI will likely depend on CBD's bioavailability (123) which varies as a function of consumption method [for a detailed review of CBD's pharmacokinetics in humans, see Millar et al. (123)]. Pulmonary, sublingual, and intranasal absorption of CBD promote the highest level of bioavailability, but these administration routes have not been systematically assessed for combating the secondary injury cascade. Nonetheless, as we discuss below, CBD's neuroprotective efficacy during secondary injury has been demonstrated in several preclinical models accompanying restoration of TBI-impaired molecular, chemical, and physiological mechanisms that would be theoretically predictive of an effective TBI pharmacological strategy in humans. These mechanisms include, but are not limited to, increasing ECS signaling and reducing glutamate excitotoxicity, promoting neurogenesis, dampening neuroinflammation, scavenging reactive oxygen species, reducing TBI-induced BBB permeability, and regulating cerebral blood flow, all discussed in further detail below (Figure 2). Alleviating these consequences is beneficial for protecting against cognitive, mood, and motor changes and helping to better restore function following TBI.
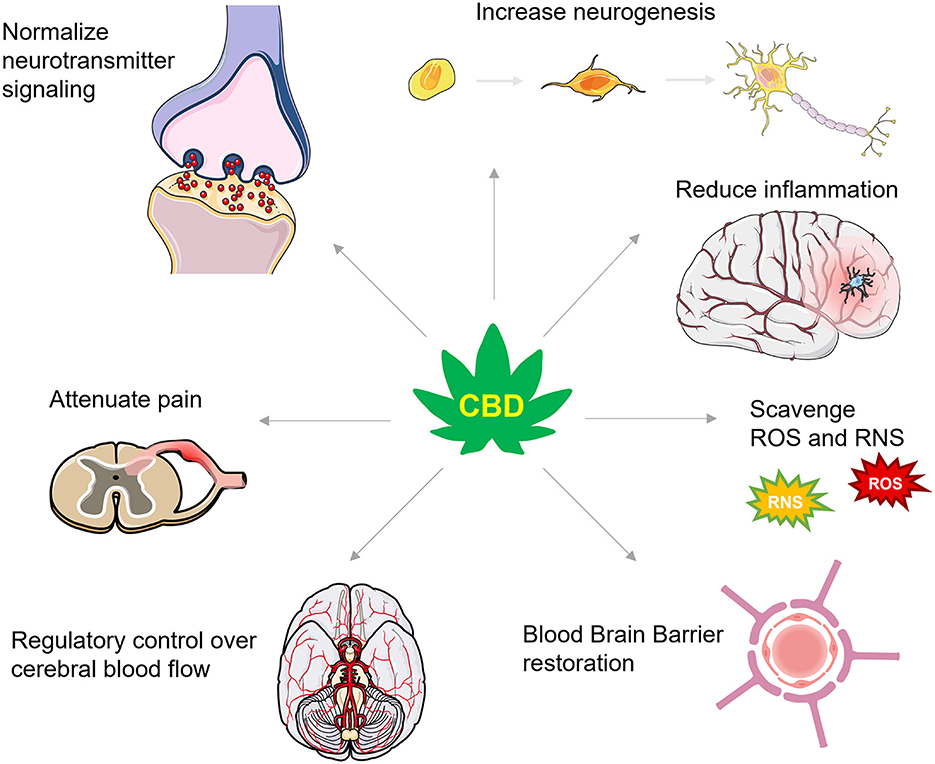
Figure 2. A summary of CBD's actions in TBI. CBD has numerous actions that are proposed to protect against secondary injury and support recovery from TBI. These actions include effects on numerous neurotransmitter systems that increase levels of brain derived neurotrophic factor and enhance neurogenesis, dampen inflammatory signaling cascades, scavenge for reactive oxygen and nitrogen species (ROS and RNS, respectively), restore the integrity of the blood brain barrier, improve control over cerebral blood flow, and attenuate inflammatory and neuropathic pain.
One important consideration regarding CBD's neuroprotective efficacy is whether CBD needs to have reached the brain prior to the primary injury or if it can be effective if administered during the secondary injury response. Addressing the time course of CBD's efficacy will be a critical experimental consideration moving forward. There is currently insufficient evidence to draw any definitive conclusions because in many cases, CBD is administered prior to the primary injury [e.g., (12)], after [e.g., (13)], or both before and after [e.g., (56)], and often not compared across administration period. Pretreatment may lead to lower mortality rates and improved sensorimotor function (12), but targeting at least some of CBD's mechanisms, like the ECS, within a “window of opportunity” of at least 15 min following the primary injury may also confer substantial neuroprotective benefits (124). Given that the window of opportunity for CBD administration is not clearly defined or consistently tested, we discuss the following neuroprotective effects across a range of administration methods and temporal relationships to the primary injury.
CBD's effects on neurotransmitter systems after TBI
CBD's neuroprotective effects likely stem from action at both ECS and non-ECS targets. CBD indirectly activates CB1 and CB2 receptors by acting as a competitive substrate for the FAAH enzyme that's responsible for the primary hydrolysis of the endocannabinoid anandamide (125). CBD may also increase 2-AG, but this is debated and may only occur regionally such as in the periaqueductal gray (126, 127). CBD may have some action directly on the ECS receptors, themselves. In vitro assessments suggest that CBD has low binding affinity for CB1 receptors and can act as a weak positive allosteric modulator of CB2 receptors (128). Together, CBD can stimulate ECS receptors by elevating anandamide levels and potentially by directly acting upon them to reduce neuroinflammation during secondary injury processes (111, 129, 130).
Pre-clinical models suggest that CBD may normalize the glutamatergic and GABAergic signaling imbalance following TBI. CBD administered prior to TBI reduced the increase in cortical glutamate release (12). This benefit was maintained with continued CBD administration for at least 30 days post-TBI. In this case, CBD's effects were observed at moderate (50 mg/kg) and high (100 mg/kg, 200 mg/kg) oral doses, but the 100 mg/kg dose was optimal suggesting that CBD's ability to restore normal glutamate levels may follow a similar inverted U dose-response curve to other conditions [e.g., (110)], albeit one with a much higher dose tolerance. Additionally, CBD blocks glutamate toxicity in cultured cortical neurons independent of inhibiting AMPA, NMDA, or kainite receptors (131). More direct dampening of glutamatergic excitotoxicity may occur through CBD stimulation of adenosine A2a signaling and resulting reduction in glutamate release (132–134). CBD's multi-target pharmacological mechanism may end up being safer and more efficacious than glutamate receptor antagonists which are insufficient for preventing neuronal dysfunction and resulting clinical symptoms (135), and may have adverse consequences such as increased risk for tumor growth (136). In addition to dampening excessive glutamate signaling, CBD increases GABAergic signaling via positive allosteric modulation of non-benzodiazepine binding sites on α-containing GABAARs, with preference for the α2-containing receptor subtype and the β2 or β3 subunit (137). Together, CBD effects restoration of the excitatory/inhibitory balance through direct and indirect actions on GABAergic and glutamatergic signaling, which may promote improved cognitive and pain symptoms that result from TBI.
Many of the alterations in mood following TBI may stem from impaired dopaminergic and serotonergic signaling (90, 138–140). The primary TBI injury can cause mechanical damage and shearing to dopaminergic axonal projections, which can additionally cause oxidative stress (90). Sustained damage to this neurotransmitter system can impair dopamine synthesis and metabolism (86). CBD aids in the neuroprotection of dopaminergic pathways, as it is a partial agonist at D2 and D3 receptors (141, 142) in rat striatal tissue, and reduces dopamine uptake (14, 143), which aids in attenuating the loss of dopaminergic neurons and microglial activation (134). The neuroprotective properties of CBD further aid in protecting the remaining neurons, dendrites, and cellular structures in dopaminergic pathways (144, 145).
Furthermore, CBD is a direct agonist of 5HT1A receptors and a partial agonist on 5HT2A receptors (14, 146–148), decreasing anxiety, pain and headaches associated with TBIs via interaction with the orthosteric binding site (141, 149–151). In vitro studies demonstrate that CBD 5HT1AR binding increases GTP binding to the 5HT1AR coupled G protein, Gi, confirming its role as an agonist. However, it should be noted that 5HT2AR agonism is only applicable in the presence of high concentrations of CBD (147). Via this action, CBD increases serotonergic and glutamate cortical signaling, as well as inhibits adenosine uptake (152). Rebalancing these neurotransmitter systems is important for TBI recovery, as reducing serotonin and adenosine reuptake results in decreased oxidative stress, excitotoxicity, and inflammation, which are hallmarks of TBI secondary injury and the main focus of pharmacological intervention (153, 154). SSRIs have been shown to improve common depressive symptoms post-TBI, which arise partly due to decreased serotonergic signaling (153). Increasing extracellular serotonin after damage has been shown as beneficial for improving mood disturbances, however it is ineffective in improving cognition (155). Therefore, it is important to use a pharmacological approach that addresses both mood and cognitive symptoms that arise from TBI.
CBD also antagonizes the G protein-coupled receptor GPR55 (156), which results in enhanced GABAergic neurotransmission in the mouse hippocampus due to increases in inhibitory neuron excitability, and ultimately decreases the excitation/inhibition ratio (120). Inhibiting GPR55 reduces Ca2+ release, and therefore aids in further reduction of glutamate release and excitotoxicity (14).
Together, CBD has widespread effects attributed to interactions with non-ECS targets, such as PPARγ (157), 5-hydroxytryptamine (5-HT1A) receptors (158), adenosine receptors (159, 160), vanilloid receptor 1 (TRPV1) (161, 162), G-protein coupled receptors (i.e., GPR18 and GPR55) (120, 141, 156, 163), GABAA receptors (137), and glutamate receptors (131, 164). CBD's effects on these various neurotransmitter mechanisms and the restoration of the excitatory/inhibitory balance are proposed to improve mood and pain-related symptoms of TBI, and limit long-term damage caused by excitotoxicity and inflammation. Notably, the net consequence of CBD's actions on these different pharmacological targets will need to be evaluated in humans with TBI since research into CBD's effect on the brain of adults with autism spectrum disorder show differential effects as a function of disease state and pathological etiology (165, 166).
CBD's effect on neurogenesis
As part of the natural healing process following TBI, neurogenesis is increased in the hippocampus, cerebral cortex, and white matter in rodents (167, 168). Additionally, neural stem/progenitor cell protein markers naturally increase in humans post-TBI, indicating that neurogenesis may be induced in humans in the same way as in an animal TBI model (167). This generation of new cells aids in cognitive and spatial TBI recovery (169). CBD can further promote neurogenesis through multiple mechanisms such as increasing CB1 (170) or PPARγ receptor signaling (171) to regulate stem cell proliferation and differentiation (172) in the granule cell layer of the hippocampus. Therefore, CBD's actions on ECS and non-ECS mechanisms may regulate neurogenesis (173), but they may do so in a dose-dependent and condition-dependent manner. In unstressed animals, CBD's pro-neurogenic effects appear to be dose-dependent, with lower doses promoting neurogenesis and higher doses suppressing it (174). The impairment to neurogenesis at high doses was postulated to result from desensitization of CB1 receptors. However, in chronically-stressed animals, higher doses retained pro-neurogenic effects (60). Therefore, the dose-dependency of CBD's effects on neurogenesis may depend on symptom etiology where different CBD targets mediate its effects. At this time, it is unknown how CBD's targets and dose-dependent mechanisms are affected by TBI across the range of severities. Further work is needed to clarify the dose-dependency of CBD on neurogenesis in TBI and identify whether these benefits also exist from CBD treatment in humans. Even though plasma CBD concentration was positively associated with hippocampal volume in heavy cannabis users (175), its direct impact on human neurogenesis remains undetermined.
Together, CBD's actions on formal ECS and related targets counteract the pro-inflammatory cascade and may enhance the neurogenic response to TBI. Although CBD is often considered in light of its effects on ECS signaling, its numerous non-ECS targets likely contributes to the range of effects it can have on secondary injury in TBI.
CBD and inflammation
Microglia play an important neuroprotective role in the healthy brain by serving as the first responders to injury or disease (176). They are responsible for clearing cellular waste and plaques and aid in neutralizing infectious agents. However, their chronic activation can damage healthy cells and is thought to contribute to numerous neuropathologies including the damage that occurs during the secondary injury cascade of TBI. Dampening microglia activation following TBI may be a targetable mechanism for limiting the extent of secondary injury damage.
Primary TBI damage ruptures axons and their myelin sheaths releasing ATP that binds to damage-associated molecular pattern (DAMP) receptors on microglia and activates their M1 pro-inflammatory state (28, 177). This proportion of polarized microglia in their M1 state is positively correlated with the severity of white matter injury (177). Microglia in their M1 state release toxic substances such as inflammatory cytokines that are important for neuroprotection from minor injury or infection (34, 178–180). Microglia that persist in this M1 state can trigger excessive and pathological synapse degradation and can induce neurotoxicity due to the release of neurotoxic mediators and pro-inflammatory factors, which creates cycles of microglial-mediated neurodegeneration (34, 180–182).
