 Noor Mary Abi Rached1
Noor Mary Abi Rached1 Oluwabukola T. Gbotosho2
Oluwabukola T. Gbotosho2 David R. Archer3
David R. Archer3 Jayre A. Jones3Morgan S. Sterling3
Jayre A. Jones3Morgan S. Sterling3 Hyacinth I. Hyacinth2*
Hyacinth I. Hyacinth2*- 1Neuroscience and Behavioral Biology Undergraduate Program, Emory University, Atlanta, GA, United States
- 2Department of Neurology and Rehabilitation Medicine, University of Cincinnati College of Medicine, Cincinnati, OH, United States
- 3Aflac Cancer and Blood Disorders Center, Emory University Department of Pediatrics and Children's Healthcare of Atlanta, Atlanta, GA, United States
Cerebrovascular abnormalities are a common feature of sickle cell disease that may be associated with risk of vaso-occlusive pain crises, microinfarcts, and cognitive impairment. An activated endothelium and adhesion factors, VCAM-1 and P-selectin, are implicated in sickle cell vasculopathy, including abnormal hemodynamics and leukocyte adherence. This study examined the association between cerebral expression of these adhesion factors and cortical microvascular blood flow dynamics by using in-vivo two-photon microscopy. We also examined the impact of blood transfusion treatment on these markers of vasculopathy. Results showed that sickle cell mice had significantly higher maximum red blood cell (RBC) velocity (6.80 ± 0.25 mm/sec, p ≤ 0.01 vs. 5.35 ± 0.35 mm/sec) and more frequent blood flow reversals (18.04 ± 0.95 /min, p ≤ 0.01 vs. 13.59 ± 1.40 /min) in the cortical microvasculature compared to controls. In addition, sickle cell mice had a 2.6-fold (RFU/mm2) increase in expression of VCAM-1 and 17-fold (RFU/mm2) increase in expression of P-selectin compared to controls. This was accompanied by an increased frequency in leukocyte adherence (4.83 ± 0.57 /100 μm/min vs. 2.26 ± 0.37 /100 μm/min, p ≤ 0.001). We also found that microinfarcts identified in sickle cell mice were 50% larger than in controls. After blood transfusion, many of these parameters improved, as results demonstrated that sickle cell mice had a lower post-transfusion maximum RBC velocity (8.30 ± 0.98 mm/sec vs. 11.29 ± 0.95 mm/sec), lower frequency of blood flow reversals (12.80 ± 2.76 /min vs. 27.75 ± 2.09 /min), and fewer instances of leukocyte adherence compared to their pre-transfusion imaging time point (1.35 ± 0.32 /100 μm/min vs. 3.46 ± 0.58 /100 μm/min). Additionally, we found that blood transfusion was associated with lower expression of adhesion factors. Our results suggest that blood transfusion and adhesion factors, VCAM-1 and P-selectin, are potential therapeutic targets for addressing cerebrovascular pathology, such as vaso-occlusion, in sickle cell disease.
Introduction
Sickle cell disease (SCD) is caused by a single point mutation in the beta-globin gene, resulting in the substitution of valine for glutamic acid in the resulting β-globin peptide. In turn, this leads to polymerization of deoxyhemoglobin, forming sickled erythrocytes (1). SCD impacts an estimated 100,000 individuals in the United States, and the incidence of SCD among African Americans is approximately 1 in 360 newborns (2, 3). SCD involves clinical complications that impact multiple organ systems, with pain being the most frequent and results from vaso-occlusion. As a common symptom of SCD, vaso-occlusive pain crises result from sickle shaped erythrocytes and leukocytes blocking blood flow, particularly in small vessels, resulting in ischemia of organs and thus, pain (4, 5). Additionally, these vaso-occlusive events (VOEs) can be triggered by processes such as inflammation, thrombosis, increased aggregation of cells, and adhesion of blood cells to the vascular endothelium, ultimately leading to blockages that deprive the tissues of nutrients and oxygen (6–9). This results in tissue death and infarction in several organ systems including the spleen, liver, kidney, and lungs (4, 10). Another possible consequence of VOEs is silent cerebral infarction (SCI) or cerebral microinfarctions, which are small ischemic lesions that may occur without overt neurological symptoms. Studies have shown that SCIs or cerebral microinfarcts may be linked to the development of cognitive decline, and are associated with vascular and hemodynamic abnormalities such as cerebral macro and/or micro-vasculopathy, hypoperfusion, or obstructions to blood flow (11–13). Magnetic resonance imaging (MRI) studies from clinical cohorts in the Cooperative Study of Sickle Cell Disease showed that children with silent cerebral microinfarcts also have a higher risk of stroke (14, 15). Further studies from this cohort showed that school-aged children with SCD and silent infarcts experienced difficulties with neuropsychological functions (16). In mouse models, a recent study from our laboratory showed that sickle cell (SS) mice had 2.5 times more cortical microinfarcts than controls. Additionally, these mice had significantly higher prevalence of evidence of spontaneous cerebral vasculopathies, such as higher red blood cell (RBC) velocity, more tortuous capillary vessels, and more frequent capillary occlusive events (12).
Vaso-occlusive events occur more frequently in the low flow “micro” vessels such as, the pre-capillary arterioles, capillaries, and post-capillary venules (17, 18). Studies in non-sickle cell mouse models showed that blockage or experimental occlusion of the principal (penetrating) cortical venules (PCV) lead to stagnant flow in the upstream arterioles. This impairs blood flow into the cortex, thus, highlighting the importance of the entire cerebral microvascular tree, in the etiology of cerebral microinfarcts (11). However, it is important to note that SS RBCs typically pass through the capillary bed prior to hemoglobin polymerization, suggesting that additional factors may also be involved in the pathology of VOEs in SCD (19, 20). For instance, endothelial activation has been shown to play a role in the pathogenesis of VOEs in SCD (8, 18, 21). This is shown by the demonstration of significantly higher levels of circulating endothelial cells in patients with SCD compared with matched controls (21). In vitro studies also demonstrated the capacity for the more rigid sickle erythrocytes to mechanically activate endothelial cells, leading to an increase in expression of cellular adhesion molecules (markers of endothelial activation) which in turn propagates further adhesion and vaso-occlusion in a vicious cycle (8, 22, 23). The role of these cellular adhesion molecules [such as Intercellular Adhesion Molecule 1 (ICAM-1), P- and E-selectins and vascular cell adhesion molecule 1 (VCAM-1)] and thus endothelial activation in VOEs is further supported by a recent report of higher levels of E-selectin, VCAM-1, and ICAM-1 in SCD patients compared to controls (18). One study demonstrated significantly higher levels of these molecules, suggesting endothelial activation among SCD patients presenting with complications and even higher levels among those presenting with an active vaso-occlusive pain crises, compared to steady state (24). It is already well demonstrated that individuals with SCD have elevated leukocyte counts, which in the setting of increased VCAM-1 expression, results in increased endothelial interaction and thus arrest (25, 26). Our lab reported a strong relationship between serum soluble VCAM-1 (sVCAM-1), P-selectin and ICAM-1 levels, and risk of stroke in patients (children) with SCD. In the same study, we also demonstrated that lower levels of these cell adhesion markers were associated with stroke free survival as well as use of blood transfusion therapy for stroke prevention in patients with SCD (27). A paradigm proposed by Frenette et al. (28) suggests a multi-step model of vaso-occlusion whereby sickle cells induce endothelial activation, creating an environment where adherent leukocytes can interact with both RBCs and the endothelium to hinder blood flow, and subsequently create blockages (28, 29). In addition, it has been documented that among patients with sickle cell disease, those with higher cerebral blood flow as a compensatory mechanism for lack of brain oxygenation, performed more poorly on tests of cognitive function (30). This highlights the importance of exploring how adhesion factors may relate to this abnormal blood flow in SCD. Taken together we reasoned that the level of expression (or deposition) of cellular adhesion molecules in the cerebral microvascular endothelium will play a role in cerebral microvascular hemodynamics and could be a physiological mechanism for the cortical microinfarct or SCIs observed in mouse models of SCD and adults/children with SCD respectively (12, 25, 31, 32).
Vascular cell adhesion molecule-1 (i.e., VCAM-1), is expressed on blood vessels (endothelial cells) after activation by chemical (such as cytokines and chemokines) and/or mechanical stimulation, resulting in cytokine release. It is involved in adhesion of lymphocytes, monocytes, eosinophils, and basophils to the endothelium (33). According to Stuart and Setty (34), in a state of hypoxia, sickled red blood cells adhere to endothelial VCAM-1 using the very late activation antigen-4 (VLA4) ligand (34). P-selectin has also been implicated in the pathophysiology of SCD and is currently the target of a newly developed anti-vaso-occlusive crisis (VOC) drug (35–42). The chronic inflammatory milieu of SCD results in a persistently elevated serum level as well as endothelial expression of P-selectin, which is necessary for the initial binding of leukocytes to the vascular endothelium (43, 44). However, in the setting of SCD, P- selectin expression is likely to result in disruption of normal hemodynamics from excess/aberrant leukocyte-endothelial interaction (45, 46). While studies have shown that excessive endothelial expression of VCAM-1 and P-selectin results in VOEs and therefore pain in the periphery, the impact of such excess adhesion molecule expression on leukocyte-endothelial interaction and therefore cerebral microvascular hemodynamics is not known. Our study investigated this relationship given its potential implication for cerebral/cortical infarction and therefore neurological complications such as cognitive impairment. We also examined the relationship between the deposition (expression) of the two most well documented cellular adhesion molecules, VCAM-1, and P-selectin, associated with SCD complications and cerebral microvascular hemodynamics. Finally, we examined the impact of packed red blood cell transfusion on these adhesion molecules and therefore cerebral hemodynamics.
Methods
Animal preparation
The Institutional Animal Care and Use Committees (IACUC) of Emory University and the Medical University of South Carolina approved this study, and all research was conducted in accordance with the National Research Council and National Institutes of Health Guide for the Care and Use of Laboratory Animals 8th Edition (47). Figure 1 provides an overview of the experiments.
