 Hongyi Zheng
Hongyi Zheng Wenbin Zheng
Wenbin Zheng Hongkun Liu2
Hongkun Liu2 Yuelin Guo
Yuelin Guo- 1Department of Radiology, The Second Affiliated Hospital, Medical College of Shantou University, Shantou, China
- 2Department of Radiology, Huizhou City Center People's Hospital, Huizhou, China
- 3Department of Radiology, Shenzhen Hospital of Integrated Traditional Chinese and Western Medicine, Shenzhen, China
Aims: This study adopted the Glutamate Chemical Exchange Saturation Transfer (GluCEST) imaging technique to quantitatively analyze cranial glutamate and discussed the effectiveness of GluCEST values in identifying the pathogenesis of encephalopathy after CO poisoning.
Methods: The routine MRI and functional MRI scans of two cohorts of subjects (CO group, n = 29; Control group, n = 21) were performed. Between-group comparisons were conducted for GluCEST% in regions of interest (ROI), including the basal ganglia, the thalamus, the frontal lobe, the occipital lobe, the genu of corpus callosum, the cingulate gyrus, and the cuneus. Moreover, an age-stratified subgroup analysis was devised, and a correlational analysis was performed for GluCEST% in each ROI, including the time in coma, Simple Mini-Mental State Examination Scale (MMSE) score, Hamilton Anxiety Scale score, and blood COHb%.
Results: As compared to the healthy control, the CO group led to significantly increasing GluCEST% in the basal ganglia, the occipital lobe, the genu of the corpus callosum, the cingulate gyrus, and the cuneus (p < 0.05). In the subgroup analysis for age, adult patients had higher GluCEST% in the basal ganglia, the thalamus, the occipital lobe, the cingulate gyrus, and the cuneus compared to healthy adults (p < 0.05). In addition, the correlational analysis of CO-poisoned patients revealed a statistical association between the GluCEST% and the MMSE in the thalamus and the genu of the corpus callosum.
Conclusion: The GluCEST technique is superior to routine MRI in that it can identify the cerebral biochemical changes sooner after acute CO poisoning, which is significant for our understanding of the role of neurotransmitters in the pathological basis of this disease. Brain injury caused by CO poisoning may be different in adults and children.
1. Introduction
Carbon monoxide (CO), which is tasteless, colorless, and odorless, is absorbed into the human body across the lining of the lungs (1). It is the main cause of death related to poisoning in multiple countries, and more importantly, it may lead to fetal poisoning in over half of the global population (2). People surviving CO poisoning still display long-term neurocognitive sequelae associated with cerebral injury and may present with symptoms including memory decline, cognitive impairment, depression, anxiety, and/or vestibular and motor dysfunctions (3). Given the affinity to CO is 250 times higher than oxygen (4), hemoglobin (Hb) in blood circulation can bind with CO to form carbonylhemoglobin (COHb). In that way, the Hb fails to carry more oxygen and the degradation of oxygenated hemoglobin will be blocked, leading to severe tissue hypoxia. The decreasing oxygen levels and mitochondrial oxidative phosphorylation will cause ischemic and hypoxic cerebral injuries that finally result in cognitive impairment (5). Ischemic brain injury might be a result of excitotoxicity, acidosis, ion imbalance, depolarization, oxidative stress, nitrosative stress, inflammation, and cell apoptosis (6). Excess CO activates the platelets and amplifies the inflammatory effects by triggering neutrophil activation, adhesion, and degranulation. These inflammatory effects are ongoing long after the initial CO poisoning and may dominate the clinical expression (7, 8). Adenosine triphosphate is the direct energy source of the organism. When its content decreases, intracellular protease and lipase will be activated accordingly to induce mitochondrial membrane depolarization, cell death, and release of neurotransmitters (especially, glutamate) (6, 9). Glutamate is a core player in excitatory neurotransmission. In the cases of impaired homeostasis, it will induce the production of neurotoxins or excitotoxins and activate the pathways of neuron death (10). In the presence of acute ischemic or toxic injury and chronic neurodegeneration, the glutamate receptor will be activated excessively, which is the key to the generation of excitotoxicity and incidence of cell death (11). Previous studies have reported that the most common manifestations of MR after CO poisoning are high signals in the bilateral globus pallidus and the white matter region of the brain (12, 13). Conventional MRI could show the involvement of globus pallidus with CO exposure as it is at risk for damage due to the hypotension-hypoxia processes because of poor collateral blood flow or from CO binding to heme iron in the globus pallidus, where the highest concentration of iron in the brain is located (1). The abnormal finding detected on MR imaging in the white matter is more responsible for the chronic symptoms than the gray matter (14). The centrum semiovale and periventricular white matter are the most common regions of white matter to be affected after CO poisoning (15, 16). The white matter damage can be observed in various other regions rather than the centrum semiovale and the periventricular white matter, such as the temporal lobe, the occipital lobe, the parietal lobe, and the corpus callosum (12, 17–19).
Chemical exchange saturation transfer (CEST) is a novel MRI technique. It uses frequency-selective radiofrequency pulses to ensure magnetization saturation from some exchangeable protons in solutes and characterize the microenvironment of the tested solution by metabolite concentrations, temperature, and pH values by calculating the proportional change in water signals in the bulk water pool (20–22). Comparatively, CEST is superior to magnetic resonance spectroscopy with higher specificity and spatial resolution (23). Given the advantages, CEST is widely adopted in the examination of multiple molecules, such as glucose with GlucoCEST (24), adenosine triphosphatase in polypeptides, tissue mobile proteins with amide proton transfer (25), and glutamate with GluCEST (26, 27). The asymmetric magnetization transfer ratio (MTRasym) can be calculated to represent the signal intensity on CEST, majorly by the difference in the values of the bilateral resonance frequency of the solvent water protons (ΔMTRasym). It is reported that GluCEST has high signal intensity at ΔMTRasym (3 ppm) (23) and ΔMTRasym. Therefore, quantitative calculation of the changes in glutamate concentration can be performed. Currently, there were some studies on magnetic resonance imaging (MRI) for cerebral metabolites after acute CO poisoning (28, 29), while less is devoted to the cranial glutamate changes (30, 31). Glutamate chemical exchange saturation transfer (GluCEST) imaging is a non-invasive quantitative technique and its application values in patients with acute CO poisoning are not yet reported.