One of CBD's proposed protective mechanisms involves biasing the polarization of these microglia from its damage-causing pro-inflammatory (M1) to the anti-inflammatory (M2) activation state (111, 183). This action is mediated through indirect action at A2A adenosine receptors or by enhancing AEA-mediated CB2 receptor signaling (133, 184, 185). CBD's ability to shift the active state of microglia is an important protective mechanism against the ongoing damage to synaptic integrity that occurs during the secondary injury cascade.
CBD and oxidative/nitrative stress
TBI increases free radical synthesis at a rate higher than can be suppressed by the endogenous scavenger system resulting in oxidative stress (186, 187). The accumulation of these free radicals during secondary injury damages nearby cells and contributes to cellular damage as they attempt to supplement their missing electron through oxidation or harmful scavenging of electrons from nearby proteins, membrane, and DNA. Activated microglia in their M1 state are a primary source of these free radicals as they increase reactive oxygen species and reactive nitrogen species as part of the proinflammatory response to neural injury (188).
One facet of CBD's neuroprotective mechanisms may stem from its potent antioxidant and antinitrative effects in addition to its indirect ability to dampen ROS generation (189, 190). CBD's molecular structure confers strong antioxidant properties; it has electrophilic-hydroxyl groups in its aromatic phenol ring which allows it to be readily oxidized (131, 189, 191). It can also prevent the formation of superoxide radicals and ROS through numerous mechanisms including blocking oxidase activity, chelating transition metal ions (192), and affecting the levels and activity of antioxidants (189). CBD can further affect the redox balance by elevating AEA levels and reducing oxidative stress through a CB2-dependent mechanism (193, 194). Together, CBD's antioxidant properties serve as an additional neuroprotective mechanism that protects cellular function, stability, and prevents the initiation of apoptotic cascades (192).
CBD and BDNF
BDNF is an important neurotrophin involved in the recovery process from TBI. Serum BDNF levels on the day-of-injury for adults with non-severe forms of TBIs can be used as a prognostic indicator, as high levels predict better recovery (195, 196). BDNF is a secreted autocrine factor that promotes the regeneration, synaptogenesis, axonal sprouting, and survival of neurons (195, 197, 198), which are critical factors in effective neurogenesis. It is involved in reducing the secondary cascade of injury in TBI by restoring functional connectivity and providing further neuroprotection (199, 200). BDNF is found in higher concentrations in the cortex and hippocampus immediately post-TBI but decreases in concentration within the first 24 h due to a molecular cascade involving phosphorylation of PERK (a protein-kinase in the endoplasmic reticulum activated by stress) and increased activation of CREB, which downregulates BDNF (201). Impairments in dopamine signaling are partly responsible for the sustained reduction in BDNF levels that follow and contributes to memory, cognitive, and depressive symptoms (55, 94).
Boosting BDNF levels may have utility in combating secondary injury. In animal models, a positive correlation has been shown between increased BDNF expression and improved functional outcomes in terms of motor, memory, behavior, and cognitive responses after TBI. A decrease in BDNF levels during secondary injury is associated with worse outcomes (195). In human studies, acute serum BDNF levels are associated with chronic memory impairments, functional cognitive limitations, and depressive symptom severity, with an inverse correlation between BDNF levels and cognitive impairment (95, 202, 203). These beneficial effects of BDNF are likely mediated through TrkB receptors, resulting in intracellular signaling cascades that enhance relevant protein synthesis (202).
CBD's ability to increase BDNF levels proposes an additional neuroprotective mechanism against secondary injury. CBD has been shown to increase synthesis of BDNF in the prefrontal cortex and hippocampus in mice (202), two regions implicated in the cognitive, memory, and mood-related impairments following TBI (204–206). However, it's unclear if CBD can rescue cognitive deficits and mood changes following TBI if these changes are associated with tissue damage and not merely altered neurotransmitter signaling (204, 205). CBD may be more effective in rescuing mood and memory deficits caused by shifts in hippocampal signaling from TBI-induced volume reduction (206) or increased amygdala activity (207). For instance, the increase in BDNF levels by CBD protected against neurotransmitter alterations and coinciding depression-like symptoms in a mouse model of brain ischemia (208). However, this neuroprotective mechanism remains speculative since there are currently no known studies to have directly assessed the role of CBD-induced upregulation of BDNF in TBI and whether this neuroprotection extends to TBI with severe tissue damage from the primary injury.
CBD and the blood brain barrier
The BBB is a specialization of the blood vessels that vascularize the CNS. It regulates the movement of ions, molecules, and cells between the blood and the brain to prevent the brain's exposure to neurotoxins and infectious agents. Tight junction networks that are established by capillary endothelial cells in the CNS protect neural tissue from bacteria, viruses, toxins, and pathogens (209). Disruption increases toxin exposure, elevates immune and inflammation responses, and disturbs the brain's biochemical environment (56).
Both the primary TBI and secondary TBI cascade enhance the BBB's permeability. The shearing force of the primary injury can cause injury to endothelial cells and a loss of blood flow resulting in decreased BBB integrity that enables the passage of potential toxins into the CNS (209). This breakdown triggers leukocyte recruitment that promotes the release of proinflammatory cytokines or ROS, activates M1 microglia, and increases neuroinflammation and apoptosis (210–214). During the secondary injury cascade, increases in oxidative stress, transforming growth factor beta 1 (TGF-β1), and TNF-α promote a further reduction in the integrity of the BBB through suppressing the tight-junction protein, claudin-5 (215, 216). This leads to protracted hyperpermeability of the BBB and neuroinflammation.
Pre-clinical studies have exposed CBD's potential to suppress BBB permeability through several protective mechanisms (Figure 3). CBD can protect against the suppression of tight-junction proteins, claudin-5 and occludin (56). These actions may be mediated, in part, through PPARγ and 5-HT1A receptors that have been shown to maintain function (217). Further, CBD decreases TNF-α which prevents further BBB damage and limits the inflammatory response (152, 218). Additional CBD targets, such as TRPV1, GPR18, and 5-HT1A are involved in regulating cerebral blood flow and conferring further protection against cerebrovascular impairments from TBI (58, 219–221). The combinatorial effects of CBD acting on numerous targets may be a promising strategy of protecting the BBB's integrity and limiting feed-forward damage that occurs during secondary injury.
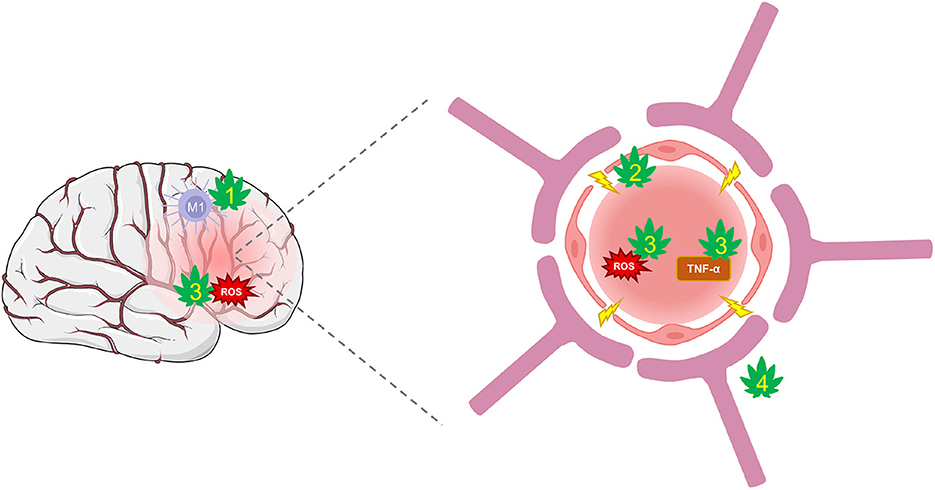
Figure 3. CBD protection against damage from BBB disruption. TBI disrupts cerebral blood flow and damages the integrity of the BBB. Hyperpermeability resulting from damaged tight-junctions and endothelial cells leads to increased inflammation and oxidative stress. (1) CBD shifts the polarization of macrophages from their pro-inflammatory M1 type to anti-inflammatory M2 type via activation of A2A adenosine receptors or by enhancing AEA-mediated CB2 receptor signaling. (2) CBD may improve BBB integrity and prevent hyperpermeability by suppressing TBI's damaging effects on tight-junction proteins via action on PPARγ and 5-HT1A receptors. (3) CBD is a potent antioxidant that reduces ROS and protects against oxidative damage to neurons and the BBB. It also reduces levels of TNF-α and other inflammatory markers that reduce the integrity of the BBB. (4) CBD may regulate cerebral blood flow to enhance reperfusion following injury via activation of GPR18, GPR55, and 5-HT1A receptors.
CBD and cerebral blood flow
Adequate and controlled cerebral blood flow (CBF) is essential to neurological function and is an important clinical consideration following TBI (222). Various clinical studies have demonstrated an acute transient decrease in CBF following TBI, which returns to baseline during the recovery process (223–225). Large reductions in CBF are associated with worse neurological outcomes post-injury (226, 227), while acutely increasing CBF following TBI aids recovery (228).
A variety of local chemical mediators of CBF are altered after TBI, including an increased release of K+ into the extracellular space, excessive release of excitatory amino acids, increased acidity of cerebral pH, and increased interstitial adenosine. The resulting impairment in CBF autoregulation causes endothelial dysfunction and vasospasm while boosting release of free radicals (229–233). Disturbances to the autoregulatory mechanisms may increase vulnerability to additional secondary injury cascades and further dysregulate CBF and sensitivity to hypotension (222).
CBD action on GPCR18 (an endothelial cannabinoid receptor) and GPR55 may aid in recovering regulatory control over CBF via vasomotor control (55, 234). CBD administration to mice before and after an experimental middle cerebral artery occlusion protected against CBF impairment via action on 5-HT1A receptors, thereby illustrating CBD's potential efficacy to enhance reperfusion following injury (235, 236). Therefore, there's emerging and supporting pre-clinical evidence that CBD may act as a cerebroprotectant by protecting against the dysregulated CBF from occlusion or injury.
CBD and pain
Given the physical trauma of the primary TBI, inflammation-induced nociception is usually a primary symptom of injury. CBD acts both at the site of injury (see discussion above) and centrally to achieve its pain-relieving effects in pre-clinical models. For instance, microinjection of CBD directly into the nucleus accumbens attenuated pain responses during both the early and late phases of the formalin test but were strongest in the late phase (237). Since plasticity in the dorsal horn generated from excessive C-fiber input is believed to contribute to late phase nociception (238), these findings suggest that CBD may dampen inflammatory and neuropathic pain. Others have demonstrated the importance of brain 5-HT1A (239) and D1 and D2 dopamine receptors (240, 241), as well as TRPV1 receptors in the spinal cord (162) in promoting these centrally mediated pain-reducing effects. Nonetheless, CBD's efficacy as a pain treatment in humans may vary due to route of consumption, dose, or pain etiology. At this time, there's no empirical evidence that CBD is effective at treating posttraumatic headache. However, CBD's ability to regulate inflammation may help prevent the sensitization of the trigeminal pain circuit (242) that underlies posttraumatic headache (243). Further research is warranted for specifically investigating CBD's potential as a pain treatment from TBI etiology.
Discussion
Current limitations
Identifying safe and effective dosing strategies remains a challenge for both pre-clinical and clinical investigation of cannabinoids. Given the multitude of synergistic and competing mechanisms (14), proper and consistent dosing remains a major challenge. Current studies of CBD's effects on TBI suggest that CBD dosing and achieved concentrations are critical for achieving therapeutic benefits [e.g., (56)]. Other characteristics such as method of administration [e.g., (13)], duration of exposure, and temporal proximity to the TBI [e.g., (12)] further complicate efforts for achieving a clear understanding of optimal therapeutic strategies.
Beyond dosing, sex-based differences in the ECS expression, cannabinoid metabolism, and energy homeostasis regulation may impact CBD's effects, and thereby, highlight the need for further investigation into biological sex and hormone-based differences in the response to CBD (244–246). For instance, estrogen levels may affect CBD action by altering ECS-related pharmacodynamics (247) and should be considered when assessing efficacy of CBD in treating TBI (56). Furthermore, pre-clinical studies have revealed sex-dependent differences in CB1 receptor expression and efficacy between male and female rats: males have more, but less efficient, CB1 receptors than females (244, 248, 249). Since CB1 receptors are involved in regulating inflammation, these differences may impact the regulation of microglia in response to injury. These differences can affect the severity of the neuroinflammatory response as well as the effectiveness of CBD in treating neuroinflammation during secondary injury.
One of CBD's promising characteristics for its treatment potential is that it's generally well-tolerated (122, 250, 251). Adults can consume an acute dose of several thousand milligrams without substantial adverse effects, as demonstrated by a formal single ascending and multiple dose pharmacokinetic trial of cannabidiol oral solution. In adults, single doses of up to 6,000 mg CBD were well-tolerated, as were multiple doses of up to 1,150 mg twice daily for 6 days (252).
