 Yalan Wan1
Yalan Wan1 Yanyan Jiang2
Yanyan Jiang2 Zhiying Xie1
Zhiying Xie1 Chen Ling1
Chen Ling1 Kang Du1Ran Li3
Kang Du1Ran Li3 Yun Yuan1
Yun Yuan1 Zhaoxia Wang1Wei Sun1*†Haiqiang Jin1*†
Zhaoxia Wang1Wei Sun1*†Haiqiang Jin1*†- 1Department of Neurology, Peking University First Hospital, Beijing, China
- 2Department of Neurology, The First Affiliated Hospital of Zhengzhou University, Zhengzhou, China
- 3Department of Neurology, Huoguosi TCM Hospital Affiliated to Beijing University of Chinese Medicine, Beijing, China
Background: PLA2G6-associated neurodegeneration (PLAN) is a heterogeneous group of neurodegenerative diseases caused by biallelic PLA2G6 mutations, covering diseases such as infantile neuroaxonal dystrophy (INAD), atypical neuroaxonal dystrophy (ANAD), dystonia parkinsonism (DP), and autosomal recessive early-onset parkinsonism (AREP). The study aims to report the clinical and genetic features of a series of PLAN patients.
Methods: The clinical and radiological findings of five Chinese patients from three families were collected. Whole-exome next generation sequencing (NGS) was applied to identify the genetic causes. Co-segregation analysis of the detected candidate variants were performed in their families. The pathogenicity of identified novel variants was predicted by in silico analysis.
Results: NGS revealed compound heterozygous variants of PLA2G6 gene in all five patients. There were six PLA2G6 variants identified, including two known variants (c.116G>A, c.238G>A) and four novel variants (c.2120dupA, c.2071C>G, c.967G>A, c1534T>A). ACMG predicts c.2120dupA to be pathogenic, c.2071C>G and c.1534T>A to be likely pathogenic, and c1534T>A to be of uncertain significance. Clinically, four patients fell into the diagnosis of ANAD, and 1 into the diagnosis of AREP. Brain imaging revealed cerebellar atrophy, iron deposition in bilateral globus pallidus, and substantia nigra in three cases.
Conclusions: Four novel pathogenic variants were discovered and the pathogenic variant spectrum of the PLA2G6 gene was expanded.
Introduction
The PLA2G6 gene was initially cloned in two unrelated Israeli infantile neuroaxonal dystrophy (INAD) families in 2006 (1). Later PLA2G6 pathogenic variants were identified in atypical neuroaxonal dystrophy (ANAD) and parkinsonian syndrome including adult onset dystonia parkinsonism (DP) and autosomal recessive early-onset parkinsonism (AREP) (2). Thus PLA2G6-associated neurodegeneration (PLAN) appears as a heterogeneous group of neurodegenerative diseases including INAD, ANAD, DP, and AREP.
INAD, first described by Seitelberger in 1952, emerges with developmental regression, hypotonia, and spastic tetraparesis, with an onset between 6 months and 3 years (3). ANAD, first described by Nardocci et al. (4) in 1999, is of a milder form compared with INAD, with a later onset (up to 6 years) and a more protracted course (4). The main symptoms of ANAD include cerebellar signs, gait abnormalities, speech delay or regression, and diminished social interaction which may lead to a misdiagnosis of autism before the occurrence of other neurological signs (5). In 2009, Paisa'n-Ruiz et al. (6) identified PLA2G6 pathogenic variants in patients suffering from early-onset levodopa responsive dystonia-parkinsonism and broadened the phenotypic spectrum of PLAN. The phenotype of AREP, first reported by Shi CH, et al. (7) in 2011, is characterized by extrapyramidal signs, cognitive decline, dystonia, dysarthria/dysphonia, swallowing problems, limb tremors, and abnormal gait, sensitive to dopaminergic agents. In recent years, considering common features of DP, AREP, and sporadic early onset parkinsonism (EOP), scholars have proposed the concept of phenotypic continuum to describe PLA2G6-related parkinsonism, the most common phenotype in late-onset PLAN (8, 9).
PLAN has been reported worldwide, but is still not a commonly detected disease. Up to now, there are only 21 ANAD and 86 PLA2G6-related parkinsonism patients (8) reported in literature. Herein this study reported the clinical features and PLAN pathogenic variants in 4 Chinese patients with ANAD and 1 with AREP (Table 1), carrying 6 different mutations, 4 of which are novel. Furthermore, the reported ANAD and adult onset AREP in literature were reviewed, with their clinical features further analyzed.
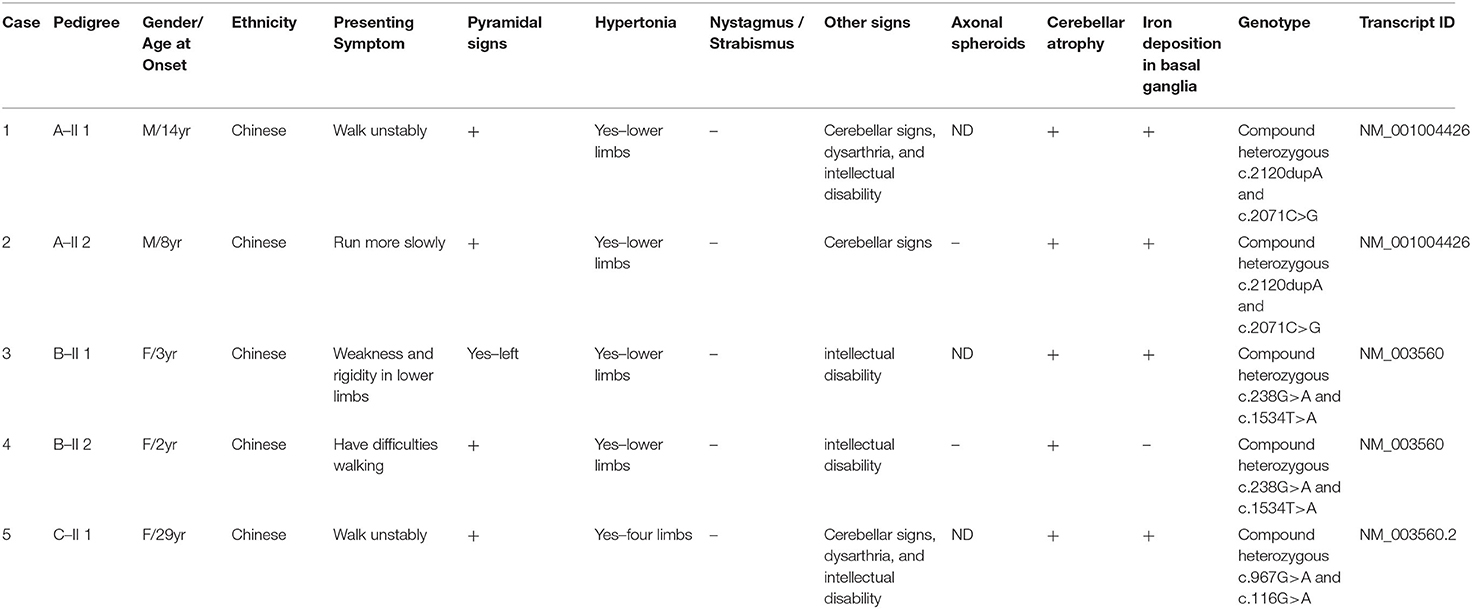
Table 1. Clinical features of the five PLAN patients in this article.
