
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
CASE REPORT article
Front. Neurol. , 28 July 2022
Sec. Pediatric Neurology
Volume 13 - 2022 | https://doi.org/10.3389/fneur.2022.928334
This article is part of the Research Topic Pediatric Neurology – Case Report Collection 2022 View all 7 articles
 Xingmiao Liu1†
Xingmiao Liu1† Xinquan Liu2†Wenxuan Fan1†
Xinquan Liu2†Wenxuan Fan1† Zhongbin Zhang1Peiyuan Zhang1Xiaojun Liu1
Zhongbin Zhang1Peiyuan Zhang1Xiaojun Liu1 Meifang Lei1
Meifang Lei1 Qing Li1
Qing Li1 Xiaoli Yu1*Dong Li1*
Xiaoli Yu1*Dong Li1*Background: Isovaleric acidaemia (IVA), characterized by an acute metabolic crisis and psychomotor delay, is a rare inherited metabolic disease caused by a deficiency in isovaleryl-CoA dehydrogenase (IVD).
Methods: We report the case of a Chinese patient with IVA who was admitted to Tianjin Children's Hospital and followed up for 8 years. Genetic analysis of the patient and his parents was conducted using the whole-exome sequencing and Sanger sequencing. We searched for similar reported cases in the PubMed and Wanfang databases using the term “isovaleric acidaemia,” reviewed the related literature to obtain a summary of the clinical and genetic characteristics, and analyzed the genotype–phenotype correlations.
Results: The patient presented with encephalopathic symptoms, such as vomiting, lethargy, and somnolence. We identified compound heterozygous variants of the IVD gene, including the unreported variant c.224A>G (p.Asn75Ser) and the reported variant c.1195G>C (p.Asp399His). The child was prescribed a low-protein diet supplemented with L-carnitine. During the 8-year follow-up, no metabolic disorder or encephalopathic symptoms recurred. At present, the child is 11 years of age and has normal mental and motor performance. Another 154 cases identified in 25 relevant references were combined with this case, resulting in a sample of 155 patients, including 52 asymptomatic patients, 64 with neonatal onset, and 39 with the chronic intermittent disease with onset from ages of 1 month to 10 years (median age, 2 years). Among articles that reported sex, the male-to-female ratio was 1:1.06. The cardinal symptoms included vomiting, lethargy, “sweaty foot” odor, poor feeding, developmental delay, and epilepsy. The proportion of variants in regions 123–159 and 356–403 of the IVD protein was greater in symptomatic patients than in asymptomatic patients. Conversely, in asymptomatic patients, the proportion of variants in the 282–318 region was greater than in symptomatic patients.
Conclusion: This case report describes an unreported variant c.224A>G (p.Asn75Ser) of the IVD gene, and summarizes previously reported cases. Furthermore, the correlation between the genotype and clinical phenotype of IVA is analyzed to improve the understanding of this disease.
Isovaleric acidaemia (IVA) is a rare inherited metabolic disease caused by a deficiency in isovaleryl-CoA dehydrogenase (IVD) (1), which catalyses the oxidation of isovaleryl-CoA to 3-methylcrotonyl-CoA during the third step of leucine catabolism (2). The clinical manifestations of IVA include paroxysmal vomiting, lethargy or altered mental status, epilepsy, poor feeding, developmental delay, severe metabolic acidosis, hyperammonaemia, ketosis, hyper- or hypoglycaemia, and cytopenia. A typical “sweaty foot” odor is often found in the acute phase. The absence or delay of treatment can result in death or developmental delay. IVA can be categorized into three subtypes: (1) Acute neonatal type: disease presents in the first 2 weeks after birth, including severe metabolic acidosis and encephalopathy, often accompanied by leukopenia, neutropenia, thrombocytopenia, electrolyte disturbances, and hypo- or hyperglycaemia. The patient may experience coma or death if not properly treated (3). (2) Chronic intermittent type: findings include psychomotor delay and paroxysmal metabolic disorder. During the metabolic crisis, clinical manifestations are similar to those of the acute neonatal type (4). (3) Asymptomatic type: findings include mild biochemical abnormalities (5).
Isovaleryl-CoA dehydrogenase is a mitochondrial flavinase and member of the acyl-CoA dehydrogenase family. Its precursor is encoded within the nuclear genome, and the precursor peptides are synthesized in the cytoplasm and then transported into the mitochondria, where the leading N-terminal sequence is removed by proteolysis to form a mature homologous tetramer. Each monomer contains a noncovalently but tightly bound flavin adenine dinucleotide (FAD) molecule (6). The lengths of its precursor protein and mature sequence are 424 and 394 amino acid residues, respectively (7). The human IVD gene is located on chromosome 15q14-q15 and is 15 kb in length, consisting of 12 exons and 11 introns (8). In previous reports, more than 130 variants have been described; however, the relationship between genotype and phenotype remains unclear. Few studies have been conducted in China, and most cases have been reported without systematic analysis of disease characteristics. This paper reports the case of a child with IVA admitted to Tianjin Children's Hospital and followed up for 8 years. The clinical phenotype, genetic characteristics, and laboratory examination findings of the child are summarized, and the correlation between the genotype and clinical phenotype of IVA is analyzed through a literature review to deepen our understanding of this genetic disease.
The patient, an 11-year-old boy with non-consanguineous Chinese parents, was born at term through an uneventful delivery and with normal birth parameters. The boy was born from a first pregnancy, and his birth weight was 3.35 kg. Developmental milestones were normal. When he was 3 years of age, after 2 days of fever, he presented with severe and frequent vomiting, up to 15 times per day, accompanied by lethargy and somnolence. The family history included no specific genetic diseases. A physical examination revealed no abnormalities. Psychomotor development was normal.
Laboratory analysis revealed metabolic acidosis with a blood pH of 7.28 (reference range, 7.35–7.45); bicarbonate, 10.3 mmol/L (26–32 mmol/L); blood ammonia, 160 μg/dl (25–94 μg/dl); blood glucose, 6.5 mmol/L; and uric acid, 1,246 μmol/L (90–420 μmol/L). The pressure, routine test, and biochemistry indicators of CSF were normal, and the test results of the bacterium, fungi, virus, mycoplasma of CSF were negative. Magnetic resonance imaging (MRI) revealed scattered high signal intensity in the bilateral frontal and parietal periventricular white matter (Figure 1). An electroencephalogram (EEG) showed diffuse slow wave activity with mainly 2–4 Hz moderate- and high-potential δ/θ activity, approximately symmetrical (Figure 2). Tandem mass spectrometry (MS/MS) revealed increased blood levels of C5 acylcarnitine, and Gas chromatography–mass spectrometry (GC/MS) revealed increased levels of urinary 3-hydroxyisovaleric acid (3HIV), isovalerylglycine, and urinary ketone bodies, confirming the diagnosis of IVA.