Glutamate is a type of excitatory neurotransmitter most abundant in the brain and involved in a variety of physiological functions in the nervous system (32, 33). It is known that glutamate is important in cerebral injury after CO poisoning. In an animal experiment, for instance, increases in glutamate release and hydroxyl free radical production were observed during and after the hypoxia is induced by CO absorption in rats, which were believed to be the cause of ischemic cerebral injuries (30). Other studies revealed that glutamate could aggravate cellular dysfunction and cell apoptosis by activating the N-methyl-D-aspartate receptor (NMDAR) (6), and the NMDAR antagonist could alleviate the CO-induced neurodegeneration (34). We assumed that GluCEST may be effective as a precise diagnostic approach for encephalopathy during the acute phase after CO poisoning.
As a non-invasive and quantitative imaging technique, GluCEST was previously applied to multiple diseases of the nervous system (35–37). Currently, there is no report on its application in encephalopathy after CO exposure. Herein, we used a 3.0-Tesla MR scanner to assess the value of the GluCEST technique in diagnosing cerebral injuries after CO poisoning and in understanding the pathogenesis.
2. Methods
2.1. Subjects
Overall, there are 29 patients with acute CO poisoning included and categorized into the CO group. These patients were treated for acute CO poisoning in our hospital at different time points from December 2020 to March 2021. There were 12 men and 17 women, aged between 6 and 48 years (average, 19.4 ± 12.5 years). The inclusion criteria (38) included (1) a history of exposure to high concentrations of CO; (2) symptoms and signs of acute central nervous system injuries; (3) timely blood COHb content conforming to the national diagnostic criteria. For MRI, the time interval to be exposed to high concentrations of CO was required to be smaller than 3 days. Hyperbaric oxygen and correction for electrolyte disturbance treatment were performed upon admission. Adult patients with CO poisoning were treated with hormone therapy for 3 days after admission to prevent immune inflammatory responses. The patients received 1 h hyperbaric oxygen therapy (0.2 MPa) 1–2 h after admission and then received hyperbaric oxygen therapy (0.2 MPa) once a day, 1 H each time. The duration was generally 7 days. After treatment, the patient still showed clinical symptoms and cognitive decline. Although the clinical symptoms recovered, the patient did not meet the criteria for recovery. In the meantime, each patient was subjected to a Mini-Mental State Examination (MMSE), and the period in coma and COHb concentration were recorded (Supplementary Table 1). At 1 month follow-up, onsite and telephone interviews were conducted and the Hamilton Anxiety Scale (HAMA) score was obtained (Supplementary Table 1). Healthy volunteers matched for gender, age, and educational level were recruited from our hospital as the control group (control group, n = 21), including 9 men and 12 women aged between 8 and 30 years (average, 20.7 ± 7.4 years). In this population, there was no history of cerebral injury, psychiatric disorder, alcohol abuse/substance dependence, or diseases of the nervous system (including stroke, seizure, and somatic disease). Table 1 shows the detailed demographic and clinical information. All patients gave written informed consent. The project was approved by the Ethics Committee of The Second Affiliated Hospital of Shantou University Medical College.
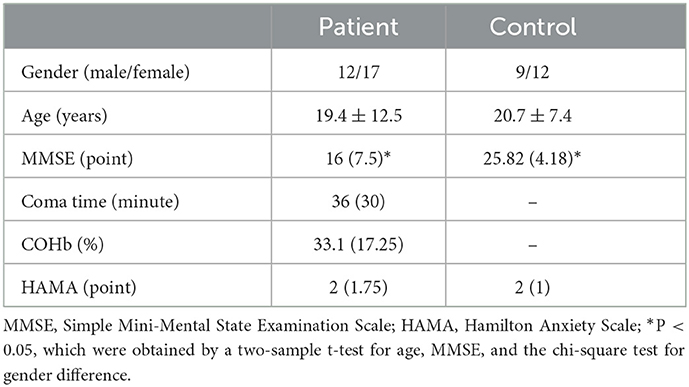
Table 1. Participant clinical characteristics.
2.2. MRI imaging
The structural MRI and CEST MRI data were collected using a 3.0 Tesla MRI scanner (Sigma; GE Healthcare, Milwaukee, WI, USA), using an 8-channel phased-array head coil. The sponge padding was used to limit head motion. The T2-weighted images (T2WI) [repetition time (TR) = 4,600 ms, echo time (TE) = 120 ms, 20 slices, and acquisition time: 1 min 35 s], the T2WI fluid-attenuated inversion recovery images (TR = 8,600 ms, TE = 155 ms, inv. time = 2,100 ms, 20 slices, and acquisition time: 1 min 53 s), and diffusion-weighted images (TR = 5,200 ms; TE = minimum; b = 1,000, 20 slices, and acquisition time: 42 s) were obtained to acquire the information of the brains of all subjects.
The CEST scan was based on an MT-prepared gradient echo MRI sequence with the following settings: TR = 50 ms, TE = 3.1 ms, field of view = 240 × 240 mm2, matrix = 128 × 128, 1 slice, slice thickness = 5 mm, and bandwidth = 15.63 kHz. The MT saturation pulse was a Fermi pulse with a 20 ms width and a B1 of 1.95 μT (39). The CEST imaging was performed on the brain slice shown in Figure 1. The regions of interest (ROI) consisted of seven bilateral standard regions in all patients with acute CO poisoning and healthy controls which are basal ganglia, thalamus, the genu of the corpus callosum, the frontal lobe, the occipital lobe, the cuneus, and the cingulate gyrus (Figure 1). Except for the basal ganglia region and the thalamus, the ROI was placed in the gray matter region, and for other brain regions, the ROI was placed in the white matter region of the genu of the corpus callosum, the frontal lobe, the occipital lobe, the cuneus, and the cingulate gyrus. The images were interpreted independently by two experienced radiologists who were blinded to the neurological manifestations and the results of the analyses.
