
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
CASE REPORT article
Front. Neurol., 16 September 2022
Sec. Epilepsy
Volume 13 - 2022 | https://doi.org/10.3389/fneur.2022.995513
This article is part of the Research TopicEpilepsy – Case Report Collection 2022View all 13 articles
 Hui Chen†
Hui Chen† Xiaoyan Li†Huaping Wu†Xiaolan SunYuanyuan Che
Xiaoyan Li†Huaping Wu†Xiaolan SunYuanyuan Che Jian ZhaRuiyan WangXiongying YuYong Chen*
Jian ZhaRuiyan WangXiongying YuYong Chen* Jianmin Zhong*
Jianmin Zhong*Benign convulsions with mild gastroenteritis (CwG) is characterized by afebrile convulsions accompanied by mild gastroenteritis, and it can be considered after central nervous system infection, hypoglycemia, electrolyte disturbance, and moderate and severe dehydration are excluded. Previous studies have suggested that genetics may be involved in CWG. Herein, we reported a novel de novo variant of SCN8A in a child with CwG. This is the first report that SCN8A may be associated with CwG. Our report may provides evidence for the genetic etiology of CwG and expands the phenotypic and genetic spectrum of SCN8A-related disorders, which previously included severe developmental and epileptic encephalopathy (DEE) phenotype, benign epilepsy phenotype, spectrum of intermediate epilepsies, and patients with cognitive and/or behavioral disturbances without epilepsy. Phenotype of CwG has a good prognosis, and it does not require long-term antiepileptic therapy. Overtreatment should be avoided clinically. However, the conclusion needs to be further defined by long-term follow-up and similar clinical reports. In spite of this, our clinical observation provides possible evidence for future studies on the relationship between SCN8A and CwG.
Benign convulsions with mild gastroenteritis (CwG) was first reported by Morooka in 1982 in Japan (1), which was later repeatedly reported (2, 3). Most of the CwG cases occurs during the winter and early spring months. This clinical condition is characterized by afebrile convulsions accompanied by mild gastroenteritis in previously healthy infants. Besides, it can be considered after central nervous system infection, hypoglycemia, electrolyte disturbance, and moderate and severe dehydration are excluded, and usually has a good prognosis (4).
CwG has been more frequently described in East Asian countries, which suggests that the genetic characteristics of the host may play a role in the development of CwG (4). In addition to this, Okumura et al. found that CwG occurred in identical twins during the course of gastroenteritis, and their convulsions occurred almost simultaneously (5). These studies suggest that CwG may be related to genetic factors. However, in recent years, several scholars have tested SCN1A, SCN1B, and PRRT2 genes in CwG patients, but no positive results were found (6–8). Finally, in 2020, Terrone et al. found heterozygous missense mutation of SPTAN1 gene by next generation sequencing (NGS) in a family, including a 13-year-old sister, an 8-year-old brother and their 39-year-old mother (9). The clinical observation suggests for the first time that variants in SPTAN1 gene might be involved in the etiology of CwG. Herein, we reported a novel de novo variant of SCN8A in a child with CwG. This is the first report that SCN8A may be associated with CwG. Our report may provides evidence for the genetic etiology of CwG and expands the phenotypic and genetic spectrum of SCN8A-related disorders.