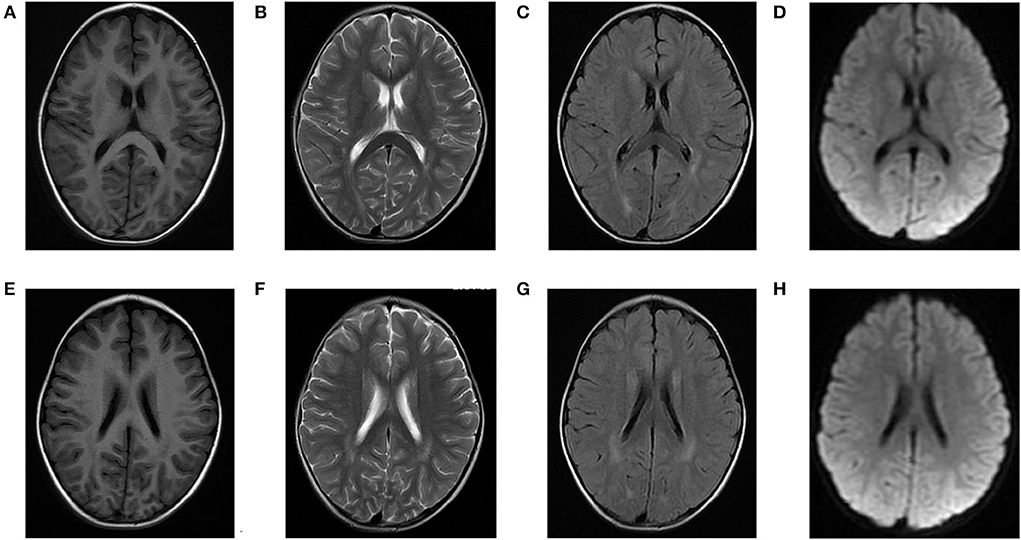
Figure 1. Brain magnetic resonance (MR) images of the patients. (A–D) and (E–H) show scattered high signal intensity in the bilateral frontal and parietal periventricular white matter.
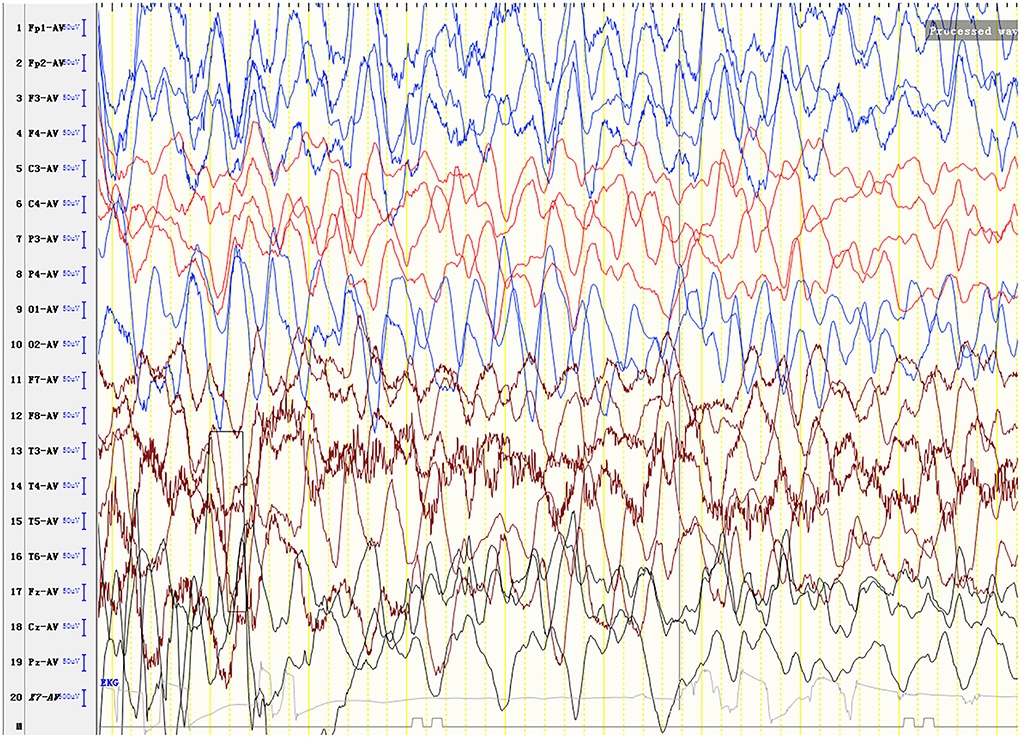
Figure 2. An electroencephalogram (EEG) shows diffuse slow wave activity with mainly 2–4 Hz moderate- and high-potential δ/θ activity, approximately symmetrical.
For genetic analysis, DNA was extracted from the peripheral blood of the patient and his parents using the QIAamp DNA Blood Mini Kit (QIAGEN). A total of 3 μg of patient DNA was cleaved into 200–300 base-pair fragments. The whole-exome enrichment was performed using an Agilent SureSelect Target Enrichment System (Agilent Technologies). Coding exons and flanking intronic regions were enriched using the Agilent SureSelect Human All-Exon V6 reagent (Agilent Technologies) according to the manufacturer's protocol. The captured libraries were loaded onto a HiSeq 2500 platform (Illumina). Base calling and assessments of sequence read quality were performed using Illumina Sequence Control Software (SCS; Illumina). Reads with average quality scores of <25 were removed, and bases with quality scores of <20 were trimmed. The mean read depth for each sample was 100× . The reads were aligned to the human reference genome (UCSC GRCh37/hg19) using the Burrows–Wheeler Aligner BWA-MEM algorithm (BWA v0.7.15). Reads with low mapping quality scores were excluded. Presumed PCR duplicates were identified and removed using the Picard MarkDuplicates. Local alignment optimisation and base quality recalibration were performed using the Genome Analysis Toolkit. Single-nucleotide variants and InDels were identified using Mutect2 and saved in a variant call format. Variants were functionally annotated and filtered using ANNOVAR (http://annovar.openbioinformatics.org/en/latest), which provides built-in public databases (OMIM, InterVar, ClinVar, HGMD, Cosmic70, dbSNP, 1000G, ESP, ExAC, and gnomAD) and the HGMD professional database. We identified two compound heterozygous variants of the IVD gene, the unreported variant c.224A>G (p.Asn75Ser) and the reported variant c.1195G>C (p.Asp399His). The gene variations were validated using Sanger sequencing (Figure 3). The conservation of the identified amino acids across multiple species and the 3D protein modeling of the variants are shown in Figure 4. The pathogenicity scores were determined using online tools, such as Polyphen2, SIFT, Mutation Taster, and ACMG (American College of Medical Genetics) classification (Table 1).