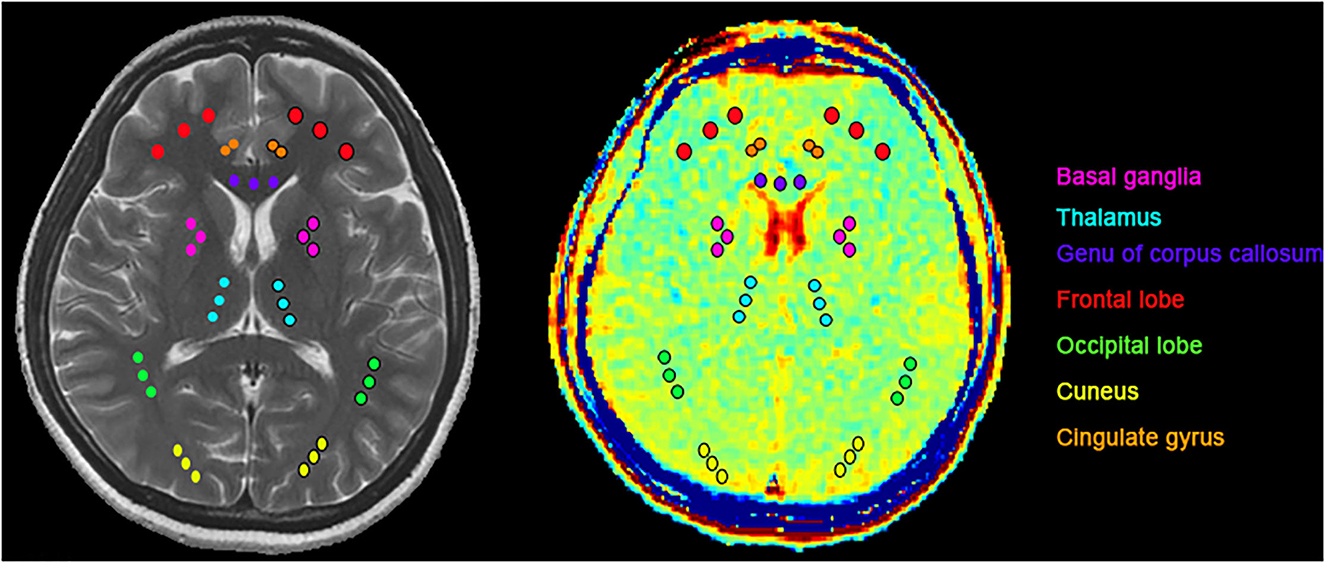
Figure 1. ROIs for T2WI and GluCEST: basal ganglia (pink), thalamus (light blue), the genu of the corpus callosum (purple), frontal lobe (red), occipital lobe (green), cuneus (yellow), cingulate gyrus (orange). Except for the basal ganglia region and the thalamus, the ROI was placed in the gray matter region, and for other brain regions, the ROI was placed in the white matter region.
2.3. Data processing
All CEST image processing was performed using software routines written in Matlab 7 (Mathworks, Natick, MA, USA). The acquired images were corrected for B0 inhomogeneity using a water saturation shift referencing map. The corresponding correction algorithm referred to a previous detailed discussion (40). Then, the GluCEST contrast map was generated using the following equation (41).
where S (−3 ppm) and S (+3 ppm) are the images obtained at −3 ppm and +3 ppm, respectively. The Z-spectra were obtained from the normalized CEST images. The MTRasym maps were computed using the equation (41).
2.4. Statistical analysis
Data processing and analyses were completed with SPSS 20.0 (SPSS 20.0, IBM, Armonk, NY). Comparative results, MMES score, time in coma, COHb concentration, and postoperative HAMA score were represented by mean (interquartile range). Age (continuous variable) was displayed in mean ± standard deviation. The categorical variables are shown in integers. The normal distribution of continuous parameters was determined by the Shapiro–Wilk test, followed by the Kruskal–Wallis H-test for data comparison.
The average GluCEST% in the bilateral ROI in the two groups was measured by GluCEST, and the data that did not conform to normal distribution were tested by the Mann–Whitney U-test. A comparison of the age and educational levels were completed by the two-sample t-test and that for gender using the chi-square test. A Spearman correlational analysis was performed to calculate the association of GluCEST% with the time in coma, MMSE score, COHb concentration, and postoperative HAMA score. A p < 0.05 demonstrates a statistically significant difference.
3. Results
3.1. Clinical data
The reason for CO poisoning in coma patients was the long bathing time in an enclosed bathroom using the gas stove. There were no statistically significant differences between the patients and the healthy volunteers in terms of gender (χ2 = 0.011 and P = 0.917) and age (P = 0.65, two-sample t-test). The MMSE score was lower in patients with CO poisoning (P < 0.001, two-sample t-test). In the acute phase, patients presented with symptoms, including coma, headache, dizziness, fatigue, and memory decline after a lucid interval. Upon follow-up 1 month after the discharge, the HAMA scores between the two groups were not remarkably different. There was no evident sequela, except for one who still had dizziness and memory decline. The demographic and clinical information of the subjects of the study are shown in Table 1. There were no statistical differences between the subgroups of age regarding COHb concentration, the time in coma, and the HAMA score upon follow-up.
3.2. MRI manifestations
In routine MRI, abnormal images were shown only in two adult patients, majorly bilateral globus pallidus on T2WI (Figure 2) and signal enhancement on T2flair and DWI. One of them also showed abnormal images in the white matter of the right occipital lobe. No abnormality was observed in the other patients. Higher GluCEST imaging parameters were generally observed in patients with CO poisoning vs. healthy volunteers (Figure 2). In terms of the GluCEST% in each ROI, it was higher in patients in the basal ganglia (Z = 3.253), the occipital lobe (Z = 4.142), the genu of the corpus callosum (Z = 2.323), the cingulate gyrus (Z = 1.96), and the cuneus (Z = 2.849) as compared to the healthy controls (P < 0.05, Tables 2, 3). In the subgroup analysis for age, higher GluCEST% was observed in the basal ganglia (Z = 3.956), the thalamus (Z = 2.11), the occipital lobe (Z = 3.429), the cingulate gyrus (Z = 2.506), and the cuneus (Z = 3.165) in adult patients, when compared to the healthy adult volunteers (P < 0.05, Tables 2, 3). In children, higher GluCEST% was also observed in multiple ROIs in patients, while the differences among those in healthy children were not statistically significant. Further correlational analysis revealed that the increasing GluCEST% in the thalamus and the genu of the corpus callosum in patients with CO poisoning was remarkably associated with the MMSE score (Figure 3). There was no statistical association between the GluCEST% in each ROI and the time in coma, COHb concentration, and postoperative HAMA score.
