
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Neurol., 19 July 2022
Sec. Neurocritical and Neurohospitalist Care
Volume 13 - 2022 | https://doi.org/10.3389/fneur.2022.963562
This article is part of the Research TopicMultimodal and auto-regulation monitoring in neuro-ICUView all 8 articles
 Giacomo Coppalini1†
Giacomo Coppalini1† Elie Duvigneaud1†
Elie Duvigneaud1† Alberto Diosdado1
Alberto Diosdado1 Ernesto Migliorino1Sophie Schuind2Jacques Creteur1
Ernesto Migliorino1Sophie Schuind2Jacques Creteur1 Fabio Silvio Taccone1
Fabio Silvio Taccone1 Elisa Gouvêa Bogossian1*
Elisa Gouvêa Bogossian1*Introduction: Tissue hypoxia and insufficient energy delivery is one of the mechanisms behind the occurrence of several complications in acute brain injured patients. Several interventions can improve cerebral oxygenation; however, the effects of inotropic agents remain poorly characterized.
Methods: Retrospective analysis including patients suffering from acute brain injury and monitored with brain oxygen pressure (PbtO2) catheter, in whom inotropic agents were administered according to the decision of the treating physician's decision; PbtO2 values were collected before, 1 and 2 h after the initiation of therapy from the patient data monitoring system. PbtO2 “responders” were patients with a relative increase in PbtO2 from baseline values of at least 20%.
Results: A total of 35 patients were included in this study. Most of them (31/35, 89%) suffered from non-traumatic subarachnoid hemorrhage (SAH). Compared with baseline values [20 (14–24) mmHg], PbtO2 did not significantly increase over time [19 (15–25) mmHg at 1 h and 19 (17–25) mmHg at 2 h, respectively; p = 0.052]. A total of 12/35 (34%) patients were PbtO2 “responders,” in particular if low PbtO2 was observed at baseline. A PbtO2 of 17 mmHg at baseline had a sensibility of 84% and a specificity of 91% to predict a PbtO2 responder. A significant direct correlation between changes in PbtO2 and cardiac output [r = 0.496 (95% CI 0.122 to 0.746), p = 0.01; n = 25] and a significant negative correlation between changes in PbtO2 and cerebral perfusion pressure [r = −0.389 (95% CI −0.681 to −0.010), p = 0.05] were observed.
Conclusions: In this study, inotropic administration significantly increased brain oxygenation in one third of brain injured patients, especially when tissue hypoxia was present at baseline. Future studies should highlight the role of inotropic agents in the management of tissue hypoxia in this setting.
Cerebral hypoxia is a major cause of secondary brain injury that can be observed in a relevant proportion of patients with acute brain injury (ABI), including patients with traumatic brain injury (TBI), subarachnoid hemorrhage (SAH) and intracerebral hemorrhage (ICH) (1–5). Indeed, low brain tissue oxygenation (PbtO2), which can be measured at bedside using an intraparenchymal probe placed into the “at-risk” areas, has been associated with cerebral anaerobic metabolism, as well as with an increased risk of mortality and poor functional outcome in these patients (6–12).
Low PbtO2 values have been observed in different conditions, such as reduced cerebral blood flow (CBF) and/or cerebral perfusion pressure (CPP), intracranial hypertension (IH), hypoxemia, anemia, altered microcirculation or excessive cellular metabolism (13). As such, several therapeutic strategies have been developed to optimize brain oxygenation into a complex and protocolized algorithm, which encompasses vasopressors (i.e., to increase CPP), treatments of IH (i.e., osmotic therapy), red blood cells transfusions or increased inspired oxygen fraction (FiO2), temperature control and sedation (i.e., to decrease cerebral oxygen consumption) (14–18).
In case of reduced CBF, one of the most commonly used intervention is the increase in CPP (i.e., induced hypertension) with normovolemia; in patients suffering from SAH, this intervention can increase regional perfusion in the presence of cerebral vasospasm and neurological deterioration (19). Among TBI patients, this strategy can increase tissue oxygenation and reduce intracranial pressure, if cerebral autoregulation is preserved (14, 15, 20). In patients with ICH, although it is generally recommended to control blood pressure to avoid hematoma expansion (21), increased CPP was associated with higher PbtO2 values in those patients with poor clinical condition at presentation (18, 22).
Interestingly, the effects of inotropes, such as dobutamine (a β1 agonist) and milrinone (a phosphodiesterase-3 inhibitor) on brain oxygenation remains unknown. These drugs have been proposed as “second-line” agents to improve CBF and CPP in acute brain injury patients (19, 23), or as a support therapy in those suffering from neurogenic heart failure (24–26). Therefore, the aim of this study was to assess the effect of inotropic agents on PbtO2 after an acute brain injury.
This is a single center retrospective analysis of prospectively collected data conducted at Erasme University Hospital, Brussels, Belgium. All adults patients (>18 years old) admitted to the Intensive Care Unit of Erasme hospital from January 2015 to February 2022, due to acute brain injury (SAH, TBI or ICH), who survived at least 24 h were eligible for inclusion. The study protocol was approved by the local ethics committees (SRB2022006). Data collection and analyses were carried out in accordance with relevant scientific and ethical guidelines and regulations.