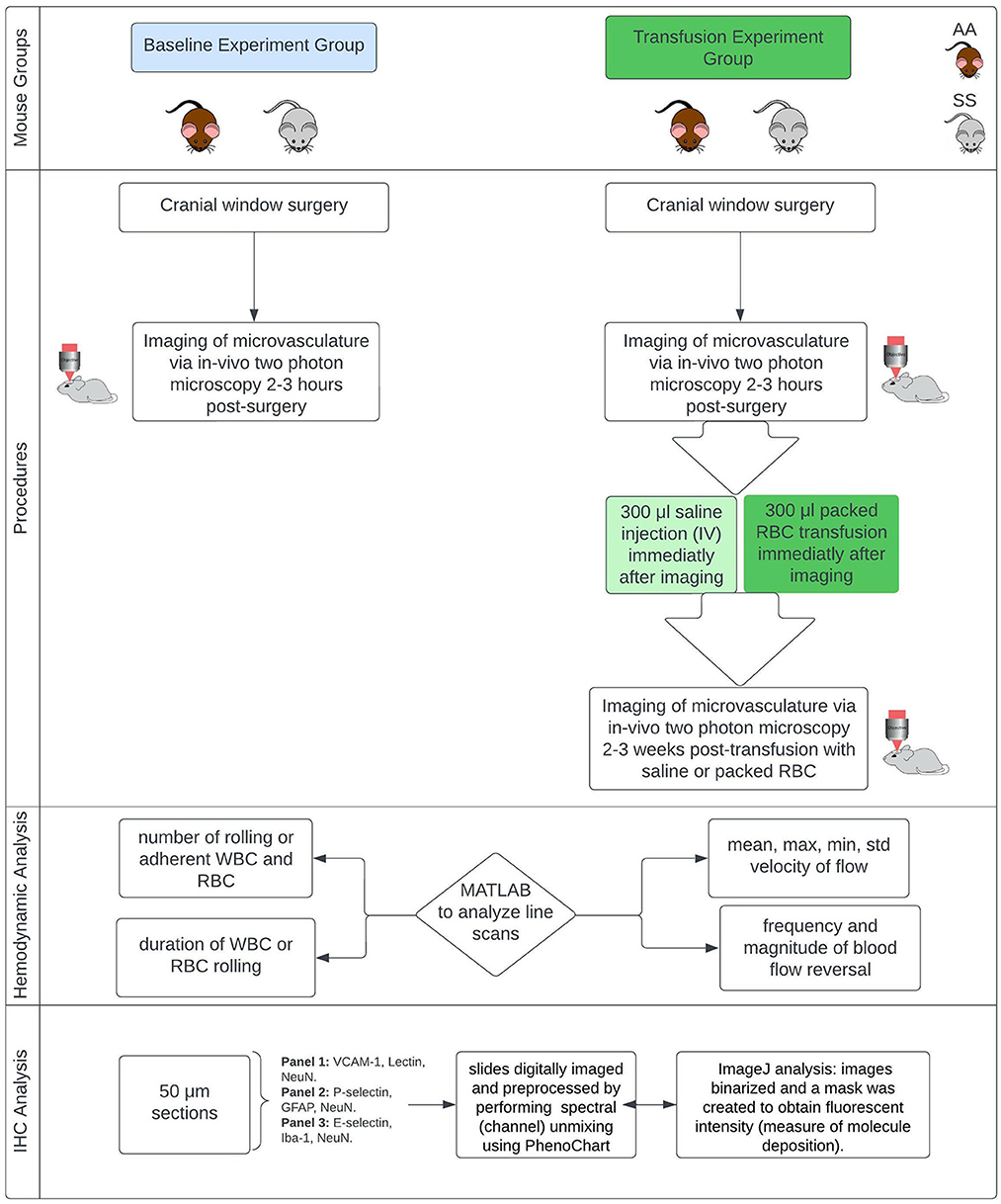
Figure 1. Overview of methods. Townes mouse model, a humanized sickle cell (with HbSS) and control (HbAA) mouse model (12–13 months). IHC, immunohistochemistry.
This study used the Townes mouse model, a humanized sickle cell mouse model (with HbSS) and corresponding humanized control group (with HbAA). Mice are male and ~13 months old at the time of starting the experiments. They were divided into two main groups. Group one was used to examine changes in microvascular hemodynamics while group two was used to examine the impact of red blood cell transfusion on microvascular hemodynamics. Blood transfusion therapy is a primary treatment option for primary and secondary stroke prevention for children and adults with SCD and has been shown to reduce hemoglobin S concentration as well as reducing the risk of stroke and silent cerebral infarct (cerebral microinfarcts) (48–50).
In our study, all mice in both the baseline group and the transfusion experiment group underwent implantation of a cranial window (placed over the somatosensory cortex) under anesthesia to obtain optical access to the intracranial space. The procedures for anesthetizing mice and performing the cranial window surgery have been described in our prior publications (12, 51). Two to three hours following surgery, the mice in the blood transfusion group underwent pre-transfusion two-photon laser scanning microscopy (TPLSM) imaging. Afterwards (i.e., immediately following imaging and prior to recovery from anesthesia), sickle cell mice received blood transfusions with 300 μl of packed red blood cells from humanized Townes HbAA mice while control (HbAA) mice received 300 μl intravenous (IV) saline. All infusions were performed slowly over 1 min. The goal of the packed RBC transfusion was to raise the hemoglobin level by at least 1g/dL. Mice in the baseline group underwent post-surgery imaging, but they did not receive any transfusion fusions and were sacrificed immediately following the two-photon (2 Photon) imaging. The mice that received blood or saline infusion, underwent a second 2 Photon imaging 2–3 weeks after the infusion. It is important to note that, in the transfusion experiment each group of mice served as their own treatment control, thus the pre and post transfusion imaging represent the same group of mice at different time points. The AA mice also served an additional purpose of being a control for the impact of handling as well as to enable us to show that any positive effect of packed RBC transfusion observed in the HbSS mice, is not due to the passage of time and thus resolution of the initial stress from surgery.
In vivo imaging procedure
To examine hemodynamic parameters, in-vivo images of cortical capillaries, precapillary arterioles, and post capillary venules (low flow cortical microvascular hemodynamics) were obtained using 2 Photon microscopy based on the schedule described above. Imaging was performed with a Sutter Moveable Objective Microscope (MOM) and a Coherent Ultra II Ti: Sapphire laser source. Methods for animal preparation during imaging and for measurement of cerebral hemodynamic and microvascular parameters via in vivo 2 Photon blood flow imaging have been previously described and published (12, 52, 53). about 2–5 min prior to commencing imaging, fluoresceine conjugated dextran (FITC-dextran 2kD to label the plasma) and Rhodamine 6G (to label leukocytes) are administered to the mice mouse IV, in that order.
Hemodynamic analysis
To examine hemodynamic parameters, we used custom MATLAB codes to analyze line scans of cerebral capillary, pre-capillary and post-capillary blood vessel images acquired using 2 Photon microscopy from SS and control (AA) mice. Using the MATLAB scripts, we were able to determine the following: mean and standard deviation of RBC flow velocity, maximum and minimum velocity of RBC flow, frequency (per minute) and magnitude of microvascular RBC/blood flow reversal and leukocyte (WBC) rolling on the endothelium. Microvascular RBC or blood flow reversal is a change in the original direction of blood flow relative to the direction of the line scan and could be multiple transient changes or a single change that last the duration of the line scan. A rolling or adherent WBC is defined as WBC stagnation lasting two or more seconds. We normalized the number of leukocyte rolling or leukocyte adherence events to a fixed vessel segment (100 μm) per unit time (1 min). This is to ensure reproducibility of our findings (54). All image analysis was performed by members of our laboratory who were blinded to the genotype or transfusion status of the mice.
Immunohistochemistry
At the end of 2 Photon imaging, the mice were sacrificed, and the brain extracted for immunohistochemistry (IHC) analysis to examine the microvascular deposition of adhesion factors, possible presence and size (area) of cerebral (cortical) microinfarcts, and possible presence of gliosis at the site of infarcts. The immunohistochemical protocol/approach has been previously described and published (12). For the baseline mice, the IHC was performed immediately following the imaging. For the mice that were transfused either with saline (HbAA) or packed RBC (HbSS), all IHC was performed after the post-transfusion 2 Photon imaging, and there was no pre-transfusion IHC analysis. We used the following primary antibody combinations to label 50-micron brain tissue sections from SS and AA mice. Panel 1 contained VCAM-1, Lectin (to localize and stain the vasculature), and NeuN (to localize and stain neuronal nuclei). Panel 2 contained P-selectin, GFAP (to localize and stain reactive astrocytes), and NeuN. Panel 3 contained E-selectin, Iba-1 (to localize and stain microglia), and NeuN. These slides are digitally imaged using a PerkinElmer digital slide scanner (Akoya Biosciences), and then images were preprocessed by performing spectral (channel) unmixing using PhenoChart (Akoya Biosciences). IHC Images were analyzed using ImageJ, with standard parameters for each fluorophore. Briefly, after spectral unmixing, the images from each fluorescent channel were transferred to image J, where they were binarized and a mask was created. The mask was then applied to the source images to obtain fluorescent intensity as an indication of the level of deposition (expression) of the molecule e.g., VCAM-1 of interest. The threshold values for creating the masks as well as analysis parameters were kept constant between images and between genotype (i.e., HbSS and HbAA) mice for each fluorophore/fluorescent channel and their adhesion molecule of interest. The resulting intensity was then normalized to a unit (mm2) image size and expressed in relative fluorescence unit (RFU) per mm2.
Statistical analysis
We performed data analysis for comparison between sickle cell and control mice using GraphPad Prism software (GraphPad Software Inc, La Jolla, CA). We checked our data for normality using the Shapiro-Wilk test, and then we used the Welch corrected t-test for comparison of differences between sickle cell and control mice because of the heteroscedasticity in our data based on Levene's F-test for equality of variance. Our study minimum sample size was 3 mice per genotype group and was based on our prior experiments and publications using this mouse model (55–57). Quantitative results are presented using bar plots with means and standard error of means (SEM), comparing sickle to control mice and with a p-value of < 0.05 considered statistically significant. Qualitative data are presented as representative array of histochemical images.