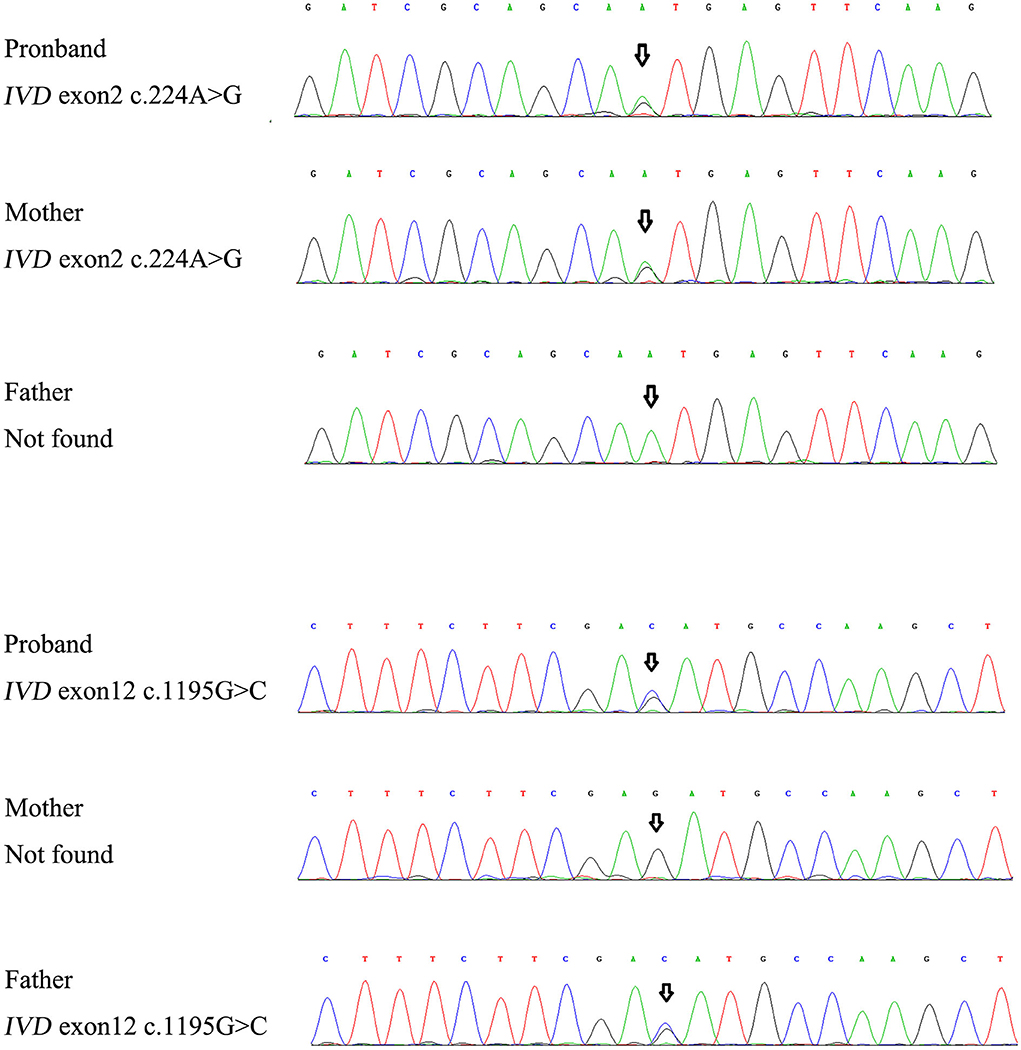
Figure 3. The gene variations were validated using Sanger sequencing.
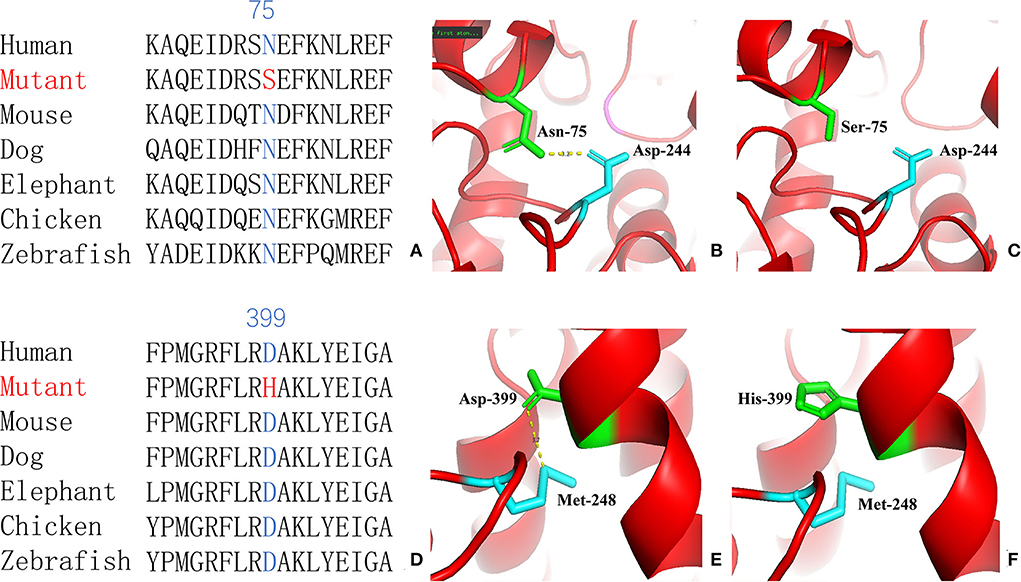
Figure 4. Conservation of the identified amino acids across diverse species (A, D) and the 3D modeling for the IVD-wild and identified missense variant of site 75 (B, C) and site 399 (E, F) in our patient with isovaleric acidaemia (IVA).
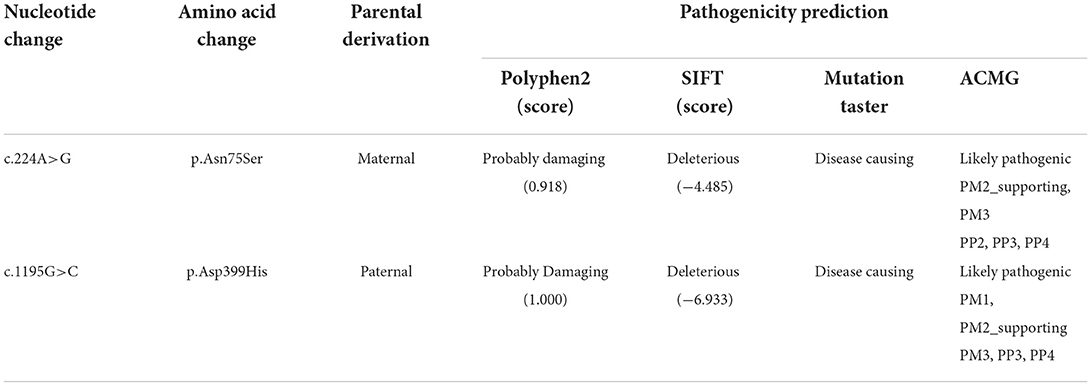
Table 1. The pathogenicity predicted using online tools.