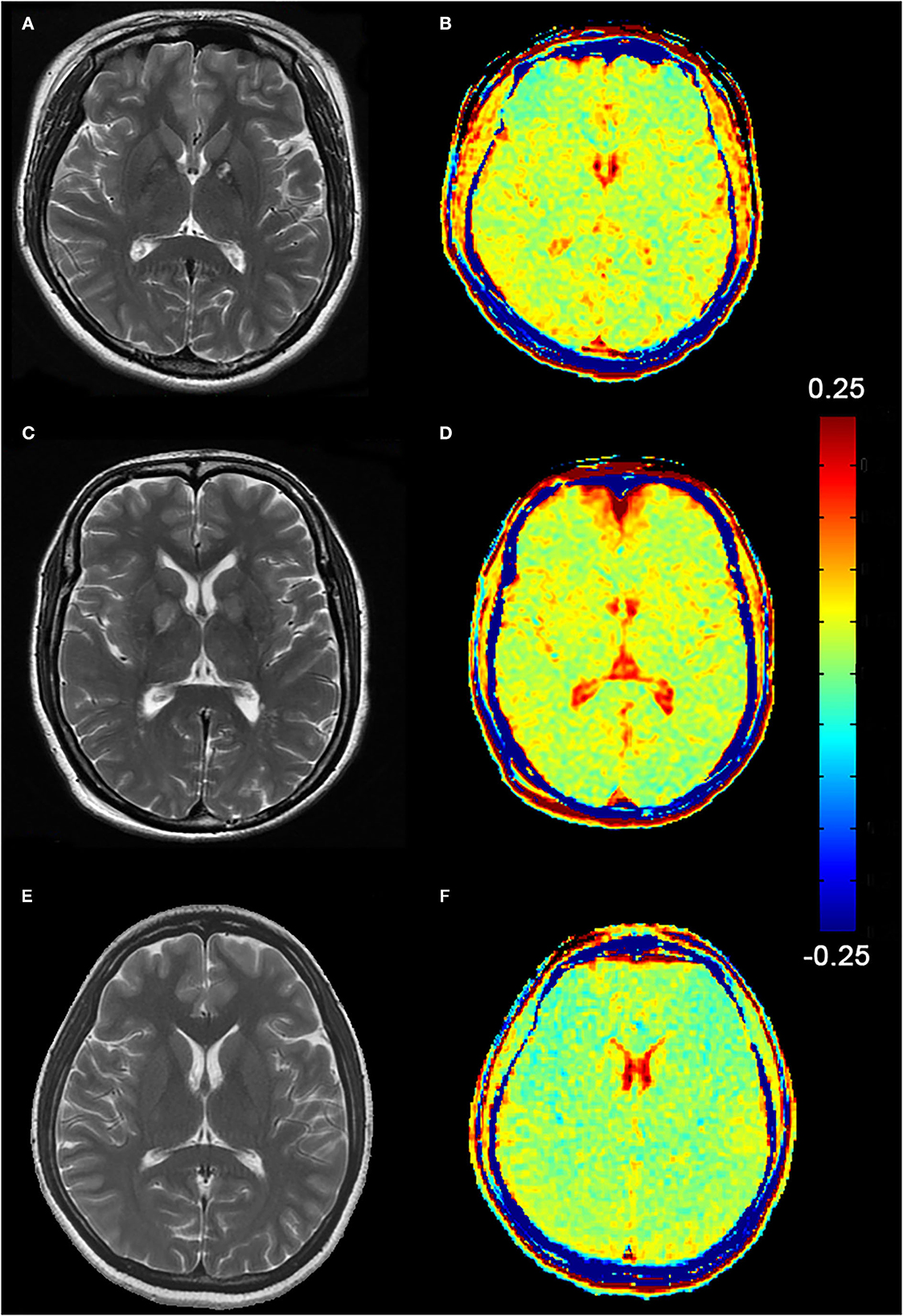
Figure 2. (A, C) Abnormal T2WI images of two female patients (Patient A, 24-year-old and Patient C, 47-year-old), mainly manifested in the bilateral globus pallidus. (B, D) Higher signals on GluCEST images present in the globus pallidus and the cerebral white matter region. (E, F) T2WI and GluCEST images of a healthy adult (female, 24-year-old).
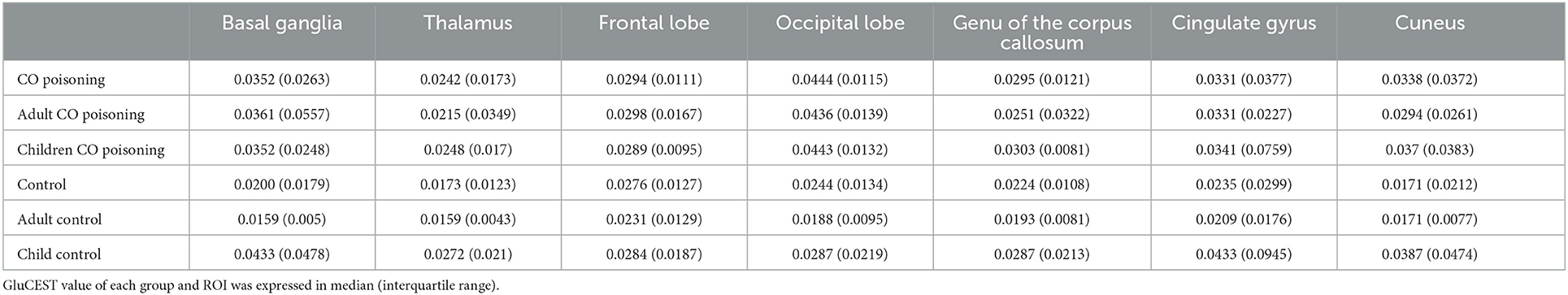
Table 2. GluCEST in each region of interest.
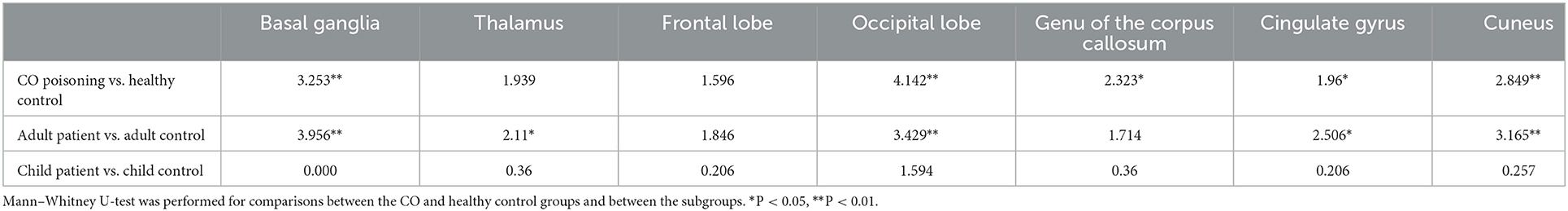
Table 3. Two-sample Mann–Whitney U-test in each group.