Clinical trials of pediatric epilepsies provide insight into the adverse effects that result from months of repeated exposure to high doses (around 10–30 mg/kg/day) (122, 253, 254). Although CBD is associated with increased risk for adverse effects such as somnolence, sedation, and impaired liver function, it's unclear if these result from CBD's interactions with other medications and therefore, it's unclear what adverse effects, if any, chronic CBD use has as a monotherapy (255). Notably, CBD's competitive inhibition of numerous cytochrome P450 liver enzymes (256) highlight the need for caution when combining CBD with additional medications that rely on metabolic breakdown by the P450 enzymes. Health-care providers, or patients concerned about discussing their off-label cannabis use with a physician, can access services such as the Cannabinoid Drug Interaction Review (CANN-DIR) (257) to identify possible interactions between CBD with concomitantly prescribed medications. Furthermore, the inconsistencies in CBD administration methods and dosing have precluded a consistent and predictable dose-response relationship that may inform TBI intervention.
Future directions
There are currently no known blinded and randomized human clinical trials for CBD's neuroprotective effects in TBI, however, an upcoming phase II clinical trial will investigate the effects of CBD with or without Δ9-THC in patients with TBI (258). Additionally, the amalgamation of pre-clinical reports has piqued the interest among organizations with high-risk athletes. Two major sports organizations in North America whose athletes are at high risk for TBI, the National Hockey League (NHL) and the National Football League (NFL), are investigating CBD's protective and restorative effects in their athletes (259, 260). At the time of this publication, the NHL Alumni Association is moving toward a double-blind, randomized study of over 100 retired NHL players to assess the benefit of CBD on recovery from concussions (260). The NFL has also awarded funding for two clinical studies involving cannabinoids, one assessing the effects on pain and recovery from sports-related injuries and the other assessing the role of cannabinoids in pain management and neuroprotection from concussion in contact sports (259). Additionally, a separate study is recruiting participants to study CBD as a treatment for PTSD and PTSD comorbid with TBI (261), which has great relevance for those who have experienced military combat. Together, these emerging clinical trials will reveal if the promising findings from pre-clinical studies extend to at-risk human populations.
Repeated TBIs, whether from sport, combat, or other causes, increases the risk for developing chronic traumatic encephalopathy (CTE). The total number of TBIs and their severity are positively correlated with CTE risk (262), which has been neuropathologically diagnosed in 110 of the 111 NFL veterans who were tested (263). It is characterized by extensive brain atrophy, astrogliosis, myelinated axonopathy, microvascular injury, perivascular neuroinflammation, and phosphorylated tau protein pathology. The exact mechanism underlying CTE is unknown, however it emerges years after one or more TBIs. Cis P-tau is positively correlated with axonal injury in CTE (264) and, for example, may be a targetable substrate by CBD for CTE treatment (265, 266). It is hypothesized that by mediating the effects of TBI with CBD, it may be possible to prevent the development of CTE, or at least decrease the severity of the disorder. Although more research into CTE and its underlying mechanisms is needed, the investment into clinical trials by these professional sport organizations is warranted.
Conclusions
TBI is a public health epidemic with inconsistent clinical diagnostic criteria. Due to its complex mechanism of injury (primary and secondary) and varying severity, there is currently no single effective pharmacological treatment for TBI. CBD targets many of the cellular, molecular, and biochemical changes associated with TBI by mediating the regulation of neurotransmitters, restoring the E/I balance, preventing BBB permeability, increasing BDNF and CBF, and decreasing both ROS/NOS and microglial inflammatory responses. To accomplish this, CBD indirectly activates CB1R and CB2R while also targeting PPARγ, 5HT1AR, TRPV1, GPR18, and GPR55. It functions to regulate Ca2+ homeostasis, prevent apoptotic signaling, reduce neuroinflammation, and serve as a neuroprotectant/cerebroprotectant. Via a variety of targets, CBD appears to reduce cognitive (changes in memory, attention, and mood) and physiological symptoms associated with TBI, and lessen TBI-induced nociception.
There is strong mechanistic support that CBD could be an effective pharmacological intervention for TBIs, however the current state of the research field is mostly derived from rodent studies. The upcoming clinical trials will be especially informative for determining CBD's efficacy as a TBI treatment.
Author contributions
All authors listed have made a substantial, direct, and intellectual contribution to the work and approved it for publication.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher's note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Dewan MC, Rattani A, Gupta S, Baticulon RE, Hung YC, Punchak M, et al. Estimating the global incidence of traumatic brain injury. J Neurosurg. (2018) 130:1080–97. doi: 10.3171/2017.10.JNS17352
2. James SL, Bannick MS, Montjoy-Venning WC, Lucchesi LR, Dandona L, Dandona R, et al. Global, regional, and national burden of traumatic brain injury and spinal cord injury, 1990-2016: a systematic analysis for the global burden of disease study 2016. Lancet Neurol. (2019) 18:56–87. doi: 10.1016/S1474-4422(18)30415-0
3. Langlois JA, Rutland-Brown W, Wald MM. The epidemiology and impact of traumatic brain injury: a brief overview. J Head Trauma Rehabil. (2006) 21:375–8. doi: 10.1097/00001199-200609000-00001
4. Finkelstein EA, Corso PS, Miller TR. The Incidence and Economic Burden of Injuries in the United States. New York, NY: Oxford University Press (2006). doi: 10.1093/acprof:oso/9780195179484.001.0001
5. Nguyen R, Fiest KM, McChesney J, Kwon CS, Jette N, Frolkis AD, et al. The international incidence of traumatic brain injury: a systematic review and meta-analysis. Can J Neurol Sci. (2016) 43:774–85. doi: 10.1017/cjn.2016.290
6. Rusnak M. Giving voice to a silent epidemic. Nat Rev Neurol. (2013) 9:186–7. doi: 10.1038/nrneurol.2013.38
7. Yu S, Kaneko Y, Bae E, Stahl CE, Wang Y, van Loveren H, et al. Severity of controlled cortical impact traumatic brain injury in rats and mice dictates degree of behavioral deficits. Brain Res. (2009) 1287:157–63. doi: 10.1016/j.brainres.2009.06.067
8. Diaz-Arrastia R, Kochanek PM, Bergold P, Kenney K, Marx CE, Grimes CJB, et al. Pharmacotherapy of traumatic brain injury: state of the science and the road forward: report of the department of defense neurotrauma pharmacology workgroup. J Neurotrauma. (2014) 31:135–58. doi: 10.1089/neu.2013.3019
9. Hiskens MI. Targets of neuroprotection and review of pharmacological interventions in traumatic brain injury. J Pharmacol Exp Ther. (2022) 382:149–66. doi: 10.1124/jpet.121.001023
10. Pirrung MC. Synthetic access to cannabidiol and analogs as active pharmaceutical ingredients. J Med Chem. (2020) 63:12131–6. doi: 10.1021/acs.jmedchem.0c00095
11. Friedman LK, Peng H, Zeman RJ. Cannabidiol reduces lesion volume and restores vestibulomotor and cognitive function following moderately severe traumatic brain injury. Exp Neurol. (2021) 346:113844. doi: 10.1016/j.expneurol.2021.113844
12. Santiago-Castañeda C, Huerta de la Cruz S, Martínez-Aguirre C, Orozco-Suárez SA, Rocha L. Cannabidiol reduces short- and long-term high glutamate release after severe traumatic brain injury and improves functional recovery. Pharm. (2022) 14:1609. doi: 10.3390/pharmaceutics14081609
13. Belardo C, Iannotta M, Boccella S, Rubino RC, Ricciardi F, Infantino R, et al. Oral cannabidiol prevents allodynia and neurological dysfunctions in a mouse model of mild traumatic brain injury. Front Pharmacol. (2019) 10:352. doi: 10.3389/fphar.2019.00352
14. Ibeas Bih C, Chen T, Nunn AVW, Bazelot M, Dallas M, Whalley BJ. Molecular targets of cannabidiol in neurological disorders. Neurotherapeutics. (2015) 12:699–730. doi: 10.1007/s13311-015-0377-3
15. Velayudhan L, McGoohan K, Bhattacharyya S. Safety and tolerability of natural and synthetic cannabinoids in adults aged over 50 years: a systematic review and meta-analysis. PLoS Med. (2021) 18:e10003524. doi: 10.1371/journal.pmed.1003524
16. Dahlgren MK, Lambros AM, Smith RT, Sagar KA, El-Abboud C, Gruber SA. Clinical and cognitive improvement following full-spectrum, high-cannabidiol treatment for anxiety: open-label data from a two-stage, phase 2 clinical trial. Commun Med. (2022) 2:139. doi: 10.1038/s43856-022-00202-8
17. Larsen C, Shahinas J. Dosage, efficacy and safety of cannabidiol administration in adults: a systematic review of human trials. J Clin Med Res. (2020) 12:129–41. doi: 10.14740/jocmr4090
18. Kaur P, Sharma S. Recent advances in pathophysiology of traumatic brain injury. Curr Neuropharmacol. (2018) 16:1224. doi: 10.2174/1570159X15666170613083606
19. Barkhoudarian G, Hovda DA, Giza CC. The molecular pathophysiology of concussive brain injury. Clin Sports Med. (2011) 30:33–48. doi: 10.1016/j.csm.2010.09.001
20. Dekosky ST, Asken BM. Brain injury injury cascades in TBI-related neurodegeneration injury cascades in TBI-related neurodegeneration. Brain Inj. (2017) 31:1177–82. doi: 10.1080/02699052.2017.1312528
21. Pearn ML, Niesman IR, Egawa J, Sawada A, Almenar-Queralt A, Shah SB, et al. Pathophysiology associated with traumatic brain injury: current treatments and potential novel therapeutics. Cell Mol Neurobiol. (2017) 37:571–85. doi: 10.1007/s10571-016-0400-1
22. Xiong K, Zhu Y, Zhang Y, Yin Z, Zhang J, Qiu M, et al. White matter integrity and cognition in mild traumatic brain injury following motor vehicle accident. Brain Res. (2014) 1591:86–92. doi: 10.1016/j.brainres.2014.10.030
23. Sulhan S, Lyon KA, Shapiro LA, Huang JH. Neuroinflammation and blood–brain barrier disruption following traumatic brain injury: pathophysiology and potential therapeutic targets. J Neurosci Res. (2020) 98:19–28. doi: 10.1002/jnr.24331
24. Guerriero RM, Giza CC, Rotenberg A. Glutamate and GABA imbalance following traumatic brain injury. Curr Neurol Neurosci Rep. (2015) 15:27. doi: 10.1007/s11910-015-0545-1
25. McGuire JL, Ngwenya LB, McCullumsmith RE. Neurotransmitter changes after traumatic brain injury: an update for new treatment strategies. Mol Psychiatry. (2018) 24:995–1012. doi: 10.1038/s41380-018-0239-6
26. Simon DW, McGeachy MJ, Baylr H, Clark RSB, Loane DJ, Kochanek PM. The far-reaching scope of neuroinflammation after traumatic brain injury. Nat Rev Neurol. (2017) 13:171–91. doi: 10.1038/nrneurol.2017.13
27. Santhakumar V, Ratzliff ADH, Jeng J, Toth Z, Soltesz I. Long-term hyperexcitability in the hippocampus after experimental head trauma. Ann Neurol. (2001) 50:708–17. doi: 10.1002/ana.1230
28. Guley NM, Del Mar NA, Ragsdale T, Li C, Perry AM, Moore BM, et al. Amelioration of visual deficits and visual system pathology after mild TBI with the cannabinoid type-2 receptor inverse agonist SMM-189. Exp Eye Res. (2019) 182:109–24. doi: 10.1016/j.exer.2019.03.013
29. Hsieh CL, Kim CC, Ryba BE, Niemi EC, Bando JK, Locksley RM, et al. Traumatic brain injury induces macrophage subsets in the brain. Eur J Immunol. (2013) 43:2010–22. doi: 10.1002/eji.201243084
30. Khatri N, Thakur M, Pareek V, Kumar S, Sharma S, Datusalia AK. Oxidative stress: major threat in traumatic brain injury. CNS Neurol Disord Drug Targets. (2018) 17:689–95. doi: 10.2174/1871527317666180627120501
31. Chaban V, Clarke GJB, Skandsen T, Islam R, Einarsen CE, Vik A, et al. Systemic inflammation persists the first year after mild traumatic brain injury: results from the prospective trondheim mild traumatic brain injury study. J Neurotrauma. (2020) 37:2120–30. doi: 10.1089/neu.2019.6963
32. Faden AI, Wu J, Stoica BA, Loane DJ. Progressive inflammation-mediated neurodegeneration after traumatic brain or spinal cord injury. Br J Pharmacol. (2016) 173:681–91. doi: 10.1111/bph.13179
33. Kokiko-Cochran ON, Godbout JP. The inflammatory continuum of traumatic brain injury and Alzheimer's disease. Front Immunol. (2018) 9:672. doi: 10.3389/fimmu.2018.00672
34. Donat CK, Scott G, Gentleman SM, Sastre M. Microglial activation in traumatic brain injury. Front Aging Neurosci. (2017) 9:208. doi: 10.3389/fnagi.2017.00208
35. Smith DH, Chen XH, Pierce JES, Wolf JA, Trojanowski JQ, Graham DI, et al. Progressive atrophy and neuron death for one year following brain trauma in the rat. J Neurotrauma. (1997) 14:715–27. doi: 10.1089/neu.1997.14.715
36. Petras JM, Bauman RA, Elsayed NM. Visual system degeneration induced by blast overpressure. Toxicology. (1997) 121:41–9. doi: 10.1016/S0300-483X(97)03654-8
37. Shitaka Y, Tran HT, Bennett RE, Sanchez L, Levy MA, Dikranian K, et al. Repetitive closed-skull traumatic brain injury in mice causes persistent multifocal axonal injury and microglial reactivity. J Neuropathol Exp Neurol. (2011) 70:551. doi: 10.1097/NEN.0b013e31821f891f
38. Mckee AC, Daneshvar DH. The neuropathology of traumatic brain injury. Handb Clin Neurol. (2015) 127:45. doi: 10.1016/B978-0-444-52892-6.00004-0
39. Tenovuo O, Diaz-Arrastia R, Goldstein LE, Sharp DJ, Van Der Naalt J, Zasler ND. Clinical medicine assessing the severity of traumatic brain injury-time for a change? J Clin Med. (2021) 10:148. doi: 10.3390/jcm10010148
40. Cifu DX, Grammer GG. VA/DoD Clinical Practice Guideline For The Management Of Concussion-Mild Traumatic Brain Injury The Management of Concussion-mild Traumatic Brain Injury Working Group. (2016). Available online at: www.tricare.mil (accessed December 28, 2022).