The child was prescribed a low-protein diet (0.5 g/kg/day) supplemented with L-carnitine (10 mg/kg/day). During the 8-year follow-up, despite the need to take daily medication and resist gourmet deals, no metabolic disorder or encephalopathic symptoms recurred. At present, the child is 11 years of age and has normal mental and motor performance at present. GC/MS revealed increased levels of isovalerylglycine, and no other products were found.
We searched for reported cases in the PubMed database and the Wanfang database of China using the term “isovaleric acidaemia”. A total of 154 cases from 25 relevant references were combined with the present case (3, 4, 9–31), resulting in a sample of 155 patients, comprising 52 asymptomatic individuals and 103 symptomatic individuals (including 64 with neonatal onset, and 39 with chronic intermittent disease) with onset from ages from 1 month to 10 years (median age, 2 years).
Clinical manifestations included vomiting (50.5%, 52/103), lethargy (42.7%, 44/103), “sweaty foot” odor (37.9%, 39/103), poor feeding (29.1%, 30/103), developmental delay (27.2%, 28/103), epilepsy (9.8%, 10/103), dystonia (7.8%, 8/103), and tachypnoea (9.8%, 10/103). The prevalence of clinical manifestation of different clinical types is listed in Table 2, with no gender difference. The prevalence of vomiting in the chronic intermittent type was higher than that in the acute neonatal type [69.0 (27/39) vs. 39.0% (25/64)], and the prevalence of sweaty foot odor in the chronic intermittent type was lower than that in the acute neonatal type [23.1 (9/39) vs. 46.9% (30/64)]. There was no statistical difference in other clinical manifestations between the two types.
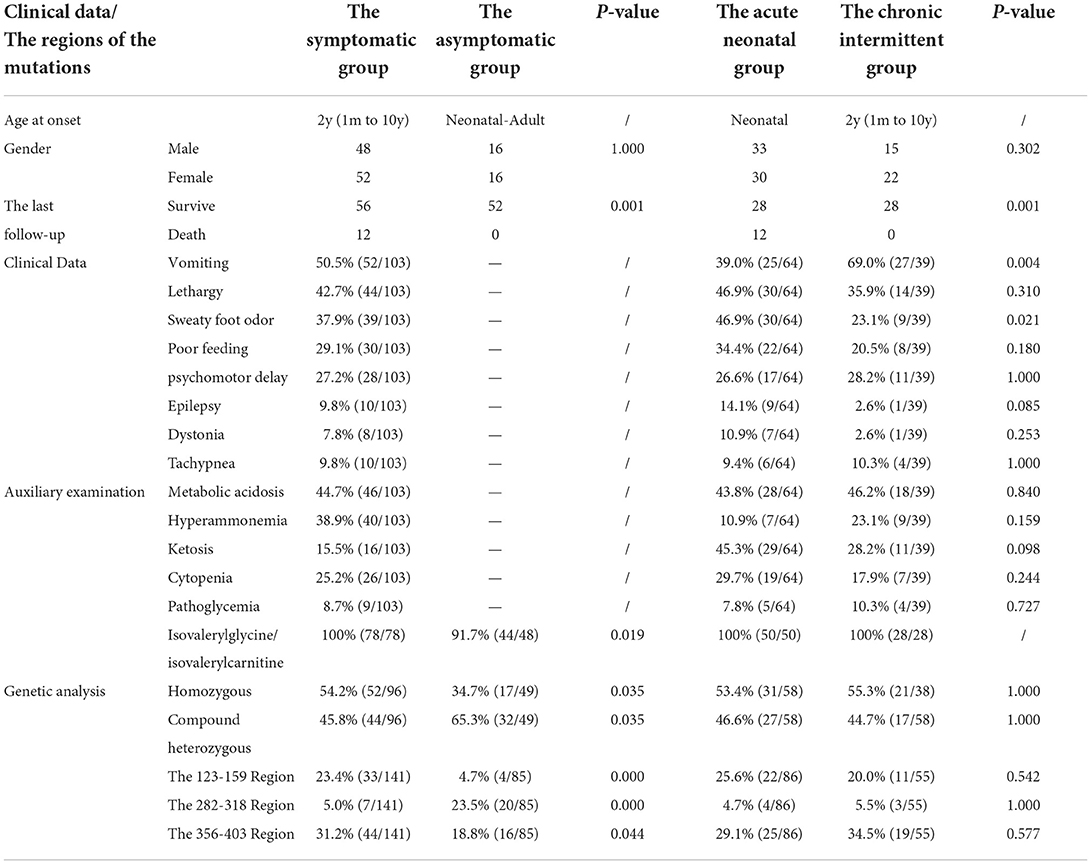
Table 2. The clinical characteristics of 155 patients with isovaleric acidaemia (IVA).
On auxiliary examination, abnormal findings included metabolic acidosis (44.7%, 46/103), hyperammonaemia (38.9%, 40/103), ketosis (15.5%, 16/103), cytopenia (25.2%, 26/103), and abnormal blood glucose levels (8.7%, 9/103). Isovalerylglycine/isovalerylcarnitine/3-hydroxyisovaleric acid eluted at 97% (122/126), and the C5-carnitine level was increased in 100% (112/112). There was no statistical difference in the auxiliary examination between the acute neonatal type and the chronic intermittent type (Table 2).
At the last follow-up, 68 patients were mentioned, including 56 survived and 12 died. The 56 surviving patients had a median age of 8 years (range, 1–25 years) and a median follow-up of 8 years (range, 2–22 years). In total, 34 cases developed normally without other complications; 22 cases had developmental/mental impairment, other neurological disorders, and paroxysmal metabolic crises; and 12 died, all of whom were acute neonatal type, and seven of them died during the neonatal period. There was no death in the chronic intermittent type, and the difference was statistically significant (p < 0.05) (Table 2).
Compound heterozygous and homozygous variants were present in 52.4 (76/145) and 47.6% (69/145), respectively. A total of 226 IVD variants were identified, including 213 missense variants, 7 deletion variants (2 large fragment deletions), 4 insertion variants, and 2 insertion-deletion variants. There were 197 exon variants and 29 intron variants. The proportion of homozygous mutations in symptomatic type (such as acute neonatal type and chronic intermittent type) was higher than that in asymptomatic type [54.2 (52/96) vs. 34.7% (17/49)], and the proportion of compound heterozygous mutations in the asymptomatic type was higher than that in the symptomatic type [65.3 (32/49) vs. 45.8% (44/96)]. The difference was statistically significant (p < 0.05), but there was no statistical difference between the acute neonatal type and the chronic intermittent type (Table 2).