We included eligible patients who were monitored with intracranial pressure (ICP) and PbtO2 and who received inotropic treatment (i.e., either by dobutamine or milrinone) during monitoring, according to the treating physician's decision. We excluded patients who had malfunctioning/unreliable PbtO2 readings or unavailable data.
Current guidelines for the management of TBI (20), SAH (27), and ICH (21) were implemented in clinical practice; invasive multimodal neuro-monitoring, including PbtO2, was included according to a recent consensus (28) and considered as “standard of care” for acute brain injury patients with a Glasgow Coma Score (GCS) <9 and requiring intracranial pressure (ICP) monitoring. PbtO2 catheter was preferably placed into the “at risk” area, i.e., close to the injured/contused area in TBI and ICH patients and in the region at risk (vascular territory of the artery harboring the aneurysm) or with demonstrated initial or delayed hypoperfusion on brain imaging for SAH.
Physiological variables as well as ICP and PbtO2, were measured in real-time and collected prospectively. Cerebral perfusion pressure (CPP) was calculated as the difference between mean arterial pressure (MAP) and ICP; MAP was zeroed at level of left atrium. Intracranial hypertension was defined by the observation of ICP values above 20 mmHg for at least 5 min at any time. Brain tissue hypoxia was defined as a PbtO2 <20 mmHg. At baseline (immediately before the start of inotropic therapy), 1 and 2 h after the start of inotropes, the 20-min mean value of the following variables was collected: PbtO2, MAP, heart rate (HR), ICP, CPP, body temperature, arterial oxygen saturation (SaO2), central venous oxygen saturation (SvcO2), cardiac output (CO), pH, PaCO2, PaO2, blood lactate and blood glucose levels. Vasopressors and inotropic doses were also recorded. We also calculated the ΔPbtO2 defined as the difference between PbtO2 values at 2 h and at baseline; similarly, the ΔCO, ΔICP, ΔCPP, and ΔPaCO2 were also computed.
We also recorded demographics and the presence of comorbidities, the Sequential Organ Failure Assessment (SOFA) (29) and the Acute Physiology and Chronic Health Evaluation (APACHE) II scores (30) on admission, as well as the GCS score (31) on admission, the use of mechanical ventilation, vasopressor and renal replacement therapy (RRT). The occurrence of neurological complications, such as epilepsy, hydrocephalus, rebleeding, delayed cerebral ischemia and intracranial hypertension, was collected.
Hospital mortality and the Glasgow Outcome Scale (GOS) (32) at 6 months was also reported, either collected from the medical charts or via the general practitioner. Unfavorable neurological outcome was defined as GOS of 1–3.
The primary outcome of the study was the changes in PbtO2 within 2 h after the initiation of inotropic therapy. PbtO2 increase was calculated as the difference between the highest mean PbtO2 values, measured at 1 or 2 h after the initiation of inotropic therapy, and the baseline PbtO2 value; PbtO2 “decrease” was identified as a difference value <0, “stable” PbtO2 as a difference = 0 and “increase” as a difference >0. A “significant” increase was defined as > 20% from the baseline. Patients that experienced a significant increase were defined as “responders.” Secondary outcomes included the identification of factors associated with a significant PbtO2 increase.
Descriptive statistics were computed for all variables. Normality was assessed using the Kolmogorov-Smirnov test. Non-gaussian continuous variables were described as median and interquartile range [IQRs] and compared using Mann-Whitney test (independent variables) or Friedman/Wilcoxon test (repeated measures of related variables); normally distributed variables were expressed as mean (±SD) and compared using Student t-test. Categorical variables were described as proportions (%) and compared using Chi square or Fisher's exact test. A logistic regression of physiological variables at baseline was conducted to assess factors associated with a significant PbtO2 increase after inotropic therapy. The independence of errors, presence of multicollinearity and of influential outlier assumptions were checked; none were violated. The area under the receiver operator characteristic (AUROC) was computed to identify the optimal cut-off value (i.e., best sensitivity and specificity—Youden's index) of baseline PbtO2, baseline ICP and baseline CPP to predict a significant increase of PbtO2 after inotrope infusion. We performed a correlation analysis using Spearman's test between ΔPbtO2 and ΔCO, ΔICP, ΔCPP, and ΔPaCO2. The R coefficient and the 95% confidence interval were computed for all correlations. We also performed a linear regression analysis with ΔPbtO2 as the dependent variable and ΔCO, ΔICP, ΔCPP, and ΔPaCO2 as covariates. All statistical analyses were performed using SPSS 27.0 for MacIntosh. A p-value <0.05 was considered significant.
On a total of 157 patients monitored over the study period, 35 patients were eligible for our study. The characteristics of the study population are shown in Supplementary Table 1. Most patients presented with SAH (31/35, 89%); SAH patients had predominantly a poor grade status at presentation [WFNS 4 or 5: 23/31(74%)] and a high radiological scale [mFisher 3 or 4: 29 (94%)]. The median age was 55 (46–62) years, with a slight predominance of male patients (18/35, 51%). The median GCS on admission was 9 (3–13) and the most common complication was the occurrence of intracranial hypertension. Ninety-one percent of patients experienced at least one episode of brain tissue hypoxia over the ICU stay. All patients received dobutamine as the studied inotrope except one patient who received milrinone. Overall, in-hospital mortality was 57%; 71% of patients had an unfavorable neurological outcome at 6 months.