Methods
Subjects
Five patients from three Chinese families with PLAN were recruited from the Peking University First Hospital, whose clinical presentations including medical history, physical examination, brain magnetic resonance imaging, as well as biological sample with peripheral blood were collected from all subjects. This study received ethical approval from Peking University First Hospital.
Genetic Analysis
Genomic DNA was extracted via standard procedures from peripheral blood samples taken from all 5 patients. Sequence variants were detected by whole exome sequencing (10). All the exomes were sequenced by an Illumina (novaseq 6000) platform. Filtering strategies were the same as what were adopted in a previous study. Co-segregation analysis of the detected candidate variants were performed in their families (10). To predict the potential pathogenicity of genetic variants, in silico prediction analysis was performed in accordance with the guideline stipulated by the American college of medical genetics and genomics (ACMG) (11). In silico algorithms including Mutation Taster (http://www.mutationtaster.org) (12), PolyPhen-2 (http://genetics.bwh.harvard.edu/pph2/), and SIFT (http://sift.jcvi.org) (13) are applied.
Literature Review
PubMed (03/20/2021) for ANAD and adult onset PLA2G6-related parkinsonism in genetically or neuropathology confirmed PLAN were systematically searched with the following terms: atypical neuroaxonal dystrophy, ANAD, PLAN, and parkinsonism. An in-depth review was carried out over all previously published case reports about ANAD and PLA2G6 related and adult-onset parkinsonism, Only cases carrying biallelic PLA2G6 mutations with individual information were included. In parkinsonism type, childhood onset cases were excluded. Corresponding demographic, clinical, genetic, and radiological results were summarized as well.
Results
Case Descriptions
Patient A-II 1, the proband in family A, was a 14-year-old male who was asymptomatic until age 5, when he showed unstable gait without obvious weakness. Over the next 4 years, the symptoms progressed to rigidity in four limbs, and also caused decline in academic performance. At the age of 11, he could not walk by himself. Even the adoption of L-dopa failed to effectively improve these symptoms. Physical examinations revealed neck and trunk dystonia, dysarthria, and mental impairment. He got incomplete ophthalmoplegia. Muscle tone in both lower limbs were significantly increased such that he got spasticity. His incordination movements were impaired. Deep tendon reflexes were increased and bilateral Babinski's signs were positive. The maximum improvement rate of levodopa challenge test was 16.7%. Laboratory tests were normal, with no Kayser–Fleischer rings observed. Brain magnetic resonance imaging (Figure 1F) showed cerebellar atrophy; susceptibility weighted imaging sequencing (Figure 1A) demonstrated iron deposition in basal ganglia and substantia nigra.
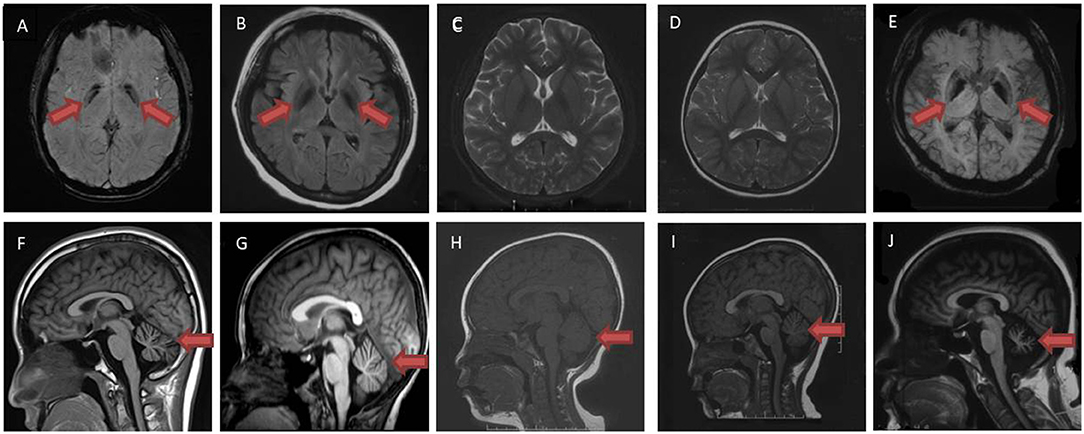
Figure 1. Brain magnetic resonance imaging examination of patient A II-1 (A,F), A II-2 (B,G), B-II 1 (C,H), B-II 2 (D,I) and C-II 1 (E,J). Susceptibility weighted imaging sequences (A,E) and T2 flair sequences (B) demonstrated iron deposition in bilateral globus pallidus (red arrow). No iron deposition was shown in T2 sequence (C,D) in patient B-II 1, B-II 2. T1 sequences demonstrated cerebellar atrophy in patient A II-1, A II-2, B-II 1, B-II 2, C-II 1 (F–J) (red arrow).