41. Gerberding JL, Binder S. Report to Congress on Mild Traumatic Brain Injury in the United States: Steps to Prevent a Serious Public Health Problem. Atlanta, GA (2003). Available online at: https://www.cdc.gov/traumaticbraininjury/pdf/mtbireporta.pdf (accessed December 28, 2022).
42. Holm L, Cassidy JD, Carroll LJ, Borg J. Summary of the WHO collaborating centre for neurotrauma task force on mild traumatic brain injury. J Rehabil Med. (2005) 37:137–41. doi: 10.1080/16501970510027321
43. Stein SC. Minor head injury: 13 is an unlucky number. J Trauma. (2001) 50:759–60. doi: 10.1097/00005373-200104000-00032
44. Rajajee V. Management of Acute Moderate and Severe Traumatic Brain Injury—UpToDate. (2022). Available online at: https://www.uptodate.com/contents/management-of-acute-moderate-and-severe-traumatic-brain-injury (accessed January 5, 2023).
45. Brasure M, Lamberty GJ, Sayer NA, Nelson NW, MacDonald R, Ouellette J, et al. Multidisciplinary postacute rehabilitation for moderate to severe traumatic brain injury in adults. Comp Eff Rev. (2012) 1–49.
46. Silverberg ND, Iverson GL, Arciniegas DB, Bayley MT, Bazarian JJ, Bell KR, et al. Expert panel survey to update the american congress of rehabilitation medicine definition of mild traumatic brain injury. Arch Phys Med Rehabil. (2021) 102:76–86. doi: 10.1016/j.apmr.2021.02.003
47. Vos PE, Battistin L, Birbamer G, Gerstenbrand F, Potapov A, Prevec T, et al. EFNS guideline on mild traumatic brain injury: report of an EFNS task force. Eur J Neurol. (2002) 9:207–19. doi: 10.1046/j.1468-1331.2002.00407.x
48. Cassidy JD, Carroll LJ, Peloso PM, Borg J, von Holst H, Holm L, et al. Incidence, risk factors and prevention of mild traumatic brain injury: results of the WHO collaborating centre task force on mild traumatic brain injury. J Rehabil Med. (2004) 36:28–60. doi: 10.1080/16501960410023732
49. Symptoms of Mild TBI and Concussion. Concussion, Traumatic Brain Injury, CDC Injury Center. Available online at: https://www.cdc.gov/traumaticbraininjury/concussion/symptoms.html (accessed December 28, 2022).
50. Evans RW, Whitlow CT. Acute Mild Traumatic Brain Injury (Concussion) in Adults. Uptodate.com (2021). Available online at: https://www.uptodate.com/contents/acutemild-traumatic-brain-injury-concussion-in-adults (accessed December 28, 2022).
51. Hoffer ME, Gottshall KR, Moore R, Balough BJ, Wester D. Characterizing and treating dizziness after mild head trauma. Otol Neurotol. (2004) 25:135–8. doi: 10.1097/00129492-200403000-00009
52. Abbassi E, Sirmon-Taylor B. Recovery progression and symptom resolution in sport-related mild traumatic brain injury. Brain Inj. (2017) 31:1667–73. doi: 10.1080/02699052.2017.1357834
53. Abdelmalik PA, Draghic N, Ling GSF. Management of moderate and severe traumatic brain injury. Transfusion. (2019) 59:1529–38. doi: 10.1111/trf.15171
54. Alvis-Miranda H, Milena Castellar-Leones S, Rafael Moscote-Salazar L, Rafael Moscote L. Decompressive craniectomy and traumatic brain injury: a review. Bull Emerg Trauma. (2013) 1:60.
55. Singh J, Neary JP. Neuroprotection following concussion: the potential role for cannabidiol. Can J Neurol Sci. (2020) 47:289–300. doi: 10.1017/cjn.2020.23
56. Jiang H, Li H, Cao Y, Zhang R, Zhou L, Zhou Y, et al. Effects of cannabinoid (CBD) on blood brain barrier permeability after brain injury in rats. Brain Res. (2021) 1768:147586. doi: 10.1016/j.brainres.2021.147586
57. Dearborn JT, Nelvagal HR, Rensing NR, Takahashi K, Hughes SM, Wishart TM, et al. Effects of chronic cannabidiol in a mouse model of naturally occurring neuroinflammation, neurodegeneration, and spontaneous seizures. Sci Rep. (2022) 12:11286. doi: 10.1038/s41598-022-15134-5
58. Alexandre De Souza Crippa J, Zuardi AW, Garrido GE, Wichert-Ana L, Guarnieri R, Ferrari L, et al. Effects of cannabidiol (CBD) on regional cerebral blood flow. Neuropsychopharmacology. (2004) 29:417–26. doi: 10.1038/sj.npp.1300340
59. Patra PH, Barker-Haliski M, White HS, Whalley BJ, Glyn S, Sandhu H, et al. Cannabidiol reduces seizures and associated behavioral comorbidities in a range of animal seizure and epilepsy models. Epilepsia. (2019) 60:303–14. doi: 10.1111/epi.14629
60. Campos AC, Ortega Z, Palazuelos J, Fogaça M V, Aguiar DC, Díaz-Alonso J, et al. The anxiolytic effect of cannabidiol on chronically stressed mice depends on hippocampal neurogenesis: involvement of the endocannabinoid system. Int J Neuropsychopharmacol. (2013) 16:1407–19. doi: 10.1017/S1461145712001502
61. Nilsson P, Hillered L, Olsson Y, Sheardown MJ, Hansen AJ. Regional changes in interstitial K+ and Ca2+ levels following cortical compression contusion trauma in rats. J Cereb Blood Flow Metab. (1993) 13:183–92. doi: 10.1038/jcbfm.1993.22
62. Fineman I, Hovda DA, Smith M, Yoshino A, Becker DP. Concussive brain injury is associated with a prolonged accumulation of calcium: a45Ca autoradiographic study. Brain Res. (1993) 624:94–102. doi: 10.1016/0006-8993(93)90064-T
63. Weber JT. Altered calcium signaling following traumatic brain injury. Front Pharmacol. (2012) 3:60. doi: 10.3389/fphar.2012.00060
64. Ikematsu K, Tsuda R, Kondo T, Nakasono I. The expression of excitatory amino acid transporter 2 in traumatic brain injury. Forensic Sci Int. (2002) 130:83–9. doi: 10.1016/S0379-0738(02)00344-4
65. Van Landeghem FKH, Weiss T, Oehmichen M, Von Deimling A. Decreased expression of glutamate transporters in astrocytes after human traumatic brain injury. J Neurotrauma. (2006) 23:1518–28. doi: 10.1089/neu.2006.23.1518
66. Palmer AM, Marion DW, Botscheller ML, Swedlow PE, Styren SD, DeKosky ST. Traumatic brain injury-induced excitotoxicity assessed in a controlled cortical impact model. J Neurochem. (1993) 61:2015–24. doi: 10.1111/j.1471-4159.1993.tb07437.x
67. Ng SY, Lee AYW. Traumatic brain injuries: pathophysiology and potential therapeutic targets. Front Cell Neurosci. (2019) 13:528. doi: 10.3389/fncel.2019.00528
68. Bu W, Ren H, Deng Y, Mar N Del, Guley NM, Moore BM, et al. mild traumatic brain injury produces neuron loss that can be rescued by modulating microglial activation using a cb2 receptor inverse agonist. Front Neurosci. (2016) 10:449. doi: 10.3389/fnins.2016.00449
69. Spaethling JM, Klein DM, Singh P, Meaney DF. Calcium-permeable AMPA receptors appear in cortical neurons after traumatic mechanical injury and contribute to neuronal fate. J Neurotrauma. (2008) 25:1207–16. doi: 10.1089/neu.2008.0532
70. Hardingham GE, Fukunaga Y, Bading H. Extrasynaptic NMDARs oppose synaptic NMDARs by triggering CREB shut-off and cell death pathways. Nat Neurosci. (2002) 5:405–14. doi: 10.1038/nn835
71. Schumm SN, Vigilante NF, Meaney DF. Nmda receptor alterations after mild traumatic brain injury induce deficits in memory acquisition and recall. Neural Comput. (2021) 33:67–95. doi: 10.1162/neco_a_01343
72. Raible DJ, Frey LC, Cruz Del Angel Y, Russek SJ, Brooks-Kayal AR. GABAA receptor regulation after experimental traumatic brain injury. J Neurotrauma. (2012) 29:2548. doi: 10.1089/neu.2012.2483
73. Lund V I, Hu Y, Raol YSH, Benham RS, Faris R, Russek SJ, et al. BDNF selectively regulates GABAA receptor transcription by activation of the JAK/STAT pathway. Sci Signal. (2008) 1:ra9. doi: 10.1126/scisignal.1162396
74. Raible DJ, Frey LC, Del Angel YC, Carlsen J, Hund D, Russek SJ, et al. JAK/STAT pathway regulation of GABAA receptor expression after differing severities of experimental TBI. Exp Neurol. (2015) 271:445–56. doi: 10.1016/j.expneurol.2015.07.001
75. Roberts DS, Hu Y, Lund I V., Brooks-Kayal AR, Russek SJ. Brain-derived neurotrophic factor (BDNF)-induced synthesis of early growth response factor 3 (Egr3) controls the levels of type A GABA receptor α4 subunits in hippocampal neurons. J Biol Chem. (2006) 281:29431–5. doi: 10.1074/jbc.C600167200
76. De Beaumont L, Tremblay S, Poirier J, Lassonde M, Théoret H. Altered bidirectional plasticity and reduced implicit motor learning in concussed athletes. Cereb Cortex. (2012) 22:112–21. doi: 10.1093/cercor/bhr096
77. Saija A, Hayes RL, Lyeth BG, Edward Dixon C, Yamamoto T, Robinson SE. The effect of concussive head injury on central cholinergic neurons. Brain Res. (1988) 452:303–11. doi: 10.1016/0006-8993(88)90034-0
78. Abe K, Shimada R, Okada Y, Kibayashi K. Traumatic brain injury decreases serotonin transporter expression in the rat cerebrum. Neurol Res. (2016) 38:358–63. doi: 10.1080/01616412.2015.1110402
79. Dixon CE, Bao J, Johnson KM, Yang K, Whitson J, Clifton GL, et al. Basal and scopolamine-evoked release of hippocampal acetylcholine following traumatic brain injury in rats. Neurosci Lett. (1995) 198:111–4. doi: 10.1016/0304-3940(95)11979-7
80. Lyeth BG, Jiang JY, Delahunty TM, Phillips LL, Hamm RJ. Muscarinic cholinergic receptor binding in rat brain following traumatic brain injury. Brain Res. (1994) 640:240–5. doi: 10.1016/0006-8993(94)91879-1
81. Gorman LK, Fu K, Hovda DA, Murray M, Traystman RJ. Effects of traumatic brain injury on the cholinergic system in the rat. J Neurotrauma. (2009) 13:457–63. doi: 10.1089/neu.1996.13.457
82. Ma S, Hangya B, Leonard CS, Wisden W, Gundlach AL. Dual-transmitter systems regulating arousal, attention, learning and memory. Neurosci Biobehav Rev. (2018) 85:21–33. doi: 10.1016/j.neubiorev.2017.07.009
83. Scholten AC, Haagsma JA, Cnossen MC, Olff M, Van Beeck EF, Polinder S. Prevalence of and risk factors for anxiety and depressive disorders after traumatic brain injury: a systematic review. J Neurotrauma. (2016) 33:1969–94. doi: 10.1089/neu.2015.4252
84. Kobori N, Clifton GL, Dash PK. Enhanced catecholamine synthesis in the prefrontal cortex after traumatic brain injury: implications for prefrontal dysfunction. J Neurotrauma. (2006) 23:1094–102. doi: 10.1089/neu.2006.23.1094
85. Wong VS, Langley B. Epigenetic changes following traumatic brain injury and their implications for outcome, recovery and therapy. Neurosci Lett. (2016) 625:26–33. doi: 10.1016/j.neulet.2016.04.009
86. Jenkins PO, Mehta MA, Sharp DJ. Catecholamines and cognition after traumatic brain injury. Brain. (2016) 139:2345–71. doi: 10.1093/brain/aww128
87. Shin SS, Bray ER, Zhang CQ, Dixon CE. Traumatic brain injury reduces striatal tyrosine hydroxylase activity and potassium-evoked dopamine release in rats. Brain Res. (2011) 1369:208–15. doi: 10.1016/j.brainres.2010.10.096
88. Fujinaka T, Kohmura E, Yuguchi T, Yoshimine T. The morphological and neurochemical effects of diffuse brain injury on rat central noradrenergic system. Neurol Res. (2013) 25:35–41. doi: 10.1179/016164103101201094
89. Elder GA, Cristian A. Blast-related mild traumatic brain injury: mechanisms of injury and impact on clinical care. Mt Sinai J Med A J Transl Pers Med. (2009) 76:111–8. doi: 10.1002/msj.20098
90. Chen YH, Huang EYK, Kuo TT, Miller J, Chiang YH, Hoffer BJ. Impact of traumatic brain injury on dopaminergic transmission. Cell Transplant. (2017) 26:1156–68. doi: 10.1177/0963689717714105