Results
Cerebral hemodynamic properties at baseline for sickle cell mice compared to age-matched controls
Analysis of the 2 Photon microscopy imaging data for cerebral microvascular hemodynamic measurements revealed that sickle cell mice had significantly higher maximum RBC velocity (6.80 ± 0.25 mm/sec vs. 5.35 ± 0.35 mm/sec, p = 0.0009) compared to age-matched controls (Figure 2A). In addition, we noted that that sickle mice have a higher frequency of cerebral microvascular blood flow reversal (18.04/min vs. 13.59/min, p = 0.008) compared to controls (Figure 2B). Further analysis revealed that the velocity of blood flow reversal was also significantly higher among sickle cell mice (0.84 ± 0.14 mm/sec vs. 0.52 ± 0.06 mm/sec, p = 0.03) compared to controls (Figure 2C). Also, blood flow reversal parameters are used to identify how often and at what velocity blood flow in a vessel is disturbed (58–60). This suggests that blood flow is abnormal in sickle cell mice, and this may be related to VOEs.
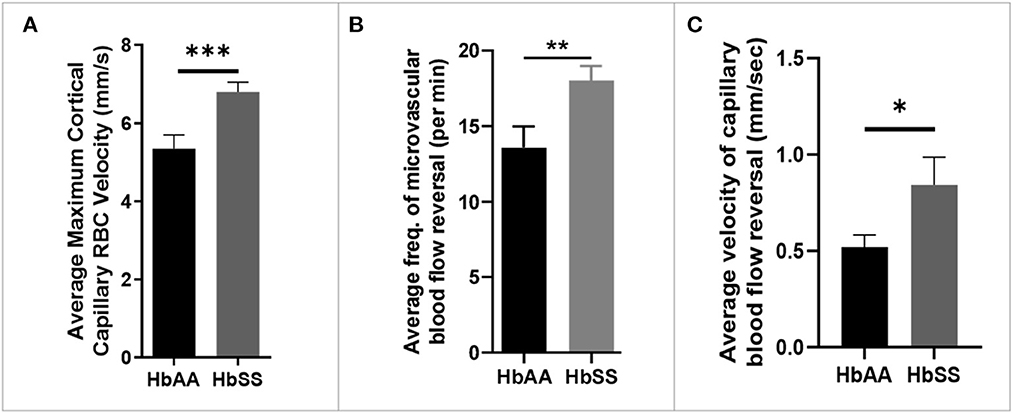
Figure 2. Altered capillary hemodynamics in 13-month-old sickle cell mice compared to age-matched controls. (A) average maximum capillary RBC velocity in millimeters per second. (B) average frequency of capillary blood flow reversal per minute (C) average velocity of capillary blood flow reversal in millimeters per second (AA: n = 5, average of ~194 vessel segments; SS: n = 7, average of ~390 vessel segments). Error bars are standard error of means (SEM). *p ≤ 0.05; **p ≤ 0.01; ***p ≤ 0.001. Mean comparisons done using Welch's corrected t-test. Results here are only from the mice in the baseline group.
Sickle cell mice have elevated expression/deposition of endothelial adhesion factors (VCAM-1 and P-selectin) compared to controls
To better understand the potential underlying factors responsible for the disturbed hemodynamics observed above, we performed immunohistochemical (IHC) analysis of the brains from the sickle cell and control mice, after completion of 2 Photon imaging. The IHC analysis focused mainly on VCAM-1 and P-selectin for reasons already mentioned in the background section (25, 27, 36, 37). Our analysis showed that sickle cell mice had significantly larger area of VCAM-1 coverage expressed per μm2 (p < 0.0001), as well as expression/deposition (intensity) measured in RFU/mm2 (p < 0.0001) in the cerebral microvasculature compared to controls at baseline (Figures 3A–C). Considering both the intensity and coverage parameters for expression, high intensity areas of fluorescence are not necessarily localized to one specific brain area. Similarly, and to large extent not surprising, we also noted that sickle cell mice had significantly larger area of P-selectin coverage per μm2 (p < 0.0001) and expression/deposition (intensity) measured in RFU/mm2 (p < 0.0001) in the cerebral microvasculature compared to controls (Figures 3D–F). Using images from 2 Photon microscopy and custom MATLAB scripts as already described, we quantified leukocyte adherence in the cortical microvasculature. The result indicates a significantly higher frequency of leukocyte adherence events in sickle cell mice (4.83 ± 0.57 /100μm/min vs. 2.26 ± 0.37 /100μm/min; p = 0.002) compared to controls at baseline (Figure 3G). This result is unsurprising given the well-established role of elevated P-selectin and VCAM-1 in as markers of endothelial activation as well as mediators of leukocytes rolling (37, 61–65).
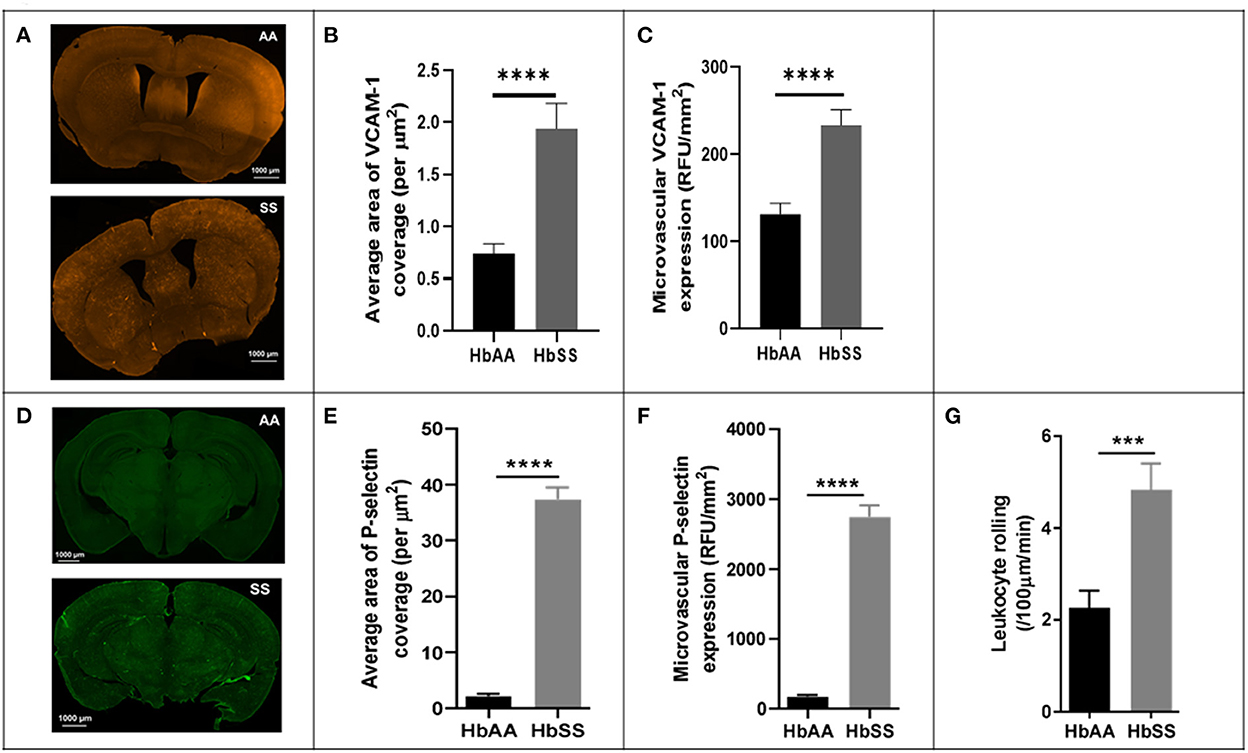
Figure 3. Adhesion factor expression and the rate of leukocyte adherence are elevated in the brains of sickle cell mice. Fluorophore of interest was isolated to show fluorescent areas representing VCAM-1 deposition along the microvasculature. (A) Representative image of stained tissue sections (50 um thick) from SS (bottom) and AA (top) mice. Areas of fluorescence indicate VCAM-1 deposition. These images were taken after spectral unmixing but prior to transferring to ImageJ for binarizing and overlaying masks. Adhesion factor expression was measured by analyzing fluorescence intensity and reported as relative fluorescence units (RFU) per millimeter squared of brain tissue. (B) Average area of VCAM-1 coverage per μm2 (C) VCAM-1 expression compared to controls. (D) Representative image of stained tissue sections (50 um thick) from SS (bottom) and AA (top) mice. Fluorophore of interest was isolated to show fluorescent areas representing P-selectin deposition along the microvasculature. Areas of fluorescence indicate P-selectin deposition. (E) Average area of P-selectin coverage per μm2. (F) microvascular P-selectin expression (RFU/mm2). (G) Leukocyte adherence (defined as lasting two seconds or more) per 100 μm length of vessel per minute was higher in sickle cell mice (p < 0.001) (AA: n = 5; SS: n = 7). Error bars are standard error of means (SEM). ***p ≤ 0.001; ****p ≤ 0.0001. Mean comparisons done using Welch's corrected t-test. Results here are only from the mice in the baseline group. For VCAM-1 analysis, AA = 184 brain slices and SS = 215 brain slices, while for P-selectin analysis, AA = 189 brain slices and SS = 165 brain slices. For leukocyte rolling, AA = average of ~194 vessel segments; SS = average of ~390 vessel segments.