Patient A-II 2 was an 8-year-old male who had ran slower than his peers since childhood. He walked slowly and could not run when he was 6, then he had difficulties squatting at the age of 7. Physical examinations showed his muscle tone in both lower limbs were significantly increased such that he got spasticity. His hand rotations were clumsy. In addition his deep tendon reflexes were increased. Bilateral Babinski's signs and Rossolimo's signs were positive. Brain magnetic resonance imaging (Figure 1G) showed cerebellar atrophy, and T2 Flair sequencing (Figure 1B) demonstrated iron deposition in basal ganglia and substantia nigra. The symptoms slightly improved after L-dopa administration.
Patient B-II 1, the proband in family B, was a 10-year-old female. Her four limbs became rigid and she had difficulties walking since age 3. When she was 5, she got dysarthria and mental decline, and she got dysphagia at 7. Physical examinations revealed dystonia in her face, neck, and hands. Her cognitive function was impaired. Muscle tone in lower limbs was increased. Deep tendon reflexes were increased in lower limbs. Left Babinski's sign was positive. Brain magnetic resonance imaging showed no obvious iron deposition in T2 sequence (Figure 1C), and cerebellar atrophy was specific (Figure 1H).
Patient B-II 2, the little sister of patient 3, was a 5-year-old female. When she was 2, she got dysarthria and mental decline. She walked in a weird posture at 3 years old and the symptoms progressed to rigidity in 4 limbs, which stopped her from walking at 4.5 years old. She and her elder sister felt no improvement after the administration of L-dopa. She got large amplitude dystonia in the trunk and neck during physical examination, but no dysphagia. Her cognitive function was impaired. Sensory examinations were normal. In lower limbs, muscle tone and deep tendon reflexes were increased. Bilateral Babinski's signs were positive. Brain magnetic resonance imaging when she was 3 years old showed no iron deposition in T2 sequence (Figure 1D), but there was cerebellar atrophy in T1 sequence (Figure 1I).
Patient C-II 1 was a 39 year-old female who was asymptomatic until 29 years old, when she began to walk unstably and felt weakness and rigidity in her lower limbs. The symptoms progressed to frequent falls and partly improved after the administration of L-dopa. Physical examinations revealed dysarthria and her cognitive functions were impaired. She got incomplete ophthalmoplegia. Muscle strength was decreased in her lower limbs, but was normal in upper limbs. Muscle tone in four limbs was increased, especially in upper limbs. Hand rotations were clumsy. Deep tendon reflexes in both lower limbs disappeared. Bilateral Babinski's signs were negative, while bilateral Chaddock signs were positive. Kayser–Fleischer rings were not observed, and ceruloplasmin was normal. Brain magnetic resonance imaging (Figure 1J) showed cerebellar atrophy, while susceptibility weighted imaging (Figure 1E) demonstrated iron deposition in bilateral globus pallidus. Electroencephalogram showed Paroxysmal slow waves in all leads. FDG PET/CT showed glucose metabolism was slightly reduced in her left temporal lobe and moderately to severely reduced in bilateral cerebellar hemisphere. Brain magnetic resonance imaging (Figure 1J) showed cerebellar atrophy, while susceptibility weighted imaging sequencing (Figure 1E) demonstrated iron deposition in basal ganglia and substantia nigra.
Genetic Analysis
In patient A-II 1 and patient A-II 2, the whole genome sequencing revealed compound heterozygous variants of c.2120dupA resulting in p.Asn707fs transition, and c.2071C>G resulting in p.Arg691Gly transition (Figures 2A,B). Their father and mother were proven heterozygous for c.2071C>G and c.2120dupA separately. The variant c.2120dupA and c.2071C>G have not been reported in ClinVar and Pubmed Database.
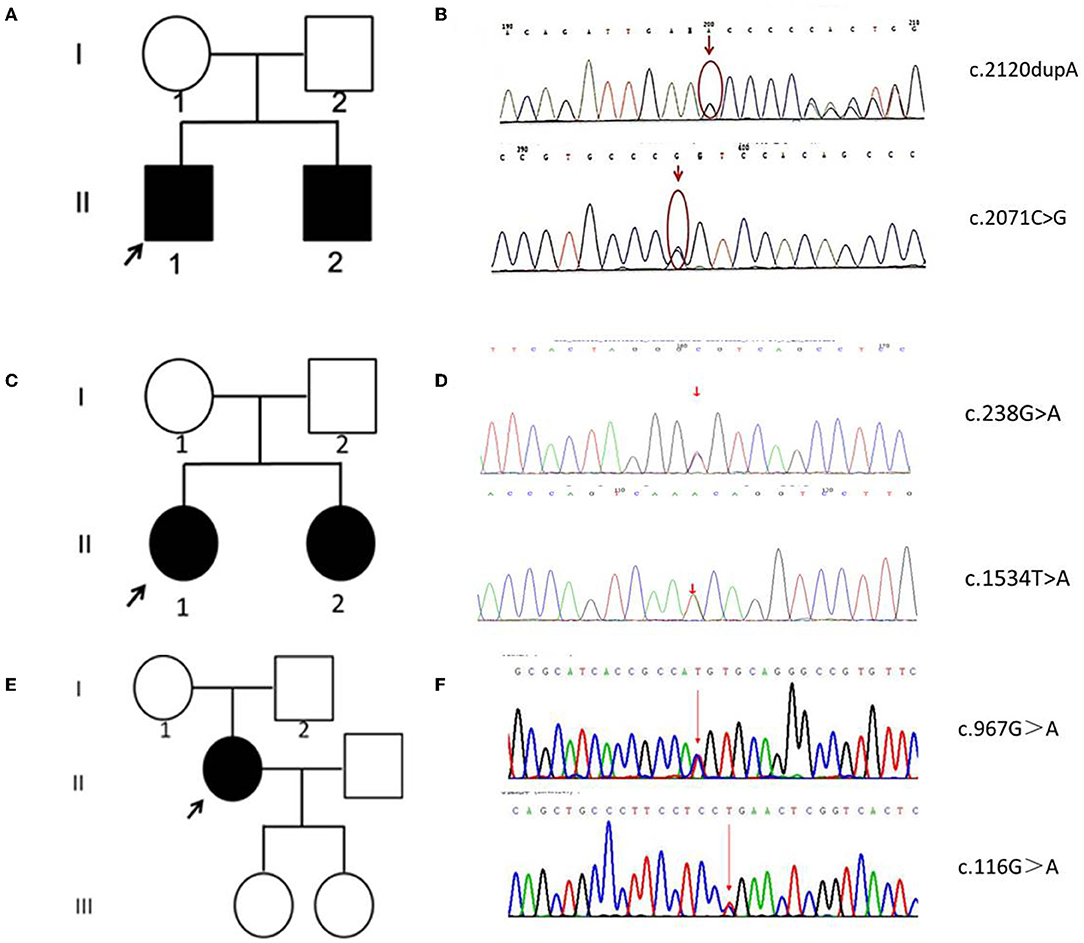
Figure 2. Family pedigrees and Sanger sequencing data of five patients. (A) The pedigree chart of family A. (B) The Sanger sequence chromatogram of family A. (C) The pedigree chart of family B. (D) The Sanger sequence chromatogram of family B. (E) The pedigree chart of family C. (F) The Sanger sequence chromatogram of family C. Open symbol: unaffected; filled symbol: affected; black arrows: proband. Red arrows: mutation sites.