As illustrated in Figure 5, pathogenic variants were non-uniformly distributed along the IVD sequence. Therefore, to further determine the correlation between the genotype and phenotype of IVD, we applied cumulative distribution functions for the first time to analyze whether the pathogenic variants were clustered in any specific regions.
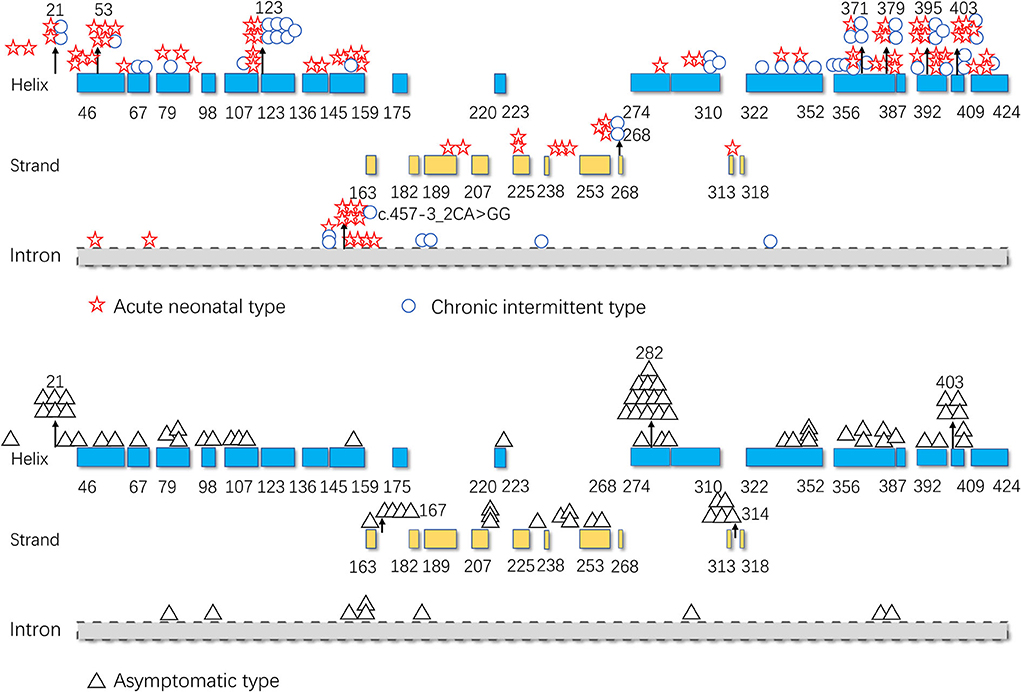
Figure 5. Distribution of the isovaleryl-CoA dehydrogenase (IVD) variants. Blue: the α-helical N-terminal domain and the second α-helical C-terminal domain of IVD; yellow: the β-sheet domain; and gray: intron region. Numbers represent the corresponding amino acid sites.
Figures 6A,B shows the cumulative distribution of symptomatic patients (including acute neonatal and chronic intermittent types) and asymptomatic, acute neonatal, and chronic intermittent patients, respectively. In Figure 6A, three steep regions were revealed: (a) 123–159; (b) 282–318; and (c) 356–403. In each of the three steep regions, we compared the difference in the number of variants between symptomatic and asymptomatic patients, and the results showed that the proportion of variants was higher in symptomatic patients than in asymptomatic patients in regions 123–159 and 356–403 (p < 0.05 and p < 0.05). In the 282–318 region, the proportion of asymptomatic patients was greater than that of symptomatic patients (p < 0.05) (Table 2). In Figure 6B, two steep areas can be observed: (a) 123–159 and (b) 356–403. Within each steep region, we observed no difference in the ratio of the number of variants in patients with the acute neonatal type and in those with the chronic intermittent type (p > 0.05 and p > 0.05) (Table 2). The 3D modeling and conservation of amino acid sequences among different species in areas 123–159 and 356–403 are shown in Figures 7, 8, respectively.
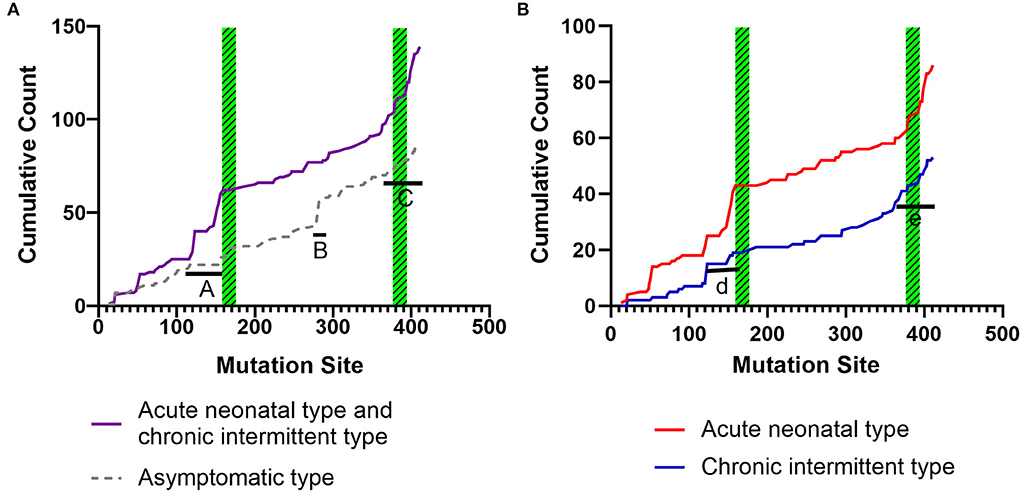
Figure 6. Cumulative distribution of the IVD variants, comparing variant distribution in symptomatic patients (including, acute neonatal and chronic intermittent type) with that of asymptomatic patients (A), and the acute neonatal type vs. the chronic intermittent type (B). Green striped boxes: FAD binding sites. FAD, flavin adenine dinucleotide.