Microinfarct frequency and area in sickle cell mice compared to age matched controls at baseline
Using a separate cohort of mice (male AA and SS mice that were 13 months old) that were neither transfused with packed red blood cells (pRBC) nor had cranial window implanted, we assessed the frequency and size (area) of microinfarcts (≥ 50 μM in diameter) in brain slices of sickle cell and control mice by examining all brain sections from sickle cell (N = 7) and control (N = 6) mice, encompassing ~20 sections per mouse spanning the entire cerebrum. Surprisingly, our analysis revealed that there was no significant difference in the frequency (22.60 ± 5.53 vs. 21.14 ± 4.0) for every 20 brain slices (Figure 4A). On the other hand, we noted that the cortical microinfarcts were significantly larger in sickle cell [0.2 ± 0.03 cm2 vs. 0.1 ± 0.02 cm2, p = 0.02] compared to the control mice (Figures 4B–D). This recapitulates data in our previous study (12).
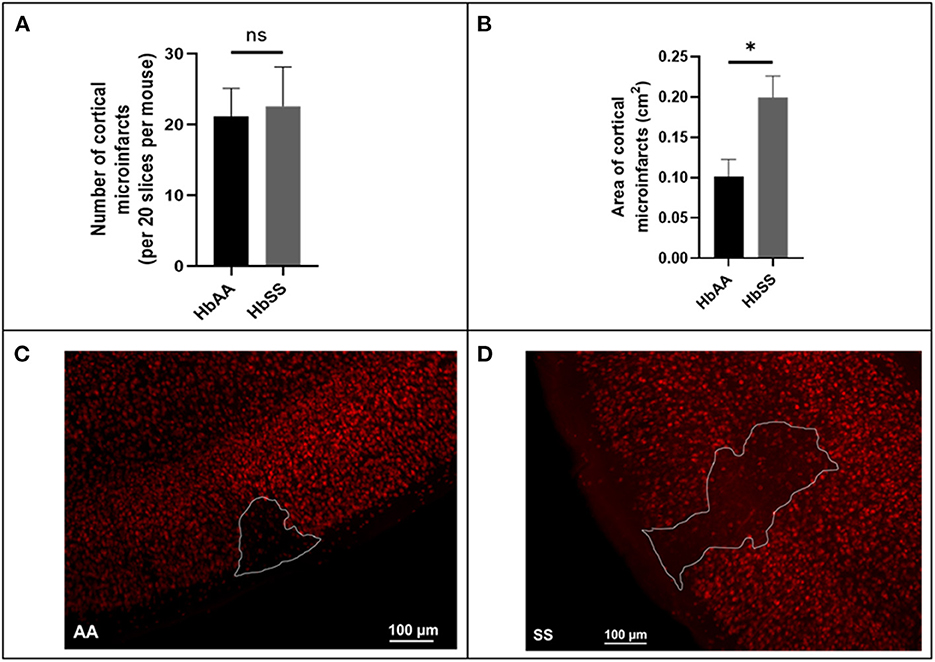
Figure 4. Baseline microinfarct data from 13-month-old mice. (A) Frequency of cortical infarcts per was not significantly different between HbAA and HbSS. (B) area of cortical infarcts in cm2. Error bars are standard error of means (SEM) (AA: n = 5; SS: n = 5). *p < 0.05. (C) Representative image of AA mouse microinfarct area. (D) Representative image of SS mouse microinfarct area. Mean comparisons done using Welch's corrected t-test.
A single packed red blood cell transfusion improved cerebral microvascular hemodynamic measures in sickle cell mice
Given that blood transfusion therapy is still one of the most effective ways to prevent cerebrovascular complications such as stroke or recurrent stroke in SCD, (66–69) we decided to offer a single packed RBC transfusion to the sickle cell mice as described earlier. To accomplish this, we obtained whole blood from Townes humanized HbAA mice, spun it down, removed the plasma, resuspended the pellet in sterile cold PBS, centrifuged again (to wash), and then resuspended in sterile PBS at room temperature before proceeding immediately to transfuse. Sickle cell mice received 300 μL of packed RBC IV (with a goal of raising the hemoglobin by 1 g/dL), immediately following the pre-transfusion (PrT) 2 Photon microscopy session as described above. Control (HbAA) mice received 300 μL of normal saline about the same as the sickle cell mice. 2–3 weeks post transfusion, the mice underwent a second (post-transfusion) 2 Photon microscopy imaging. It is important to note, that while control mice received saline infusion, they did not receive blood transfusions and the PrT and PT designations are meant to indicate the fact that they were imaged at two time points that align with pre- and post-transfusion imaging time points in sickle cell mice.
The 2 photon microscopy images generated from the pre- and post-transfusion images were analyzed as described earlier. The result from our analysis showed that sickle cell mice that received packed RBC transfusion had a 36% (p = 0.03) reduction in maximum cortical microvascular RBC velocity, compared to their pre-transfusion values. On the other hand, we noted a 43% increase (p = 0.04) in maximum cortical microvascular RBC velocity in the control mice compared to their pre-transfusion values (Figure 5A). Also, compared to pre-transfusion levels, we noted significant lower frequency of microvascular blood flow reversal (Figure 4B, p < 0.0001) as well as decreased velocity (0.62 ± 0.06 mm/sec vs. 0.46 ± 0.06 mm/sec, p = 0.04) of cerebral microvascular blood flow reversal (Figure 4C) in sickle cell mice that were transfused with packed RBC. This suggests that blood transfusion treatment significantly improves hemodynamic abnormalities in sickle cell mice.
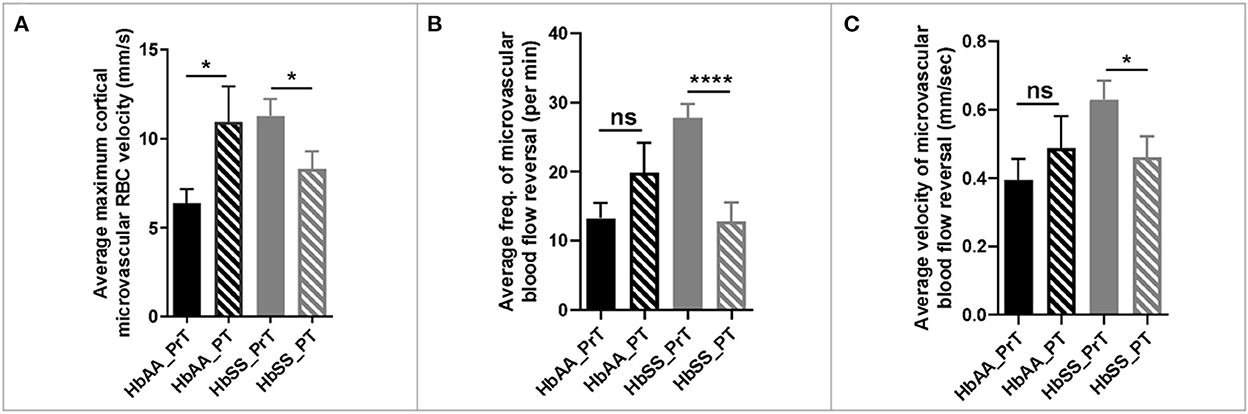
Figure 5. Hemodynamic analysis in sickle cell and control mice before and after blood or saline transfusion. (A) average maximum capillary RBC velocity in millimeters per second. (B) average frequency of capillary blood flow reversal per minute (C) average velocity of capillary blood flow reversal in millimeters per second (AA: n = 3–5; SS: n = 5–7). Error bars are standard error of means (SEM). NS, Not significant; *p ≤ 0.05; ****p ≤ 0.0001. Mean comparisons done using Welch's corrected t-test (HbAA_PrT was compared to HbAA_PT and HbSS_PrT was compared to HbSS_PT. There was no cross-genotype comparison since AA mice were not transfused with packed RBC. Prt, Pre-transfusion; PT, Post-transfusion. AA = average of ~73 vessel segments pre-transfusion and 23 vessel segments post-transfusion; SS = average of ~90 vessel segments pre-transfusion and 54 vessel segments post-transfusion.
Packed RBC transfusion decreases endothelial activation in cerebral microvasculature by decreasing expression/deposition of VCAM-1 and P-selectin, and leukocyte adherence in sickle cell mice
As stated earlier (Figure 3B), packed RBC transfusion is one of the mainstays for the prevention of stroke and neurovascular complications of SCD. Thus, as reported earlier, we used IHC to examine whether the single bolus of packed RBC transfusion had any impact on endothelial activation and therefore expression/deposition of VCAM-1 and/or P-selectin. Analysis of the IHC images, showed that sickle cell mice had significantly lower coverage (0.25 ± 0.02 /μm vs. 0.91 ± 0.14 /μm, p < 0.0001) compared with control mice. Also, when compared to the values obtained for sickle cell mice at baseline (Figure 3B), sickle cell mice that were transfused with packed RBC had a more than 7-fold lower VCAM-1 coverage. Additionally, microvascular VCAM-1 expression/deposition was significantly lower in sickle cell mice post transfusion (33.45 ± 3.44 RFU/mm2 vs. 168.90 ± 20.71 RFU/mm2, p < 0.0001) compared with control mice. And, when compared with values measured at baseline for sickle cell mice that were not transfused (Figure 3C), sickle cell mice that were transfused with packed RBC had an ~7-fold lower VCAM-1 expression/deposition in the cerebral microvasculature (Figures 6A,B). We also examined P-selectin coverage and expression/deposition as described earlier and observed that microvascular P-selectin coverage (12.45 ± 0.94 /μm vs. 4.85 ± 0.33 /μm, p < 0.0001) and expression/deposition (1165.00 ± 109.20 RFU/mm2 vs. 302.30 ± 24.87 RFU/mm2, p < 0.0001) were higher among sickle cell mice that were transfused with packed RBC, compared with controls (Figures 3C,D). Notwithstanding, when compared to values measured at baseline for sickle cell mice (Figures 3E,F), sickle cell mice that received packed RBC transfusion, had an approximately 3-fold (12.45 ± 0.94 /μm vs. 37.34 ± 2.16 /μm) and 2.4-fold (1165.00 ± 109.20 RFU/mm2 vs. 2742.00 ± 169.70 RFU/mm2) lower P-selectin coverage and expression/deposition respectively (Figures 6C,D). Finally, we examined pre- and post- packed RBC (for sickle cell mice) and saline (for controls) transfusion leukocyte adherence events. We noted that there was no significant difference in leukocyte adherence events between both time points for the control mice. However, sickle cell mice transfused with packed RBC had a significant reduction in leukocyte adherence events (1.35 ± 0.32 /100μm/min vs. 3.46 ± 0.58 /100μm/min; p = 0.0017) compared to pre-transfusion levels (Figure 6E). The data here suggests that a potential underlying benefit of transfusion for reduction of VOE could be via mitigating endothelial activation and therefore leukocyte adherence events.