The patient was a 36 months old boy. The Chinese boy was born full term with normal birth weight. She had no history of transient hypoxia at birth. There was no family history of neurologic disorders. He developed acute gastroenteritis symptoms at 19 months, presenting with 3 times of vomiting and diarrhea (3 times daily). A day later, the boy began with 3 generalized tonic-clonic seizures (GTCS) without fever in 1 day. The duration of each convulsion was 2–3 min. Since then, phenobarbital was given (5 mg/kg, intravenous injection). However, he still had two more similar convulsions. After treatment of diazepam (0.3 mg/kg, intravenous injection), there was no further convulsion. Vomiting resolved within 2 days, while diarrhea recurred over 5 days (2–4 times a day) and then resolved. Dehydration did not occur throughout the course of the disease. In the interictal phase, the child's mental state was normal and his consciousness was clear. However, 10 days later, he developed symptoms of gastroenteritis again, including diarrhea and vomiting, and had one GTCS a day later. The duration of the convulsion was 2 min. The symptoms of the mild gastroenteritis lasted for 4 days, and the convulsions only occurred once during the course of the gastroenteritis. We did not give the child long-term antiepileptic treatment. After a long-term follow-up of 17 months, he never had another seizure. The child's motor and intellectual milestones were the same as those of normal children. Physical examination at the age of 19 months showed a height of 83 cm (50–85th centile), weight of 13.5 kg (85–97th centile) and head circumference of 47 cm (25–50th centile). His consciousness was clear. Cardiac auscultation was normal. Muscle strength and tone were normal. Knee reflex were normal and babinski signs were negative. EEG showed slow background activity during wakefulness in interictal periods, and no epileptic discharges were observed (Figure 1). Rotavirus of stool was positive. Other investigations were non-diagnostic including liver function tests, renal function, electrolytes, glucose, blood white cells, blood red cells, platelets, ammonia, creatine kinase, plasma lactate, white blood cells and red blood cells in stool, electrocardiogram, and brain MRI. Therefore, he was eventually diagnosed as suffering from CwG. Due to recurrent convulsions, the child's parents wanted to further understand the genetic causes, so we performed NGS on him. He was found to have a de novo heterozygous mutations c.5503 (exon27) C>G in SCN8A gene that was not detected in either parent. The variant led to protein changes p.Pro1835Ala (NM_014191.4). The variant (p.Pro1835Ala) was a de novo heterozygous variant (PS2), which affected highly conserved amino acid region. It was not found in the normal control population in multiple databases, such as gnomAD, ExAC, 1,000 Genomes, ESP6500 (PM2_Supporting). Various statistical methods predicted that the variation would have deleterious effects on gene products (PP3), such as SIFT (Damaging), MutationTaster (Disease_causing), FATHMM (Damaging), PROVEAN (Damaging), DANN (Damaging), CADD (Damaging) and Eigen (Damaging). In addition to this, the missense mutation of SCN8A gene had a high pathogenic possibility, and the Z-score was 7.64 in gnomAD database (PP2). Hence, the variant was classified as likely pathogenic in accordance with the ACMG guideline (PS2 + PM2_Supporting + PP2 + PP3).
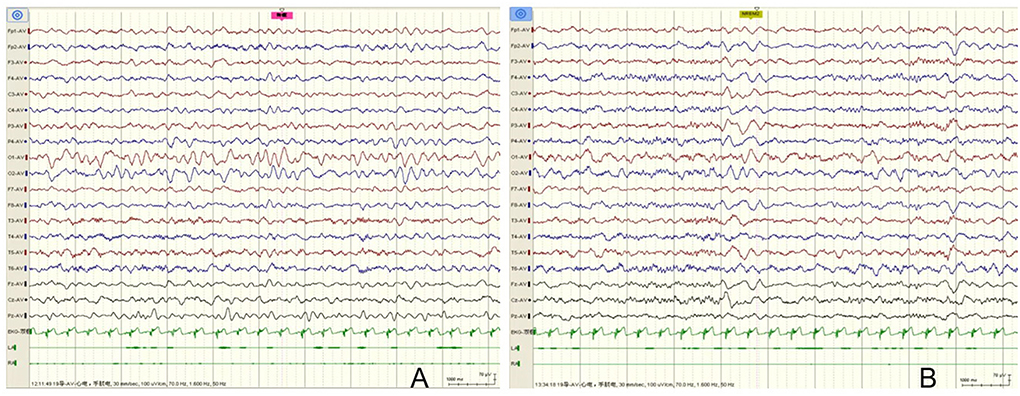
Figure 1. EEG of the case. (A) EEG showed slow background activity during wakefulness in interictal periods, and no epileptic discharges were observed. (B) No epileptic discharges were observed during sleep stages.
In our case report, the patient presented with multiple afebrile convulsions accompanied by mild gastroenteritis, and he did not present with electrolyte disturbances, dehydration, hypoglycemia, or central nervous system infection. In addition, he did not receive long-term antiepileptic medication. After a long-term follow-up of 17 months, he did not develop convulsions again, and his motor and cognitive development was normal. he has a good prognosis. In the first course of acute gastroenteritis, vomiting resolved within 2 days, while diarrhea resolved within 5 days. Ten days later, he developed symptoms of gastroenteritis again, including diarrhea and vomiting. The symptoms of the mild gastroenteritis lasted for 4 days. The convulsions only occurred during the course of the gastroenteritis. During a 10-day period, there were no symptoms of gastroenteritis or convulsions. Therefore, I think it was two episodes. Recent study has shown that children with CwG have a possibility of recurrence, with a recurrence rate of about 6.3% (10). Therefore, child of the case might suffer from recurrent CwG according to its diagnostic criteria (4, 10). However, because the interval time is too short (10-day period), it needs to be differentiated from epilepsy. The diagnosis of recurrent CwG still needs to be further defined by long-term follow-up and similar clinical reports. CwG is similar to an epileptic syndrome within benign infantile seizures in the classification set by the International League Against Epilepsy (ILAE) because of its clinical features of afebrile convulsions. However, until now, CwG has not been fully recognized as an epileptic syndrome by ILAE. Because convulsion of CwG is accompanied by mild gastroenteritis, it has been suggested that CwG might be termed as situation-related seizures (4). In conclusion, CwG is a separate disease which is different from epilepsy.