91. Feng Y, Lu Y. Immunomodulatory effects of dopamine in inflammatory diseases. Front Immunol. (2021) 12:987. doi: 10.3389/fimmu.2021.663102
92. Ruttimann Y, Schutz Y, Jequier E, Lemarchand T, Chiolero R. Thermogenic and metabolic effects of dopamine in healthy men. Crit Care Med. (1991) 19:1030–6. doi: 10.1097/00003246-199108000-00010
93. Williams SN, Undieh AS. Dopamine D1-like receptor activation induces brain-derived neurotrophic factor protein expression. Neuroreport. (2009) 20:606. doi: 10.1097/WNR.0b013e32832a0a98
94. Gustafsson D, Klang A, Thams S, Rostami E. The role of bdnf in experimental and clinical traumatic brain injury. Int J Mol Sci. (2021) 22:3582. doi: 10.3390/ijms22073582
95. Failla MD, Conley YP, Wagner AK. Brain-derived neurotrophic factor (BDNF) in traumatic brain injury-related mortality: interrelationships between genetics and acute systemic and central nervous system BDNF profiles. Neurorehabil Neural Repair. (2016) 30:83–93. doi: 10.1177/1545968315586465
96. Shin SS, Dixon CE. Alterations in cholinergic pathways and therapeutic strategies targeting cholinergic system after traumatic brain injury. J Neurotrauma. (2015) 32:1429. doi: 10.1089/neu.2014.3445
97. Mufson EJ, Perez SE, Nadeem M, Mahady L, Kanaan NM, Abrahamson EE, et al. Progression of tau pathology within cholinergic nucleus basalis neurons in chronic traumatic encephalopathy: a chronic effects of neurotrauma consortium study. Brain Inj. (2016) 30:1399–413. doi: 10.1080/02699052.2016.1219058
98. Mattson AJ, Levin HS. Frontal lobe dysfunction following closed head injury. A review of the literature. J Nerv Ment Dis. (1990) 178:282–91. doi: 10.1097/00005053-199005000-00002
99. Mayeux J, Katz P, Edwards S, Middleton JW, Molina PE. Inhibition of endocannabinoid degradation improves outcomes from mild traumatic brain injury: a mechanistic role for synaptic hyperexcitability. J Neurotrauma. (2017) 34:436. doi: 10.1089/neu.2016.4452
100. Fucich EA, Mayeux JP, McGinn MA, Stoulig PJ, Romero LM, Edwards S, et al. Inhibition of endocannabinoid degradation improves behavioral effects of traumatic brain injury that are associated with amygdalar hyperexcitability in rats. FASEB J. (2018) 32:877.5. doi: 10.1096/fasebj.2018.32.1_supplement.877.5
101. Katz PS, Sulzer JK, Impastato RA, Teng SX, Rogers EK, Molina PE. Endocannabinoid degradation inhibition improves neurobehavioral function, blood-brain barrier integrity, and neuroinflammation following mild traumatic brain injury. J Neurotrauma. (2014) 32:297–306. doi: 10.1089/neu.2014.3508
102. Witkamp R, Meijerink J. The endocannabinoid system: an emerging key player in inflammation. Curr Opin Clin Nutr Metab Care. (2014) 17:130–8. doi: 10.1097/MCO.0000000000000027
103. Mackie K. Cannabinoid receptors: where they are and what they do. J Neuroendocrinol. (2008) 20:10–4. doi: 10.1111/j.1365-2826.2008.01671.x
104. Gaffal E, Cron M, Glodde N, Bald T, Kuner R, Zimmer A, et al. Cannabinoid 1 receptors in keratinocytes modulate proinflammatory chemokine secretion and attenuate contact allergic inflammation. J Immunol. (2013) 190:4929–36. doi: 10.4049/jimmunol.1201777
105. Kendall DA, Yudowski GA, Wang H, Cannizzaro C, Dowd E. Cannabinoid receptors in the central nervous system: their signaling and roles in disease. Front Cell Neurosci. (2017) 10:294. doi: 10.3389/fncel.2016.00294
106. De Giacomo V, Ruehle S, Lutz B, Häring M, Remmers F. Differential glutamatergic and GABAergic contributions to the tetrad effects of Δ9-tetrahydrocannabinol revealed by cell-type-specific reconstitution of the CB1 receptor. Neuropharmacology. (2020) 179:108287. doi: 10.1016/j.neuropharm.2020.108287
107. Rey AA, Purrio M, Viveros M-P, Lutz B. Biphasic effects of cannabinoids in anxiety responses: CB1 and GABAB receptors in the balance of GABAergic and glutamatergic neurotransmission. Neuropsychopharmacology. (2012) 37:2624–2634. doi: 10.1038/npp.2012.123
108. Fortin DA, Levine ES. Differential effects of endocannabinoids on glutamatergic and GABAergic inputs to layer 5 pyramidal neurons. Cereb Cortex. (2007) 17:163–74. doi: 10.1093/cercor/bhj133
109. Turcotte C, Blanchet MR, Laviolette M, Flamand N. The CB2 receptor and its role as a regulator of inflammation. Cell Mol Life Sci. (2016) 73:4449. doi: 10.1007/s00018-016-2300-4
110. Cabral GA, Raborn ES, Griffin L, Dennis J, Marciano-Cabral F. CB2 receptors in the brain: role in central immune function. Br J Pharmacol. (2008) 153:240. doi: 10.1038/sj.bjp.0707584
111. Braun M, Khan ZT, Khan MB, Kumar M, Ward A, Achyut BR, et al. Selective activation of cannabinoid receptor-2 reduces neuroinflammation after traumatic brain injury via alternative macrophage polarization. Brain Behav Immun. (2018) 68:224. doi: 10.1016/j.bbi.2017.10.021
112. Belen Lopez-Rodriguez A, Siopi E, Finn DP, Marchand-Leroux C, Garcia-Segura LM, Jafarian-Tehrani M, et al. CB 1 and CB 2 cannabinoid receptor antagonists prevent minocycline-induced neuroprotection following traumatic brain injury in mice. Cereb Cortex. (2013) 25:35–45. doi: 10.1093/cercor/bht202
113. Selvaraj P, Tanaka M, Wen J, Zhang Y. The novel monoacylglycerol lipase inhibitor MJN110 suppresses neuroinflammation, normalizes synaptic composition and improves behavioral performance in the repetitive traumatic brain injury mouse model. Cells. (2021) 10:3454. doi: 10.3390/cells10123454
114. Zhang J, Teng Z, Song Y, Hu M, Chen C. Inhibition of monoacylglycerol lipase prevents chronic traumatic encephalopathy-like neuropathology in a mouse model of repetitive mild closed head injury. J Cereb Blood Flow Metab. (2015) 35:443–53. doi: 10.1038/jcbfm.2014.216
115. Sigel E, Baur R, Racz I, Marazzi J, Smart TG, Zimmer A, et al. The major central endocannabinoid directly acts at GABAA receptors. Proc Natl Acad Sci USA. (2011) 108:18150–5. doi: 10.1073/pnas.1113444108
116. Brickley SG, Mody I. Extrasynaptic GABAA receptors: their function in the CNS and implications for disease. Neuron. (2012) 73:23. doi: 10.1016/j.neuron.2011.12.012
117. Burston JJ, Wiley JL, Craig AA, Selley DE, Sim-Selley LJ. Regional enhancement of cannabinoid CB1 receptor desensitization in female adolescent rats following repeated Δ9-tetrahydrocannabinol exposure. Br J Pharmacol. (2010) 161:103. doi: 10.1111/j.1476-5381.2010.00870.x
118. Martin BR, Sim-Selley LJ, Selley DE. Signaling pathways involved in the development of cannabinoid tolerance. Trends Pharmacol Sci. (2004) 25:325–30. doi: 10.1016/j.tips.2004.04.005
119. Zuardi AW, Rodrigues NP, Silva AL, Bernardo SA, Hallak JEC, Guimarães FS, et al. Inverted U-shaped dose-response curve of the anxiolytic effect of cannabidiol during public speaking in real life. Front Pharmacol. (2017) 8:259. doi: 10.3389/fphar.2017.00259
120. Kaplan JS, Stella N, Catterall WA, Westenbroek RE. Cannabidiol attenuates seizures and social deficits in a mouse model of Dravet syndrome. Proc Natl Acad Sci USA. (2017) 114:11229–34. doi: 10.1073/pnas.1711351114
121. Nedelescu H, Wagner GE, De Ness GL, Carroll A, Kerr TM, Wang J, et al. Cannabidiol produces distinct U-shaped dose-response effects on cocaine-induced conditioned place preference and associated recruitment of prelimbic neurons in male rats. Biol psychiatry Glob open Sci. (2022) 2:70–8. doi: 10.1016/j.bpsgos.2021.06.014
122. Devinsky O, Cross JH, Laux L, Marsh E, Miller I, Nabbout R, et al. Trial of cannabidiol for drug-resistant seizures in the Dravet syndrome. N Engl J Med. (2017) 376:2011–20. doi: 10.1056/NEJMoa1611618
123. Millar SA, Stone NL, Yates AS, O'Sullivan SE. A systematic review on the pharmacokinetics of cannabidiol in humans. Front Pharmacol. (2018) 9:1365. doi: 10.3389/fphar.2018.01365
124. Panikashvili D, Simeonidou C, Ben-Shabat S, Hanuš L, Breuer A, Mechoulam R, et al. An endogenous cannabinoid (2-AG) is neuroprotective after brain injury. Nature. (2001) 413:527–31. doi: 10.1038/35097089
125. Elmes MW, Kaczocha M, Berger WT, Leung KN, Ralph BP, Wang L, et al. Fatty acid-binding proteins (FABPs) are intracellular carriers for Δ9-tetrahydrocannabinol (THC) and cannabidiol (CBD). J Biol Chem. (2015) 290:8711–21. doi: 10.1074/jbc.M114.618447
126. Maione S, Piscitelli F, Gatta L, Vita D, De Petrocellis L, Palazzo E, et al. Non-psychoactive cannabinoids modulate the descending pathway of antinociception in anaesthetized rats through several mechanisms of action. Br J Pharmacol. (2011) 162:584–96. doi: 10.1111/j.1476-5381.2010.01063.x
127. McPartland JM, Duncan M, Di Marzo V, Pertwee RG. Are cannabidiol and Δ9-tetrahydrocannabivarin negative modulators of the endocannabinoid system? A systematic review. Br J Pharmacol. (2015) 172:737. doi: 10.1111/bph.12944
128. Martínez-Pinilla E, Varani K, Reyes-Resina I, Angelats E, Vincenzi F, Ferreiro-Vera C, et al. Binding and signaling studies disclose a potential allosteric site for cannabidiol in cannabinoid CB 2 receptors. Front Pharmacol. (2017) 8:744. doi: 10.3389/fphar.2017.00744
129. Campos AC, Brant F, Miranda AS, Machado FS, Teixeira AL. Cannabidiol increases survival and promotes rescue of cognitive function in a murine model of cerebral malaria. Neuroscience. (2015) 289:166–80. doi: 10.1016/j.neuroscience.2014.12.051
130. Shohami E, Cohen-Yeshurun A, Magid L, Algali M, Mechoulam R. Endocannabinoids and traumatic brain injury. Br J Pharmacol. (2011) 163:1402. doi: 10.1111/j.1476-5381.2011.01343.x
131. Hampson AJ, Grimaldi M, Axelrod J, Wink D. Cannabidiol and (–)Δ9-tetrahydrocannabinol are neuroprotective antioxidants. Proc Natl Acad Sci USA. (1998) 95:8268. doi: 10.1073/pnas.95.14.8268
132. Stollenwerk TM, Pollock S, Hillard CJ. Contribution of the adenosine 2A receptor to behavioral effects of tetrahydrocannabinol, cannabidiol and PECS-101. Molecules. (2021) 26:5354. doi: 10.3390/molecules26175354
133. Aso E, Fernández-Dueñas V, López-Cano M, Taura J, Watanabe M, Ferrer I, et al. Adenosine A2A-cannabinoid CB1 receptor heteromers in the hippocampus: cannabidiol blunts δ9-tetrahydrocannabinol-induced cognitive impairment. Mol Neurobiol. (2019) 56:5382–91. doi: 10.1007/s12035-018-1456-3
134. Maroon J, Bost J. Review of the neurological benefits of phytocannabinoids. Surg Neurol Int. (2018) 9:91. doi: 10.4103/sni.sni_45_18
135. Ikonomidou C, Turski L. Why did NMDA receptor antagonists fail clinical trials for stroke and traumatic brain injury? Lancet Neurol. (2002) 1:383–6. doi: 10.1016/S1474-4422(02)00164-3
136. Lindenschmidt RC, Tryka AF, Goad ME, Witschi HP. The effects of dietary butylated hydroxytoluene on liver and colon tumor development in mice. Toxicology. (1986) 38:151–60. doi: 10.1016/0300-483X(86)90116-2
137. Bakas T, van Nieuwenhuijzen PS, Devenish SO, McGregor IS, Arnold JC, Chebib M. The direct actions of cannabidiol and 2-arachidonoyl glycerol at GABAA a receptors. Pharmacol Res. (2017) 119:358–70. doi: 10.1016/j.phrs.2017.02.022
138. Lan YL, Li S, Lou JC, Ma XC, Zhang B. The potential roles of dopamine in traumatic brain injury: a preclinical and clinical update. Am J Transl Res. (2019) 11:2616–31.