In patient B-II 1 and patient B-II 2, the whole genome sequencing revealed compound heterozygous variants of c.238G>A resulting in p.Ala80Thr transition, and c.1534T>A resulting in p.Phe512Ile (Figures 2C,D). Their father and mother were proven heterozygous for c.238G>A and c.1534T>A separately. The variant c.1534T>A has not been reported in ClinVar and Pubmed Database, while c.238G>A has been reported in INAD.
In patient C-II 1, the whole genome sequencing revealed compound heterozygous variants of c.967G>A resulting in p.Val323Met transition, and c.116G>A resulting in p.Arg39Gln transition (Figures 2E,F). Their father and mother were proven heterozygous for c.967G>A and c.116G>A separately. The variant c.967G>A has not been reported in ClinVar and Pubmed Database, while c.116G>A has been reported in INAD.
The predicting results of ACMG standard, Mutation Taster, and SIFT of the four novel sites were listed in the Supplementary Table.
Literature Review
For ANAD, we screened 9 references for eligibility from PubMed search results (n = 3) and their reference lists. We found 7 references, and predefined data were extracted. The summary of the demographic, clinical, genetic, and radiological data of the reported 21 patients in literature (Table 2) (4, 5, 14–18) was shown in Table 1, where among the 21 ANAD patients, 86% (18/21) have iron deposition, 62% (13/21) parkinsonism, 71% (15/21) cerebellar atrophy, 67% (14/21) pyramidal signs, 52% (11/21) cognitive impairment, and 29% (6/21) nystagmus or Strabismus. Genetic analysis had been conducted on eleven of the reported ANAD patients, and c.238G>A was found to be the most popular mutation, which was detected in two unrelated pedigrees.
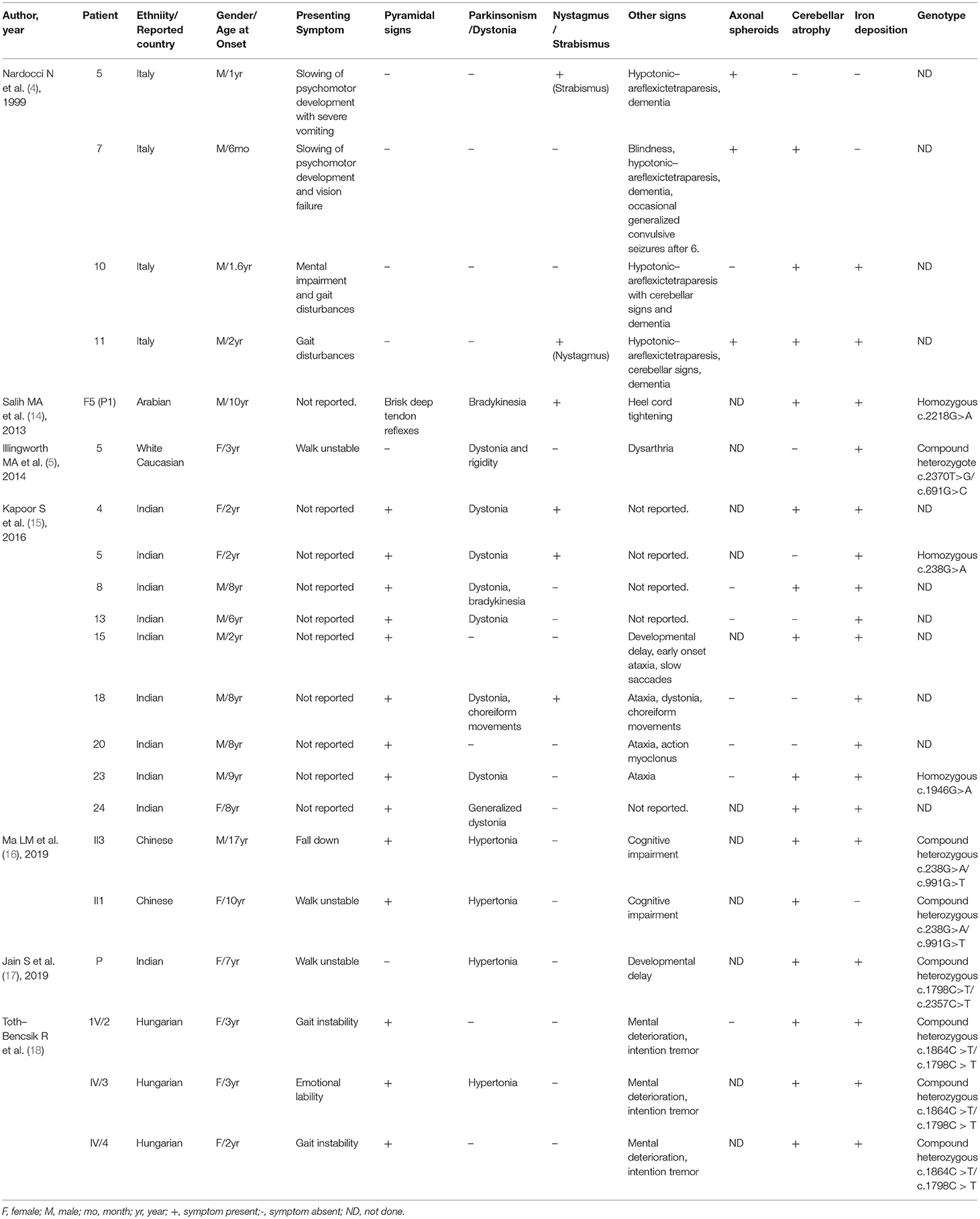
Table 2. Summary of demographic and clinical findings of reported 21 ANAD patients.