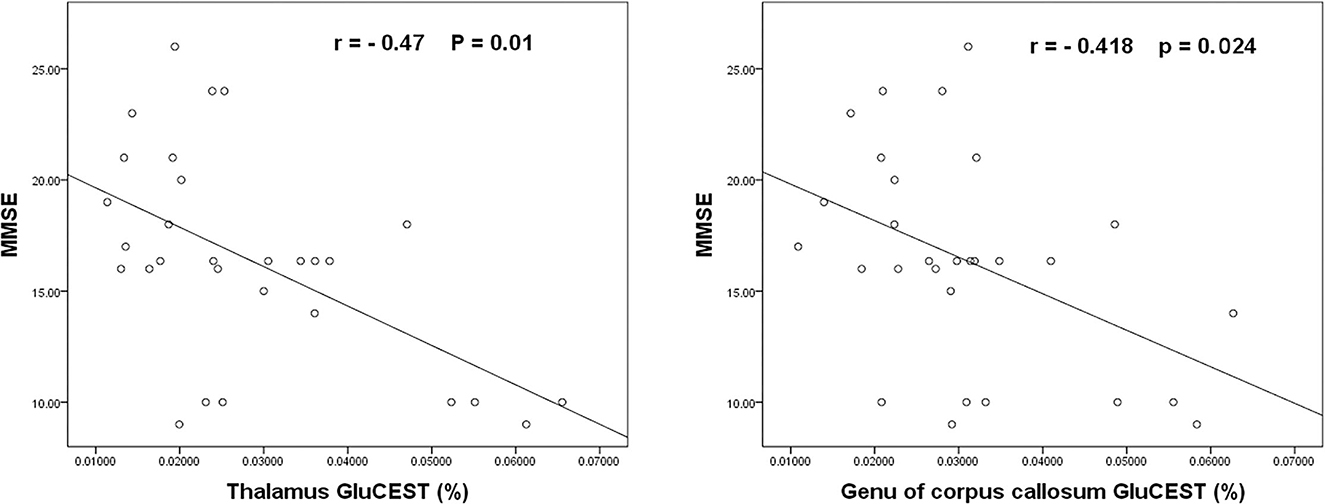
Figure 3. The negative association of the GluCEST% in the thalamus and the genu of the corpus callosum with the MMSE scores of patients with CO poisoning.
3.3. Discussion
CEST is a relatively new contrast mechanism of MRI. It uses the specific MR frequency (chemical shift) at the molecular level and the standard MRI technique to generate images of good spatial resolution. Some CEST techniques, such as GluCEST, can indirectly examine molecules by combining the specificity and the spatial resolution of MRI (42, 43). GluCEST is effective in measuring the glutamate content in the brain, which is generally higher than the concentration of other metabolites in the brain (42). In the current study, changes in cranial glutamate levels were examined in the acute phase after CO poisoning to study the potential effect on cranial neurotransmitters, which was not previously reported.
In a previous animal experiment, hypoxia was induced in rats by CO inhalation, and instantly, the release of glutamate and the production of hydroxyl free radicals increased in the cerebral cortex and the hippocampus, which were considered to be the cause of ischemic brain injury after CO poisoning (30). Other studies revealed that glutamate could aggravate cellular dysfunction and cell apoptosis by activating NMDAR (6). Here, we found that patients with CO poisoning had remarkably increasing GluCEST% in the basal ganglia, the occipital lobe, the corpus callosum, the cingulate gyrus, and the cuneus, indicative of more cranial excitatory neurotransmitters and a tendency of accumulation in the above brain regions. Autopsy findings suggested that the major pathological changes after CO poisoning were the necrosis of globus pallidus and demyelination in the cerebral white matter area (44–46), which were mainly attributed to cerebral ischemia and edema. Given the hyperactivity and sufficient blood supply of neurons in the gray matter regions, such as the deep brain nucleus, the gray matter is more susceptible to poisoning-induced brain ischemia than the white matter. In the subacute phase, DWI manifestations are a hypointense signal and a high ADC value in the globus pallidus and a hyperintense signal and a low ADC value in the cerebral white matter (47). This suggests that the injury in cerebral white matter is a result of both ischemic globus pallidus and progressive demyelination changes (48). In the present study, only two patients exhibited abnormal images in MRI and no abnormality was noted in the other patients. It is tempting to assume that the GluCEST technique can show early changes in cranial neurotransmitters after hypoxia induced by CO exposure, and the changes can involve the deep brain nucleus and white matter simultaneously. It is reported that the bilateral basal ganglia, especially the globus pallidus, is the most susceptible region to CO poisoning (45). Controversially, some researchers report that the changes in the striatum after CO poisoning were independent of glutamate receptor activation induced by the increasing extracellular glutamate content (49). Tambasco et al. (10) revealed that CO poisoning leads to impaired glutamate homeostasis and significantly affects neurons by producing neurotoxins or excitotoxins and activating the pathways of neuronal death. In this context, the specific mechanism of the action of glutamate elevation in the basal ganglia requires further research. Herein, we also found that the GluCEST% was abnormally increased in the bilateral occipital lobe areas, except for the common bilateral basal ganglia. The occipital lobe is the visual center and when an injury occurs, visual impairment will be developed. A previous study found that COHb >30% could result in visually evoked abnormal potentials and that CO poisoning came with a series of clinical symptoms, including ocular discomfort, blurred vision, and visual field defects, which are not entirely consistent with the findings in ophthalmic examinations (50). This is in line with our findings. In the injured areas of the cerebral white matter, the semioval center and the white matter next to the lateral ventricle are the most susceptible, and the corpus callosum can also be affected. Research revealed that corpus callosum could present with generalized atrophy after CO poisoning (18), which was once reported to be associated with neuropsychological presentations in other diseases (51). Our results showed that the GluCEST% in the genu of the corpus callosum was statistically different between the patients and healthy volunteers. In addition, the GluCEST% in the thalamus and the genu of the corpus callosum were negatively associated with the MMSE score, which was weak. This infers that the changes in glutamate levels in the genu of corpus callosum may indicate the disease condition in patients to some extent. The thalamus is key to cognitive tasks, consciousness, and awakening. Impairment of the thalamus and the connections may cause damage to a wide range of neurological functions, which might be clinically translated into significant cognitive, physical, and psychic disorders (52). We also noted that there was no correlation with the MMSE score in the other ROIs. The possible explanation might be that the MMSE score mainly targets the directional and verbal functions. There is a certain rate of false negative score when applied for moderate to critical cognitive disorders, and it is readily affected by the speech and educational levels of the patients.
