 Yong Mo1†
Yong Mo1† Weilin Xu2†
Weilin Xu2† Kaijing Fu1
Kaijing Fu1 Hainan Chen1Jing Wen3Qianrong Huang1Fangzhou Guo1
Hainan Chen1Jing Wen3Qianrong Huang1Fangzhou Guo1 Ligen Mo1*
Ligen Mo1* Jun Yan1*‡
Jun Yan1*‡- 1Department of Neurosurgery, Guangxi Medical University Cancer Hospital, Nanning, China
- 2Department of Neurosurgery, Second Affiliated Hospital, School of Medicine, Zhejiang University, Hangzhou, China
- 3Department of Rheumatism, First Affiliated Hospital of Guangxi Medical University, Nanning, China
Stroke is the leading cause of disability and death worldwide, with ischemic stroke occurring in ~5% of the global population every year. Recently, many studies have been conducted on the inflammatory response after stroke. Microglial/macrophage polarization has a dual function and is critical to the pathology of ischemic stroke. Microglial/macrophage activation is important in reducing neuronal apoptosis, enhancing neurogenesis, and promoting functional recovery after ischemic stroke. In this review, we investigate the physiological characteristics and functions of microglia in the brain, the activation and phenotypic polarization of microglia and macrophages after stroke, the signaling mechanisms of polarization states, and the contribution of microglia to brain pathology and repair. We summarize recent advances in stroke-related microglia research, highlighting breakthroughs in therapeutic strategies for microglial responses after stroke, thereby providing new ideas for the treatment of ischemic stroke.
Introduction
Stroke is the leading cause of mortality worldwide (1, 2) with poor curative effect, high lethality, and poor prognosis. Among all types of stroke, ischemic stroke caused by the occlusion of blood vessels represents the majority (3). Previous research has indicated that brain injury is caused not only by the hematoma mass effect and potential hematoma expansion (which are the main causes of primary brain injury) but also by secondary brain injury (SBI) (4). Cerebral ischemia can lead to a series of pathological processes including excitatory toxicity, calcium overload, oxygen free radical damage, inflammatory responses, necrosis/apoptosis, and blood–brain barrier (BBB) destruction, which ultimately lead to irreparable neuronal damage (5). It is now proposed that injury after stroke is a complex pathophysiological process involving several genes and signaling pathways. The BBB is important, and its permeability appears to follow a heterogeneous pattern of different stroke stages associated with different biological substrates. In the hyperacute phase, sudden hypoxia damages the BBB, leading to cytotoxic edema and increased permeability; in the acute phase, neuroinflammatory responses exacerbate BBB damage, leading to higher permeability and subsequent risk, which can be stimulated by reperfusion therapy; and in the subacute phase (1–3 weeks), repair mechanisms, particularly neovascularization, occur. BBB leakage occurs in immature vessels, but this permeability is associated with improved clinical recovery. In the chronic phase (>6 weeks), an increase in the BBB restoration factor causes the barrier to begin to reduce its permeability (6). Manipulation of microglial polarization is a potential treatment strategy for patients with ischemic stroke, but small- and medium-sized glial cells in the potential molecular mechanisms of the polarization in ischemic stroke are still controversial. Despite the simplicity of the experiment, more work and clinical trials are needed to fully understand the mechanisms of microglial polarization (7). Evaluating the best time to intervene with microglia and monocyte/macrophage therapeutic strategies against ischemic stroke, as well as determining how to stimulate cells and to polarize their states, as well as the role of microRNAs (miRNA) and transplanted stem cells in mediating microglial activation and polarization during cerebral ischemia, are all important topics for future research (8, 9). Targeting specific miRNAs may provide major restorative therapy, and microglia-based therapy for ischemic stroke may become a future research area.
Recent studies have shown that there are still no effective therapeutic targets to improve the neurological function of patients after stroke, and potential treatment methods for SBI remain a hot point of research. Currently, an effective treatment for ischemic stroke is mainly intravenous thrombolysis and mechanical thrombectomy. However, these treatment options are limited by the recommended treatment window (10, 11). In addition, a series of reperfusion injuries caused by inflammation and oxidative stress may occur after ischemia-reperfusion (12); oxidative stress can induce inflammation (13, 14). There is increasing evidence that, during cerebral infarction, persistent neuroinflammation damages neurons and the BBB, leading to tissue destruction and impaired function (15–17). Neuroinflammation plays a crucial role in ischemic stroke-induced brain injury and affects disease prognosis. Future research will focus on controlling stroke-induced inflammation by targeted drugs and will be challenging.
Microglia are the permanent substantial macrophages in the central nervous system (CNS), and activated microglia typically behave “amoeba-like,” primed for action (18). Several findings showed that almost five different types of microglia morphology were identified in control and experimental status epilepticus (SE) tissues, and were categorized as follows: (1) ramified; (2) hypertrophic; (3) bushy; (4) amoeboid; and (5) rod-shaped (19) (Figure 1). Microglial polarization plays a major role in promoting brain injury and nerve recovery (20). As the main source of inflammatory cells in ischemic brain injury, microglia play a key role in the inflammatory response after stroke (21). After stroke, microglia are polarized to the classical pro-inflammatory type (M1-like) or the alternative protective type (M2-like) under optimal conditions (8). Classical M1-like microglia are related to the induction of pro-inflammatory molecules, while other M2-like microglial activations are related to neuroprotection (22). In this review, advances in microglia and ischemic stroke, including the dual functions of phenotypic polarization of microglia/macrophages and polarization-related signaling pathways, have been studied. Future ischemic stroke treatments may target microglial polarization in the future.

Figure 1. Representative images of microglial/macrophage cells (brown) with different morphologicalphenotypes observed in the control and status epilepticus (SE) groups, including (1) ramified, (2) hypertrophic, (3) bushy (cell indicated by arrows), (4) amoeboid, and (5) rod-shaped. Images were taken from the hippocampus of control or SE animals. Nissl-stained nuclei are indicated in blue [the figure is reproduced (19)].