SCN8A encodes Nav1.6, which is one of four voltage-gated sodium channels expressed in the mammalian brain. Nav1.6 is found in the central and peripheral nervous system with a predominant expression in excitatory, but also in inhibitory neurons (11, 12). The first pathogenic variants in SCN8A have been described in an affected individual with developmental and epileptic encephalopathy (DEE) (13). In recent years, a wide clinical spectrum of neurodevelopmental phenotypes has been reported, including severe DEE phenotype, benign epilepsy phenotype, spectrum of intermediate epilepsies, generalized epilepsy, unclassifiable epilepsy and patients with cognitive and/or behavioral disturbances without epilepsy (12, 14–17). Most pathogenic variants in SCN8A are missense. Previous functional studies of selected epilepsy-associated SCN8A variants have revealed a gain of function (GoF) pathogenic mechanism, which cause ultimately hyperactivity of the ion channel. On the contrary, variants causing loss of function (LoF) are related with patients with cognitive and/or behavioral disturbances without epilepsy, Such as ID, ASD, myoclonus, and ataxia (14). However, this view is no longer true, a recent study showed generalized epilepsy with absence seizures was the main epilepsy phenotype of LOF variant carriers and the extent of the electrophysiological dysfunction of the GOF variants was a main determinant of the severity of the clinical phenotype in focal epilepsies including severe DEE phenotype, benign epilepsy phenotype, spectrum of intermediate epilepsies. However, a few functional studies of SCN8A variants in DEE were LOF (12). Our clinical observation is the first report that SCN8A may be associated with CwG. Our clinical observation is only a case report, which need to be supported by more case reports. Moreover, relevant functional studies will be needed to further clarify the related mechanisms. This is the limitation of our clinical observation.
In conclusion, our clinical observation suggests that variants in SCN8A gene might be involved in pathogenesis of CwG. Phenotype of CwG has a good prognosis. It does not require long-term antiepileptic therapy, and overtreatment should be avoided clinically. However, the conclusion needs to be further defined by long-term follow-up and similar clinical reports. In spite of this, our clinical observation provides possible evidence for future studies on the relationship between SCN8A and CwG.
The raw data supporting the conclusions of this article will be made available by the authors, without undue reservation.
The studies involving human participants were reviewed and approved by the Research Ethics Committee of Children's Hospital of Jiangxi Province. Written informed consent to participate in this study was provided by the participants' legal guardian/next of kin. Written informed consent was obtained from the individual(s), and minor(s)' legal guardian/next of kin, for the publication of any potentially identifiable images or data included in this article.
All authors listed have made a substantial, direct, and intellectual contribution to the work and approved it for publication.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
1. Morooka K. Convulsions and mild diarrhea [in Japanese]. Shonika. (1982) 23:131–7. doi: 10.1536/ihj.23.697
2. Ma X, Luan S, Zhao Y, Lv X, Zhang R. Clinical characteristics and follow-up of benign convulsions with mild gastroenteritis among children. Medicine. (2019) 98:e14082. doi: 10.1097/MD.0000000000014082
3. Hao XS, Liang JM, Wu XM, Hao YP, Wang JT, Liu SY. Clinical characteristics, treatment, and long-term outcomes in children suffering from benign convulsions with mild gastroenteritis: a retrospective study. BMC Pediatr. (2020) 20:516. doi: 10.1186/s12887-020-02406-0
4. Castellazzi L, Principi N, Agostoni C, Esposito S. Benign convulsions in children with mild gastroenteritis. Eur J Paediatr Neurol. (2016) 20:690–5. doi: 10.1016/j.ejpn.2016.05.014
5. Okumura A, Kato T, Hayakawa F, Kuno K, Watanabe K. Convulsion associated with mild gastroenteritis: occurrence in identical twins on the same day. No To Hattatsu. (1999) 31:59–62.