139. Robert S. Traumatic brain injury and mood disorders. Ment Heal Clin. (2020) 10:335–80. doi: 10.9740/mhc.2020.11.335
140. Bales JW, Wagner AK, Kline AE, Dixon CE. Persistent cognitive dysfunction after traumatic brain injury: a dopamine hypothesis. Neurosci Biobehav Rev. (2009) 33:981–1003. doi: 10.1016/j.neubiorev.2009.03.011
141. De Almeida DL, Devi LA. Diversity of molecular targets and signaling pathways for CBD. Pharmacol Res Perspect. (2020) 8:e00682. doi: 10.1002/prp2.682
142. Seeman P. Cannabidiol is a partial agonist at dopamine D2High receptors, predicting its antipsychotic clinical dose. Transl Psychiatry. (2016) 6:e920. doi: 10.1038/tp.2016.195
143. Poddar MK, Dewey WL. Effects of cannabinoids on catecholamine uptake and release in hypothalamic and striatal synaptosomes. J Pharmacol Exp Ther. (1980) 214:63–7.
144. García C, Palomo-Garo C, García-Arencibia M, Ramos JA, Pertwee RG, Fernández-Ruiz J. Symptom-relieving and neuroprotective effects of the phytocannabinoid Δ9-THCV in animal models of Parkinson's disease. Br J Pharmacol. (2011) 163:1495–506. doi: 10.1111/j.1476-5381.2011.01278.x
145. Peres FF, Lima AC, Hallak JEC, Crippa JA, Silva RH, Abílio VC. Cannabidiol as a promising strategy to treat and prevent movement disorders? Front Pharmacol. (2018) 9:482. doi: 10.3389/fphar.2018.00482
146. Russo EB. The case for the entourage effect and conventional breeding of clinical cannabis: no “strain,” no gain. Front Plant Sci. (2019) 9:1969. doi: 10.3389/fpls.2018.01969
147. Russo EB, Burnett A, Hall B, Parker KK. Agonistic properties of cannabidiol at 5-HT1a receptors. Neurochem Res. (2005) 30:1037–43. doi: 10.1007/s11064-005-6978-1
148. De Gregorio D, McLaughlin RJ, Posa L, Ochoa-Sanchez R, Enns J, Lopez-Canul M, et al. Cannabidiol modulates serotonergic transmission and reverses both allodynia and anxiety-like behavior in a model of neuropathic pain. Pain. (2019) 160:136–50. doi: 10.1097/j.pain.0000000000001386
149. Martínez-Aguirre C, Carmona-Cruz F, Velasco AL, Velasco F, Aguado-Carrillo G, Cuéllar-Herrera M, et al. Cannabidiol acts at 5-HT1A receptors in the human brain: relevance for treating temporal lobe epilepsy. Front Behav Neurosci. (2020) 14:611278. doi: 10.3389/fnbeh.2020.611278
150. Linge R, Jiménez-Sánchez L, Campa L, Pilar-Cuéllar F, Vidal R, Pazos A, et al. Cannabidiol induces rapid-acting antidepressant-like effects and enhances cortical 5-HT/glutamate neurotransmission: role of 5-HT1A receptors. Neuropharmacology. (2016) 103:16–26. doi: 10.1016/j.neuropharm.2015.12.017
151. Campos AC, Guimarães FS. Involvement of 5HT1A receptors in the anxiolytic-like effects of cannabidiol injected into the dorsolateral periaqueductal gray of rats. Psychopharmacology. (2008) 199:223–30. doi: 10.1007/s00213-008-1168-x
152. Carrier EJ, Auchampach JA, Hillard CJ. Inhibition of an equilibrative nucleoside transporter by cannabidiol: a mechanism of cannabinoid immunosuppression. Proc Natl Acad Sci USA. (2006) 103:7895–900. doi: 10.1073/pnas.0511232103
153. Yue JK, Burke JF, Upadhyayula PS, Winkler EA, Deng H, Robinson CK, et al. Selective serotonin reuptake inhibitors for treating neurocognitive and neuropsychiatric disorders following traumatic brain injury: an evaluation of current evidence. Brain Sci. (2017) 7:93. doi: 10.3390/brainsci7080093
154. Pazos MR, Mohammed N, Lafuente H, Santos M, Martínez-Pinilla E, Moreno E, et al. Mechanisms of cannabidiol neuroprotection in hypoxic–ischemic newborn pigs: role of 5HT1A and CB2 receptors. Neuropharmacology. (2013) 71:282–91. doi: 10.1016/j.neuropharm.2013.03.027
155. Shinoda J, Yano H, Nakayama N. Altered biphasic serotonin discharge hypothesis in mild traumatic brain injury. Concussion. (2021) 6:CNC94. doi: 10.2217/cnc-2021-0001
156. Ryberg E, Larsson N, Sjögren S, Hjorth S, Hermansson N-O, Leonova J, et al. The orphan receptor GPR55 is a novel cannabinoid receptor. Br J Pharmacol. (2007) 152:1092–101. doi: 10.1038/sj.bjp.0707460
157. Pandelides Z, Aluru N, Thornton C, Watts HE, Willett KL. Transcriptomic changes and the roles of cannabinoid receptors and PPARγ in developmental toxicities following exposure to δ9-tetrahydrocannabinol and cannabidiol. Toxicol Sci. (2021) 99:366–94. doi: 10.1093/toxsci/kfab046
158. Landucci E, Mazzantini C, Lana D, Davolio PL, Giovannini MG, Pellegrini-Giampietro DE. Neuroprotective effects of cannabidiol but not δ9-tetrahydrocannabinol in rat hippocampal slices exposed to oxygen-glucose deprivation: studies with cannabis extracts and selected cannabinoids. Int J Mol Sci. (2021) 22:9773. doi: 10.3390/ijms22189773
159. Oláh A, Tóth BI, Borbíró I, Sugawara K, Szöllõsi AG, Czifra G, et al. Cannabidiol exerts sebostatic and antiinflammatory effects on human sebocytes. J Clin Invest. (2014) 124:3713. doi: 10.1172/JCI64628
160. Oláh A, Bíró T. Targeting cutaneous cannabinoid signaling in inflammation - a “high”-way to heal? EBioMedicine. (2017) 16:3–5. doi: 10.1016/j.ebiom.2017.01.003
161. Costa B, Giagnoni G, Franke C, Trovato AE, Colleoni M. Vanilloid TRPV1 receptor mediates the antihyperalgesic effect of the nonpsychoactive cannabinoid, cannabidiol, in a rat model of acute inflammation. Br J Pharmacol. (2004) 143:247–50. doi: 10.1038/sj.bjp.0705920
162. Anand U, Jones B, Korchev Y, Bloom SR, Pacchetti B, Anand P, et al. CBD effects on TRPV1 signaling pathways in cultured drg neurons. J Pain Res. (2020) 13:2269–78. doi: 10.2147/JPR.S258433
163. Console-Bram L, Brailoiu E, Brailoiu GC, Sharir H, Abood ME. Activation of GPR18 by cannabinoid compounds: a tale of biased agonism. J Pharmacol. (2014) 171:3908–17. doi: 10.1111/bph.12746
164. Yu Y, Yang Z, Jin B, Qin X, Zhu X, Sun J, et al. Cannabidiol inhibits febrile seizure by modulating AMPA receptor kinetics through its interaction with the N-terminal domain of GluA1/GluA2. Pharmacol Res. (2020) 161:105128. doi: 10.1016/j.phrs.2020.105128
165. Pretzsch CM, Voinescu B, Mendez MA, Wichers R, Ajram L, Ivin G, et al. The effect of cannabidiol (CBD) on low-frequency activity and functional connectivity in the brain of adults with and without autism spectrum disorder (ASD). J Psychopharmacol. (2019) 33:1141–8. doi: 10.1177/0269881119858306
166. Pretzsch CM, Freyberg J, Voinescu B, Lythgoe D, Horder J, Mendez MA, et al. Effects of cannabidiol on brain excitation and inhibition systems; a randomised placebo-controlled single dose trial during magnetic resonance spectroscopy in adults with and without autism spectrum disorder. Neuropsychopharmacology. (2019) 9:313. doi: 10.1038/s41398-019-0654-8
167. Zheng W, Zhuge Q, Zhong M, Chen G, Shao B, Wang H, et al. Neurogenesis in adult human brain after traumatic brain injury. J Neurotrauma. (2013) 30:1872–80. doi: 10.1089/neu.2010.1579
168. Ernst C, Christie BR. Temporally specific proliferation events are induced in the hippocampus following acute focal injury. J Neurosci Res. (2005) 83:349–61. doi: 10.1002/jnr.20724
169. Xu N-J, Lagace D, Santhakumar V, Danzer SC, Ngwenya LB. Impact of traumatic brain injury on neurogenesis. Front Neurosci. (2019) 12:1014. doi: 10.3389/fnins.2018.01014
170. Wolf SA, Bick-Sander A, Fabel K, Leal-Galicia P, Tauber S, Ramirez-Rodriguez G, et al. Cannabinoid receptor CB1 mediates baseline and activity-induced survival of new neurons in adult hippocampal neurogenesis. Cell Commun Signal. (2010) 8:12. doi: 10.1186/1478-811X-8-12
171. Esposito G, Scuderi C, Valenza M, Togna GI, Latina V. Cannabidiol reduces ab-induced neuroinflammation and promotes hippocampal neurogenesis through PPARc involvement. PLoS ONE. (2011) 6:28668. doi: 10.1371/journal.pone.0028668
172. Yi JH, Park SW, Brooks N, Lang BT, Vemuganti R. PPARgamma agonist rosiglitazone is neuroprotective after traumatic brain injury via anti-inflammatory and anti-oxidative mechanisms. Brain Res. (2008) 1244:164–72. doi: 10.1016/j.brainres.2008.09.074
173. Luján M, Valverde O. The pro-neurogenic effects of cannabidiol and its potential therapeutic implications in psychiatric disorders. Front Behav Neurosci. (2020) 14:109. doi: 10.3389/fnbeh.2020.00109
174. Schiavon AP, Bonato JM, Milani H, Guimarães FS, Weffort de Oliveira RM. Influence of single and repeated cannabidiol administration on emotional behavior and markers of cell proliferation and neurogenesis in non-stressed mice. Prog Neuro-Psychopharmacology Biol Psychiatry. (2016) 64:27–34. doi: 10.1016/j.pnpbp.2015.06.017
175. Beale C, Broyd SJ, Chye Y, Suo C, Schira M, Galettis P, et al. Prolonged cannabidiol treatment effects on hippocampal subfield volumes in current cannabis users. Cannabis Cannabinoid Res. (2018) 3:94. doi: 10.1089/can.2017.0047