Origin and function of microglial cells
Derived from primitive yolk sac progenitor cells, microglia are a type of fixed macrophages (9). The number of microglia showed a steady increase in the first 2 weeks after birth, and gradually decreased to 50% of the level at birth between 3 to 6 weeks later, after which the density gradually stabilized. A decrease in the rate of proliferation accompanied by an increase in apoptosis results in a decrease in the overall number of microglia, and mature microglia maintain their numbers in the CNS by self-renewal (23, 24). In the CNS, microglial cells in the brain of healthy adults are renewed to maintain their number and local expansion (25). In the physiological state, microglial cells present a typical branch-like state of small cell body and long branches and are referred to as “resting microglial cells.” The protrusions have high mobility and can carry out extensive and continuous monitoring of the surrounding environment. In the pathological state, microglial cells are changed from the resting state to the active state. Polarization refers to the fact that microglia are affected by exogenous substances to achieve a specific phenotype, and there are one or more molecular markers and significant changes in molecular distribution (26). M1-like and M2-like microglia are essential in tissue damage and repair, respectively. Polarization of M1-like and M2-like microglia is also considered a functional manifestation of CNS disease, which is specifically manifested in the release of CNS disease-related inflammatory factors and the role of neuroinflammatory responses (27–29).
Microglial polarization
Polarization of the M1-like phenotype
M1-like microglia can secrete a variety of pro-inflammatory factors and chemokines, which can cause a neuroinflammatory response and induce neuronal apoptosis (30). As for the classic excitation type, it is mainly induced by interferon-γ (INF-γ), lipopolysaccharides (LPS), and tumor necrosis factor-alpha (TNF-α), which is characterized by the production of several pro-inflammatory cytokines, such as interleukin-1β (IL-1β), IL-6, stromal cell-derived factor-1 (SDF-1/CXCL12), IL-1β, IL-12, and IL-23, and can be detected using cell surface markers such as cluster of differentiation 16 (CD16), CD32, major histocompatibility complex class II (MHCII), CD86, TNF-α, inducible nitric oxide synthase (iNOS), etc. During ischemia/hypoxia, nuclear factor-κB (NF-κB) is activated in microglia and transferred from the cytoplasm to the nucleus. This activates the release of pro-inflammatory cytokines that lead to SBI (31–33), such as IL-1β, IL-6, TNF-α, and iNOS. In addition, TNF-α secreted by M1-like microglia was identified to increase endothelial necrosis and BBB leakage after ischemic stroke in middle cerebral artery occlusion (MCAO) model mice. This further promotes neuroinflammation and cerebral edema, leading to poor outcomes (34, 35). Classically activated microglia can perform pro-inflammation, phagocytosis, cytotoxicity, present antigens, and kill intracellular pathogens to maintain the homeostasis of the microenvironment (36, 37). Notably, the M1-like phenotype of microglia is usually associated with protection during the early acute stages of infection, but it can also be detrimental to the host in case of its persistence for a longer time. Changes in the expression of corresponding proteins also follow a similar course node (38–40). If homeostasis is destroyed or stimulation persists, inflammatory cascades can be induced, resulting in the massive release of inflammatory factors and neurotoxic substances, aggravating the inflammatory response, and inducing neuronal death (36, 37) (Table 1).

Table 1. Characteristics of M1 microglia and M2 microglia in ischemic stroke.
Polarization of the M2-like phenotype
According to the unique functions of microglia/macrophages and their gene expression profiles, there are four different types of polarized activation states of M2-like activated phenotype: M2a microglia, M2b microglia, M2c microglia, and M2d microglia (41). There are many markers in M2-like microglia, such as CD206, increased arginase 1 (Arg-1), TGF-β, insulin growth factor 1 (IGF-1), IL-10, and others, and they secrete anti-inflammatory cytokines and neurotrophic factors, such as IL-10β, brain- and glial cell-derived neurotrophic factors, and Arg-1, the expression of factors such as IGF-1, thereby inhibiting inflammation (42), involved in tissue repair, cell debris removal, tissue remodeling, the provision of nutritional factors, and the maintenance of tissue dynamics after infection or injury (43, 44). In general, M2-like microglia can be identified by CD206 and Arg1, IGF-1, among other markers (45). M2a is produced by IL-4 and IL-13 stimulation and inhibits NF-κB signal transduction and the anti-inflammatory phenotype of activated B cells. Moreover, M2a is involved in parasite immunity, T helper 2 cell recruitment, tissue repair, and growth stimulation. M2b is produced by stimulating immune complexes and LPSs to secrete anti-inflammatory cytokines (such as IL-10, MHC II, and co-stimulatory CD86). This subset exhibits both pro- and anti-inflammatory features and is associated with adaptive immunity. M2c is activated by IL-10 and TGF- β. Arg-1, CD163, and CD206 are the markers of M2c cells, which mainly function in scavenging cell debris during the repair process and is related to immunosuppression and tissue remodeling (46–48). M2c is different from the M2-like subtypes described earlier and is produced by activating the activation state of adenosine A2a receptor (A2aR) in M1 pro-inflammatory cells. M2d is different from M2-like subtypes described earlier and is produced by activating the activation state of A2aR in M1 pro-inflammatory cells (49). Peroxisome proliferator-activated receptor γ (PPARγ), a transcription factor with anti-inflammatory properties, is activated in microglia and translocated from the nucleus to the cytoplasm under ischemia/hypoxia conditions (50). This leads to the activation of M2-like microglia, which release anti-inflammatory cytokines and improve stroke outcomes. In addition, Choi et al. (51) demonstrated that M2-like microglia promoted the proliferation and neuronal differentiation of nerve stem/progenitor cells in the ipsilateral subventricular region after ischemic stroke by upregulating the TGF-α expression level, which may provide an effective treatment for neurogenesis (Figure 2).