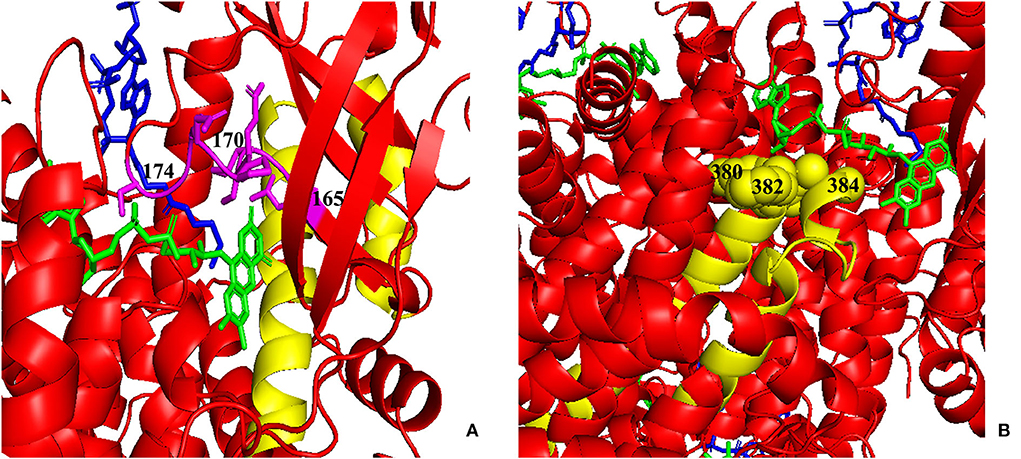
Figure 7. Three-dimensional (3D) modeling of areas 123–159 and 356–403. (A) Yellow: the 123–159 region; green: FAD; blue: CoA Persulfide; purple: FAD binding sites (numbered). (B) Yellow: the 356–403 region; green: FAD; blue: CoA Persulfide; yellow balls: FAD binding sites (numbered). FAD, flavin adenine dinucleotide.

Figure 8. Conservation of amino acid sequences in areas 123–159 and 356–403 across diverse species.
The hot region of the distribution of IVD variants in Chinese patients with IVA was 356–403, and the hotspots were 53, 120, 214, 339, 371, 395, 398, and 403 (Figure 9).
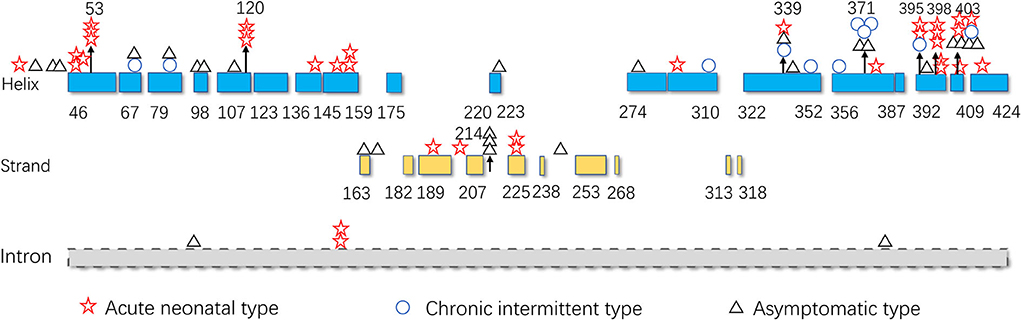
Figure 9. The IVD variant distribution in Chinese patients with IVA.
Isovaleric acidaemia is one of four typical organic acidaemias: propionic acidaemia, methylmalonic acidaemia, glutaric acidaemia, and IVA. Currently, there are no data on the overall prevalence of IVA in China. The prevalence of neonatal IVA is 1:400,000 in the neonatal screening data of the Shanghai Institute of Pediatrics (25). The overall prevalence of IVA is 1:365,000 in Taiwan (9), 1:250,000 in the United States (26), and 1–9/100,000 in other Western countries (21).
The structure of IVD was first elucidated in 1966, and the enzyme was first isolated and purified from rat-liver mitochondria in 1983 (2). It is a homologous tetramer that belongs to an important family of flavin proteins. Each monomer consists of three domains: an α-helical domain at the N-terminus, a β-sheet domain, and a second α-helical domain at the C-terminus. The α-helical N-terminal domain consists of six α-helices labeled A–F from the N-terminus toward the C-terminus. The β-sheet domain comprises seven β-sheets labeled 1–7. The C-terminal α-helical domain consists of five α-helices designated G–K. Each monomer contains an FAD molecule (also known as active vitamin B2) that catalyses the α- and β-dehydrogenation of various thioester substrates. The opening of the CoA binding pocket, where the adenosine diphosphate portion of CoA binds, is exposed to the solvent, while the bottom of this pocket, which surrounds the fatty acyl segment of the CoA thioester substrate, is mainly composed of hydrophobic amino acid residues of the polypeptide and the isoalloxazine ring of FAD. The residues that line the substrate binding pocket and the active site cavity are located on the loops connecting β-strands 1 and 2, β-strands 4 and 5, and the loops between helices J and K, H and I, and E and G (2).
IVD catalyses the oxidation of isovaleryl-CoA to 3-methylcrotonyl-CoA during the third step of leucine catabolism. IVD deficiency disrupts leucine metabolism, leading to the accumulation of upstream isovaleric acid and its metabolites 3-hydroxyisovaleric acid, isovaleryl (C5)-carnitine, and isovalerylglycine (11), resulting in the metabolic acidosis and functional impairment of the brain, liver, kidneys, bone marrow, and other organs.
To date, the correlation between phenotype and genotype in patients with IVA has been unclear, and siblings carrying the same variant have shown very different phenotypes (17). Therefore, in this study, the data from 155 patients were summarized, and the correlation between phenotype and genotype was further analyzed. The results showed that the two hot regions of the distribution of pathogenic variants in symptomatic patients (including, acute neonatal type and chronic intermittent type) were located in the 123–159 and 356–403 regions, while the distribution in asymptomatic patients was mainly located in the 282–318 region. The c.932C>T (p.Ala282Val) and c.941C>T (p.Ala314Val) variants were common in asymptomatic and neonatal screening patients who remained asymptomatic during the follow-up period. This indicates that the variants in the first two regions are highly pathogenic, whereas the concentrated distribution of variants with an asymptomatic phenotype indicates that the amino acid variations are pathogenically milder.
As shown in Figures 5, 7, the 123–159 and 356–403 regions were mainly in the α-helix of the N-terminal domain near the center of the monomer and in the α-helix at the end of the C-terminal domain, respectively, which are relatively conserved across diverse species (Figure 8). The C-terminal amino acids of IVD are necessary for tetramer stability and subunit interaction. According to the Uniprot database, regions 165–174 and 380–384 are FAD binding sites. FAD has an extended conformation and is located between the middle and C-terminal domains of one monomer and the C-terminal domain of the adjacent monomer (2). As shown in Figure 6, the aggregated regions of the pathogenic variant distribution were essentially near the FAD binding sites, with partially overlapping regions. Therefore, variants in these two regions may affect the binding between amino acid residues and FAD molecules, destroy the protein structure, affect the stability of the tetramer, and further affect protein function. Asymptomatic hot regions are located at 282–318, which is far from the FAD binding sites; therefore, they may have little influence on protein function.