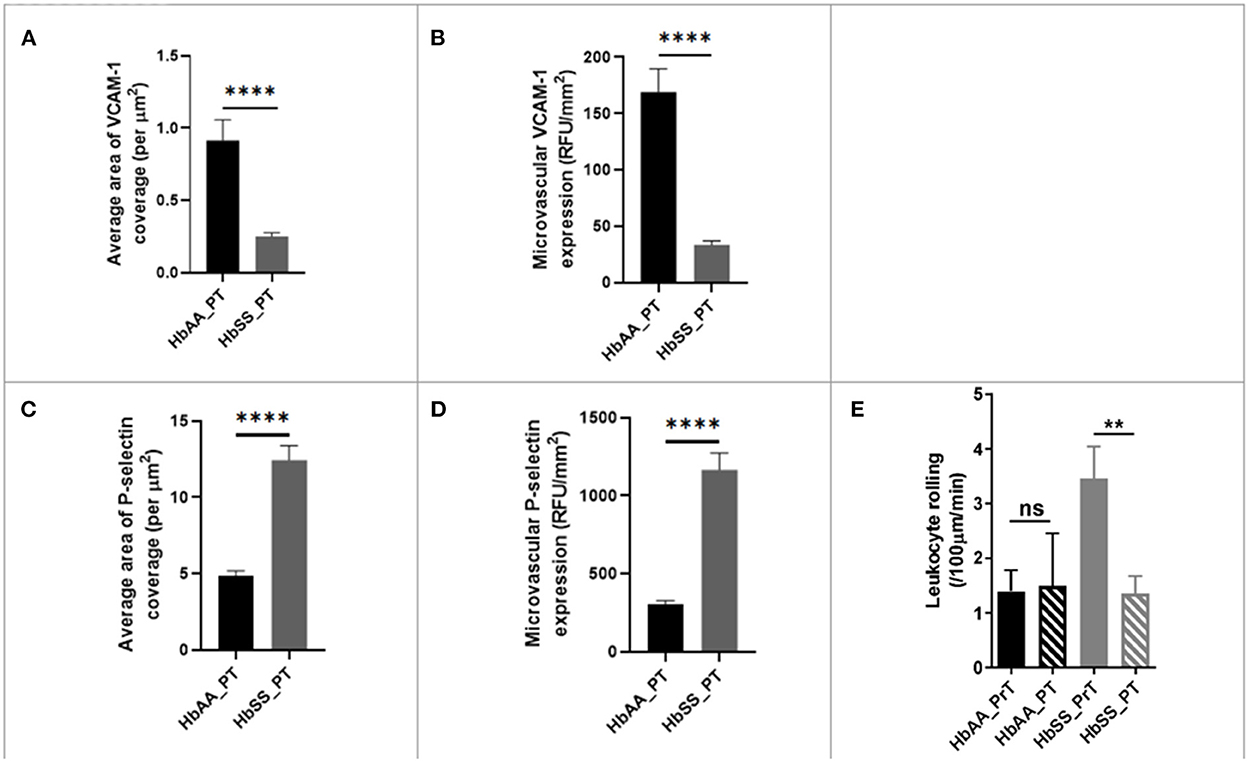
Figure 6. Blood transfusion improves cerebral vascular endothelium in sickle cell mice. (A) Average area of VCAM-1 coverage per μm2 in AA mice compared to SS. (B) Microvascular VCAM-1 expression in RFU/mm2 in AA mice compared to SS. (C) Average area of P-selectin coverage per μm2 (p < 0.0001). (D) Microvascular P-selectin expression (RFU/mm2). (E) Leukocyte adherence in AA and SS mice pre- and post-transfusion. AA: n = 3–5; SS: n = 5-7. Error bars are SEM. NS, Not significant; **p ≤ 0.01; ****p ≤ 0.0001. Mean comparisons done using Welch's corrected t-test. For VCAM-1 analysis, AA = 73 brain slices and SS = 147 brain slices, while for P-selectin analysis, AA = 162 brain slices and SS = 164 brain slices. For leukocyte rolling, AA = average of ~73 vessel segments pre-transfusion and 23 vessel segments post-transfusion; SS = average of ~90 vessel segments pre-transfusion and 54 vessel segments post-transfusion.
Discussion
Cerebrovascular abnormalities, including strokes and microinfarcts, have been well-documented in sickle cell disease and are associated with cognitive impairment (14, 70). In addition, high levels of adhesion factors have been implicated in SCD related complications, where they act as mediators of cellular (especially leukocyte) endothelial interactions and therefore vaso-occlusion (71, 72). Taken together, these events are crucial in the understanding of neurological pathology in SCD. The goal of this study was to examine the role of endothelial adhesion molecules (VCAM-1 and P-selectin) in cerebral microvascular hemodynamics, as well as the impact of blood transfusion treatment on these parameters (hemodynamics and expression of adhesion molecules). This study showed that compared to age-matched controls, aged Townes sickle cell mice have an abnormal cerebral microvascular hemodynamic profile, which is also associated with increased leukocyte adherence that seems to be mediated by a higher expression of adhesion factors (VCAM-1 and P-selectin). We also observed that these abnormalities were reduced, and in some instances, reversed, by blood transfusion treatment.
We observed a significantly higher average maximum microvascular RBC velocity in sickle cell mice compared to controls (Figure 2A). This finding corresponds with that from a previous study that showed higher capillary RBC velocity in aged sickle cell mice (12) as well as with observations in children and adults with SCD (25). The average maximum cortical blood flow velocity may be an indication of a compensatory mechanism for poor cerebral perfusion from downstream narrowing or obstruction and/or anemia. It is important to note that we are unable to precisely report capillary RBC velocity because we did not stain for smooth muscle actin, which would have enabled us to discriminate capillaries from pre-capillary arterioles and/or post capillary venules. Thus, our 2 Photon imaging may have included precapillary arterioles and post-capillary venules, and we refer to this throughout the paper as “microvasculature”. Notwithstanding, it is well known that besides the cortical capillaries, other segments of the cerebral vascular tree, such as the arteries and arteriole, carotid, and vertebral arteries, are affected in SCD, (73, 74) and the large vessel changes might reflect a later manifestation of microvascular abnormalities such as documented in this study. Furthermore, sickle cell mice had greater instances and higher velocity of blood flow reversal (Figures 2B,C). The exact mechanism for these reversals is unclear, however, studies (75, 76) suggest that experimental occlusion of either arterioles or venules results in cortical microvascular blood flow reversals as documented here. Thus, our finding suggests that the spontaneous blood flow reversals observed in our study could be due to spontaneous cerebral microvascular VOEs. While peripheral VOEs which are a hallmark of SCD and are well documented, cerebral microvascular VOEs have not been documented until recently (12) and are potentially manifesting in our study as blood flow reversals. These disturbances/turbulence in flow may also lead to endothelial activation. Overall, our report of a high velocity of flow, is corroborated by a recent report among patients with SCD, where using multiple-inflow-time arterial spin labeling, they showed a significantly higher cerebral blood flow in patients with SCD compared to controls (77).
Another important observation from our study is the elevated baseline expression/deposition of cerebral microvascular VCAM-1 and P-selectin in sickle cell mice compared to controls (Figures 3B–F). This data suggests that endothelial dysfunction may play a large role in SCD-related cerebral micro vasculopathy and thus neurovascular complications. The high maximum velocity and frequent flow reversals found in these mice may constitute some of the mechanical forces that trigger the expression of these endothelial adhesion factors (25, 78, 79). A recent study documented increased expression of VCAM-1 in the endothelium of aortic valve leaflets when they were exposed to shear stress (80). Thus, in concert with the prior stated mechanical damage to the endothelium, resulting from the physical contact with sickle RBC, these mechanical forces contribute to the promotion of microvascular thrombus generation from increased released of tissue factors, exposure of platelets to subendothelial tissues and therefore formation of platelet aggregates. This further promotes and increases the likelihood of cerebral microvascular VOEs and highlights the value of closely examining the role of endothelial adhesion factors in cerebral microvasculopathy (81).
Given the observations (Figures 3B–F), it is therefore no surprise for us to see a significantly higher frequency of leukocyte adherence in sickle cell mice compared to controls. This increased frequency of leukocyte adherence may also be associated with the higher expression of adhesion factors observed in Figures 3B–F. For example, VCAM-1 is the primary means by which leukocytes bind to the endothelium, using VLA-4, while P-selectin is one of the primarily means via which neutrophils interacts with the endothelium. Several studies in both humans and mice have shown that P-selectin and VCAM-1 are heavily implicated in SCD vascular dysfunction (26, 78, 82). These studies suggest that leukocyte-endothelial interactions (possibly mediated by VCAM-1 and/or P-selectin) are predominantly occurring in the post-capillary venules (83–86). Furthermore, recent clinical trials with crizanlizumab, a humanized P-selectin monoclonal antibody, have shown incredible promise, with patients with SCD who were treated with the drug, experiencing significantly fewer vaso-occlusive pain crises compared to patients on placebo, although these studies have no cerebral endpoints (38, 87). Another in-vitro study of Crizanlizumab showed inhibition of leukocyte adherence to P-selectin under physiologic blood flow conditions (44). Although the long-term benefits of crizanlizumab are yet to be determined, based on these previous studies and our results, it is tempting to predict that long-term use of crizanlizumab may attenuate some features of cerebral microvasculopathy and possibly SCD-related neurovascular pathologies, via reduction occurrence of VOEs in in the cerebral microvasculature (38, 44, 87).