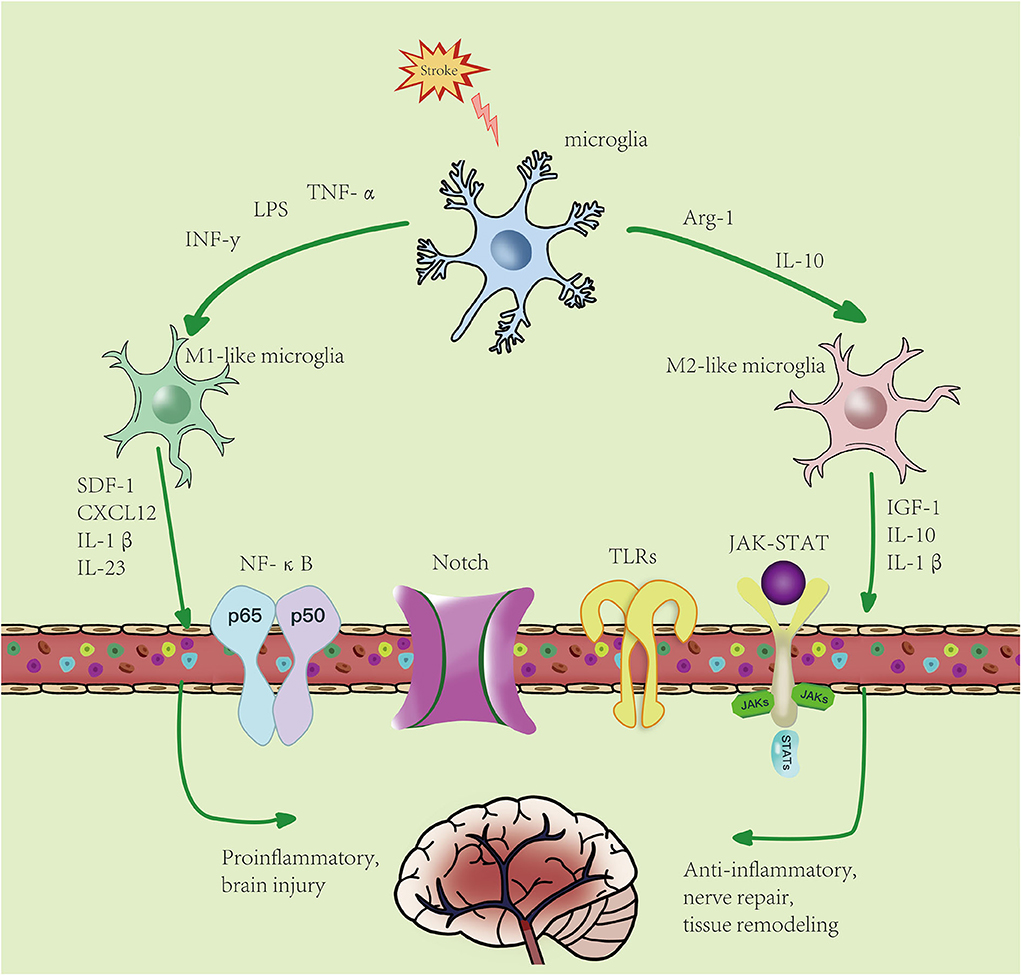
Figure 2. Microglial polarization after stroke: microglial activation is divided into two phenotypes: M1-like and M2-like microglia. M1 microglia can be induced by lipopolysaccharides (LPS), interferon-γ (IFN-γ), etc., resulting in an increase in pro-inflammatory factors. M2-like microglia can be induced by IL-4, IL-13, etc., resulting in an increase in anti-inflammatory factors. Activated microglia pass through NF-κB, JAK-STAT, Notch, TLRs, and other signaling pathways. M1-like type promotes the inflammatory response and kills intracellular substances, while M2-like type plays an anti-inflammatory, neuroprotective, and repairing role in tissues.
The transition between M1 and M2
A shift from M2 to M1 has been observed in models of traumatic brain injury and ischemic stroke; however, it re mains to be determined whether this transformation is caused by phenotypic transformation of individual microglia or by the migration and infiltration of M2-like microglia (52). Studies have shown that in ischemic stroke, activated microglia express M2-like microglia markers in the acute phase. However, within ~1 week, a gradual transition to M1-like phenotypes occurs and persists for several weeks after injury, this phenotypic transition may be due to the recruitment of M1-like microglia to the injury site and the transformation of locally activated microglia from M2 to M1 cells (53). Therefore, selective neuro-immunomodulatory therapies, which largely focus on suppressing M1-like phenotypes and shifting microglia from the M1-like phenotype to the M2-like phenotype, have been proposed as neuroprotective strategies for stroke (54). Furthermore, an experiment showed that the silencing of NF-κB p65 downregulated the expression of M1-like biomarkers and promoted the expression of M2-like biomarkers in the in vitro and in vivo model of cerebral ischemia (55).
Recently, a mouse model of transient focal cerebral ischemia has been used to study the temporal dynamics of microglial/macrophage polarization after stroke. Research results suggest that microglia/macrophages respond dynamically to ischemic injury, experiencing an early “healthy” M2-like phenotype, followed by a transition to a “sick” M1-like phenotype (56). In vivo temporal distribution of increased iNOS and chitinase-like protein 3 (Chil3; Ym 1) promoter activity in the mouse brain (57). The relatively low iNOS signal in healthy brain increased ~3-fold within 3 days of stroke induction, whereas Ym1 signal reached a maximum at 11–13 days after stroke induction and then declined over the following week. Hu et al. found an early increase of iNOS messenger RNA (mRNA) levels as well as of other pro-inflammatory markers, such as CD16, CD32, CD86, and CD11b beginning at 3 dps and continuing up to 14 dps, with the exception of CD86. They also reported that chitinase-3-like protein 3 (Ym1/2) mRNA levels peaked on day 3 and then declined up to 14 dps (56).
Mechanism of microglial polarization in ischemic stroke
Signaling pathways that regulate microglial polarization
NF-κB signaling pathways
Studies have shown that in mammals there are all five members of the NF-κB family including NF-κB1 (p105/p50), RelA (P65), and NF-κB2 (p100/p52), which are composed of homo- and heterodimers. In contrast to the c-NF-κB dimers, the p65/p50 heterodimer RelB is the most classical form of existence. It exists in most cell types and plays the most important role as an effective transcription factor. The activation of NF-κB is required for transcriptional induction of many pro-inflammatory mediators, such as IL-6, iNOS, intercellular adhesion molecule 1 (ICAM1), matrix metalloproteinase 9 (MMP-9), and cyclooxygenase-2 (COX-2), which are involved in innate immunity. A previous study demonstrated that the NF-κB signaling pathway was overactivated in microglia after ischemic stroke. Therefore, the activation of NF-κB was responsible for the polarization of M1 and M2 in microglia (58, 59). In p50 KO mice, NF-κB activation exacerbated ischemic neuronal damage, especially in microglia. NF-κB p65 and p50 form heterodimers to initiate pro-inflammatory responses, thereby enhancing M1-like activation and attenuating microglial M2-like responses (60). The activation of the NF-κB signaling pathway promotes the conversion of microglia to M1-like type, and effective inhibition of the activation of the signaling pathway is more conducive to the conversion of microglia to M2-like type (61). Studies have shown that the inhibition of the NF-κB signaling pathway or the expression of NF-κBp65 and IκBα or interference with the nuclear metastasis of NF-κB can inhibit the activation of microglia and the expression of the M1-like phenotype, reduce the expression of inflammatory factors such as IL-1β, IL-6, TNF-α in microglia, and have neuroprotective effects (62, 63). Therefore, the suppression of neuroinflammation and the amelioration of brain injury by inhibiting the expression and activity of NF-κB in microglia after ischemic stroke has become a breakthrough target for therapeutic strategies (Table 2).