An increase in PbtO2 was observed in 22/35 patients (63%); of those, 12 (55% of patients with PbtO2 increase and 34% of the whole cohort) experienced a significant PbtO2 increase from baseline (responders). In 4 patients, PbtO2 did not change while it decreased in 9 patients (26%).
There was a non-significant change in PbtO2 after the initiation of the inotropic agents, as shown in Table 1 and Figure 1 (p = 0.052). Also, a significant increase in heart rate and in cardiac output was observed after inotropic agents' administration; no changes in CPP values were observed. The doses of dobutamine are reported in Table 2; milrinone was given at 0.375 mcg/kg*min.
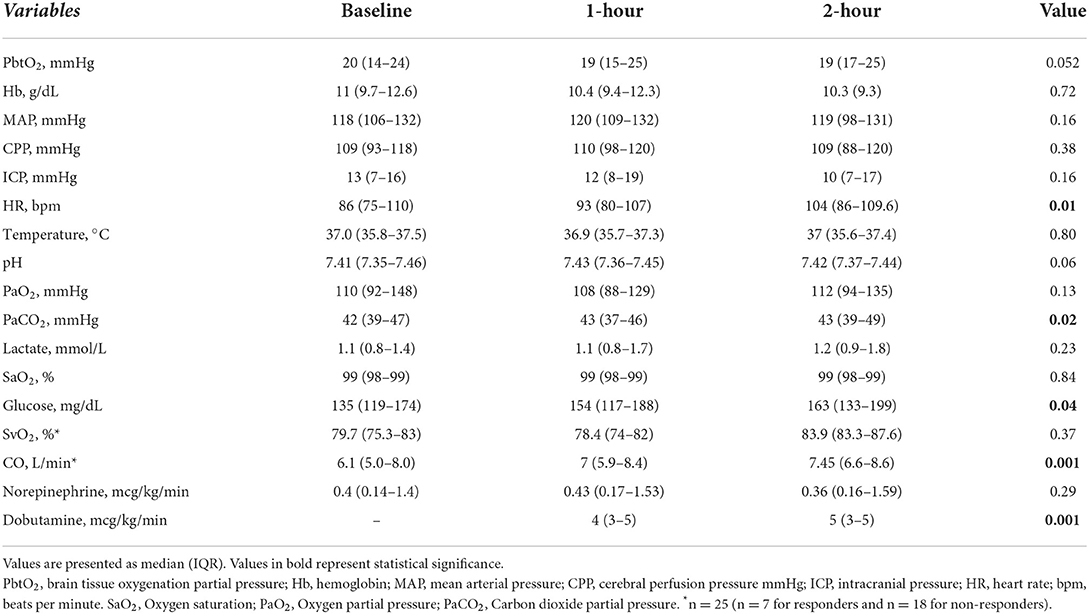
Table 1. Changes in main variables from baseline values after inotropic agents administration.
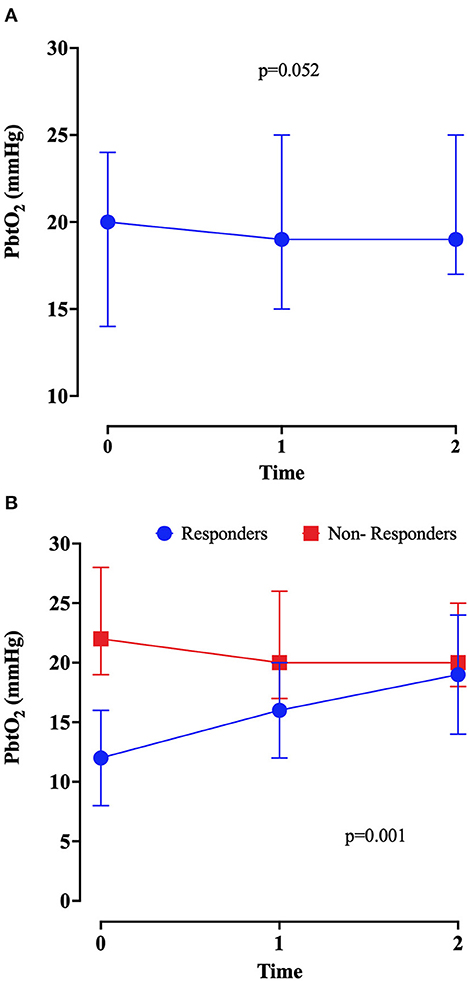
Figure 1. Brain tissue oxygenation (PbtO2) values at baseline (before initiation of inotropes—T0), at 1 (T1) and 2 (T2) hours after the introduction of inotropic continuous infusion in the overall population (A) and in responders and non-responders (B). P-value was calculated by Friedmann test.

Table 2. Logistic regression multivariable analysis to predict a significant PbtO2 increase after inotropic treatment.