Additionally, we noted that at baseline (without any intervention) the frequency of spontaneous microinfarct was not significantly different. The size (area) was significantly larger among sickle cell mice compared to controls (Figure 4B). The exact mechanism behind this is not yet clear. In a prior study, Luo et al. (88) reported that following middle cerebral artery occlusion, Townes sickle cell mice had significantly larger cerebral infarct size, compared to controls (88). Based on the rest of our data, it is plausible to reason that the growth of the infarct might be a self-propagating process (89) in the background of increased endothelial expression of adhesion molecules, increased leukocyte adherence and therefore higher frequency of VOEs as reported earlier in this section. This is a mechanism that warrants further investigation, and our lab is actively looking into this as it could also represent a therapeutic target for reducing the well described SCD-related cognitive decline which occurs in children and adults in the absence of an overt cerebral injury (90, 91).
Our observation of an apparent “normalization” of hemodynamic parameters/measured with packed RBC transfusion, compared to pre-transfusion levels in sickle cell mice and compared to controls (Figures 5A–C) was an intriguing albeit unsurprising finding. This result is exciting as it represents the first-time demonstration of a possible underlying mechanism of the benefits of blood transfusion. The dramatic change in maximum cerebral microvascular blood flow velocity could imply an improved ability to effectively perfuse the brain tissue without needing additional output velocity as a compensatory mechanism. According to this data, it seems that blood transfusion might have as a benefit, the normalization of cerebral microvascular dysfunction as a mechanism for the reported benefit in stroke prevention reported in children with SCD (48). Additionally, it is also possible that the reduction in flow velocity could be due to a decreased in VOEs, as we also observed a reduction in frequency (Figure 5B) and velocity (Figure 5B) of blood flow reversal. Furthermore, we also noted that compared to pre-transfusion or baseline levels, there was a significantly less microvascular expression and deposition of VCAM-1 and P-selectin, as well as significant decrease in leukocyte adherence in the cerebral of sickle cell mice. Taken together, the significant difference in expression and deposition of adhesion molecules combined with the reduction in leukocyte adherence, might account for some if not all the improvement seen in cerebral hemodynamic parameters. Due to the short duration of the blood transfusion therapy in addition to the fact that the assessment of cortical microinfarcts were performed post-mortem, we were unable to examine the impact of blood transfusion on frequency or size of the infarct as a function of the lower expression/deposition of adhesion endothelial adhesion molecules and/or leukocyte adherence. However, this will be the subject of future investigations in our laboratory.
A limitation of our study was that the expression of adhesion factors was quantified in the whole brain while hemodynamic changes were measured in cortical microvasculature. Nevertheless, our study corroborates the findings of several in vivo and in vitro studies by showing evidence of leukocyte-endothelial interactions, likely promoted by increased expression of adhesion factors expression. As mentioned earlier, we were also not able to reliably evaluate the impact of blood transfusion on frequency or size of cortical microinfarcts due to the short duration of time (2 weeks) from packed RBC transfusion to imaging and sacrifice of the mice. Future study designs have already worked out ways around this limitation using longitudinal approach to imaging. Due to equipment availability and other logistical reasons, we were also not able to access the post-transfusion hemoglobin levels and as such we are unable to make a firm statement with regards to how much the hemoglobin levels of the sickle cell mice went up post transfusion. Finally, we are not able to directly infer from our current data, a causal relationship between the improvement of hemodynamic parameters and lower expression of adhesion factors. However, ongoing studies in our lab using bone marrow chimera as well as sickle cell mice null for P-selectin and VCAM-1, should enable us to make such inference in the future.
Conclusion
By examining hemodynamics and adhesion factors using two-photon laser microscopy and post-mortem immunohistochemistry in both pre- and post- transfusion sickle cell mice, we were able to document evidence of cerebral microvasculopathy in sickle cell mice. Additionally, we were able to show that blood transfusion might exert its benefit of preventing neurovascular complications by mitigating cerebral microvascular endothelial activation and the underlying mechanism of VOEs. Thus, our study potentially highlights one of the mechanisms that may be contributing to how blood transfusion prevents stroke and other neurovascular pathologies. The significant decrease in VCAM-1 and P-selectin expression in the brain following blood transfusion offers a particular new avenue for investigation, as well as therapeutic targets.
Data availability statement
The raw data supporting the conclusions of this article will be made available by the authors, without undue reservation.
Ethics statement
The animal study was reviewed and approved by Institutional Animal Care and Use Committees (IACUC) of Emory University and the Medical University of South Carolina.
Author contributions
HH designed the experiment and performed the final critical review. NA, DA, JJ, MS, and HH performed experiments and data analysis. NA, OTG, and HH wrote the manuscript. DA, JJ, and MS provided critical review. All authors endorsed the submission of this manuscript.
Funding
This study was supported by grants from the National Institutes of Health, R01HL138423, R01HL156024, and R01AG072592 to HH.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher's note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Pauling L, Itano HA, Singer SJ, Wells IC. Sickle cell anemia, a molecular disease. Science. (1949) 110:543–8. doi: 10.1126/science.110.2865.543
2. Wang Y, Kennedy J, Caggana M, Zimmerman R, Thomas S, Berninger J, et al. Grosse SD. Sickle cell disease incidence among newborns in New York State by maternal race/ethnicity and nativity. Gen Med. (2013) 15:222–8. doi: 10.1038/gim.2012.128
3. Hassell K. Population estimates of sickle cell disease in the US. Am J Prev Med. 38:S512–21. 2010. doi: 10.1016/j.amepre.2009.12.022
4. Kato GJ, Piel FB, Reid CD, Gaston MH, Ohene-Frempong K, Krishnamurti L, et al. Costa FF. Sickle cell disease nature reviews disease. Primers. (2018) 4:1–22. doi: 10.1038/nrdp.2018.10
5. Manwani D, Frenette PS. Vaso-occlusion in sickle cell disease: pathophysiology and novel targeted therapies. Blood. (2013) 122:3892–8. doi: 10.1182/blood-2013-05-498311
6. Sundd P, Gladwin MT, Novelli EM. Pathophysiology of sickle cell disease. Annu Rev Pathol. (2019) 14:263–92. doi: 10.1146/annurev-pathmechdis-012418-012838
7. Zhang D, Xu C, Manwani D, Frenette PS. Neutrophils, platelets, and inflammatory pathways at the nexus of sickle cell disease pathophysiology. Blood. (2016) 127:801–9. doi: 10.1182/blood-2015-09-618538
8. Telen MJ. Role of adhesion molecules and vascular endothelium in the pathogenesis of sickle cell disease. Hematol Am Soc Hematol Educ Program. (2007) 2007:84–90. doi: 10.1182/asheducation-2007.1.84
9. Horton RE. Microfluidics for investigating vaso-occlusions in sickle cell disease. Microcirculation. (2017) 24:e12373. doi: 10.1111/micc.12373
10. Ofori-Acquah SF. Sickle cell disease as a vascular disorder. Expert Rev Hematol. (2020) 13:645–53. doi: 10.1080/17474086.2020.1758555
11. Hartmann DA, Hyacinth HI, Liao FF, Shih AY. Does pathology of small venules contribute to cerebral microinfarcts and dementia? J Neurochem. (2018) 144:517–26. doi: 10.1111/jnc.14228
12. Hyacinth HI, Sugihara CL, Spencer TL, Archer DR, Shih AY. Higher prevalence of spontaneous cerebral vasculopathy and cerebral infarcts in a mouse model of sickle cell disease. J Cereb Blood Flow Metab. (2019) 39:342–51. doi: 10.1177/0271678X17732275
13. Guilliams KP, Fields ME, Ragan DK, Chen Y, Eldeniz C, Hulbert ML, et al., Ford AL. Large-vessel vasculopathy in children with sickle cell disease: a magnetic resonance imaging study of infarct topography and focal atrophy. Pediatr Neurol. (2017) 69:49–57. doi: 10.1016/j.pediatrneurol.2016.11.005
14. Ohene-Frempong K, Weiner SJ, Sleeper LA, Miller ST, Embury S, Moohr JW, et al. Gill FM. Cerebrovascular accidents in sickle cell disease: rates and risk factors. Blood. (1998) 91:288–94.