For adult onset ANAD, we screened 24 references for eligibility from PubMed search results (n = 19) and their reference lists. The demographic, clinical, genetic, and radiological data of the 34 patients described in those articles (Table 3) (6, 8, 19–29) were summarized Table 3. Childhood onset cases and cases that didn't conform to the phenotype of AREP were excluded. Among the 34 adult onset PLA2G6-related parkinsonism patients, 76% (26/34) had pyramidal signs, 38% (13/34) displayed cerebellar atrophy, and 26% (9/34) iron deposition. Genetic analysis had been conducted on all of the 34 patients, and c. 2222G > A was found to be the most popular mutation. Eleven patients had c. 2222G > A homozygous mutation. Moreover, patients with homozygous 2222G > A mutation showed mostly neuropsychiatric symptoms as initial symptoms. And, inconsistent with previous reports (30), c.991G > T mutation was almost exclusively found in Chinese patients.
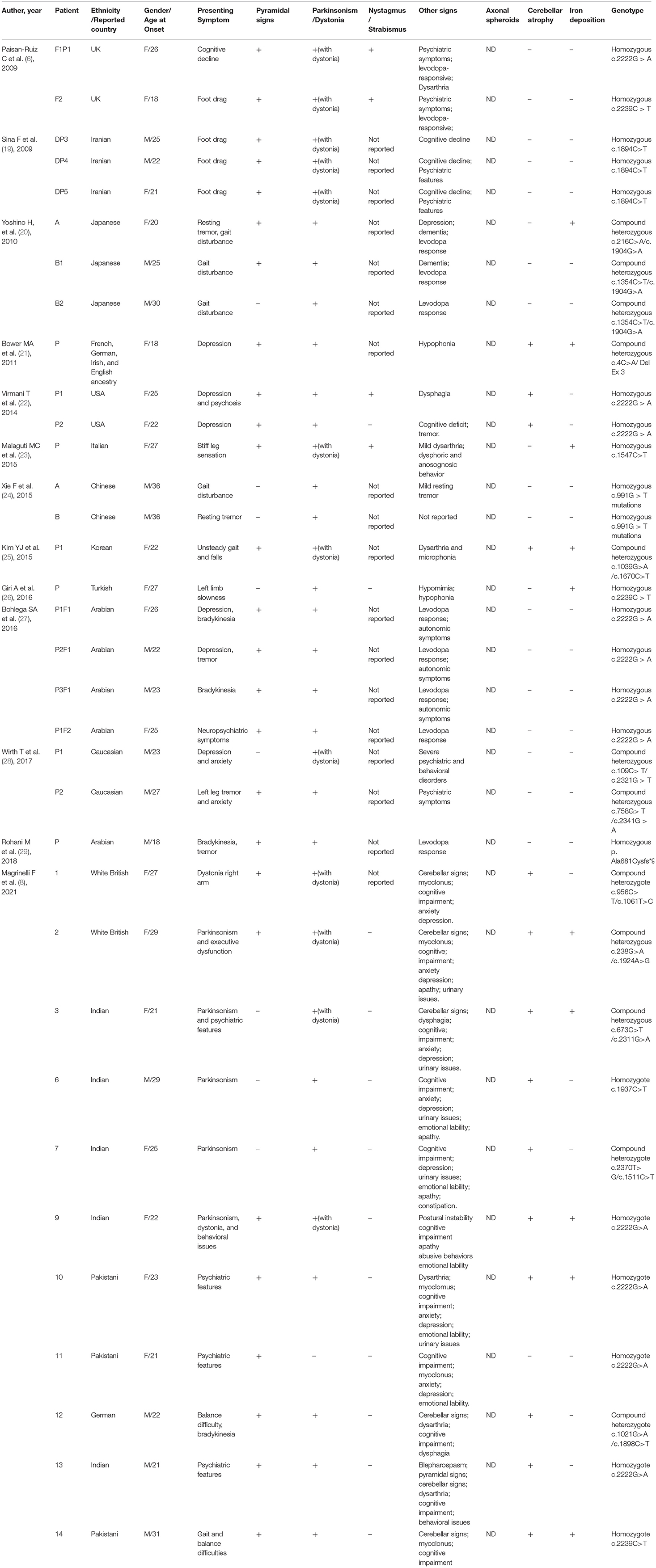
Table 3. Summary of demographic and clinical findings of reported 34 adult onset PLA2G6-related parkinsonism patients.
Discussion
PLA2G6-associated neurodegeneration (PLAN) is a heterogenous group of neurodegenerative diseases caused by mutations of PLA2G6. According to the onset age and clinical features, PLAN can be mainly classified into four subtypes, i.e., infantile onset INAD, childhood onset ANAD, adult-onset DP, and AREP. Clinically, PLAN belongs to complex movement disorders, since extrapyramidal manifestations (parkinsonism/dystonia) are shared by all subtypes, with the combination of psychomotor deterioration, ataxia, pyramidal signs, etc. In this study, four patients (Patient 1–4) presenting psychomotor decline, rigidity, ataxia, bradykinesia, dystonia, and autosomal recessive inheritance, with their onset ages between 2–6 years old, were clinically diagnosed as ANAD. The symptoms of Patient 5 were consistent with AREP based on adolescent-onset parkinsonism accompanied with pyramidal signs. Magnetic resonance imaging showed cerebellar atrophy and iron deposition in globus pallidus in all five patients. Finally next generation sequencing confirmed pathogenic variants in PLA2G6, and excluded other possible genetic causes. PLA2G6 gene is located on chromosome 22q13.1, with 17 exons included. To date, the Human Gene Mutation Database (http://www.hgmd.cf.ac.uk/ac/gene.php?gene =PLA2G6) has reported a total of 238 PLA2G6 mutations, including 178 missense mutations, 14 splicing-site mutations, 32 small indels, 11 gross deletions, and 4 gross insertions. The reported variants are widely distributed in the gene. Here, 6 variants of PLA2G6 were identified, which included 4 novel and 2 known variants (https://preview.ncbi.nlm.nih.gov/clinvar/variation/437465). Co-segregation analysis on the parents of the patients confirmed all five patients carrying compound heterozygous mutations. Among the 4 novel variants, c.2120dupA was predicted to be pathogenic and c.2071C>G, c.967G>A, and c.1534T>A, to be likely pathogenic, in accordance with ACMG standards (11).