There was a significant direct correlation between ΔPbtO2 and ΔCO [r = 0.496 (95% CI 0.122 to 0.746), p = 0.01; n = 25] and a significant negative correlation between ΔPbtO2 and ΔCPP [r = −0.389 (95% CI −0.681 to −0.010), p = 0.05], as shown in Supplementary Figure 1. There was a weak non-significant correlation between ΔPbtO2 and ΔICP [r = 0.26 (95% CI −0.151 to 0.597)]. Linear regression analysis described the association of ΔPbtO2 and ΔCPP [beta coefficient −0.274 (95% CI −0.130 to 0.041)], adjusted for ΔICP [beta coefficient 0.018 (95% CI −0.139 to 0.148)], ΔPaCO2 [beta coefficient −0.038 (95% CI −0.026 to 0.021)] and ΔCO [beta coefficient 0.371 (95% CI −0.054 to 1.306)].
Main differences in collected variables at baseline between responders and non-responders are presented in Supplementary Table 2. Responders had more frequently seizures and rebleeding than non-responders; also, PbtO2 and CPP were significantly lower, while ICP higher, than non-responders at baseline. No significant changes in measured variables over time were observed between groups, except a higher requirement in norepinephrine in the “responders” group when compared to the others (Supplementary Table 3). No statistically significant difference in hospital mortality and neurological outcome at 6 months between responders and non-responders was observed (Supplementary Table 2); no multivariable analysis was performed due to the limited sample size. Among survivors (n = 15), responders [GOS 4 (4, 5)] had numerically higher GOS at 6 months than non-responders [GOS 4 (3, 4)—p = 0.30]. No differences in mFisher and WFNS score on admission between responders and no responders were observed (Supplementary Table 2).
In a multivariable model, lower baseline PbtO2 was associated with higher probability of a significant PbtO2 increase after inotropic treatment (Table 2). The AUROC for baseline PbtO2 (Figure 2) to predict a significant PbtO2 increase was 0.85 (95% CIs 0.70–1.00). The baseline PbtO2 cut-off that better discriminated between responders and non-responders was 17 mmHg, with a sensibility of 84% and a specificity of 91% to predict a significant PbtO2 increase. The AUROC for CPP and ICP and response to predict a significant PbtO2 increase were 0.72 (95% CIs 0.50–0.94—best cut-off of 99 mm Hg) and 0.76 (95% CIs 0.60–0.92—best cut-off of 14 mmHg), respectively. In SAH patients, a multivariable model adjusted for poor grade on admission (WFNS 4 or 5) and mFisher, lower baseline PbtO2 was associated with a higher chance of response to inotropes (Supplementary Table 4).
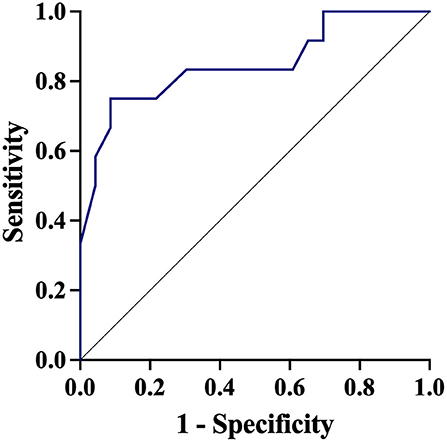
Figure 2. Receiver Operator Curve (ROC) of baseline brain oxygen pressure (PbtO2) to predict a significant PbtO2 increased after therapy.
In this study including mostly SAH patients, we observed that the administration of inotropic agents was associated with an increase in brain oxygenation in 63% of patients; however, only half of those experienced a significant PbtO2 increase from baseline. The response to inotropic agents was greater in patients with lower PbtO2 values at baseline. There was a positive correlation between ΔPbtO2 and ΔCO and a negative correlation between ΔPbtO2 and ΔCPP.
The relationship between cardiac output and cerebral blood flow is complex and difficult to study in clinical practice (33). The use of inotropes to improve brain hemodynamics and oxygenation comes from the hypothesis of a so-called “cardio-cerebral coupling” (34, 35), where an increase in cardiac output could promote a proportional increase in CBF, in particular in case of impaired autoregulation and ischemia (36), both being common features of acute brain injury (37). We observed a positive correlation of PbtO2 changes with CO changes, suggesting the higher was the effects on global hemodynamics, the highest the changes in brain oxygenation. Also, CPP was reduced by the administration of dobutamine, probably due to its vasodilatory via β2 receptors, but this did not result in impaired cerebral hemodynamics. This might suggest the importance of systemic hemodynamics monitoring to adequately understand the effects of increasing CO on brain oxygenation (38) and the relevance of an “hyperdynamic” approach, rather than “hypertensive” one, to manipulate cerebral hemodynamics in ABI patients.