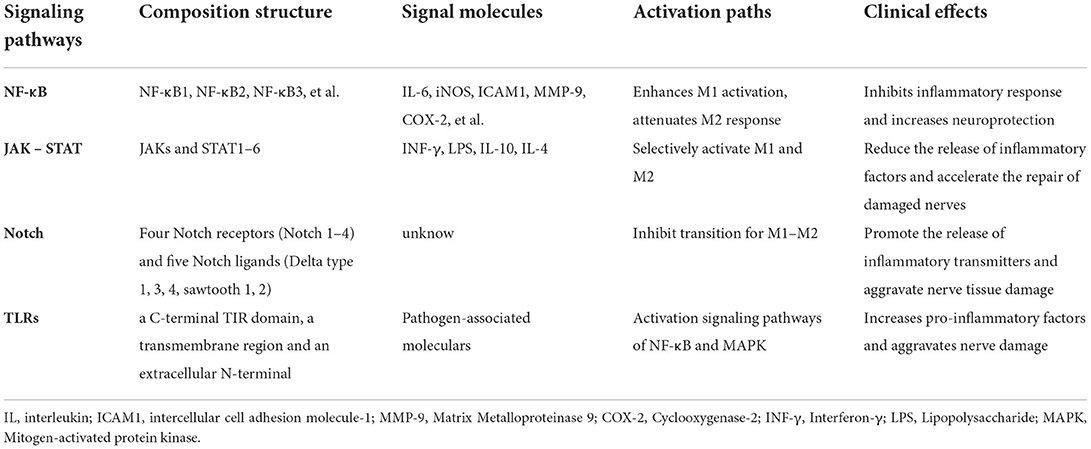
Table 2. Overview of signaling pathways and their associated molecules.
Janus kinase/signal transducer and activator of transcription pathway
Signal transducer and activator of transcription (STAT) is phosphorylated by Janus kinase (JAK), dimerized, and then transported to the nucleus through the nuclear membrane to regulate the expression of related genes. This pathway is termed the JAK/STAT signaling pathway (64). STAT plays a key role in signal activation and transcription. The STAT family in the cytoplasm is a downstream target of JAKs, which is one of the most crucial cytokine-activated transcription factors in the process of immune response. It is composed of seven members, namely STAT1, STAT2, STAT3, STAT4, STAT5A, STAT5B, and STAT6 (65). STAT1, STAT3, and STAT6 members of the STAT family are involved in the polarization of microglia. IFN-γ can induce microglia to polarize toward M1-like type through the STAT1/STAT3 pathway, and release inflammatory factors such as iNOS/nitrous oxide (NO) at a higher level than normal cells (66). STAT1 responds to M1-like microglial polarization signals (INF-γ and LPS), while STAT3 and STAT6 are selectively activated by M2-like microglial polarization cytokines (IL-10, IL-4, etc.). Thus, the release of inflammatory factors can be reduced and the injured nerve repair can be accelerated (67). Considering the data related to JAK/STAT and autoimmune diseases, this method is extremely attractive to the pharmaceutical industry, which is also one of its goals.
Notch signaling pathway
The extracellular domain of Notch is composed of epidermal growth factors (EGFs) like repeats, the number of which varies among species and different Notch receptors. Two functional domains are present in the extracellular region, the ligand-binding domain (EGF 11–12), which mediates the interaction with ligands, and the Abruptex domain (EGF 24–29), whose function remains unclear. The extracellular region is followed by the negative regulatory region (NRR), which masks a cleavage site (S2) important for Notch activation, the heterodimerization domain (HD), and the transmembrane spanning region of the receptor (64, 68). The Notch signal pathway is an important signal transduction pathway that begins with the binding of the Notch receptor and ligand and then forms a transcriptional activation complex through interactions with transcription factors, which activates the target genes of the transcriptional suppressor family (e.g., HES, HEY, NERP, etc.) to play a transcriptional inhibitory role. In mammals, there are four Notch receptors (Notch 1–4) and five Notch ligands (Delta type 1, 3, 4, sawtooth 1, 2). Notch signaling can regulate the differentiation and development of cells, tissues, and associated cells. These cells include neurons, oligodendrocytes, astrocytes, and microglia. In the pathological state, the Notch pathway can promote the release of inflammatory transmitters and aggravate tissue damage by activating microglia and inhibiting the transformation of M1-like to M2-like microglia (69). It was confirmed in experiments of BV2 microglia-related cells that the release of inflammatory mediators from M1-like microglia decreased and converted to M2-like microglia after the use of a Notch signaling antagonist. Meanwhile, there is an increase in anti-inflammatory cytokines released by M2-like microglia. This confirms the involvement of the Notch pathway in the inflammatory response following microglial activation (70).
Toll-like receptor signaling pathway
Toll-like receptors, named after the Toll proteins in Drosophila melanogaster (13), are a class of inherent immune recognition receptors that detects microbial pathogens associated with molecular patterns to induce an immune response (71). TLR is expressed on neurons in glial cells (microglia, astrocytes, and oligodendrocytes), the CNS, and the peripheral nervous system (PNS) (72). TLRs are type I transmembrane proteins composed of a C-terminal TIR domain, a transmembrane region, and an extracellular N-terminal. An extracellular N-terminal mainly recognizes extracellular pathogens and tissue damage signals. TLR4 in human microglia can recognize pathogen-associated molecular models and activate nonspecific immunity in ischemic brain injury via a myeloid differentiation factor pathway, and both NF-κB and mitogen-activated protein kinase (MAPK) signaling pathways are activated and participate in the inflammatory response. TLR4 receptors can repeatedly recognize different pathogen-related molecular patterns through extracellular leucine (73), and ultimately lead to the production of NF-κB and an increase in pro-inflammatory factors, and the secretion of serotonin may aggravate nerve damage (74).