PLA2G6 gene encodes a protein named Ca2+-independent phospholipase A2 (iPLA2), an enzyme causing the release of free fatty acids and lysophospholipids by catalyzing the hydrolysis of the sn-2 fatty acyl bond of phospholipids (31), which plays an essential role in phospholipid remodeling, signal transduction, cell proliferation, endoplasmic reticulum stress-mediated apoptosis, and ferroptosis (32, 33). Multiple studies have verified that iPLA2 deficiency may alter membrane permeability, fluidity, and ion homeostasis, thereby causing mitochondrial abnormalities (24). The PLA2G6 (iPLA2-VIA) protein harbors various domains from the N-terminus to C-terminus, including ankyrin repeats, patatin-like phospholipase, and two binding sites for calmodulin (https://www.uniprot.org/). Previous studies have also revealed that mutation sites in different domains may lead to different enzyme activities, which could be a key factor for the phenotypic variability (34). For example, mutations in the ankyrin repeat domain causes significant decreases in enzymatic activity, and results in INAD phenotype (34). In this study, there was only one mutation located in patatin-like phospholipase domain among the four mutation sites of the four ANAD patients, while the others were located in regions between domains; the simplex AREP patient had 1 mutation in the ankyrin repeats, and another one was in regions between domains (Figure 3), which was in line with results of other studies. Most of the mutations reported in these studies were not located in domains, which may explain the reason why the patients in question presented milder clinical features than INAD.
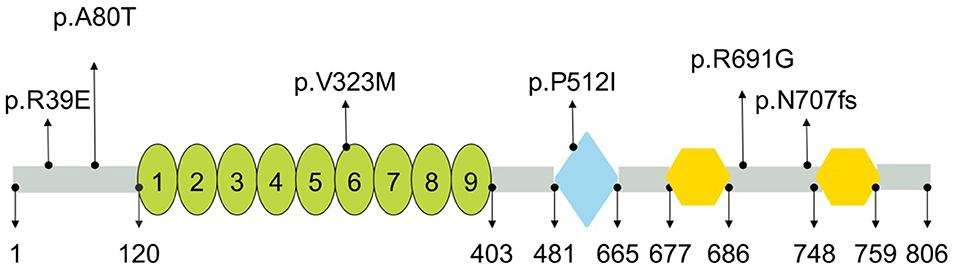
Figure 3. Schematic representation of PLA2G6 and location of mutations identified in present study. PLA2G6 consisted of nine ankyrin repeats (oval), patatin-like phospholipase (diamond), and two binding sites for calmodulin (hexagon). Numbers shown below were the amino acid positions (https://www.uniprot.org/).
The case series in this study highlighted the classical, atypical clinical, and neuroimaging features of PLA2G6 gene related ANAD/AREP. A review of all ANAD patients reported so far showed that the most common features of ANAD were iron deposition of globus pallidus, cerebellar atrophy, pyramidal signs, and parkinsonism, accounting for 86, 71, 67, and 62%, respectively, which also appeared in four ANAD patients in this study. Notably, all of the four ANAD patients in question presented prominent spastic rigidity in lower limbs in their early stage, making them misdiagnosed as complex hereditary spastic paraplegia (HSP) for a long time. Burcak Ozes et al. reported two affected Turkish siblings presenting HSP with PLA2G6 c.2239C>T homozygous missense (35). And C19ORF12 (SPG43) gene was also reported to be responsible for both HSP and NBIA (36). However, patients in question also showed significant cerebellar signs, cognitive impairment, parkinsonism, and dystonia, indicating they are different from those suffering from pure HSP. Importantly, imaging showed 3 of the 5 patients had iron deposition and cerebellar atrophy, which was consistent with the literature where 86% reported ANAD cases got iron deposition. As the disease developed, the iron deposition and cerebellar atrophy became severe, when physicians were reminded to follow up magnetic resonance imaging when considering the diagnosis of PLAN. However, inconsistent with literature review, c.238G>A was proven to be the most popular mutation in ANAD patients and had been reported in two Chinese patients and one Indian patient.
Literature review of PLA2G6-AREP patients showed that, in addition to early-onset parkinsonism, pyramidal signs (76%), cerebellar atrophy (38%), and iron deposition of globus pallidus (26%) are also common in these patients. Different from those with ANAD, the present Patient 5, who was sporadic, showed dopa-responsive parkinsonism with additional pyramidal signs. Neither of the mutations had been reported in PLA2G6-related parkinsonism, making it hard to distinguish AREP from other early onset genetic parkinsonism only by these clinical features. Again, iron deposition and cerebellar atrophy on magnetic resonance imaging could give helpful clues to the underlying genetic causes, while NGS could help make a definite diagnosis.
In conclusion, great clinical heterogeneity was shown in PLA2G6-neurodegeneration, making these patients prone to misdiagnosis or delay diagnosis. Brain magnetic resonance imaging could provide helpful diagnostic clues and next generation sequencing played an important role in diagnosis. The four novel pathogenic variants (c.2120dupA, c.2071C>G, c.967G>A, c.1534T>A) identified in this study enriched the mutational spectrum of PLA2G6-associated neurodegeneration.
Data Availability Statement
The datasets which support the findings of the present article are included in the manuscript/supplementary material. Queries should be directed to the corresponding author(s).