Previous studies conducted in SAH patients found that fluid boluses to improve CO yielded a positive correlation between changes in cardiac output and PbtO2 (39–41); to the best of our knowledge, this is the first study to assess the impact of inotropic agents on brain oxygenation in ABI patients. In experimental model of SAH, administration of dobutamine increased global CBF, which was initially reduced because of an induced brain injury, and resulted into a lower prevalence of DCI and better functional outcomes (42). Moreover, dobutamine may provide, through its effects on □2-receptors, some vasodilatory effects and protect vascular smooth cell muscle against the occurrence of vasospasm in an in vitro study (43). Furthermore, adding dobutamine and nimodipine to the management of cerebral vasospasm in SAH patients compared to nimodipine alone tended to improve the outcome of these patients (43).
Similarly, a study by Levy et al. found that dobutamine administration was able to clinically reverse vasospasm in 78% of patients, together with a significant increase in cardiac output (44). Also, in SAH patients with cerebral vasospasm, dobutamine effectively and significantly increased CBF, measured with Xenon-CT scan, independently from changes in blood pressure and similarly to norepinephrine (45). As for dobutamine, intravenous milrinone can also improve cardiac function after SAH (46) and reverse symptoms due to clinical deterioration caused by DCI in most cases (47), although no data on cerebral oxygenation are available.
It has been suggested that cerebral hemodynamics after an acute brain injury could be assessed using non-invasive monitoring, such as cerebral blood flow velocities or cerebral autoregulation indices. Nevertheless, cerebral autoregulation or CBF velocities cannot evaluate the adequacy of tissue oxygenation. As CPP target is routinely based on PbtO2 or other monitoring tools using vasopressors, our findings suggest that tissue hypoxia can be present in some ABI patients in the absence of elevated ICP and that systemic targets, including CPP or CO, should be individualized according to cerebral demand (15). As inotropic agents are not superior to norepinephrine alone to improve outcome in ABI patients at risk of cerebral ischemia (48), our results suggest that inotropic agents could be considered as a “second-line” therapy to improve brain oxygenation when initial CPP optimization with vasopressors has failed. Interestingly, the more significant effects on tissue oxygenation were observed in those patients with baseline tissue hypoxia, as reported for CPP augmentation or red blood cells transfusions (15, 17, 39).
This study has several limitations. First, this study has an important selection bias since the decision to start inotropes was based on the attending physician' opinion in the absence of an established protocol and due to its retrospective nature we could not determine what were the criteria used. Moreover, we included a very small number of patients, since inotropic treatment were used only in selected cases; therefore, this study is only hypothesis generating. Second, this is a single center study, which limits the generalization of our results. Third, we had only one patient who received milrinone and no comparison between the effects of different inotropes was possible. Additionally, milrinone was used in a lower dose than usually recommended for the treatment of refractory DCI (bolus of 0.1–0.2 mg/kg and continuous infusion of 0.75 mcg/kg/min up to 2.5 mcg/kg/min) (49, 50); however, in the case included in our cohort, this dose was sufficient to increase CO and improve PbtO2. Forth, we did not assess the clinical impact of inotropic administration, only its physiological effects on brain oxygenation; whether optimizing PbtO2 in TBI patients improves outcome remains to be demonstrated. Fifth, we did not take into consideration the exact positioning of the probes, i.e., some of those might have been placed into normally appearing areas, where no potential benefits from hemodynamic manipulation would have been expected. Sixth, we could not assess the correlation between cardiac output and PbtO2 changes in the subgroup analysis because of several missing CO values. Seventh, responders required higher doses of epinephrine after the start of inotropic treatment; therefore, we cannot exclude that the improvement in PbtO2 was due to the increased dose of vasopressors in addition to inotropic agents. Similarly, there was a slight increase in CO2 concomitant to the inotropic administration, which can also induce vasodilation and acts as important confounder. Finally, most of patients had SAH; whether this strategy is effective also in other forms of acute brain injuries remains to be further studied.
In this study, the administration of inotropic agents could increase brain tissue oxygenation in nearly 65% of patients; however, only half of them had a significant PbtO2 increase. Baseline tissue hypoxia could predict this significant PbtO2 increase after the initiation of therapy. The effects on brain oxygenation were proportional to changes in cardiac output. Further prospective studies are needed to explore the impact of inotropic therapy on brain oxygenation and outcome in this setting.
The original contributions presented in the study are included in the article/supplementary material, further inquiries can be directed to the corresponding author/s.
The studies involving human participants were reviewed and approved by Erasme Hospital Ethics Committee. The Ethics Committee waived the requirement of written informed consent for participation.