15. Pegelow CH, Macklin EA, Moser FG, Wang WC, Bello JA, Miller ST, et al. Kinney TR. Longitudinal changes in brain magnetic resonance imaging findings in children with sickle cell disease. Blood. (2002) 99:3014–8. doi: 10.1182/blood.V99.8.3014
16. Wang W, Enos L, Gallagher D, Thompson R, Guarini L, Vichinsky E, et al. Neuropsychologic performance in school-aged children with sickle cell disease: a report from the cooperative study of sickle cell disease. J Pediatr. (2001) 139:391–7. doi: 10.1067/mpd.2001.116935
17. Shih AY, Hyacinth HI, Hartmann DA, Van Veluw SJ. Rodent models of cerebral microinfarct and microhemorrhage. Stroke. (2018) 49:803–10. doi: 10.1161/STROKEAHA.117.016995
18. Kato GJ, Martyr S, Blackwelder WC, Nichols JS, Coles WA, Hunter LA, et al. Levels of soluble endothelium-derived adhesion molecules in patients with sickle cell disease are associated with pulmonary hypertension, organ dysfunction, and mortality. Br J Haematol. (2005) 130:943–53. doi: 10.1111/j.1365-2141.2005.05701.x
19. Pathare A, Kindi SA, Daar S, Dennison D. Cytokines in sickle cell disease. Hematology. (2003) 8:329–37. doi: 10.1080/10245330310001604719
20. Hofrichter J, Ross PD, Eaton WA. Kinetics and mechanism of deoxyhemoglobin S gelation: a new approach to understanding sickle cell disease. Proc Natl Acad Sci U S A. (1974) 71:4864–8. doi: 10.1073/pnas.71.12.4864
21. Solovey A, Lin Y, Browne P, Choong S, Wayner E, Hebbel RP. Circulating activated endothelial cells in sickle cell anemia. N Engl J Med. (1997) 337:1584–90. doi: 10.1056/NEJM199711273372203
22. Brown MD, Wick TM, Eckman JR. Activation of vascular endothelial cell adhesion molecule expression by sickle blood cells. Pediatr Pathol Mol Med. (2001) 20:47–72. doi: 10.3109/15513810109168817
23. Shiu YT, Udden MM, McIntire LV. Perfusion with sickle erythrocytes up-regulates ICAM-1 and VCAM-1 gene expression in cultured human endothelial cells. Blood. (2000) 95:3232–41. doi: 10.1182/blood.V95.10.3232.010k16_3232_3241
24. Antwi-Boasiako C, Donkor ES, Sey F, Dzudzor B, Dankwah GB, Otu KH, et al. Levels of soluble endothelium adhesion molecules and complications among sickle cell disease patients in Ghana. Diseases. (2018) 6:29. doi: 10.3390/diseases6020029
25. Connes P, Verlhac S, Bernaudin F. Advances in understanding the pathogenesis of cerebrovascular vasculopathy in sickle cell anaemia. Br J Haematol. (2013) 161:484–98. doi: 10.1111/bjh.12300
26. White J, Lancelot M, Gao XF, McGraw BJ, Tabb C, Hines P. Cross-sectional analysis of adhesion in individuals with sickle cell disease using a standardized whole blood adhesion bioassay to VCAM-1. Blood Cells Mol Dis. (2020) 81:102397. doi: 10.1016/j.bcmd.2019.102397
27. Hyacinth HI, Adams RJ, Voeks JH, Hibbert JM, Gee BE. Frequent red cell transfusions reduced vascular endothelial activation and thrombogenicity in children with sickle cell anemia and high stroke risk. Am J Hematol. (2014) 89:47–51. doi: 10.1002/ajh.23586
28. Frenette PS. Sickle cell vaso-occlusion: multistep and multicellular paradigm. Curr Opin Hematol. (2002) 9:101–6. doi: 10.1097/00062752-200203000-00003
29. Barabino GA, Platt MO, Kaul DK. Sickle cell biomechanics. Annu Rev Biomed Eng. (2010) 12:345–67. doi: 10.1146/annurev-bioeng-070909-105339
30. Prussien KV, Compas BE, Siciliano RE, Ciriegio AE, Lee CA, Kassim AA, et al. Jordan LC. Cerebral hemodynamics and executive function in sickle cell anemia. Stroke. (2021) 52:1830–4. doi: 10.1161/STROKEAHA.120.032741
31. Jordan LC, Casella JF, DeBaun MR. Prospects for primary stroke prevention in children with sickle cell anaemia. Br J Haematol. (2012) 157:14–25. doi: 10.1111/j.1365-2141.2011.09005.x
32. Kassim AA, DeBaun MR. The case for and against initiating either hydroxyurea therapy, blood transfusion therapy or hematopoietic stem cell transplant in asymptomatic children with sickle cell disease. Expert Opin Pharmacother. (2014) 15:325–36. doi: 10.1517/14656566.2014.868435
33. Barreiro O, Yanez-Mo M, Serrador JM, Montoya MC, Vicente-Manzanares M, Tejedor R, et al. Dynamic interaction of VCAM-1 and ICAM-1 with moesin and ezrin in a novel endothelial docking structure for adherent leukocytes. J Cell Biol. (2002) 157:1233–45. doi: 10.1083/jcb.200112126
34. Stuart MJ, Setty BN. Sickle cell acute chest syndrome: pathogenesis and rationale for treatment. Blood. (1999) 94:1555–60. doi: 10.1182/blood.V94.5.1555.417k36_1555_1560
35. Kutlar A, Ataga KI, McMahon L, Howard J, Galacteros F, Hagar W, et al. Embury SH. A potent oral P-selectin blocking agent improves microcirculatory blood flow and a marker of endothelial cell injury in patients with sickle cell disease. Am J Hematol. (2012) 87:536–9. doi: 10.1002/ajh.23147
36. Wood K, Russell J, Hebbel RP, Granger DN. Differential expression of E- and P-selectin in the microvasculature of sickle cell transgenic mice. Microcirculation. (2004) 11:377–85. doi: 10.1080/10739680490437559
37. Wood KC, Hebbel RP, Granger DN. Endothelial cell P-selectin mediates a proinflammatory and prothrombogenic phenotype in cerebral venules of sickle cell transgenic mice. Am J Physiol Heart Circ Physiol. (2004) 286:H1608–14. doi: 10.1152/ajpheart.01056.2003
38. Ataga KI, Kutlar A, Kanter J, Liles D, Cancado R, Friedrisch J, et al. Crizanlizumab for the prevention of pain crises in sickle cell disease. N Engl J Med. (2017) 376:429–39. doi: 10.1056/NEJMoa1611770
39. Blair HA. Crizanlizumab: first approval. Drugs. (2020) 80:79–84. doi: 10.1007/s40265-019-01254-2
40. Slomski A. Crizanlizumab prevents sickle cell pain crises. JAMA. (2017) 317:798. doi: 10.1001/jama.2017.0355
41. Stevens DL, Hix M, Gildon BL. Crizanlizumab for the prevention of vaso-occlusive pain crises in sickle cell disease. J Pharm Technol. (2021) 37:209–15. doi: 10.1177/87551225211008460
42. Yu Z, Blankenship L, Jaiyesimi I. Crizanlizumab in sickle cell disease. N Engl J Med. (2017) 376:1795–6. doi: 10.1056/NEJMc1703162
43. Matsui NM, Borsig L, Rosen SD, Yaghmai M, Varki A, Embury SH. P-selectin mediates the adhesion of sickle erythrocytes to the endothelium. Blood. (2001) 98:1955–62. doi: 10.1182/blood.V98.6.1955
44. Man Y, Goreke U, Kucukal E, Hill A, An R, Liu S, et al. Leukocyte adhesion to P-selectin and the inhibitory role of Crizanlizumab in sickle cell disease: A standardized microfluidic assessment. Blood Cells Mol Dis. (2020) 83:102424. doi: 10.1016/j.bcmd.2020.102424
45. Lawrence MB, Springer TA. Leukocytes roll on a selectin at physiologic flow rates: distinction from and prerequisite for adhesion through integrins. Cell. (1991) 65:859–73. doi: 10.1016/0092-8674(91)90393-D
46. Robinson SD, Frenette PS, Rayburn H, Cummiskey M, Ullman-Cullere M, Wagner DD, et al. Multiple, targeted deficiencies in selectins reveal a predominant role for P-selectin in leukocyte recruitment. Proc Natl Acad Sci U S A. (1999) 96:11452–7. doi: 10.1073/pnas.96.20.11452
47. Clark JD, Gebhart GF, Gonder JC, Keeling ME, Kohn DF. The 1996 guide for the care and use of laboratory animals. ILAR J. (1997) 38:41–8. doi: 10.1093/ilar.38.1.41
48. Adams RJ, McKie VC, Hsu L, Files B, Vichinsky E, Pegelow C, et al. Prevention of a first stroke by transfusions in children with sickle cell anemia and abnormal results on transcranial Doppler ultrasonography. N Engl J Med. (1998) 339:5–11. doi: 10.1056/NEJM199807023390102
49. DeBaun MR, Gordon M, McKinstry RC, Noetzel MJ, White DA, Sarnaik SA, et al. Controlled trial of transfusions for silent cerebral infarcts in sickle cell anemia. N Engl J Med. (2014) 371:699–710. doi: 10.1056/NEJMc1411133
50. Beverung LM, Strouse JJ, Hulbert ML, Neville K, Liem RI, Inusa B, et al. Health-related quality of life in children with sickle cell anemia: impact of blood transfusion therapy. Am J Hematol. (2015) 90:139–43. doi: 10.1002/ajh.23877
51. Shih AY, Mateo C, Drew PJ, Tsai PS, Kleinfeld D. A polished and reinforced thinned-skull window for long-term imaging of the mouse brain. J Vis Exp. (2012) 61:e3742. doi: 10.3791/3742
52. Shih AY, Driscoll JD, Drew PJ, Nishimura N, Schaffer CB, Kleinfeld D. Two-photon microscopy as a tool to study blood flow and neurovascular coupling in the rodent brain. J Cereb Blood Flow Metab. (2012) 32:1277–309. doi: 10.1038/jcbfm.2011.196
53. Driscoll JD, Shih AY, Drew PJ, Cauwenberghs G, Kleinfeld D. Two-photon imaging of blood flow in the rat cortex. Cold Spring Harb Protoc. (2013) 2013:759–67. doi: 10.1101/pdb.prot076513
54. Drew PJ, Blinder P, Cauwenberghs G, Shih AY, Kleinfeld D. Rapid determination of particle velocity from space-time images using the Radon transform. J Comput Neurosci. (2010) 29:5–11. doi: 10.1007/s10827-009-0159-1
55. Capers PL, Hyacinth HI, Cue S, Chappa P, Vikulina T, Roser-Page S, et al. Body composition and grip strength are improved in transgenic sickle mice fed a high-protein diet. J Nutr Sci. (2015) 4:e6. doi: 10.1017/jns.2014.63
56. Hyacinth HI, Capers PL, Archer DR, Hibbert JM. TNF-alpha, IFN-gamma, IL-10, and IL-4 levels were elevated in a murine model of human sickle cell anemia maintained on a high protein/calorie diet Experimental biology and medicine (Maywood, NJ). (2014) 239:65–70. doi: 10.1177/1535370213508357
57. Manci EA, Hyacinth HI, Capers PL, Archer DR, Pitts S, Ghosh S, et al. High protein diet attenuates histopathologic organ damage and vascular leakage in transgenic murine model of sickle cell anemia. Exp Biol Med. (2014) 239:966–74. doi: 10.1177/1535370214531863
58. Hashimoto J, Ito S. Aortic stiffness determines diastolic blood flow reversal in the descending thoracic aorta: potential implication for retrograde embolic stroke in hypertension. Hypertension. (2013) 62:542–9. doi: 10.1161/HYPERTENSIONAHA.113.01318
59. Hashimoto J, Ito S. Aortic blood flow reversal determines renal function: potential explanation for renal dysfunction caused by aortic stiffening in hypertension. Hypertension. (2015) 66:61–7. doi: 10.1161/HYPERTENSIONAHA.115.05236
60. Tsai CL, Lee JT, Cheng CA, Liu MT, Chen CY, Hu HH, Peng GS. Reversal of ophthalmic artery flow as a predictor of intracranial hemodynamic compromise: implication for prognosis of severe carotid stenosis. Eur J Neurol. (2013) 20:564–70. doi: 10.1111/ene.12038
61. Panes J, Perry M, Granger DN. Leukocyte-endothelial cell adhesion: avenues for therapeutic intervention. Br J Pharmacol. (1999) 126:537–50. doi: 10.1038/sj.bjp.0702328
62. Gavins F, Yilmaz G, Granger DN. The evolving paradigm for blood cell-endothelial cell interactions in the cerebral microcirculation. Microcirculation. (2007) 14:667–81. doi: 10.1080/10739680701404903
63. Henninger DD, Panes J, Eppihimer M, Russell J, Gerritsen M, Anderson DC, Granger DN. Cytokine-induced VCAM-1 and ICAM-1 expression in different organs of the mouse. J Immunol. (1997) 158:1825–32.