Ethics Statement
The studies involving human participants were reviewed and approved by the Peking University First Hospital Ethics Committee. Written informed consent to participate in this study was provided by the participants' legal guardian/next of kin.
Author Contributions
Study conception and design: YW, YJ, RL, ZW, YY, WS, and HJ. Analysis and interpretation of results: YW, ZX, CL, and KD. Draft manuscript preparation: YW, HJ, CL, ZX, and WS. All authors reviewed the results and approved the final version of the manuscript.
Funding
This work was supported by the National Natural Science Foundation of China [Grant Number 82071306].
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher's Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fneur.2022.922528/full#supplementary-material
References
1. Khateeb S, Flusser H, Ofir R, Shelef I, Narkis G, Vardi G, et al. PLA2G6 mutation underlies infantile neuroaxonal dystrophy. Am J Hum Genet. (2006) 79:942–8. doi: 10.1086/508572
2. Guo YP, Tang BS, Guo JF. PLA2G6-Associated Neurodegeneration (PLAN): review of clinical phenotypes and genotypes. Front Neurol. (2018) 9:1100. doi: 10.3389/fneur.2018.01100
3. Gregory A, Westaway SK, Holm IE, Kotzbauer PT, Hogarth P, Sonek S, et al. Neurodegeneration associated with genetic defects in phospholipase A(2). Neurology. (2008) 71:1402–9. doi: 10.1212/01.wnl.0000327094.67726.28
4. Nardocci N, Zorzi G, Farina L, Binelli S, Scaioli W, Ciano C, et al. Infantile neuroaxonal dystrophy: clinical spectrum and diagnostic criteria. Neurology. (1999) 52:1472–8. doi: 10.1212/WNL.52.7.1472
5. Illingworth MA, Meyer E, Chong WK, Manzur AY, Carr LJ, Younis R, et al. PLA2G6-associated neurodegeneration (PLAN): further expansion of the clinical, radiological and mutation spectrum associated with infantile and atypical childhood-onset disease. Mol Genet Metab. (2014) 112:183–9. doi: 10.1016/j.ymgme.2014.03.008
6. Paisan-Ruiz C, Bhatia KP, Li A, Hernandez D, Davis M, Wood NW, et al. Characterization of PLA2G6 as a locus for dystonia-parkinsonism. Ann Neurol. (2009) 65:19–23. doi: 10.1002/ana.21415
7. Shi CH, Tang BS, Wang L, Lv ZY, Wang J, Luo LZ, et al. PLA2G6 gene mutation in autosomal recessive early-onset parkinsonism in a Chinese cohort. Neurology. (2011) 77:75–81. doi: 10.1212/WNL.0b013e318221acd3
8. Magrinelli F, Mehta S, Di Lazzaro G, Latorre A, Edwards MJ, Balint B, et al. Dissecting the phenotype and genotype of PLA2G6-related parkinsonism. Mov Disord. (2021) 37:148-161. doi: 10.1002/mds.28807
9. Erro R, Balint B, Kurian MA, Brugger F, Picillo M, Barone P, et al. Early ataxia and subsequent parkinsonism: PLA2G6 mutations cause a continuum rather than three discrete phenotypes. Mov Disord Clin Pract. (2017) 4:125–8. doi: 10.1002/mdc3.12319
10. Xie Z, Hou Y, Yu M, Liu Y, Fan Y, Zhang W, et al. Clinical and genetic spectrum of sarcoglycanopathies in a large cohort of Chinese patients. Orphanet J Rare Dis. (2019) 14:43. doi: 10.1186/s13023-019-1021-9
11. Richards S, Aziz N, Bale S, Bick D, Das S, Gastier-Foster J, et al. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American college of medical genetics and genomics and the association for molecular pathology. Genet Med. (2015) 17:405–24. doi: 10.1038/gim.2015.30
12. Schwarz JM, Rodelsperger C, Schuelke M, Seelow D. Mutation taster evaluates disease-causing potential of sequence alterations. Nat Methods. (2010) 7:575–6. doi: 10.1038/nmeth0810-575
13. Adzhubei IA, Schmidt S, Peshkin L, Ramensky VE, Gerasimova A, Bork P, et al. A method and server for predicting damaging missense mutations. Nat Methods. (2010) 7:248–9. doi: 10.1038/nmeth0410-248
14. Salih MA, Mundwiller E, Khan AO, AlDrees A, Elmalik SA, Hassan HH, et al. New findings in a global approach to dissect the whole phenotype of PLA2G6 gene mutations. PLoS ONE. (2013) 8:e76831. doi: 10.1371/journal.pone.0076831
15. Kapoor S, Shah MH, Singh N, Rather MI, Bhat V, Gopinath S, et al. Genetic analysis of PLA2G6 in 22 indian families with infantile neuroaxonal dystrophy, atypical late-onset neuroaxonal dystrophy and dystonia parkinsonism complex. PLoS ONE. (2016) 11:e0155605. doi: 10.1371/journal.pone.0155605
16. Ma LM, Zhao J, Shi YY, Chen ZZ, Ren ZX, Zhang JW, et al. [PLA2G6 compound complicated mutation in an atypical neuroaxonal dystrophy pedigree]. Zhonghua Yi Xue Za Zhi. (2019) 99:354–8. doi: 10.3760/cma.j.issn.0376-2491.2019.05.007
17. Jain S, Bhasin H, Romani M, Valente EM, Sharma S. Atypical childhood-onset neuroaxonal dystrophy in an Indian girl. J Pediatr Neurosci. (2019) 14:90–3. doi: 10.4103/jpn.JPN_91_18
18. Toth-Bencsik R, Balicza P, Varga ET, Lengyel A, Rudas G, Gal A, et al. New insights of phospholipase A2 associated neurodegeneration phenotype based on the long-term follow-up of a large Hungarian family. Front Genet. (2021) 12:628904. doi: 10.3389/fgene.2021.628904