ED, GC, EG, and FT contributed to conception and design of the study and wrote the first draft of the manuscript. GC, AD, EM, and ED organized the database. EG performed the statistical analysis. SS, EM, AD, and JC revised the manuscript for intellectual content and English editing. All authors contributed to manuscript revision, read, and approved the submitted version.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fneur.2022.963562/full#supplementary-material
1. Lazaridis C, Rusin CG, Robertson CS. Secondary brain injury: predicting and preventing insults. Neuropharmacology. (2019) 145:145–52. doi: 10.1016/j.neuropharm.2018.06.005
2. O'Leary R A, Nichol AD. Pathophysiology of severe traumatic brain injury. J Neurosurg Sci. (2018) 62:542–8. doi: 10.23736/S0390-5616.18.04501-0
3. Taufique Z, May T, Meyers E, Falo C, Mayer SA, Agarwal S, et al. Predictors of poor quality of life 1 year after subarachnoid hemorrhage. Neurosurgery. (2016) 78:256–64. doi: 10.1227/NEU.0000000000001042
4. Lazaridis C, Robertson CS. The role of multimodal invasive monitoring in acute traumatic brain injury. Neurosurg Clin N Am. (2016) 27:509–17. doi: 10.1016/j.nec.2016.05.010
5. Rosengart AJ, Schultheiss KE, Tolentino J, Macdonald RL. Prognostic factors for outcome in patients with aneurysmal subarachnoid hemorrhage. Stroke. (2007) 38:2315–21. doi: 10.1161/STROKEAHA.107.484360
6. Oddo M, Levine JM, Mackenzie L, Frangos S, Feihl F, Kasner SE, et al. Brain hypoxia is associated with short-term outcome after severe traumatic brain injury independently of intracranial hypertension and low cerebral perfusion pressure. Neurosurgery. (2011) 69:1037–45; discussion 1045. doi: 10.1227/NEU.0b013e3182287ca7
7. Bardt TF, Unterberg AW, Hartl R, Kiening KL, Schneider GH, Lanksch WR. Monitoring of brain tissue PO2 in traumatic brain injury: effect of cerebral hypoxia on outcome. Acta Neurochir Suppl. (1998) 71:153–6. doi: 10.1007/978-3-7091-6475-4_45
8. van den Brink WA, van Santbrink H, Steyerberg EW, Avezaat CJ, Suazo JA, Hogesteeger C, et al. Brain oxygen tension in severe head injury. Neurosurgery. (2000) 46:868–76; discussion 876–868. doi: 10.1227/00006123-200004000-00018
9. Maloney-Wilensky E, Gracias V, Itkin A, Hoffman K, Bloom S, Yang W, et al. Brain tissue oxygen and outcome after severe traumatic brain injury: a systematic review. Crit Care Med. (2009) 37:2057–63. doi: 10.1097/CCM.0b013e3181a009f8
10. Kett-White R, Hutchinson PJ, Al-Rawi PG, Gupta AK, Pickard JD, Kirkpatrick PJ. Adverse cerebral events detected after subarachnoid hemorrhage using brain oxygen and microdialysis probes. Neurosurgery. (2002) 50:1213–21; discussion 1221–1212. doi: 10.1227/00006123-200206000-00008
11. Vath A, Kunze E, Roosen K, Meixensberger J. Therapeutic aspects of brain tissue pO2 monitoring after subarachnoid hemorrhage. Acta Neurochir Suppl. (2002) 81:307–9. doi: 10.1007/978-3-7091-6738-0_78
12. Chen HI, Stiefel MF, Oddo M, Milby AH, Maloney-Wilensky E, Frangos S, et al. Detection of cerebral compromise with multimodality monitoring in patients with subarachnoid hemorrhage. Neurosurgery. (2011) 69:53–63; discussion 63. doi: 10.1227/NEU.0b013e3182191451
13. Haitsma IK, Maas AI. Advanced monitoring in the intensive care unit: brain tissue oxygen tension. Curr Opin Crit Care. (2002) 8:115–20. doi: 10.1097/00075198-200204000-00005
14. Chesnut R, Aguilera S, Buki A, Bulger E, Citerio G, Cooper DJ, et al. A management algorithm for adult patients with both brain oxygen and intracranial pressure monitoring: the Seattle International Severe Traumatic Brain Injury Consensus Conference (SIBICC). Intensive Care Med. (2020) 46:919–29. doi: 10.1007/s00134-019-05900-x
15. Kovacs M, Peluso L, Njimi H, De Witte O, Gouvea Bogossian E, Quispe Cornejo A, et al. Optimal cerebral perfusion pressure guided by brain oxygen pressure measurement. Front Neurol. (2021) 12:732830. doi: 10.3389/fneur.2021.732830
16. Gouvea Bogossian E, Diaferia D, Ndieugnou Djangang N, Menozzi M, Vincent J.-L., et al. Brain tissue oxygenation guided therapy and outcome in non-traumatic subarachnoid hemorrhage. Sci Rep. (2021) 11:16235. doi: 10.1038/s41598-021-95602-6