64. Ishikawa M, Stokes KY, Zhang JH, Nanda A, Granger DN. Cerebral microvascular responses to hypercholesterolemia: roles of NADPH oxidase and P-selectin. Circ Res. (2004) 94:239–44. doi: 10.1161/01.RES.0000111524.05779.60
65. Wood KC, Hebbel RP, Granger DN. Endothelial cell NADPH oxidase mediates the cerebral microvascular dysfunction in sickle cell transgenic mice. FASEB J. (2005) 19:989–91. doi: 10.1096/fj.04-3218fje
66. Russell MO, Goldberg HI, Hodson A, Kim HC, Halus J, Reivich M, Schwartz E. Effect of transfusion therapy on arteriographic abnormalities and on recurrence of stroke in sickle cell disease. Blood. (1984) 63:162–9. doi: 10.1182/blood.V63.1.162.bloodjournal631162
67. Pegelow CH, Adams RJ, McKie V, Abboud M, Berman B, Miller ST, et al. Risk of recurrent stroke in patients with sickle cell disease treated with erythrocyte transfusions. J Pediatr. (1995) 126:896–9. doi: 10.1016/S0022-3476(95)70204-0
68. Scothorn DJ, Price C, Schwartz D, Terrill C, Buchanan GR, Shurney W, et al. Risk of recurrent stroke in children with sickle cell disease receiving blood transfusion therapy for at least five years after initial stroke. J Pediatr. (2002) 140:348–54. doi: 10.1067/mpd.2002.122498
69. Wang WC, Kovnar EH, Tonkin IL, Mulhern RK, Langston JW, Day SW, et al. High risk of recurrent stroke after discontinuance of five to twelve years of transfusion therapy in patients with sickle cell disease. J Pediatr. (1991) 118:377–82. doi: 10.1016/S0022-3476(05)82150-X
70. Hirtz D, Kirkham FJ. Sickle cell disease and stroke. Pediatr Neurol. (2019) 95:34–41. doi: 10.1016/j.pediatrneurol.2019.02.018
71. Darbari DS, Sheehan VA, Ballas SK. The vaso-occlusive pain crisis in sickle cell disease: definition, pathophysiology, and management. Eur J Haematol. (2020) 105:237–46. doi: 10.1111/ejh.13430
72. Hebbel RP. Perspectives series: cell adhesion in vascular biology. Adhesive interactions of sickle erythrocytes with endothelium. J Clin Invest. (1997) 99:2561–4. doi: 10.1172/JCI119442
73. Detterich JA, Kato R, Bush A, Chalacheva P, Ponce D, De Zoysa M, et al. Sickle cell microvascular paradox-oxygen supply-demand mismatch. Am J Hematol. (2019) 94:678–88. doi: 10.1002/ajh.25476
74. Bush AM, Borzage MT, Choi S, Vaclavu L, Tamrazi B, Nederveen AJ, et al. Determinants of resting cerebral blood flow in sickle cell disease. Am J Hematol. (2016) 91:912–7. doi: 10.1002/ajh.24441
75. Blinder P, Shih AY, Rafie C, Kleinfeld D. Topological basis for the robust distribution of blood to rodent neocortex. Proc Nat Acad Sci U S A. (2010) 107:12670–5. doi: 10.1073/pnas.1007239107
76. Nguyen J, Nishimura N, Fetcho RN, Iadecola C, Schaffer CB. Occlusion of cortical ascending venules causes blood flow decreases, reversals in flow direction, and vessel dilation in upstream capillaries. J Cereb Blood Flow Metab. (2011) 31:2243–54. doi: 10.1038/jcbfm.2011.95
77. Kawadler JM, Hales PW, Barker S, Cox TCS, Kirkham FJ, Clark CA. Cerebral perfusion characteristics show differences in younger versus older children with sickle cell anaemia: Results from a multiple-inflow-time arterial spin labelling study. NMR Biomed. (2018) 31:e3915. doi: 10.1002/nbm.3915
78. Kaul DK, Finnegan E, Barabino GA. Sickle red cell-endothelium interactions. Microcirculation. (2009) 16:97–111. doi: 10.1080/10739680802279394
79. Breen LT, McHugh PE, Murphy BP. HUVEC ICAM-1 and VCAM-1 synthesis in response to potentially athero-prone and athero-protective mechanical and nicotine chemical stimuli. Ann Biomed Eng. (2010) 38:1880–92. doi: 10.1007/s10439-010-9959-8
80. Sucosky P, Balachandran K, Elhammali A, Jo H, Yoganathan AP. Altered shear stress stimulates upregulation of endothelial VCAM-1 and ICAM-1 in a BMP-4–and TGF-β1–dependent pathway. Arterioscler Throm Vas Biol. (2009) 29:254–60. doi: 10.1161/ATVBAHA.108.176347
81. Hollopeter G, Jantzen HM, Vincent D, Li G, England L, Ramakrishnan V, et al. Identification of the platelet ADP receptor targeted by antithrombotic drugs. Nature. (2001) 409:202–7. doi: 10.1038/35051599
82. Embury SH, Matsui NM, Ramanujam S, Mayadas TN, Noguchi CT, Diwan BA, et al. The contribution of endothelial cell P-selectin to the microvascular flow of mouse sickle erythrocytes in vivo. Blood. (2004) 104:3378–85. doi: 10.1182/blood-2004-02-0713
83. Kaul DK, Fabry ME, Nagel RL. Microvascular sites and characteristics of sickle cell adhesion to vascular endothelium in shear flow conditions: pathophysiological implications. Proc Natl Acad Sci U S A. (1989) 86:3356–60. doi: 10.1073/pnas.86.9.3356
84. Kaul DK, Fabry ME, Costantini F, Rubin EM, Nagel RL. In vivo demonstration of red cell-endothelial interaction, sickling and altered microvascular response to oxygen in the sickle transgenic mouse. J Clin Invest. (1995) 96:2845–53. doi: 10.1172/JCI118355
85. Kaul DK, Kollander R, Mahaseth H, Liu XD, Solovey A, Belcher J, et al. Robust vascular protective effect of hydroxamic acid derivatives in a sickle mouse model of inflammation. Microcirculation. (2006) 13:489–97. doi: 10.1080/10739680600778456
86. Zennadi R, Moeller BJ, Whalen EJ, Batchvarova M, Xu K, Shan S, et al. Epinephrine-induced activation of LW-mediated sickle cell adhesion and vaso-occlusion in vivo. Blood. (2007) 110:2708–17. doi: 10.1182/blood-2006-11-056101
87. Kutlar A, Kanter J, Liles DK, Alvarez OA, Cancado RD, Friedrisch JR, et al. Effect of crizanlizumab on pain crises in subgroups of patients with sickle cell disease: A SUSTAIN study analysis. Am J Hematol. (2019) 94:55–61. doi: 10.1002/ajh.25308
88. Luo W, Su EJ, Wang J, Wang H, Guo C, Pawar A, et al. Increased stroke size following MCA occlusion in a mouse model of sickle cell disease. Blood. (2014) 123:1965–7. doi: 10.1182/blood-2014-01-549717
89. Taylor ZJ, Hui ES, Watson AN, Nie X, Deardorff RL, Jensen JH, et al. Microvascular basis for growth of small infarcts following occlusion of single penetrating arterioles in mouse cortex. J Cereb Blood Flow Metab. (2016) 36:1357–73. doi: 10.1177/0271678X15608388
90. Vichinsky EP, Neumayr LD, Gold JI, Weiner MW, Rule RR, Truran D, et al. Neuropsychological dysfunction and neuroimaging abnormalities in neurologically intact adults with sickle cell anemia. JAMA. (2010) 303:1823–31. doi: 10.1001/jama.2010.562
Keywords: sickle cell disease, adhesion molecules, microvascular hemodynamics, cerebral microinfarct, two-photon microscope
Citation: Abi Rached NM, Gbotosho OT, Archer DR, Jones JA, Sterling MS and Hyacinth HI (2022) Adhesion molecules and cerebral microvascular hemodynamic abnormalities in sickle cell disease. Front. Neurol. 13:976063. doi: 10.3389/fneur.2022.976063
Received: 23 June 2022; Accepted: 21 November 2022;
Published: 07 December 2022.
Edited by:
Fenella Jane Kirkham, University College London, United KingdomReviewed by:
Mariel Kozberg, Massachusetts General Hospital and Harvard Medical School, United StatesAndria Ford, Washington University in St. Louis, United States
Copyright © 2022 Abi Rached, Gbotosho, Archer, Jones, Sterling and Hyacinth. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Hyacinth I. Hyacinth, aHlhY2luaGlAdWNtYWlsLnVjLmVkdQ==