19. Sina F, Shojaee S, Elahi E, Paisan-Ruiz C. R632W mutation in PLA2G6 segregates with dystonia-parkinsonism in a consanguineous Iranian family. Eur J Neurol. (2009) 16:101–4. doi: 10.1111/j.1468-1331.2008.02356.x
20. Yoshino H, Tomiyama H, Tachibana N, Ogaki K, Li Y, Funayama M, et al. Phenotypic spectrum of patients with PLA2G6 mutation and PARK14-linked parkinsonism. Neurology. (2010) 75:1356–61. doi: 10.1212/WNL.0b013e3181f73649
21. Bower MA, Bushara K, Dempsey MA, Das S, Tuite PJ. Novel mutations in siblings with later-onset PLA2G6-associated neurodegeneration (PLAN). Mov Disord. (2011) 26:1768–9. doi: 10.1002/mds.23617
22. Virmani T, Thenganatt MA, Goldman JS, Kubisch C, Greene PE, Alcalay RN, et al. Oculogyric crises induced by levodopa in PLA2G6 parkinsonism-dystonia. Parkinsonism Relat Disord. (2014) 20:245–7. doi: 10.1016/j.parkreldis.2013.10.016
23. Malaguti MC, Melzi V, Di Giacopo R, Monfrini E, Di Biase E, Franco G, et al. A novel homozygous PLA2G6 mutation causes dystonia-parkinsonism. Parkinsonism Relat Disord. (2015) 21:337–9. doi: 10.1016/j.parkreldis.2015.01.001
24. Xie F, Cen Z, Ouyang Z, Wu S, Xiao J, Luo W. et al. Homozygous pD331Y mutation in PLA2G6 in two patients with pure autosomal-recessive early-onset parkinsonism: further evidence of a fourth phenotype of PLA2G6-associated neurodegeneration. Parkinsonism Relat Disord. (2015) 21:420–2. doi: 10.1016/j.parkreldis.2015.01.012
25. Kim YJ, Lyoo CH, Hong S, Kim NY, Lee MS. Neuroimaging studies and whole exome sequencing of PLA2G6-associated neurodegeneration in a family with intrafamilial phenotypic heterogeneity. Parkinsonism Relat Disord. (2015) 21:402–6. doi: 10.1016/j.parkreldis.2015.01.010
26. Giri A, Guven G, Hanagasi H, Hauser AK, Erginul-Unaltuna N, Bilgic B, et al. PLA2G6 mutations related to distinct phenotypes: a new case with early-onset parkinsonism. Tremor Other Hyperkinet Mov. (2016) 6:363. doi: 10.5334/tohm.289
27. Bohlega SA, Al-Mubarak BR, Alyemni EA, Abouelhoda M, Monies D, Mustafa AE, et al. Clinical heterogeneity of PLA2G6-related Parkinsonism: analysis of two Saudi families. BMC Res Notes. (2016) 9:295. doi: 10.1186/s13104-016-2102-7
28. Wirth T, Weibel S, Montaut S, Bigaut K, Rudolf G, Chelly J, et al. Severe early-onset impulsive compulsive behavior and psychosis in PLA2G6-related juvenile Parkinson's disease. Parkinsonism Relat Disord. (2017) 41:127–9. doi: 10.1016/j.parkreldis.2017.05.014
29. Rohani M, Shahidi G, Vali F, Lang AE, Slow E, Gahl WA, et al. Oculogyric crises in PLA2G6 associated neurodegeneration. Parkinsonism Relat Disord. (2018) 52:111–2. doi: 10.1016/j.parkreldis.2018.03.010
30. Chu YT, Lin HY, Chen PL, Lin CH. Genotype-phenotype correlations of adult-onset PLA2G6-associated neurodegeneration: case series and literature review. BMC Neurol. (2020) 20:101. doi: 10.1186/s12883-020-01684-6
31. Ji Y, Li Y, Shi C, Gao Y, Yang J, Liang D, et al. Identification of a novel mutation in PLA2G6 gene and phenotypic heterogeneity analysis of PLA2G6-related neurodegeneration. Parkinsonism Relat Disord. (2019) 65:159–64. doi: 10.1016/j.parkreldis.2019.04.002
32. Winstead MV, Balsinde J, Dennis EA. Calcium-independent phospholipase A(2): structure and function. Biochim Biophys Acta. (2000) 1488:28–39. doi: 10.1016/S1388-1981(00)00107-4
33. Sun WY, Tyurin VA, Mikulska-Ruminska K, Shrivastava IH, Anthonymuthu TS, Zhai YJ, et al. Phospholipase iPLA2beta averts ferroptosis by eliminating a redox lipid death signal. Nat Chem Biol. (2021) 17:465–76. doi: 10.1038/s41589-020-00734-x
34. Wada H, Yasuda T, Miura I, Watabe K, Sawa C, Kamijuku H, et al. Establishment of an improved mouse model for infantile neuroaxonal dystrophy that shows early disease onset and bears a point mutation in Pla2g6. Am J Pathol. (2009) 175:2257–63. doi: 10.2353/ajpath.2009.090343
35. Ozes B, Karagoz N, Schule R, Rebelo A, Sobrido MJ, Harmuth F, et al. PLA2G6 mutations associated with a continuous clinical spectrum from neuroaxonal dystrophy to hereditary spastic paraplegia. Clin Genet. (2017) 92:534–9. doi: 10.1111/cge.13008
Keywords: PLA2G6 gene, iron deposition, atypical neuroaxonal dystrophy, parkinsonism, neurogenetic
Citation: Wan Y, Jiang Y, Xie Z, Ling C, Du K, Li R, Yuan Y, Wang Z, Sun W and Jin H (2022) Novel PLA2G6 Pathogenic Variants in Chinese Patients With PLA2G6-Associated Neurodegeneration. Front. Neurol. 13:922528. doi: 10.3389/fneur.2022.922528
Received: 18 April 2022; Accepted: 20 June 2022;
Published: 13 July 2022.
Edited by:
Pramod Kumar Pal, National Institute of Mental Health and Neurosciences (NIMHANS), IndiaReviewed by:
Chin-Hsien Lin, National Taiwan University Hospital, TaiwanVikram Venkappayya Holla, National Institute of Mental Health and Neurosciences, India
Copyright © 2022 Wan, Jiang, Xie, Ling, Du, Li, Yuan, Wang, Sun and Jin. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Wei Sun, sunweibjmu@163.com; Haiqiang Jin, jhq911@bjmu.edu.cn
†These authors contributed equally to this work