17. Gouvea Bogossian E, Rass V, Lindner A, Iaquaniello C, Miroz JP, Cavalcante Dos Santos E, et al. Factors associated with brain tissue oxygenation changes after RBC transfusion in acute brain injury patients. Crit Care Med. (2022) 50:e539–47. doi: 10.1097/CCM.0000000000005460
18. Bohman LE, Heuer GG, Macyszyn L, Maloney-Wilensky E, Frangos S, Le Roux PD, et al. Medical management of compromised brain oxygen in patients with severe traumatic brain injury. Neurocrit Care. (2011) 14:361–9. doi: 10.1007/s12028-011-9526-7
19. Francoeur CL, Mayer SA. Management of delayed cerebral ischemia after subarachnoid hemorrhage. Crit Care. (2016) 20:277. doi: 10.1186/s13054-016-1447-6
20. Carney N, Totten AM, O'Reilly C, Ullman JS, Hawryluk GW, Bell MJ, et al. Guidelines for the management of severe traumatic brain injury, Fourth edition. Neurosurgery. (2017) 80:6–15. doi: 10.1227/NEU.0000000000001432
21. Hemphill JC III;, Greenberg SM, Anderson CS, Becker K, Bendok BR, Cushman M, Fung GL, et al. Guidelines for the management of spontaneous intracerebral hemorrhage: a guideline for healthcare professionals from the American Heart Association/American Stroke Association. Stroke. (2015) 46:2032–60. doi: 10.1161/STR.0000000000000069
22. Ko SB, Choi HA, Parikh G, Helbok R, Schmidt JM, Lee K, et al. Multimodality monitoring for cerebral perfusion pressure optimization in comatose patients with intracerebral hemorrhage. Stroke. (2011) 42:3087–92. doi: 10.1161/STROKEAHA.111.623165
23. Haenggi M, Andermatt A, Anthamatten C, Galimanis A, Mono ML, Alfieri A, et al. CO(2)-Dependent vasomotor reactivity of cerebral arteries in patients with severe traumatic brain injury: time course and effect of augmentation of cardiac output with dobutamine. J Neurotrauma. (2012) 29:1779–84. doi: 10.1089/neu.2011.1809
24. Finsterer J, Wahbi K. CNS disease triggering Takotsubo stress cardiomyopathy. Int J Cardiol. (2014) 177:322–9. doi: 10.1016/j.ijcard.2014.08.101
25. Kilbourn KJ, Levy S, Staff I, Kureshi I, McCullough L. Clinical characteristics and outcomes of neurogenic stress cadiomyopathy in aneurysmal subarachnoid hemorrhage. Clin Neurol Neurosurg. (2013) 115:909–14. doi: 10.1016/j.clineuro.2012.09.006
26. Cheah CF, Kofler M, Schiefecker AJ, Beer R, Klug G, Pfausler B, et al. Takotsubo cardiomyopathy in traumatic brain injury. Neurocrit Care. (2017) 26:284–91. doi: 10.1007/s12028-016-0334-y
27. Connolly ES Jr, Rabinstein AA, Carhuapoma JR, Derdeyn CP, Dion J, et al. Guidelines for the management of aneurysmal subarachnoid hemorrhage: a guideline for healthcare professionals from the American Heart Association/american Stroke Association. Stroke. (2012) 43:1711–37. doi: 10.1161/STR.0b013e3182587839
28. Le Roux P, Menon DK, Citerio G, Vespa P, Bader MK, Brophy GM, et al. Consensus summary statement of the International Multidisciplinary Consensus Conference on Multimodality Monitoring in Neurocritical Care : a statement for healthcare professionals from the Neurocritical Care Society and the European Society of Intensive Care Medicine. Intensive Care Med. (2014) 40:1189–209. doi: 10.1007/s00134-014-3369-6
29. Vincent JL, Moreno R, Takala J, Willatts S, De Mendonca A, Bruining H, et al. The SOFA (Sepsis-related Organ Failure Assessment) score to describe organ dysfunction/failure. On behalf of the Working Group on Sepsis-Related Problems of the European Society of Intensive Care Medicine. Intensive Care Med. (1996) 22:707–10. doi: 10.1007/BF01709751
30. Knaus WA, Draper EA, Wagner DP, Zimmerman JE. APACHE II: a severity of disease classification system. Crit Care Med. (1985) 13:818–29. doi: 10.1097/00003246-198510000-00009
31. Teasdale G, Jennett B. Assessment of coma and impaired consciousness. A practical scale. Lancet. (1974) 2:81–4. doi: 10.1016/S0140-6736(74)91639-0
32. Jennett B, Bond M. Assessment of outcome after severe brain damage. Lancet. (1975) 1:480–4. doi: 10.1016/S0140-6736(75)92830-5
33. Steiner LA, Siegemund M. Vasoactive agents to improve brain perfusion: pathophysiology and clinical utilization. Curr Opin Crit Care. (2019) 25:110–6. doi: 10.1097/MCC.0000000000000586
34. Castle-Kirszbaum M, Parkin WG, Goldschlager T, Lewis PM. Cardiac output and cerebral blood flow: a systematic review of cardio-cerebral coupling. J Neurosurg Anesthesiol. (2021). doi: 10.1097/ANA.0000000000000768 [Epub ahead of print].