Regulatory mechanisms of microglial polarization
In addition to the influence of the abovementioned signaling pathways, there are also several regulatory mechanisms of microglial polarization: transcription factors, the regulation of gene expression, ion channels, and autophagy (75). Firstly, transcription factors, it was found that nuclear factor erythroid 2-related factor 2 (Nrf2) activation reduced the expression levels of reactive oxygen species (ROS), nucleotide-binding oligomerization domain- (NOD-) like receptor family Pyrin domain 3 (NLRP3), and IL-1 β in BV2 microglia, and played a protective role after ischemic stroke (12). In acute ischemic stroke, PPARγ is activated to directly reduce tissue damage by inhibiting the NF-κB pathway, reducing inflammation, and stimulating the Nrf2/ARE axis to reduce oxidative stress (76). IL-4 produced by neurons was determined to bind to IL-4 receptors expressed on microglia surfaces and activate M2-like microglia by modulating the PPARγ signaling pathway to reduce ischemic brain injury (77). Second, ion channel expression changes in response to voltage and pH gradients in the microenvironment, thereby inducing intracellular signal transduction. Currently, the two important ion channels Hv1 and Kv1.3 are closely related to microglial polarization. Studies have shown that Hvl can aggravate brain injury by increasing the expression levels of ROS and pro-inflammatory cytokines produced by M1-like microglia. However, it remains unclear whether Hv1 affects the polarization of M2-like microglia. The Kv1.3 inhibitor 5-(4-phenoxybutyl-psoralen) pSORalen (PAP-1) decreased the polarization of M1-like microglia and the expression level of pro-inflammatory cytokines. This also suggests that Kv1.3 may be one of the major mediators of the polarization of M1-like microglia (78, 79). Third, miRNA-155 and miRNA-124 in gene expression regulators are closely related to microglial polarization in ischemic stroke. The expression levels of miRNA-155 were significantly increased in LPS-activated microglia, which might target the inhibition of cytokine signaling to trigger M1-like microglia-mediated inflammation and aggravate brain injury (33). miRNA-124 induces neuroprotection and functional improvement by regulating M2-like microglial polarization in ischemic stroke (80). Finally, autophagy is a cellular metabolic pathway by which damaged organelles and misfolded proteins are degraded and recycled to maintain cellular homeostasis. Studies have shown that autophagy is activated in neurons, endothelial cells, microglia, and other brain cells in ischemic stroke and that interference with autophagy can aggravate brain injury. Studies have shown that autophagy may stimulate the transformation of microglia to the M1-like phenotype, thereby exacerbating cerebral ischemia. However, the role of autophagy in microglial polarization in ischemic stroke requires further investigation (81).
Treatment targets of microglial polarization in ischemic stroke
Currently, there are numerous studies on stroke treatment, including extensive research on small molecules. For example, the small molecule miRNA-124 can regulate the activation state of microglia/macrophages, thereby improving stroke recovery (82). Chemokine-like factor 1 (CKLF1) is an important mediator that skews microglia/macrophages toward the M1-like phenotype in the early stage of cerebral ischemic injury, and targeting CKLF1 may also be a novel approach for IS treatment (83). Cytokine IL-4 may improve long-term neurological outcomes after stroke by inducing the M2-like phenotype in microglia/macrophages (84). In addition, the current treatment of SBI after stroke has become more promising. The inhibition of the inflammatory response promotes M2-like microglial polarization, reduces M1-like activation, and promotes the clearance of hematoma, thus playing a therapeutic role. The role of microglia-mediated inflammation in the undamaged CNS remains a hot spot of research. The development of multi-treatment targets is likely to become an important direction for the development of new therapeutic targets for ischemic stroke (Table 3).

Table 3. Therapeutic goals and related mechanisms of drugs in ischemic stroke.
Minocycline, an antibiotic of the tetracycline family, is known for its anti-inflammatory effects in neurological disorders, and has been reported to potentially improve functional recovery in ischemic stroke (85, 86). Minocycline can cross the BBB, accumulate in CNS cells, and inhibit microglia activation and proliferation, as well as MMP concentration and activity (85). Anti-inflammatory effects of minocycline have been demonstrated in neurological diseases in experimental models of ischemia, traumatic brain injury, and neuropathic pain as well as in Alzheimer's disease, Parkinson's disease, multiple sclerosis, Huntington's disease, amyotrophic lateral sclerosis, and several neurodegenerative diseases including spinal cord injury (87–93) (Table 4). Various experimental animal models and clinical trials have shown that minocycline can effectively cross the BBB, lead to the production of ROS and apoptosis by inhibiting the activation of microglia, and play a neuroprotective role against nervous system injury (94, 95). It has been reported that minocycline partially suppressed the production of inflammatory molecules (IL-6, TNF-A, and IL-1B) induced by LPS in peripheral monocytes by inhibiting nuclear translocation of NF-κB (96). In addition, experiments have shown that minocycline regulates M1/M2 microglial polarization through the STAT1/STAT6 pathway, reduces the production of M1-like polarization genes and enhances the expression of M2-like polarization genes by regulating STAT1 and STAT6 signaling, thus achieving the treatment of ischemia (97). Minocycline can effectively inhibit the diffusion of the neuroinflammatory cytokines IL-1β and NO, thereby reducing the brain water content and alleviating early brain edema and brain injury in the early stages of stroke by reducing the M1-like polarization of microglia (98).
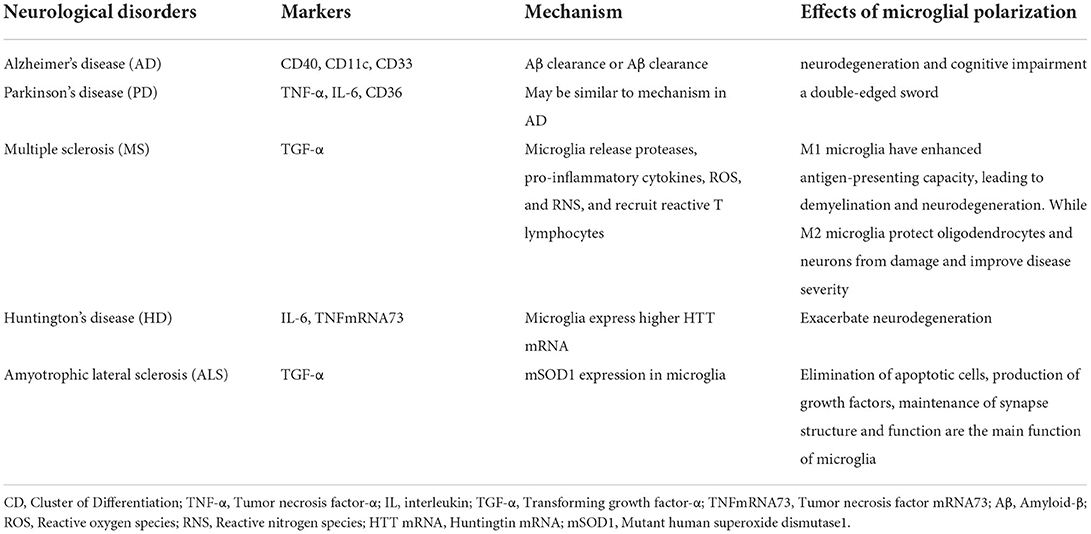
Table 4. Summary of microglial polarization in neurological disorders.