Our patient was identified as having the compound heterozygous variants c.224A>G (p.Asn75Ser) and c.1195G>C (p.Asp399His). Asn75 is located in the α-helical N-terminal domain, whereas Asp399 is located in the α-helical C-terminal domain. All these residues are embedded in the protein and interact with other residues to stabilize the protein structure. When Asn75 is replaced with serine, the hydrogen bond between Asn75 and Asp244 is predicted to be disrupted (Figure 4), thus affecting protein structure and stability. When Asp399 is replaced with histidine, an imidazole ring is added to the side chain, resulting in destroying the hydrogen bond between Asp399 and Met248 (Figure 4), which changes three-dimensional structure and may affect the binding of FAD.
The hotspots for pathogenic variants depend on ethnicity and region. According to previous reports, c.1199A>G (p.Tyr371Cys) and c.1208A>G (p.Tyr403Cys) are common among the Han Chinese population (17). The variant c.466-3_466-2CA>GG (originally reported as c.457-3_2CA>GC) is common in both Thai and Korean populations (4, 22); c.932C>T (p.Ala282Val) has been reported as a recurrent variant in White populations (26); c.367G>A (p.Gly123Arg) has been described as a common variant in Whites settled in South Africa (15); c.158G>C (p.Arg53Pro) was the most common variant in the Mexican population (27); and p.Arg395Gln may be a common variant in the UAE population (23). Based on the literature review, the hotspots in the mainland China and Taiwan are 53, 120, 214, 339, 371, 395, 398, and 403 (Figure 9).
For IVA treatment, daily protein intake should be limited, and the dietary supplementation with L-carnitine and glycine should be prescribed to convert isovaleric acid to non-toxic isovalerylcarnitine and isovalerylglycine, respectively (11).
The clinical manifestations and prognosis of IVA vary greatly, and some patients die in the neonatal period. However, many asymptomatic patients are found through family or neonatal screening and remain asymptomatic during the follow-up. In the last follow-up, 12/68 had died and 56/68 had survived, 34 of whom developed normally without other complications. In total, twenty-two cases had developmental/mental impairment, other neurological disorders, and paroxysmal metabolic disorders. Early diagnosis and treatment have been reported that cannot prevent the onset of metabolic crisis but can prevent mental impairment (21). Therefore, the correlation between genotype and phenotype can help us understand the disease and guide genetic counseling, and early screening and diagnosis as early as possible for IVA can guide treatment and improve prognosis.
The datasets presented in this article are not readily available because of ethical and privacy restrictions. Requests to access the datasets should be directed to the corresponding author/s.
The studies involving human participants were reviewed and approved by Medical Ethics Committee of Tianjin Children's Hospital/Tianjin University Children's Hospital. Written informed consent to participate in this study was provided by the participants' legal guardian/next of kin. Written informed consent was obtained from the minor(s)' legal guardian/next of kin for the publication of any potentially identifiable images or data included in this article.
XingL and WF retrieved and summarized the related literature. XinqL drew all the figures and tables. ZZ provided statistical analysis. PZ, XiaoL, ML, and QL followed up the patients. XY and DL revised the review article. All authors contributed to the article and approved the submitted version.
We thank our patient's family for allowing us to share his case and Editage for English language editing.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
1. Tanaka K, Budd MA, Efron ML, Isselbacher KJ. Isovaleric acidemia: a new genetic defect of leucine metabolism. Proc Natl Acad Sci U S A. (1966) 56:236–42. doi: 10.1073/pnas.56.1.236
2. Tiffany KA, Roberts DL, Wang M, Paschke R, Mohsen AW, Vockley J, et al. Structure of human isovaleryl-CoA dehydrogenase at 2. 6 A resolution: structural basis for substrate specificity. Biochemistry. (1997) 36:8455–64. doi: 10.1021/bi970422u
3. Wu F, Fan SJ, Zhou XH. Neonatal isovaleric acidemia in China: a case report and review of literature. World J Clin Cases. (2021) 9:436–44. doi: 10.12998/wjcc.v9.i2.436
4. Vatanavicharn N, Liammongkolkul S, Sakamoto O, Sathienkijkanchai A, Wasant P. Phenotypic and mutation spectrums of Thai patients with isovaleric acidemia. Pediatr Int. (2011) 53:990–4. doi: 10.1111/j.1442-200X.2011.03488.x
5. Vockley J, Ensenauer R. Isovaleric acidemia: new aspects of genetic and phenotypic heterogeneity. Am j med genet C Semin med genet. (2006) 142c:95–103. doi: 10.1002/ajmg.c.30089
6. Ikeda Y, Keese SM, Fenton WA, Tanaka K. Biosynthesis of four rat liver mitochondrial acyl-CoA dehydrogenases: in vitro synthesis, import into mitochondria, and processing of their precursors in a cell-free system and in cultured cells. Arch Biochem Biophys. (1987) 252:662–74. doi: 10.1016/0003-9861(87)90072-5
7. Matsubara Y, Indo Y, Naito E, Ozasa H, Glassberg R, Vockley J, et al. Molecular cloning and nucleotide sequence of cDNAs encoding the precursors of rat long chain acyl-coenzyme A, short chain acyl-coenzyme A, and isovaleryl-coenzyme A dehydrogenases. Sequence homology of four enzymes of the acyl-CoA dehydrogenase family. J Biol Chem. (1989) 264:16321–31. doi: 10.1016/S0021-9258(18)71624-4
8. Kraus JP, Matsubara Y, Barton D, Yang-Feng TL, Glassberg R, Ito M, et al. Isolation of cDNA clones coding for rat isovaleryl-CoA dehydrogenase and assignment of the gene to human chromosome 15. Genomics. (1987) 1:264–9. doi: 10.1016/0888-7543(87)90053-X
9. Lin WD, Wang CH, Lee CC, Lai CC, Tsai Y, Tsai FJ. Genetic mutation profile of isovaleric acidemia patients in Taiwan. Mol Genet Metab. (2007) 90:134–9. doi: 10.1016/j.ymgme.2006.08.011