35. Meng L, Hou W, Chui J, Han R, Gelb AW. Cardiac output and cerebral blood flow: the integrated regulation of brain perfusion in adult humans. Anesthesiology. (2015) 123:1198–208. doi: 10.1097/ALN.0000000000000872
36. Diringer MN, Axelrod Y. Hemodynamic manipulation in the neuro–intensive care unit: cerebral perfusion pressure therapy in head injury and hemodynamic augmentation for cerebral vasospasm. Curr Opin Crit Care. (2007) 13:156–62. doi: 10.1097/MCC.0b013e32807f2aa5
37. Armstead WM. Cerebral blood flow autoregulation and dysautoregulation. Anesthesiol Clin. (2016) 34:465–77. doi: 10.1016/j.anclin.2016.04.002
38. Taccone FS, Citerio G Participants Participants in the International Multi-disciplinary Consensus Conference on Multimodality M. Advanced monitoring of systemic hemodynamics in critically ill patients with acute brain injury. Neurocrit Care. (2014) 21(Suppl. 2):S38–63. doi: 10.1007/s12028-014-0033-5
39. Rass V, Bogossian EG, Ianosi BA, Peluso L, Kofler M, Lindner A, et al. The effect of the volemic and cardiac status on brain oxygenation in patients with subarachnoid hemorrhage: a bi-center cohort study. Ann Intensive Care. (2021) 11:176. doi: 10.1186/s13613-021-00960-z
40. Kurtz P, Helbok R, Ko SB, Claassen J, Schmidt JM, Fernandez L, et al. Fluid responsiveness and brain tissue oxygen augmentation after subarachnoid hemorrhage. Neurocrit Care. (2014) 20:247–54. doi: 10.1007/s12028-013-9910-6
41. Muench E, Horn P, Bauhuf C, Roth H, Philipps M, Hermann P, et al. Effects of hypervolemia and hypertension on regional cerebral blood flow, intracranial pressure, and brain tissue oxygenation after subarachnoid hemorrhage. Crit Care Med. (2007) 35:1844–51; quiz 1852. doi: 10.1097/01.CCM.0000275392.08410.DD
42. Mutoh T, Mutoh T, Nakamura K, Sasaki K, Tatewaki Y, Ishikawa T, et al. Inotropic support against early brain injury improves cerebral hypoperfusion and outcomes in a murine model of subarachnoid hemorrhage. Brain Res Bull. (2017) 130:18–26. doi: 10.1016/j.brainresbull.2016.12.009
43. Clark JF, Pyne GJ, Choutka J, Carrozzella JA, Khoury J, Broderick JP, et al. In vitro therapy with dobutamine, isoprenaline and sodium nitroprusside protects vascular smooth muscle metabolism from subarachnoid haemorrhage induced cerebral vasospasm. Acta Neurochir. (2001) 143:721–8. doi: 10.1007/s007010170052
44. Levy ML, Rabb CH, Zelman V, Giannotta SL. Cardiac performance enhancement from dobutamine in patients refractory to hypervolemic therapy for cerebral vasospasm. J Neurosurg. (1993) 79:494–9. doi: 10.3171/jns.1993.79.4.0494
45. Joseph M, Ziadi S, Nates J, Dannenbaum M, Malkoff M. Increases in cardiac output can reverse flow deficits from vasospasm independent of blood pressure: a study using xenon computed tomographic measurement of cerebral blood flow. Neurosurgery. (2003) 53:1044–51; discussion 1051–1042. doi: 10.1227/01.NEU.0000088567.59324.78
46. Naidech A, Du Y, Kreiter KT, Parra A, Fitzsimmons BF, Lavine SD, et al. Dobutamine versus milrinone after subarachnoid hemorrhage. Neurosurgery. (2005) 56:21–26l discussion 26–27. doi: 10.1227/01.NEU.0000144780.97392.D7
47. Castle-Kirszbaum M, Lai L, Maingard J, Asadi H, Danks RA, Goldschlager T, et al. Intravenous milrinone for treatment of delayed cerebral ischaemia following subarachnoid haemorrhage: a pooled systematic review. Neurosurg Rev. (2021) 44:3107–24. doi: 10.1007/s10143-021-01509-1
48. Rondeau N, Cinotti R, Rozec B, Roquilly A, Floch H, Groleau N, et al. Dobutamine-induced high cardiac index did not prevent vasospasm in subarachnoid hemorrhage patients: a randomized controlled pilot study. Neurocrit Care. (2012) 17:183–90. doi: 10.1007/s12028-012-9732-y
49. Lannes M, Teitelbaum J, del Pilar Cortes M, Cardoso M, Angle M. Milrinone and homeostasis to treat cerebral vasospasm associated with subarachnoid hemorrhage: the Montreal Neurological Hospital protocol. Neurocrit Care. (2012) 16:354–62. doi: 10.1007/s12028-012-9701-5
Keywords: hemodynamics, acute brain injury, cerebral blood flow, brain oxygenation, inotropic agents
Citation: Coppalini G, Duvigneaud E, Diosdado A, Migliorino E, Schuind S, Creteur J, Taccone FS and Gouvêa Bogossian E (2022) Effect of inotropic agents on oxygenation and cerebral perfusion in acute brain injury. Front. Neurol. 13:963562. doi: 10.3389/fneur.2022.963562
Received: 07 June 2022; Accepted: 28 June 2022;
Published: 19 July 2022.
Edited by:
Sen Sheng, Washington Regional Medical Center, United StatesReviewed by:
Bishow Mahat, University of Pittsburgh Medical Center, United StatesCopyright © 2022 Coppalini, Duvigneaud, Diosdado, Migliorino, Schuind, Creteur, Taccone and Gouvêa Bogossian. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Elisa Gouvêa Bogossian, ZWxpc2EuZ291dmVhLmJvZ29zc2lhbkB1bGIuYmU=
†These authors share first authorship
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.