Metformin, a well-known AMP-activated protein kinase (AMPK) activator, can be used in chronic post-stroke therapy to promote functional recovery after experimental stroke. Experimental evidence suggests that post-stroke metformin treatment results in a long-term elevation of M2-like signature gene expression and the suppression of M1-like signature gene expression. Metformin enhances the M2-like polarized function of microglia/macrophages involved in tissue repair and is beneficial in ischemic stroke, thereby improving post-stroke brain function recovery. Therefore, promoting the functional phenotype of microglia tilted toward M2-like polarization via AMPK activation after stroke emerges as a novel therapeutic strategy for stroke (99). Animal experiments have also shown that, in chronic ischemic stroke, metformin pretreatment inhibits the inflammatory pathway mediated by brain NF-κB, which is accompanied by a reduction in pro-inflammatory cytokines, such as TNF-α, IL-1β, IL-6, and others. This significantly reduces infarct volume and improves neurological deficits while also promoting tissue repair (100).
Peroxisome proliferator-activated receptor, a ligand-activated transcription factor belonging to the nuclear receptor superfamily, has been shown to orchestrate the macrophage phenotype switch, thus leading to the inhibition of inflammation and tissue repair. Its agonist, rosiglitazone, promotes the polarization of microglia toward M2-like phenotype with a direct and indirect effect on the white matter. It may improve white matter integrity after stroke. In addition, it can reduce cerebral infarct size and edema in different animal models of stroke through the nuclear receptor PPAR-γ, thereby protecting neurons, reducing oxidative stress, attenuating excitotoxicity, and contributing to long-term recovery from stroke (44, 101). Unfortunately, the mechanism by which rosiglitazone improves stroke prognosis is still unknown and needs to be further explored.
Dexmedetomidine (DEX) is an α-adrenergic receptor agonist with different properties, including sedative, anxiolytic, antisympathetic, and analgesic, widely used as an adjuvant in the perioperative period (102). In a model of LPS-induced inflammation, many previous studies have reported that DEX can diminish neuroinflammation in the mouse brain and to modulate cytokine-associated changes in sickness behavior (103). In addition, it has been experimentally confirmed that in microglia, LPS induces a pro-inflammatory response through activation of the MAPK and NF-κB pathways (104). However, it remains to be further explored whether microglial polarization after stroke exerts neuroprotective effects through the abovementioned pathways, and the effect of clinically relevant concentrations of DEX on microglial M1/M2 polarization remains to be further investigated.
Etifoxine, a benzoxazine-based anti-anxiety compound, is an exogenous ligand of the 18-kDa translocator protein (TSPO) with high affinity (105). TSPO principally affects microglia (106). Experiments have confirmed that etifoxine can reduce brain damage and inflammation after stroke, reduce leukocyte infiltration, control the production of pro-inflammatory cells in microglia, improve BBB integrity, and reduce nerve cell death during hemorrhagic stroke. Thus, the function of repairing damaged nerves is achieved. In addition to finding reduced brain inflammation and altered microglial responses following etifoxine treatment, this still has been confirmed in mouse experiments. Together, these results demonstrate the therapeutic potential of etifoxine to reduce neurological deficits and infarct volume, limit brain inflammation, and provide protection against ischemia/reperfusion (I/R) injury. However, the mechanism of its effect needs further investigation (107).
In recent years, some traditional Chinese medicine formulations have also greatly improved post-stroke symptoms by promoting M2-like polarization. A novel resveratrol oligomer, named malibatol A, can reduce infarct size after MCAO in ischemic stroke (108) and increases M2-like microglial polarization markers such as CD206 and YM-1, producing anti-inflammatory protection. Its neuroprotective effect is largely associated with PPARγ-dependent activation of M2-like microglial polarization (44). The results of another similar study showed that the pharmacologically active component (hyperforin) of the medicinal plant Hypericum perforatum (St. John's wort) reduced infarct volume and induced microglia from M1-like to M2-like phenotype via the inhibition of IL-17A (109). Meisoindigo, a second-generation derivative of indierythroid (110), modulates microglial/macrophage polarization by inhibiting TLR4/NF-κB, reducing ischemic stroke-induced brain injury in vivo and in vitro. In addition, Meisoindigo has a neuroprotective effect in the ischemic brain. This protective effect is attributed to the inhibition of NOD-like receptor protein 3 (NLRP3) (111) inflammasome activation and the prevention of microglia/macrophages from the pro-inflammatory M1-like phenotype to the protective M2-like phenotype to relieve the inflammation in the brain (112, 113). If the mechanism of action of these drugs is understood accurately, ischemic stroke will be treated better.
In addition, recent studies have shown that inflammasome inhibitors have also been crucial treatment targets of microglial polarization in ischemic stroke, and NLRP3 inflammasomes have been proven to play a role in ischemic stroke. JLX001, a novel compound structurally similar to cycloviral flavonoid D (CVB-D), inhibits the expression of NLRP3 and proteins associated with the NLRP3 inflammasome axis in vivo, promoting a transition to a microglial M2 phenotype, suggesting that JLX001 is a promising treatment for ischemic stroke (114). Treatment with the LPR3 inflammasome inhibitor tranilast reduces the expression of M1 markers and pro-inflammatory cytokines, while stimulating the expression of M2-like microglia markers, thereby ameliorating ischemic stroke (115). Acute treatment with NLRP3-specific drugs, such as MCC950, reduces neuroinflammation in IS and improves neurological outcomes after stroke (116). These outcomes may also provide targeted therapeutic opportunities for stroke-related inflammation; however, research on the role of inflammasome inhibitors against ischemic stroke is still a long way off.
In one study, it was found that the body's circulating steroid, dehydroepiandrosterone (DHEA), can penetrate the BBB, and the inflammatory response of microglia is regulated by phosphorylation of tropomyosin-associated kinase A (TrkA) and subsequent activation of pathways involving protein kinase B 1/protein kinase B 2 (Akt1/Akt2) cAMP response element-binding proteins. The latter induces the expression of Histone 3 Lysine 27 (H3K27) demethylase Jumonji D3 (Jmjd3), which enhances the polarization of M2-like microglia and may contribute to phenotype conversion in microglia. Thus, the expression of inflammation-related genes and microglial polarization were controlled, thus providing a platform for future therapeutic interventions in neuroinflammatory pathology (117, 118). Recent studies on single-cell analysis suggest that microglia are spatially and developmentally heterogeneous, have time-specific and region-dependent subtypes (119), and exhibit distinct genetic characteristics associated with changes in the CNS microenvironment (120). Heterogeneous subsets of microglia may provide a new pathway for microglia to target neuroinflammation (121, 122).