The age range of patients included in this study was large and ranged from childhood to middle age. However, there are no unified reports on the changes in craniocerebral metabolites at different stages of craniocerebral development, especially glutamate. Although the patients included in this study have a large age span, in addition to the measurement analysis of all patients with CO poisoning, adults and children are also divided into different subgroups for analysis, and different results are obtained. In the subgroup analysis for age, GluCEST% in both the adult and child patients increased when compared to the corresponding healthy control. Notably, there was no statistical significance in the difference in children. We reasoned that the varying collateral circulation compensation mechanism between adults and children (53) may be used to explain the less damage to the basal ganglia region in the early acute phase after CO exposure in children. Some previous literature suggested that patients with CO poisoning could be divided into mild, moderate, and severe according to the blood COHb value, symptoms, and coma state (54, 55). However, this classification was not well-suited to the patients we included in the study. First of all, according to clinical symptoms, the conditions of our patients were relatively consistent, whose state of consciousness was manifested as mild to moderate coma, and they recovered after rescue without obvious complications. Previous literature state that the degree of poisoning in patients can be determined according to the standard of blood COHb concentration. However, clinical symptoms of acute CO poisoning and their severity do not always correlate with the concentrations of CO-Hb on admission (8, 56–58). The COHb level in the clinical diagnosis of CO poisoning is not significant (58). Herein, we also found that there was no statistical correlation between the GluCEST% and COHb concentration after CO poisoning.
At follow-up, patients made a good recovery of mental status according to postoperative HAMA scores. There was no evidence of significant sequela, and the time in coma was independent of the GluCEST values. This indicates that timely treatment was achieved in patients after coma was induced by CO poisoning and the early increase in GluCEST values might not cause irreversible damage to the brain. There are two possible explanations. First, the time in coma lacks subject assessment. Here the value was obtained mainly by the bath time, which is changeable and objective possibly affecting the data correlations. Second, patients included in this study had different degrees of CO exposure and are not only from the most critically affected populations. Only in the cases of severe CO exposure, the patients might be more likely to develop higher GluCEST values and severe clinical symptoms. More significant associations might be present, while further validation is in demand.
4. Limitations
The strength of this study is that it evaluates the value of the GluCEST technique as a new method in the diagnosis and understanding of the pathogenesis of brain injury after CO poisoning. However, there are still some limitations in this study. First, due to the obvious seasonal recruitment of patients with CO poisoning in our area, the number of cases collected in this study is relatively small, which may affect the experimental results. In the future, we will increase the sample size to consolidate our experimental results. Second, the grouping used in this study was based on age. According to the clinical symptoms of the patients and the different periods of CO poisoning and prognosis, patients with CO poisoning can also be divided into other subgroups. Finally, the participants in this study included subjects with a large age span. Such samples are vastly different in terms of brain development and life trajectories and could affect the results. In the future, we will increase the sample size to further explore the ability of the GluCEST technique as a new method of diagnosis and evaluation of the prognosis of CO poisoning in different subgroup analyses.
5. Conclusion
The GluCEST technique can provide a view of the glutamate concentration in the early phase after CO poisoning. It is superior to routine MRI as it can identify cerebral biochemical changes earlier in the acute phase after CO exposure, which is significant for our understanding of the role of neurotransmitters in the pathological basis of this disease. Cerebral injuries after CO poisoning might vary among adults and children. The early glutamate concentrations of the thalamus and the corpus callosum may be of importance in the assessment of the degree of cerebral injury after CO poisoning. The GluCEST imaging technique provides a new way to understand the pathophysiological mechanisms of CO poisoning.
Data availability statement
The original contributions presented in the study are included in the article/Supplementary material, further inquiries can be directed to the corresponding author.
Ethics statement
The studies involving human participants were reviewed and approved by Ethics Committee of the Second Affiliated Hospital of Shantou University Medical College. Written informed consent to participate in this study was provided by the participants' legal guardian/next of kin.
Author contributions
HZ and WZ designed the research. HZ wrote the article. WZ contributed to the manuscript revision and reading and approved the submitted version. JZ, WL, and GZ completed the data collation. YG, GZ, and WL performed the research and analyzed the data. All authors contributed to the article and approved the submitted version.
Funding
This study was funded by the Joint Research Fund for Enterprise and Basic and Applied Basic Research Programs of Guangdong Province of China (Grant No. 2021A1515 220112), the 2019 Li Ka Shing Foundation Cross-Disciplinary Research Grant, and the 2020 Basic Research General Project of the Shenzhen Science and Technology Innovation Commission Program grant awarded to WZ, a research grant awarded by the Natural Science Foundation of Shenzhen (JCYJ20190808095403639) to YG, and the grants by the 2023 Medical Scientific Research Foundation of Guangdong Province, China and the Foundation of 2022 Medical and Health Science and Technology Project of Shantou City (Grant No. [2022]169-44), Guangdong Province, China awarded to HZ.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher's note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fneur.2023.1065490/full#supplementary-material
References