6. Weng WC, Hirose S, Lee WT. Benign convulsions with mild gastroenteritis: is it associated with sodium channel gene SCN1A mutation? J Child Neurol. (2010) 25:1521–4. doi: 10.1177/0883073810370898
7. Yamashita S, Okumura A, Yamamoto T, Shimojima K, Tanabe T, Shimizu T. SCN1B is not related to benign partial epilepsy in infancy or convulsions with gastroenteritis. Neuropediatrics. (2011) 42:135–7. doi: 10.1055/s-0031-1285837
8. Ishii A, Yasumoto S, Ihara Y, Inoue T, Fujita T, Nakamura N, et al. Genetic analysis of PRRT2 for benign infantile epilepsy, infantile convulsions with choreoathetosis syndrome, and benign convulsions with mild gastroenteritis. Brain Dev. (2013) 35:524–30. doi: 10.1016/j.braindev.2012.09.006
9. Terrone G, Pinelli M, Bernardo P, Parrini E, Imperati F, Brunetti-Pierri N, et al. Intrafamilial variability in SPTAN1-related disorder: From benign convulsions with mild gastroenteritis to developmental encephalopathy. Eur J Paediatr Neurol. (2020) 28:237–9. doi: 10.1016/j.ejpn.2020.07.008
10. Shi K, Yang J, Wu Y, Han H, Guo J, Chen W. Risk factors for the recurrence of convulsions with mild gastroenteritis in children. Seizure. (2020) 80:192–5. doi: 10.1016/j.seizure.2020.06.016
11. Bunton-Stasyshyn RKA, Wagnon JL, Wengert ER, Barker BS, Faulkner A, Wagley PK, et al. Prominent role of forebrain excitatory neurons in SCN8A encephalopathy. Brain. (2019) 142:362–75. doi: 10.1093/brain/awy324
12. Johannesen KM, Liu Y, Koko M, Gjerulfsen CE, Sonnenberg L, Schubert J, et al. Genotype-phenotype correlations in SCN8A-related disorders reveal prognostic and therapeutic implications. Brain. (2021). doi: 10.1093/brain/awab321. [Epub ahead of print].
13. Veeramah KR, O'Brien JE, Meisler MH, Cheng X, Dib-Hajj SD, Waxman SG, et al. De novo pathogenic SCN8A mutation identified by whole-genome sequencing of a family quartet affected by infantile epileptic encephalopathy and SUDEP. Am J Hum Genet. (2012) 90:502–10. doi: 10.1016/j.ajhg.2012.01.006
14. Gardella E, Møller RS. Phenotypic and genetic spectrum of SCN8A-related disorders, treatment options, and outcomes. Epilepsia. (2019) 60(Suppl. 3):S77–85. doi: 10.1111/epi.16319
15. Johannesen KM, Gardella E, Encinas AC, Lehesjoki AE, Linnankivi T, Petersen MB, et al. The spectrum of intermediate SCN8A-related epilepsy. Epilepsia. (2019) 60:830–44. doi: 10.1111/epi.14705
16. Anand G, Collett-White F, Orsini A, Thomas S, Jayapal S, Trump N, et al. Autosomal dominant SCN8A mutation with an unusually mild phenotype. Eur J Paediatr Neurol. (2016) 20:761–5. doi: 10.1016/j.ejpn.2016.04.015
Keywords: SCN8A, gene, benign, convulsions, gastroenteritis
Citation: Chen H, Li X, Wu H, Sun X, Che Y, Zha J, Wang R, Yu X, Chen Y and Zhong J (2022) Case report: A novel de novo variant of SCN8A in a child with benign convulsions with mild gastroenteritis. Front. Neurol. 13:995513. doi: 10.3389/fneur.2022.995513
Received: 16 July 2022; Accepted: 31 August 2022;
Published: 16 September 2022.
Edited by:
Fernando Cendes, State University of Campinas, BrazilReviewed by:
Elena Gardella, University of Southern Denmark, DenmarkCopyright © 2022 Chen, Li, Wu, Sun, Che, Zha, Wang, Yu, Chen and Zhong. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Yong Chen, Y2hlbjk2MjVAMTYzLmNvbQ==; Jianmin Zhong, emhvbmdqbUAxNjMuY29t
†These authors have contributed equally to this work and share first authorship
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.