As mentioned earlier, TLR2/4 on microglia are important regulators of inflammatory responses during cerebral I/R. TLR2 and TLR4 were found to be significantly elevated during reperfusion injury, which was associated with the degree of ischemic injury and inflammation (123). TLR can also interact with endogenous and exogenous molecules released during ischemia to increase tissue damage. In addition, TLR2 and TLR4 activate different downstream inflammatory signaling pathways. The relationship between neurosteroids and TLR after ischemic events may serve as a therapeutic target for stroke therapy (124). Meanwhile, inflammatory signaling of TLR2 in the ischemic brain requires the scavenger receptor CD36. It is possible to suppress inflammation by not having this receptor. These findings suggest that the TLR2-CD36 complex can act as a sensor for ischemia at the onset of death signals and is critical for inflammatory responses (72). Therefore, TLR2 inhibition may be considered as the future treatment for ischemic stroke. TLR2 and TLR4 signaling appears to be important in controlling pathogenic immune responses after stroke, and estrogen, progesterone, and vitamin D3 all regulate TLR2 and TLR4 signaling, making them therapeutic options for stroke treatment (72).
In addition, after stroke, the immune response induces inflammation, which is one of the main reasons for the progression of ischemic injury. Microglia are involved in the inflammation of the brain and have a bone marrow source (125). A focus of current research is the trigger receptor 2 (TREM2) expressed on myeloid cells. TREM2 is a cell-surface receptor, a unidirectional transmembrane receptor, belonging to the immunoglobulin-like receptor superfamily. In the CNS, it is mainly expressed on microglia (126). The activation of trigger receptors expressed on TREM2 stimulates microglial phagocytic activity and downregulates the expression of TNF-α and inducible iNOS (8). TREM2 overexpression has been shown to have the opposite effect, while TREM2 deficiency attenuates microglial phagocytic activity and exacerbates ischemic damage in experimental stroke (127). TREM2 overexpression significantly inhibits the inflammatory response and neuronal apoptosis in cerebral I/R injury (125). Docosahexaenoic acid (DHA) treatment enhances mesencephalic astrocyte-derived neurotrophic factor (MANF), reduces the expression of TREM2 and ischemic brain damage, activates neurogenesis, and promotes functional recovery after experimental ischemic stroke (128). These findings suggest that TREM2 is an attractive target for microglia regulation in the treatment of ischemic stroke, which may be a promising therapeutic strategy (129).
Conclusion
After ischemic stroke, microglia polarize toward the classical pro-inflammatory type (M1-like) or the alternative protective type (M2-like) for a certain period of time and under different conditions, respectively, to promote intracranial inflammation, exert an anti-inflammatory and nerve-repairing effect, and repair damaged nerve functions. Microglia play a dual role in the deleterious effects of ischemic stroke, by both protecting and controlling polarization through multiple signaling pathways. With the deepening of research, research hot spots of targeted drugs for microglial polarization are increasing year by year, providing a new therapeutic strategy for the treatment of ischemic stroke. We are looking forward to more drugs that will benefit patients.
Limitation
Although the two microglial polarization states are well studied, some researchers in the field have questioned this and even suggested discontinuing the M1/M2 classification. The idea is that the current nomenclature derived from the study of peripheral macrophages is applied to microglia, they argue that M1/M2 class macrophage activation is useless to organize our thinking about microglia, frankly said to be destructive (130). With the deepening of research, it was found that because microglia and macrophages are homologous, many markers of these two types of cells are the same (131), so research continues to use microglia/macrophages. In addition, from 14 May 2016 to 30 May 2022, a PubMed search for “M1 M2 microglia” retrieved 1,121 articles, and the number is increasing year by year. After that, in addition to using the original M1/M2-like microglia classification, some scholars proposed additional refined phenotypes (M1 microglia, M2a microglia, M2b microglia, and M2c microglia). If the M1/M2-like microglia classification had some flaws in the research at the time, then with more research, the M1-like, M2a-like, M2b-like, and M2c-like classifications would be more of the morphology and function of microglia, which would be exactly what this review reflected.
As mentioned earlier, there are many therapeutic targets for ischemic stroke in the microglial polarization process, but there are still many problems to be studied and solved. Firstly, experimental models and basic experiments of stroke are needed, and more experimental model data must be collected and organized to confirm the authenticity of relevant views. Secondly, the transition factors between M1-like and M2-like microglia and their processes require further studies. Finally, the homeostatic regulatory mechanisms of microglial polarization are discussed in this review, and the range of potential therapy targets needs to be further explored. Then, in the future, numerous studies on microglial polarization must be conducted.
Author contributions
YM and WX were in charge of the literature search and manuscript writing. The content of this article was made by consensus of all the authors. All the listed authors contributed substantially, directly, and intellectually to the work and approved it for publication.
Funding
This study was funded by grants from the National Natural Science Foundation of China (No. 82060225), the Guangxi Natural Science Foundation (No. 2018GXNSFAA281151 and 2020GXNSFAA297154), and the Scientific Research Project of Guangxi Health Commission (No. S2018020).
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher's note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Abbreviations
SBI, Secondary brain injury; BBB, Blood–brain barrier; CNS, Central Nervous System; INF-γ, Interferon-γ; LPS, Lipopolysaccharide; TNF- α, Tumor necrosis factor-α; IL, interleukin; Ym1 [chitinase-like protein 3 (Chil3)], ; Ym1/2, chitinase-3-like protein 3; ICAM1, intercellular adhesion molecule 1; STAT, Signal transducer and activator of transcription; JAK, janus kinase; NO, nitrous oxide; SDF-1/CXCL12, Stromal cell-derived factor-1; CD, Cluster of Differentiation; MHC II, Major histocompatibility complex class II; Arg-1, Arginase-1; TGF-β, Transforming growth factor-β; IGF-1, Insulin growth factor 1; A2a R, Adenosine A2a Receptor; iNOS, Inducible nitric oxide synthase; MCAO, Middle cerebral artery occlusion; MMP-9, Matrix Metalloproteinase 9; COX-2, Cyclooxygenase-2; EGFs, Epidermal growth factors; NRR, Negative Regulatory Region; HD, Heterodimerization domain; TLRs, Toll-like receptors; MAPK, Mitogen-activated protein kinase; MMP, Metalloproteinase; PParγ, Peroxisome proliferator-activated receptor γ; PNS, peripheral nervous system; Nrf2, Nuclear Factor erythroid 2-Related Factor 2; PPAR, Peroxisome proliferator-activated receptor; DEX, Dexmedetomidine; TSPO, Translocator protein; MCAO, Middle cerebral artery occlusion; NLRP3, Nod-like receptor pyrin domain-containing protein 3; ICAM-1, Intercellular cell adhesion molecule-1; Aβ, Amyloid-β; ROS, Reactive oxygen species; RNS, Reactive nitrogen species; HTT, Huntingtin; mSOD1, Mutant human superoxide dismutase1; DHEA, Dehydroepiandrosterone; TrkA, tropomyosin-associated kinase A; Akt, protein kinase B; Jmjd3, Histone 3 Lysine 27 (H3K27) demethylase Jumonji D3; TREM2, trigger receptor 2; DHA, Docosahexaenoic acid; MANF, Mesencephalic astromarch-derived neurotrophic factor.