10. Che F, Yang Y, Wang Z, Wang G, Wu H, Zhang L, et al. [Isovaleric acidemia due to compound heterozygous variants of IVD gene in a case]. Zhonghua Yi Xue Yi Chuan Xue Za Zhi. (2021) 38:150–3. doi: 10.3760/cma.j.cn511374-20200415-00271
11. Chinen Y, Nakamura S, Tamashiro K, Sakamoto O, Tashiro K, Inokuchi T, et al. Isovaleric acidemia: Therapeutic response to supplementation with glycine, l-carnitine, or both in combination and a 10-year follow-up case study. Mol Genet Metab Rep. (2017) 11:2–5. doi: 10.1016/j.ymgmr.2017.03.002
12. Li Y, Shen M, Jin Y, Liu Y, Kang L, He R, et al. Eight novel mutations detected from eight Chinese patients with isovaleric acidemia. Clin Chim Acta. (2019) 498:116–21. doi: 10.1016/j.cca.2019.08.019
13. Gilbert-Barness E, Barness LA. Isovaleric acidemia with promyelocytic myeloproliferative syndrome. Pediatr Dev Pathol. (1999) 2:286–91. doi: 10.1007/s100249900125
14. Kiliç M, Kaymaz N, Özgül RK. Isovaleric acidemia presenting as diabetic ketoacidosis: a case report. J Clin Res Pediatr Endocrinol. (2014) 6:59–61. doi: 10.4274/Jcrpe.1181
15. Dercksen M, Duran M, Ijlst L, Mienie LJ, Reinecke CJ, Ruiter JP, et al. Clinical variability of isovaleric acidemia in a genetically homogeneous population. J Inherit Metab Dis. (2012) 35:1021–9. doi: 10.1007/s10545-012-9457-2
16. Couce ML, Aldamiz-Echevarría L, Bueno MA, Barros P, Belanger-Quintana A, Blasco J, et al. Genotype and phenotype characterization in a Spanish cohort with isovaleric acidemia. J Hum Genet. (2017) 62:355–60. doi: 10.1038/jhg.2016.144
17. Ozgul RK, Karaca M, Kilic M, Kucuk O, Yucel-Yilmaz D, Unal O, et al. Phenotypic and genotypic spectrum of Turkish patients with isovaleric acidemia. Eur J Med Genet. (2014) 57:596–601. doi: 10.1016/j.ejmg.2014.08.006
18. Kaya N, Colak D, Al-Bakheet A, Al-Younes B, Tulbah S, Daghestani M, et al. Identification of a novel IVD mutation in a consanguineous family with isovaleric acidemia. Gene. (2013) 513:297–300. doi: 10.1016/j.gene.2012.09.097
19. Sag E, Cebi AH, Kaya G, Karaguzel G, Cakir MA. Rare cause of recurrent acute pancreatitis in a child: isovaleric acidemia with novel mutation. Pediat Gastroenterol Hepatol Nutr. (2017) 20:61–4. doi: 10.5223/pghn.2017.20.1.61
20. Sakamoto O, Arai-Ichinoi N, Mitsubuchi H, Chinen Y, Haruna H, Maruyama H, et al. Phenotypic variability and newly identified mutations of the IVD gene in Japanese patients with isovaleric acidemia. Tohoku J Exp Med. (2015) 236:103–6. doi: 10.1620/tjem.236.103
21. Szymańska E, Jezela-Stanek A, Bogdańska A, Rokicki D, Ehmke Vel Emczyńska-Seliga E, Pajdowska M, et al. Long term follow-up of polish patients with isovaleric aciduria. clinical and molecular delineation of isovaleric aciduria. Diagnostics. (2020) 10:738. doi: 10.3390/diagnostics10100738
22. Lee YW, Lee DH, Vockley J, Kim ND, Lee YK Ki CS. Different spectrum of mutations of isovaleryl-CoA dehydrogenase (IVD) gene in Korean patients with isovaleric acidemia. Mol Genet Metab. (2007) 92:71–7. doi: 10.1016/j.ymgme.2007.05.003
23. Hertecant JL, Ben-Rebeh I, Marah MA, Abbas T, Ayadi L, Ben Salem S, et al. Clinical and molecular analysis of isovaleric acidemia patients in the United Arab Emirates reveals remarkable phenotypes and four novel mutations in the IVD gene. Eur J Med Genet. (2012) 55:671–6. doi: 10.1016/j.ejmg.2012.08.001
24. Cho JM, Lee BH, Kim GH, Kim YM, Choi JH, Yoo HW. Chronic intermittent form of isovaleric aciduria in a 2-year-old boy. Korean J Pediatr. (2013) 56:351–4. doi: 10.3345/kjp.2013.56.8.351
25. Bei F, Sun JH Yu YG, Jia J, Zheng ZJ, Fu QH, et al. Two novel isovaleryl-CoA dehydrogenase gene mutations in a Chinese infant. Gene. (2013) 524:396–400. doi: 10.1016/j.gene.2013.03.139
26. Ensenauer R, Vockley J, Willard JM, Huey JC, Sass JO, Edland SD, et al. A common mutation is associated with a mild, potentially asymptomatic phenotype in patients with isovaleric acidemia diagnosed by newborn screening. Am J Hum Genet. (2004) 75:1136–42. doi: 10.1086/426318
27. Lin Y, Chen D, Peng W, Wang K, Lin W, Zhuang J, et al. Newborn screening for isovaleric acidemia in Quanzhou, China. Clin Chim Acta. (2020) 509:25–9. doi: 10.1016/j.cca.2020.06.010
28. Hu Z, Yang J, Hu L, Zhao Y, Zhang C, Yang R, et al. Screening and clinical analysis of isovaleric acidemia newborn in Zhejiang province. J Zhejiang Univ. (2020) 49:556–64. doi: 10.3785/j.issn.1008-9292.2020.10.02
29. Li X, Hua Y, Ding Y, Liu Y, Song J, Wang Q, et al. Analysis of four Chinese patients with neonatal-onset isovaleric acidemia. Chin J Perinat Med. (2015) 18:188–94. doi: 10.3760/cma.j.issn.1007-9408.2015.03.005
30. Mei S, Bai N, Hu S, Liu N, Liu L, Kong X. Analysis of genetic variation in two families with isovaleric acidemia and prenatal diagnosis of one of them. Chin J Perinat Med. (2018) 21:31–5. doi: 10.3760/cma.j.issn.1007-9408.2018.01.007
Keywords: isovaleric acidaemia, isovaleryl-CoA dehydrogenase (IVD), inherited metabolic disease, genotype-phenotype correlation, case report
Citation: Liu X, Liu X, Fan W, Zhang Z, Zhang P, Liu X, Lei M, Li Q, Yu X and Li D (2022) Analysis of the genotype–phenotype correlation in isovaleric acidaemia: A case report of long-term follow-up of a chinese patient and literature review. Front. Neurol. 13:928334. doi: 10.3389/fneur.2022.928334
Received: 25 April 2022; Accepted: 06 July 2022;
Published: 28 July 2022.
Edited by:
Kette D. Valente, University of São Paulo, BrazilReviewed by:
Roberto Chimenz, University of Messina, ItalyCopyright © 2022 Liu, Liu, Fan, Zhang, Zhang, Liu, Lei, Li, Yu and Li. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Dong Li, dGpla3NqQDE2My5jb20=; Xiaoli Yu, eXhsbml1QDE2My5jb20=
†These authors have contributed equally to this work
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.