176. Bilbo S, Stevens B. Microglia: the brain's first responders. Cerebrum Dana Forum Brain Sci. (2017) 2017:cer-14–7.
177. Madathil SK, Wilfred BS, Urankar SE, Yang W, Leung LY, Gilsdorf JS, et al. Early microglial activation following closed-head concussive injury is dominated by pro-inflammatory M-1 type. Front Neurol. (2018) 9:964. doi: 10.3389/fneur.2018.00964
178. Cao T, Thomas TC, Ziebell JM, Pauly JR, Lifshitz J. Morphological and genetic activation of microglia after diffuse traumatic brain injury in the rat. Neuroscience. (2012) 225:65. doi: 10.1016/j.neuroscience.2012.08.058
179. Patterson ZR, Holahan MR. Understanding the neuroinflammatory response following concussion to develop treatment strategies. Front Cell Neurosci. (2012) 6:58. doi: 10.3389/fncel.2012.00058
180. Loane DJ, Kumar A. Microglia in the TBI brain: the good, the bad, and the dysregulated. Exp Neurol. (2016) 275:316. doi: 10.1016/j.expneurol.2015.08.018
181. Gao X, Chen J. Moderate traumatic brain injury promotes neural precursor proliferation without increasing neurogenesis in the adult hippocampus. Exp Neurol. (2013) 239:38–48. doi: 10.1016/j.expneurol.2012.09.012
182. Qin L, Liu Y, Wang T, Wei SJ, Block ML, Wilson B, et al. NADPH oxidase mediates lipopolysaccharide-induced neurotoxicity and proinflammatory gene expression in activated microglia. J Biol Chem. (2004) 279:1415–21. doi: 10.1074/jbc.M307657200
183. Yeisley DJ, Arabiyat AS, Hahn MS. Cannabidiol-Driven alterations to inflammatory protein landscape of lipopolysaccharide-activated macrophages in vitro may be mediated by autophagy and oxidative stress. Cannabis Cannabinoid Res. (2021) 6:253–63. doi: 10.1089/can.2020.0109
184. Pinhal-Enfield G, Ramanathan M, Hasko G, Vogel SN, Salzman AL, Boons GJ, et al. An angiogenic switch in macrophages involving synergy between toll-like receptors 2, 4, 7, and 9 and adenosine A2A receptors. Am J Pathol. (2003) 163:711–21. doi: 10.1016/S0002-9440(10)63698-X
185. Mecha M, Feliú A, Iñigo PM, Mestre L, Carrillo-Salinas FJ, Guaza C. Cannabidiol provides long-lasting protection against the deleterious effects of inflammation in a viral model of multiple sclerosis: a role for A2A receptors. Neurobiol Dis. (2013) 59:141–50. doi: 10.1016/j.nbd.2013.06.016
186. O'connell KM, Littleton-Kearney MT. The role of free radicals in traumatic brain injury. Biol Res Nurs. (2013) 15:253–63. doi: 10.1177/1099800411431823
187. Gilgun-Sherki Y, Rosenbaum Z, Melamed E, Offen D. Antioxidant therapy in acute central nervous system injury: current state. Pharmacol Rev. (2002) 54:271–84. doi: 10.1124/pr.54.2.271
188. Dohi K, Ohtaki H, Nakamachi T, Yofu S, Satoh K, Miyamoto K, et al. Gp91phox (NOX2) in classically activated microglia exacerbates traumatic brain injury. J Neuroinflammation. (2010) 7:41. doi: 10.1186/1742-2094-7-41
189. Atalay S, Jarocka-karpowicz I, Skrzydlewskas E. Antioxidative and anti-inflammatory properties of cannabidiol. Antioxidants. (2020) 9:21. doi: 10.3390/antiox9010021
190. Soares RZ, Vuolo F, Dall'Igna DM, Michels M, De Souza Crippa JA, Hallak JEC, et al. Avaliação do papel do sistema canabidiol em um modelo de lesão renal por isquemia/reperfusão em animais. Rev Bras Ter Intensiva. (2015) 27:383–9. doi: 10.5935/0103-507X.20150064
191. Borges RS, Batista J, Viana RB, Baetas AC, Orestes E, Andrade MA, et al. molecules understanding the molecular aspects of tetrahydrocannabinol and cannabidiol as antioxidants. Molecules. (2013) 18:12663–74. doi: 10.3390/molecules181012663
192. Hamelink C, Hampson A, Wink DA, Eiden LE, Eskay RL. Comparison of cannabidiol, antioxidants, and diuretics in reversing binge ethanol-induced neurotoxicity. J Pharmacol Exp Ther. (2005) 314:780–8. doi: 10.1124/jpet.105.085779
193. Rajesh M, Mukhopadhyay P, Bátkai S, Haskó G, Liaudet L, Drel VR, et al. Cannabidiol attenuates high glucose-induced endothelial cell inflammatory response and barrier disruption. Am J Physiol. (2007) 293:H610–9. doi: 10.1152/ajpheart.00236.2007
194. Pan H, Mukhopadhyay P, Rajesh M, Patel V, Mukhopadhyay B, Gao B, et al. Cannabidiol attenuates cisplatin-induced nephrotoxicity by decreasing oxidative/nitrosative stress, inflammation, and cell death. J Pharmacol Exp Ther. (2009) 328:708–14. doi: 10.1124/jpet.108.147181
195. Korley FK, Diaz-Arrastia R, Wu AHB, Yue JK, Manley GT, Sair HI, et al. Circulating brain-derived neurotrophic factor has diagnostic and prognostic value in traumatic brain injury. J Neurotrauma. (2016) 33:215. doi: 10.1089/neu.2015.3949
196. Chiaretti A, Piastra M, Polidori G, Di Rocco C, Caresta E, Antonelli A, et al. Correlation between neurotrophic factor expression and outcome of children with severe traumatic brain injury. Intensive Care Med. (2003) 29:1329–38. doi: 10.1007/s00134-003-1852-6
197. Huang EJ, Reichardt LF. Neurotrophins: roles in neuronal development and function. Annu Rev Neurosci. (2007) 24:677–736. doi: 10.1146/annurev.neuro.24.1.677
198. Cohen-Cory S, Kidane AH, Shirkey NJ, Marshak S. Brain-Derived neurotrophic factor and the development of structural neuronal connectivity. Inc Dev Neurobiol. (2010) 70:271–88. doi: 10.1002/dneu.20774
199. Kaplan GB, Vasterling JJ, Vedak PC. Brain-derived neurotrophic factor in traumatic brain injury, post-traumatic stress disorder, and their comorbid conditions: role in pathogenesis and treatment. Behav Pharmacol. (2010) 21:427–37. doi: 10.1097/FBP.0b013e32833d8bc9
200. Sofroniew M V, Howe CL, Mobley WC. Nerve growth factor signaling, neuroprotection, and neural repair. Annu Rev Neurosci. (2001) 24:1217–81. doi: 10.1146/annurev.neuro.24.1.1217
201. Sen T, Gupta XR, Kaiser H, Sen N. Activation of PERK elicits memory impairment through inactivation of CREB and downregulation of PSD95 after traumatic brain injury. J Neurosci. (2017) 37:5900–11. doi: 10.1523/JNEUROSCI.2343-16.2017
202. Sales AJ, Fogaça M V, Sartim AG, Pereira VS, Wegener G, Guimarães FS, et al. Cannabidiol induces rapid and sustained antidepressant-like effects through increased BDNF signaling and synaptogenesis in the prefrontal cortex. Mol Neurobiol. (2019) 56:1070–81. doi: 10.1007/s12035-018-1143-4
203. Rosa RF, Ugolini-Lopes MR, Gandara APR, Vendramini MBG, Campanholo KR, Dutra L, et al. Cognitive dysfunction and serum levels of brain-derived neurotrophic factor (BDNF) in primary anti-phospholipid syndrome (PAPS). Rheumatology. (2021) 60:179–87. doi: 10.1093/rheumatology/keaa252
204. Robinson RG, Szetela B. Mood change following left hemispheric brain injury. Ann Neurol. (1981) 9:447–53. doi: 10.1002/ana.410090506
205. Van Der Horn HJ, Liemburg EJ, Aleman A, Spikman JM, Van Der Naalt J. Brain networks subserving emotion regulation and adaptation after mild traumatic brain injury. J Neurotrauma. (2015) 33:1–9. doi: 10.1089/neu.2015.3905
206. Jorge RE, Acion L, Starkstein SE, Magnotta V. Hippocampal volume and mood disorders after traumatic brain injury. Biol Psychiatry. (2007) 62:332–8. doi: 10.1016/j.biopsych.2006.07.024
207. Réus GZ, Stringari RB, Ribeiro KF, Luft T, Abelaira HM, Fries GR, et al. Administration of cannabidiol and imipramine induces antidepressant-like effects in the forced swimming test and increases brain-derived neurotrophic factor levels in the rat amygdala. Acta Neuropsychiatrica. (2011) 23:241–8. doi: 10.1111/j.1601-5215.2011.00579.x
208. Mori MA, Meyer E, Soares LM, Milani H, Guimarães FS, de Oliveira RMW. Cannabidiol reduces neuroinflammation and promotes neuroplasticity and functional recovery after brain ischemia. Prog Neuro Psychopharmacol Biol Psychiatry. (2017) 75:94–105. doi: 10.1016/j.pnpbp.2016.11.005
209. Thal SC, Neuhaus W. The blood-brain barrier as a target in traumatic brain injury treatment. Arch Med Res. (2014) 45:698–710. doi: 10.1016/j.arcmed.2014.11.006
210. Hu YC, Sun Q, Li W, Zhang DD, Ma B, Li S, et al. Biphasic activation of nuclear factor kappa B and expression of p65 and c-Rel after traumatic brain injury in rats. Inflamm Res. (2014) 63:109–15. doi: 10.1007/s00011-013-0677-1
211. Mantle JL, Lee KH. A differentiating neural stem cell-derived astrocytic population mitigates the inflammatory effects of TNF-α and IL-6 in an iPSC-based blood-brain barrier model. Neurobiol Dis. (2018) 119:113–20. doi: 10.1016/j.nbd.2018.07.030
212. David Y, Cacheaux LP, Ivens S, Lapilover E, Heinemann U, Kaufer D, et al. Neurobiology of disease astrocytic dysfunction in epileptogenesis: consequence of altered potassium and glutamate homeostasis? J Neurosci. (2009) 29:10588–99. doi: 10.1523/JNEUROSCI.2323-09.2009
213. Aihara N, Hall JJ, Pitts LH, Fukuda K, Noble LJ. Altered immunoexpression of microglia and macrophages after mild head injury. J Neurotrauma. (1995) 12:53–63. doi: 10.1089/neu.1995.12.53
214. Stamatovic SM, Dimitrijevic OB, Keep RF, Andjelkovic A V. Inflammation and brain edema: new insights into the role of chemokines and their receptors. Acta Neurochir. (2006) 96:444–50. doi: 10.1007/3-211-30714-1_91
215. McMillin MA, Frampton GA, Seiwell AP, Patel NS, Jacobs AN, DeMorrow S. TGFβ1 exacerbates blood-brain barrier permeability in a mouse model of hepatic encephalopathy via upregulation of MMP9 and downregulation of claudin-5. Lab Invest. (2015) 95:903. doi: 10.1038/labinvest.2015.70
216. Rochfort KD, Collins LE, Murphy RP, Cummins PM. Downregulation of blood-brain barrier phenotype by proinflammatory cytokines involves NADPH oxidase-dependent ROS generation: consequences for interendothelial adherens and tight junctions. PLoS ONE. (2014) 9:e101815. doi: 10.1371/journal.pone.0101815
217. Hind WH, England TJ, O'Sullivan SE. Cannabidiol protects an in vitro model of the blood–brain barrier from oxygen-glucose deprivation via PPARγ and 5-HT1A receptors. Br J Pharmacol. (2016) 173:815–25. doi: 10.1111/bph.13368
218. Robinson ES, Alves P, Bashir MM, Zeidi M, Feng R, Werth VP. Cannabinoid reduces inflammatory cytokines tumor necrosis factor alpha and type I interferons in dermatomyositis in vitro. J Invest Dermatol. (2017) 137:2445. doi: 10.1016/j.jid.2017.05.035
219. O'Sullivan SE. An update on PPAR activation by cannabinoids. Br J Pharmacol. (2016) 173:1899. doi: 10.1111/bph.13497
220. Tyagi S, Gupta P, Saini A, Kaushal C, Sharma S. The peroxisome proliferator-activated receptor: a family of nuclear receptors role in various diseases. J Adv Pharm Technol Res. (2011) 2:236. doi: 10.4103/2231-4040.90879
221. Wang D, Shi L, Xin W, Xu J, Xu J, Li Q, et al. Activation of PPARγ inhibits pro-inflammatory cytokines production by upregulation of miR-124 in vitro and in vivo. Biochem Biophys Res Commun. (2017) 486:726–31. doi: 10.1016/j.bbrc.2017.03.106
222. Golding EM, Robertson CS, Bryan RM. The consequences of traumatic brain injury on cerebral blood flow and autoregulation: a review. Clin Exp Hypertens. (2009) 21:299–332. doi: 10.3109/10641969909068668
223. Gobiet W, Grote W, Bock WJ. The relation between intracrainal pressure, mean arterial pressure and cerebral blood flow in patients with severe head injury. Acta Neurochir. (1975) 32:13–24. doi: 10.1007/BF01405899
224. Langfitt TW, Obrist WD, Gennarelli TA, O'Connor MJ, Weeme CA. Correlation of cerebral blood flow with outcome in head injured patients. Ann Surg. (1977) 186:411–4. doi: 10.1097/00000658-197710000-00002
225. Len TK, Neary JP. Cerebrovascular pathophysiology following mild traumatic brain injury. Clin Physiol Funct Imaging. (2011) 31:85–93. doi: 10.1111/j.1475-097X.2010.00990.x
226. Sunbul M, Sunbul EA, Kosker SD, Durmus E, Kivrak T, Ileri C, et al. Depression and anxiety are associated with abnormal nocturnal blood pressure fall in hypertensive patients. Clin Exp Hypertens. (2014) 36:354–8. doi: 10.3109/10641963.2013.827701
227. Robertson CS, Contant CF, Gokaslan ZL, Narayan RK, Grossman RG. Cerebral blood flow, arteriovenous oxygen difference, and outcome in head injured patients. J Neurol Neurosurg Psychiatry. (1992) 55:594–603. doi: 10.1136/jnnp.55.7.594
228. Kelly DF, Martin NA, Kordestani R, Counelis G, Hovda DA, Bergsneider M, et al. Cerebral blood flow as a predictor of outcome following traumatic brain injury. J Neurosurg. (1997) 86:633–41. doi: 10.3171/jns.1997.86.4.0633
229. Jünger EC, Newell DW, Grant GA, Avellino AM, Ghatan S, Douville CM, et al. Cerebral autoregulation following minor head injury. J Neurosurg. (1997) 86:425–32. doi: 10.3171/jns.1997.86.3.0425
230. Bullock R, Zauner A, Woodward JJ, Myseros J, Choi SC, Ward JD, et al. Factors affecting excitatory amino acid release following severe human head injury. J Neurosurg. (1998) 89:507–18. doi: 10.3171/jns.1998.89.4.0507
231. McCulloch J, Edvinsson L. Cerebrovascular smooth muscle reactivity: a critical appraisal of in vitro and in situ techniques. J Cereb Blood Flow Metab. (1984) 4:129–39. doi: 10.1038/jcbfm.1984.21