1. Bleecker ML. Carbon monoxide intoxication. Handb Clin Neurol. (2015) 131:191–203. doi: 10.1016/B978-0-444-62627-1.00024-X
2. Ning K, Zhou YY, Zhang N, Sun XJ, Liu WW, Han CH. Neurocognitive sequelae after carbon monoxide poisoning and hyperbaric oxygen therapy. Med Gas Res. (2020) 10:30–6. doi: 10.4103/2045-9912.279981
3. Hampson NB, Piantadosi CA, Thom SR, Weaver LK. Practice recommendations in the diagnosis, management, and prevention of carbon monoxide poisoning. Am J Respir Crit Care Med. (2012) 186:1095–101. doi: 10.1164/rccm.201207-1284CI
5. Geocadin RG, Koenig MA, Jia X, Stevens RD, Peberdy MA. Management of brain injury after resuscitation from cardiac arrest. Neurol Clin. (2008) 26:487–506, ix. doi: 10.1016/j.ncl.2008.03.015
6. Doyle KP, Simon RP, Stenzel-Poore MP. Mechanisms of ischemic brain damage. Neuropharmacology. (2008) 55:310–8. doi: 10.1016/j.neuropharm.2008.01.005
7. Weaver LK. Clinical practice. Carbon monoxide poisoning. N Engl J Med. (2009) 360:1217–25. doi: 10.1056/NEJMcp0808891
8. Rose JJ, Wang L, Xu Q, McTiernan CF, Shiva S, Tejero J, et al. Carbon monoxide poisoning: pathogenesis, management, and future directions of therapy. Am J Respir Crit Care Med. (2017) 195:596–606. doi: 10.1164/rccm.201606-1275CI
9. Kim I, Xu W, Reed JC. Cell death and endoplasmic reticulum stress: disease relevance and therapeutic opportunities. Nat Rev Drug Discov. (2008) 7:1013–30. doi: 10.1038/nrd2755
10. Tambasco N, Romoli M, Calabresi P. Selective basal ganglia vulnerability to energy deprivation: experimental and clinical evidences. Prog Neurobiol. (2018) 169:55–75. doi: 10.1016/j.pneurobio.2018.07.003
11. Vizi ES, Kisfali M, Lorincz T. Role of nonsynaptic GluN2B-containing NMDA receptors in excitotoxicity: evidence that fluoxetine selectively inhibits these receptors and may have neuroprotective effects. Brain Res Bull. (2013) 93:32–8. doi: 10.1016/j.brainresbull.2012.10.005
12. Horowitz AL, Kaplan R, Sarpel G. Carbon monoxide toxicity: MR imaging in the brain. Radiology. (1987) 162:787–8. doi: 10.1148/radiology.162.3.3809495
13. Vieregge P, Klostermann W, Blumm RG, Borgis KJ. Carbon monoxide poisoning: clinical, neurophysiological, and brain imaging observations in acute disease and follow-up. J Neurol. (1989) 236:478–81. doi: 10.1007/BF00328511
14. Beppu T. The role of MR imaging in assessment of brain damage from carbon monoxide poisoning: a review of the literature. AJNR Am J Neuroradiol. (2014) 35:625–31. doi: 10.3174/ajnr.A3489
15. O'Donnell P, Buxton PJ, Pitkin A, Jarvis LJ. The magnetic resonance imaging appearances of the brain in acute carbon monoxide poisoning. Clin Radiol. (2000) 55:273–80. doi: 10.1053/crad.1999.0369
16. Chalela JA, Wolf RL, Maldjian JA, Kasner SE. MRI identification of early white matter injury in anoxic-ischemic encephalopathy. Neurology. (2001) 56:481–5. doi: 10.1212/WNL.56.4.481
17. Bruno A, Wagner W, Orrison WW. Clinical outcome and brain MRI four years after carbon monoxide intoxication. Acta Neurol Scand. (1993) 87:205–9. doi: 10.1111/j.1600-0404.1993.tb04102.x
18. Porter SS, Hopkins RO, Weaver LK, Bigler ED, Blatter DD. Corpus callosum atrophy and neuropsychological outcome following carbon monoxide poisoning. Arch Clin Neuropsychol. (2002) 17:195–204. doi: 10.1093/arclin/17.2.195
19. Teksam M, Casey SO, Michel E, Liu H, Truwit CL. Diffusion-weighted MR imaging findings in carbon monoxide poisoning. Neuroradiology. (2002) 44:109–13. doi: 10.1007/s002340100639
20. Huang D, Li S, Dai Z, Shen Z, Yan G, Wu R. Novel gradient echo sequencebased amide proton transfer magnetic resonance imaging in hyperacute cerebral infarction. Mol Med Rep. (2015) 11:3279–84. doi: 10.3892/mmr.2015.3165
21. Wu R, Longo DL, Aime S, Sun PZ. Quantitative description of radiofrequency (RF) power-based ratiometric chemical exchange saturation transfer (CEST) pH imaging. NMR Biomed. (2015) 28:555–65. doi: 10.1002/nbm.3284
22. Sun PZ, Xiao G, Zhou IY, Guo Y, Wu R. A method for accurate pH mapping with chemical exchange saturation transfer (CEST) MRI. Contrast Media Mol Imaging. (2016) 11:195–202. doi: 10.1002/cmmi.1680
23. Cai K, Haris M, Singh A, Kogan F, Greenberg JH, Hariharan H, et al. Magnetic resonance imaging of glutamate. Nat Med. (2012) 18:302–6. doi: 10.1038/nm.2615
24. Walker-Samuel S, Ramasawmy R, Torrealdea F, Rega M, Rajkumar V, Johnson SP, et al. In vivo imaging of glucose uptake and metabolism in tumors. Nat Med. (2013) 19:1067–72. doi: 10.1038/nm.3252
25. Wylie GR, Freeman K, Thomas A, Shpaner M, M OK, Watts R, et al. Cognitive improvement after mild traumatic brain injury measured with functional neuroimaging during the acute period. PLoS ONE. (2015) 10:e0126110. doi: 10.1371/journal.pone.0126110
26. Haris M, Nath K, Cai K, Singh A, Crescenzi R, Kogan F, et al. Imaging of glutamate neurotransmitter alterations in Alzheimer's disease. NMR Biomed. (2013) 26:386–91. doi: 10.1002/nbm.2875
27. Davis KA, Nanga RP, Das S, Chen SH, Hadar PN, Pollard JR, et al. Glutamate imaging (GluCEST) lateralizes epileptic foci in nonlesional temporal lobe epilepsy. Sci Transl Med. (2015) 7:309ra161. doi: 10.1126/scitranslmed.aaa7095
28. Simonsen C, Magnusdottir SO, Andreasen JJ, Wimmer R, Rasmussen BS, Kjaergaard B, et al. Metabolic changes during carbon monoxide poisoning: an experimental study. J Cell Mol Med. (2021) 25:5191–201. doi: 10.1111/jcmm.16522
29. Yang KC, Yang BH, Lirng JF, Liu MN, Hu LY, Liou YJ, et al. Interaction of dopamine transporter and metabolite ratios underpinning the cognitive dysfunction in patients with carbon monoxide poisoning: a combined SPECT and MRS study. Neurotoxicology. (2021) 82:26–34. doi: 10.1016/j.neuro.2020.11.002
30. Piantadosi CA, Zhang J, Levin ED, Folz RJ, Schmechel DE. Apoptosis and delayed neuronal damage after carbon monoxide poisoning in the rat. Exp Neurol. (1997) 147:103–14. doi: 10.1006/exnr.1997.6584
31. Beppu T, Nishimoto H, Fujiwara S, Kudo K, Sanjo K, Narumi S, et al. 1H-magnetic resonance spectroscopy indicates damage to cerebral white matter in the subacute phase after CO poisoning. J Neurol Neurosurg Psychiatry. (2011) 82:869–75. doi: 10.1136/jnnp.2010.222422
32. Meldrum BS. Glutamate as a neurotransmitter in the brain: review of physiology and pathology. J Nutr. (2000) 130 (4S Suppl):1007S−15S. doi: 10.1093/jn/130.4.1007S
33. Zhou Y, Danbolt NC. Glutamate as a neurotransmitter in the healthy brain. J Neural Transm. (2014) 121:799–817. doi: 10.1007/s00702-014-1180-8
34. Ishimaru H, Katoh A, Suzuki H, Fukuta T, Kameyama T, Nabeshima T. Effects of N-methyl-D-aspartate receptor antagonists on carbon monoxide-induced brain damage in mice. J Pharmacol Exp Ther. (1992) 261:349–52.