232. Compton JS, Teddy PJ. Cerebral Arterial Vasospasm Following Severe Head Injury: A Transcranial Doppler Study.
233. Kontos HA, Wei EP. Superoxide production in experimental brain injury. J Neurosurg. (1986) 64:803–7. doi: 10.3171/jns.1986.64.5.0803
234. Hayakawa K, Mishima K, Nozako M, Hazekawa M, Irie K, Fujioka M, et al. Delayed treatment with cannabidiol has a cerebroprotective action via a cannabinoid receptor-independent myeloperoxidase-inhibiting mechanism. J Neurochem. (2007) 102:1488–96. doi: 10.1111/j.1471-4159.2007.04565.x
235. Resstel LBM, Tavares RF, Lisboa SFS, Joca SRL, Corrêa FMA, Guimarães FS. 5-HT 1A receptors are involved in the cannabidiol-induced attenuation of behavioural and cardiovascular responses to acute restraint stress in rats. Br J Pharmacol. (2009) 156:181–8. doi: 10.1111/j.1476-5381.2008.00046.x
236. Bloomfield MAP, Green SF, Hindocha C, Yamamori Y, Yim JLL, Jones APM, et al. The effects of acute cannabidiol on cerebral blood flow and its relationship to memory: an arterial spin labelling magnetic resonance imaging study. J Psychopharmacol. (2020) 34:981. doi: 10.1177/0269881120936419
237. Razavi Y, Rashvand M, Sharifi A, Haghparast A, Keyhanfar F, Haghparast A. Cannabidiol microinjection into the nucleus accumbens attenuated nociceptive behaviors in an animal model of tonic pain. Neurosci Lett. (2021) 762:136141. doi: 10.1016/j.neulet.2021.136141
238. Tjølsen A, Berge OG, Hunskaar S, Rosland JH, Hole K. The formalin test: an evaluation of the method. Pain. (1992) 51:5–17. doi: 10.1016/0304-3959(92)90003-T
239. Jesus CHA, Redivo DDB, Gasparin AT, Sotomaior BB, de Carvalho MC, Genaro K, et al. Cannabidiol attenuates mechanical allodynia in streptozotocin-induced diabetic rats via serotonergic system activation through 5-HT1A receptors. Brain Res. (2019) 1715:156–64. doi: 10.1016/j.brainres.2019.03.014
240. Haghparast A, Ghalandari-Shamami M, Hassanpour-Ezatti M. Blockade of D1/D2 dopamine receptors within the nucleus accumbens attenuated the antinociceptive effect of cannabinoid receptor agonist in the basolateral amygdala. Brain Res. (2012) 1471:23–32. doi: 10.1016/j.brainres.2012.06.023
241. Wang XQ, Mokhtari T, Zeng YX, Yue LP, Hu L. The distinct functions of dopaminergic receptors on pain modulation: a narrative review. Neural Plast. (2021) 2021:6682275. doi: 10.1155/2021/6682275
242. Elliott MB, Ward SJ, Abood ME, Tuma RF, Jallo JI. Understanding the endocannabinoid system as a modulator of the trigeminal pain response to concussion. Concussion. (2017) 2:CNC49. doi: 10.2217/CNC-2017-0010
243. Daiutolo B V, Tyburski A, Clark SW, Elliott MB. Trigeminal pain molecules, allodynia, and photosensitivity are pharmacologically and genetically modulated in a model of traumatic brain injury. J Neurotrauma. (2016) 33:748–60. doi: 10.1089/neu.2015.4087
244. Rubino T, Parolaro D. Sexually dimorphic effects of cannabinoid compounds on emotion and cognition. Front Behav Neurosci. (2011) 5:64. doi: 10.3389/fnbeh.2011.00064
245. Wagner EJ. Sex differences in cannabinoid-regulated biology: a focus on energy homeostasis. Front Neuroendocrinol. (2016) 40:101. doi: 10.1016/j.yfrne.2016.01.003
246. Levine A, Liktor-Busa E, Lipinski AA, Couture S, Balasubramanian S, Aicher SA, et al. Sex differences in the expression of the endocannabinoid system within V1M cortex and PAG of sprague dawley rats. Biol Sex Differ. (2021) 12:1–18. doi: 10.1186/s13293-021-00402-2
247. Cooper ZD, Craft RM. Sex-dependent effects of cannabis and cannabinoids: a translational perspective. Neuropsychopharmacology. (2018) 43:34–51. doi: 10.1038/npp.2017.140
248. Viveros MP, Llorente R, Suarez J, Llorente-Berzal A, López-Gallardo M, Rodriguez De Fonseca F. The endocannabinoid system in critical neurodevelopmental periods: sex differences and neuropsychiatric implications. J Psychopharmacol. (2012) 26:164–76. doi: 10.1177/0269881111408956
249. Xing G, Carlton J, Jiang X, Wen J, Jia M, Li H. Differential expression of brain cannabinoid receptors between repeatedly stressed males and females may play a role in age and gender-related difference in traumatic brain injury: implications from animal studies. Front Neurol. (2014) 5:161. doi: 10.3389/fneur.2014.00161
250. Iffland K, Grotenhermen F. An update on safety and side effects of cannabidiol: a review of clinical data and relevant animal studies. Cannabis cannabinoid Res. (2017) 2:139–54. doi: 10.1089/can.2016.0034
251. Bergamaschi MM, Queiroz RH, Zuardi AW, Crippa JA. Safety and side effects of cannabidiol, a cannabis sativa constituent. Curr Drug Saf. (2011) 6:237–49. doi: 10.2174/157488611798280924
252. Taylor L, Gidal B, Blakey G, Tayo B, Morrison G. A phase I, randomized, double-blind, placebo-controlled, single ascending dose, multiple dose, and food effect trial of the safety, tolerability and pharmacokinetics of highly purified cannabidiol in healthy subjects key points. CNS Drugs. (2018) 32:1053–67. doi: 10.1007/s40263-018-0578-5
253. Szaflarski JP, Bebin EM, Comi AM, Patel AD, Joshi C, Checketts D, et al. Long-term safety and treatment effects of cannabidiol in children and adults with treatment-resistant epilepsies: expanded access program results. Epilepsia. (2018) 59:1540–8. doi: 10.1111/epi.14477
254. Devinsky O, Patel AD, Cross JH, Villanueva V, Wirrell EC, Privitera M, et al. Effect of cannabidiol on drop seizures in the lennox–gastaut syndrome. N Engl J Med. (2018) 378:1888–97. doi: 10.1056/NEJMoa1714631
255. Chesney E, Oliver D, Green A, Sovi S, Wilson J, Englund A, et al. Adverse effects of cannabidiol: a systematic review and meta-analysis of randomized clinical trials. Neuropsychopharmacology. (2020) 45:1799–806. doi: 10.1038/s41386-020-0667-2
256. Nasrin S, Watson CJW, Perez-Paramo YX, Lazarus P. Cannabinoid metabolites as inhibitors of major hepatic CYP450 enzymes, with implications for cannabis-drug interactions. Drug Metab Dispos. (2021) 49:1070–80. doi: 10.1124/dmd.121.000442
257. CANNabinoid Drug Interaction Review. Available online at: https://www.canndir.psu.edu/#/ (accessed June 28, 2022).
258. Cannabinoids and Traumatic Brain Injury: A Randomized Placebo Controlled Trial. Available online at: https://clinicaltrials.gov/ct2/show/NCT05632627?term=cannabidiol&cond=injury&draw=2&rank=1 (accessed December 26, 2022).
259. NFL Awards $1 Emillion to Study Impact of Cannabis and CBD on Pain Management. Available online at: https://nflcommunications.com/Pages/NFLAWARDS-$1-MILLION-TO-STUDY-IMPACT-OF-CANNABIS-AND-CBD-ONPAIN-MANAGEMENT-.aspx (accessed June 28, 2022).
260. NHL Alumni Association Teams Up With Canopy Growth to Research Concussions and Cannabinoids. (2019). Available online at: https://www.prnewswire.com/news-releases/nhl-alumni-association-teams-up-with-canopy-growth-to-research-concussions-and-cannabinoids-300805451.html (accessed January 2, 2023).
261. Cannabidiol as a Treatment for PTSD and PTSD Comorbid With TBI. Available online at: https://clinicaltrials.gov/ct2/show/NCT04550377?term=TBI%2CCCBD&cond=PTSD&draw=2&rank=1 (accessed December 26, 2022).
262. VanItallie TB. Traumatic brain injury (TBI) in collision sports: possible mechanisms of transformation into chronic traumatic encephalopathy (CTE). Metabolism. (2019) 100S:153943. doi: 10.1016/j.metabol.2019.07.007
263. Mez J, Daneshvar DH, Kiernan PT, Abdolmohammadi B, Alvarez VE, Huber BR, et al. Clinicopathological evaluation of chronic traumatic encephalopathy in players of american football. JAMA. (2017) 318:360–70. doi: 10.1001/jama.2017.8334
264. Katsumoto A, Takeuchi H, Tanaka F. Tau pathology in chronic traumatic encephalopathy and Alzheimer's disease: similarities and differences. Front Neurol. (2019) 10:980. doi: 10.3389/fneur.2019.00980
265. Alali S, Riazi G, Ashrafi-Kooshk MR, Meknatkhah S, Ahmadian S, Ardakani MH, et al. Cannabidiol inhibits tau aggregation in vitro. Cells. (2021) 10:3521. doi: 10.3390/cells10123521
Keywords: cannabidiol, cannabis, traumatic brain injury, concussion, neuroprotection, neuroinflammation, marijuana, blood brain barrier
Citation: Aychman MM, Goldman DL and Kaplan JS (2023) Cannabidiol's neuroprotective properties and potential treatment of traumatic brain injuries. Front. Neurol. 14:1087011. doi: 10.3389/fneur.2023.1087011
Received: 01 November 2022; Accepted: 17 January 2023;
Published: 02 February 2023.
Edited by:
Wiliam Panenka, University of British Columbia, CanadaReviewed by:
Yumin Zhang, Uniformed Services University of the Health Sciences, United StatesLuisa Lilia Rocha, Instituto Politécnico Nacional de México (CINVESTAV), Mexico
Copyright © 2023 Aychman, Goldman and Kaplan. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Joshua S. Kaplan, 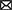 Sm9zaC5rYXBsYW5Ad3d1LmVkdQ==
Sm9zaC5rYXBsYW5Ad3d1LmVkdQ==