35. Zhao X, Wen Z, Huang F, Lu S, Wang X, Hu S, et al. Saturation power dependence of amide proton transfer image contrasts in human brain tumors and strokes at 3 T. Magn Reson Med. (2011) 66:1033–41. doi: 10.1002/mrm.22891
36. Dula AN, Pawate S, Dethrage LM, Conrad BN, Dewey BE, Barry RL, et al. Chemical exchange saturation transfer of the cervical spinal cord at 7 T. NMR Biomed. (2016) 29:1249–57. doi: 10.1002/nbm.3581
37. Zaiss M, Windschuh J, Goerke S, Paech D, Meissner JE, Burth S, et al. Downfield-NOE-suppressed amide-CEST-MRI at 7 tesla provides a unique contrast in human glioblastoma. Magn Reson Med. (2017) 77:196–208. doi: 10.1002/mrm.26100
38. Jiang W, Wu Q, Zhou C, Zhao Z, Tan Y. Gray matter nuclei damage in acute carbon monoxide intoxication assessed in vivo using diffusion tensor MR imaging. Radiol Med. (2020) 125:80–6. doi: 10.1007/s11547-019-01078-w
39. Shen Y, Xiao G, Shen Z, Zhang X, Tang X, Hu W, et al. Imaging of nuclear overhauser enhancement at 7 and 3 T. NMR Biomed. (2017) 30. doi: 10.1002/nbm.3735
40. Kim M, Gillen J, Landman BA, Zhou J, van Zijl PC. Water saturation shift referencing (WASSR) for chemical exchange saturation transfer (CEST) experiments. Magn Reson Med. (2009) 61:1441–50. doi: 10.1002/mrm.21873
41. Liu G, Song X, Chan KW, McMahon MT. Nuts and bolts of chemical exchange saturation transfer MRI. NMR Biomed. (2013) 26:810–28. doi: 10.1002/nbm.2899
42. Jones KM, Pollard AC, Pagel MD. Clinical applications of chemical exchange saturation transfer (CEST) MRI. J Magn Reson Imaging. (2018) 47:11–27. doi: 10.1002/jmri.25838
43. Goldenberg JM, Pagel MD. Assessments of tumor metabolism with CEST MRI. NMR Biomed. (2019) 32:e3943. doi: 10.1002/nbm.3943
44. Lapresle J, Fardeau M. The central nervous system and carbon monoxide poisoning. II. Anatomical study of brain lesions following intoxication with carbon monixide (22 cases). Prog Brain Res. (1967) 24:31–74. doi: 10.1016/S0079-6123(08)60181-8
45. Okeda R, Funata N, Takano T, Miyazaki Y, Higashino F, Yokoyama K, et al. The pathogenesis of carbon monoxide encephalopathy in the acute phase–physiological and morphological correlation. Acta Neuropathol. (1981) 54:1–10. doi: 10.1007/BF00691327
46. Thom SR, Bhopale VM, Fisher D, Zhang J, Gimotty P. Delayed neuropathology after carbon monoxide poisoning is immune-mediated. Proc Natl Acad Sci USA. (2004) 101:13660–5. doi: 10.1073/pnas.0405642101
47. Kim HY, Kim BJ, Moon SY, Kwon JC, Shon YM, Na DG, et al. Serial diffusion-weighted MR imaging in delayed postanoxic encephalopathy. A case study. J Neuroradiol. (2002) 29:211–5.
48. Geraldo AF, Silva C, Neutel D, Neto LL, Albuquerque L. Delayed leukoencephalopathy after acute carbon monoxide intoxication. J Radiol Case Rep. (2014) 8:1–8. doi: 10.3941/jrcr.v8i5.1721
49. Hara S, Mukai T, Kurosaki K, Kuriiwa F, Endo T. Characterization of hydroxyl radical generation in the striatum of free-moving rats due to carbon monoxide poisoning, as determined by in vivo microdialysis. Brain Res. (2004) 1016:281–4. doi: 10.1016/j.brainres.2004.05.047
50. Bi WK, Wang JL, Zhou XD, Li ZK, Jiang WW, Zhang SB, et al. Clinical characteristics of visual dysfunction in carbon monoxide poisoning patients. J Ophthalmol. (2020) 2020:9537360. doi: 10.1155/2020/9537360
51. Johnson SC, Pinkston JB, Bigler ED, Blatter DD. Corpus callosum morphology in normal controls and traumatic brain injury: sex differences, mechanisms of injury, neuropsychological correlates. Neuropsychology. (1996) 10:408–15. doi: 10.1037/0894-4105.10.3.408
52. Kipp M, Wagenknecht N, Beyer C, Samer S, Wuerfel J, Nikoubashman O. Thalamus pathology in multiple sclerosis: from biology to clinical application. Cell Mol Life Sci. (2015) 72:1127–47. doi: 10.1007/s00018-014-1787-9
53. Liu ZW, Han C, Wang H, Zhang Q, Li SJ, Bao XY, et al. Clinical characteristics and leptomeningeal collateral status in pediatric and adult patients with ischemic moyamoya disease. CNS Neurosci Ther. (2020) 26:14–20. doi: 10.1111/cns.13130
54. Hao NN, Tian C, Lian KX, Han T, Jin S. [Imaging diagnosis of 95 cases of moderate and severe acute carbon monoxide poisoning]. Zhonghua Lao Dong Wei Sheng Zhi Ye Bing Za Zhi. (2017) 35:463–7. doi: 10.3760/cma.j.issn.1001-9391.2017.06.016
55. Weaver LK. Carbon monoxide poisoning. Undersea Hyperb Med. (2020) 47:151–69. doi: 10.22462/01.03.2020.17
56. Raub JA, Mathieu-Nolf M, Hampson NB, Thom SR. Carbon monoxide poisoning–a public health perspective. Toxicology. (2000) 145:1–14. doi: 10.1016/S0300-483X(99)00217-6
57. Shimazu T. [Pathophysiology, myths and mysteries of acute carbon monoxide poisoning]. Chudoku Kenkyu. (2006) 19:23–33.
Keywords: carbon monoxide poisoning, chemical exchange saturation transfer, CO poisoning, GluCEST, glutamate
Citation: Zheng H, Zheng W, Liu H, Zhang G, Li W, Zhuang J and Guo Y (2023) Imaging of glutamate in acute carbon monoxide poisoning using chemical exchange saturation transfer. Front. Neurol. 14:1065490. doi: 10.3389/fneur.2023.1065490
Received: 11 October 2022; Accepted: 09 January 2023;
Published: 02 February 2023.
Edited by:
Jerzy Walecki, Medical University of Warsaw, PolandCopyright © 2023 Zheng, Zheng, Liu, Zhang, Li, Zhuang and Guo. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Wenbin Zheng,  aHdlbmJAMTI2LmNvbQ==
aHdlbmJAMTI2LmNvbQ==