References
1. Kuramatsu JB, Huttner HB. Management of oral anticoagulation after intracerebral hemorrhage. Int J Stroke. (2019) 14:238–46. doi: 10.1177/1747493019828555
2. Ding Q, Liu S, Yao Y, Liu H, Cai T, Han L. Global, regional, and national burden of ischemic stroke, 1990-2019. Neurology. (2022) 98:e279–90. doi: 10.1212/WNL.0000000000013115
3. Liu Y, Deng S, Song Z, Zhang Q, Guo Y, Yu Y, et al. MLIF modulates microglia polarization in ischemic stroke by targeting eEF1A1. Front Pharmacol. (2021) 12:725268. doi: 10.3389/fphar.2021.725268
4. Chen S, Yang Q, Chen G, Zhang JH. An update on inflammation in the acute phase of intracerebral hemorrhage. Transl Stroke Res. (2015) 6:4–8. doi: 10.1007/s12975-014-0384-4
5. Xu Y, Hu Y, Xu S, Liu F, Gao Y. Exosomal microRNAs as potential biomarkers and therapeutic agents for acute ischemic stroke: new expectations. Front Neurol. (2021) 12:747380. doi: 10.3389/fneur.2021.747380
6. Bernardo-Castro S, Sousa JA, Bras A, Cecilia C, Rodrigues B, Almendra L, et al. Pathophysiology of blood-brain barrier permeability throughout the different stages of ischemic stroke and its implication on hemorrhagic transformation and recovery. Front Neurol. (2020) 11:594672. doi: 10.3389/fneur.2020.594672
7. Mao JH, Xu Y, Li BW, Yang YL, Peng Y, Zhi F. Microglia polarization in ischemic stroke: complex mechanisms and therapeutic interventions. Chin Med J. (2021) 134:2415–7. doi: 10.1097/CM9.0000000000001711
8. Kanazawa M, Ninomiya I, Hatakeyama M, Takahashi T, Shimohata T. Microglia and monocytes/macrophages polarization reveal novel therapeutic mechanism against stroke. Int J Mol Sci. (2017) 18:2135. doi: 10.3390/ijms18102135
9. Ma Y, Wang J, Wang Y, Yang G-Y. The biphasic function of microglia in ischemic stroke. Prog Neurobiol. (2017) 157:247–72. doi: 10.1016/j.pneurobio.2016.01.005
10. Ozaki T, Nakamura H, Kishima H. Therapeutic strategy against ischemic stroke with the concept of neurovascular unit. Neurochem Int. (2019) 126:246–51. doi: 10.1016/j.neuint.2019.03.022
11. Thiebaut AM, Gauberti M, Ali C, Martinez De Lizarrondo S, Vivien D, Yepes M, et al. The role of plasminogen activators in stroke treatment: fibrinolysis and beyond. Lancet Neurol. (2018) 17:1121–32. doi: 10.1016/S1474-4422(18)30323-5
12. Xu X, Zhang L, Ye X, Hao Q, Zhang T, Cui G, et al. Nrf2/ARE pathway inhibits ROS-induced NLRP3 inflammasome activation in BV2 cells after cerebral ischemia reperfusion. Inflamm Res. (2018) 67:57–65. doi: 10.1007/s00011-017-1095-6
13. Azedi F, Mehrpour M, Talebi S, Zendedel A, Kazemnejad S, Mousavizadeh K, et al. Melatonin regulates neuroinflammation ischemic stroke damage through interactions with microglia in reperfusion phase. Brain Res. (2019) 1723:146401. doi: 10.1016/j.brainres.2019.146401
14. Zhuang P, Wan Y, Geng S, He Y, Feng B, Ye Z, et al. Salvianolic acids for injection (SAFI) suppresses inflammatory responses in activated microglia to attenuate brain damage in focal cerebral ischemia. J Ethnopharmacol. (2017) 198:194–204. doi: 10.1016/j.jep.2016.11.052
15. Chamorro Á, Dirnagl U, Urra X, Planas AM. Neuroprotection in acute stroke: targeting excitotoxicity, oxidative and nitrosative stress, and inflammation. Lancet Neurol. (2016) 15:869–81. doi: 10.1016/S1474-4422(16)00114-9
16. Zhou S, Zhu W, Zhang Y, Pan S, Bao J. S100B promotes microglia M1 polarization and migration to aggravate cerebral ischemia. Inflamm Res. (2018) 67:937–49. doi: 10.1007/s00011-018-1187-y
17. Zhao H, Wan L, Chen Y, Zhang H, Xu Y, Qiu S. FasL incapacitation alleviates CD4(+) T cells-induced brain injury through remodeling of microglia polarization in mouse ischemic stroke. J Neuroimmunol. (2018) 318:36–44. doi: 10.1016/j.jneuroim.2018.01.017
18. Savage JC, Carrier M, Tremblay M-È. Morphology of microglia across contexts of health and disease. Methods Mol Biol. (2019) 2034:13–26. doi: 10.1007/978-1-4939-9658-2_2
19. Wyatt-Johnson SK, Herr SA, Brewster AL. Status epilepticus triggers time-dependent alterations in microglia abundance and morphological phenotypes in the hippocampus. Front Neurol. (2017) 8:700. doi: 10.3389/fneur.2017.00700
20. Xu J, Duan Z, Qi X, Ou Y, Guo X, Zi L, et al. Injectable gelatin hydrogel suppresses inflammation and enhances functional recovery in a mouse model of intracerebral hemorrhage. Front Bioeng Biotechnol. (2020) 8:785. doi: 10.3389/fbioe.2020.00785
21. Chen S, Dong Z, Cheng M, Zhao Y, Wang M, Sai N, et al. Homocysteine exaggerates microglia activation and neuroinflammation through microglia localized STAT3 overactivation following ischemic stroke. J Neuroinflammation. (2017) 14:187. doi: 10.1186/s12974-017-0963-x
22. Plastira I, Bernhart E, Goeritzer M, Reicher H, Kumble VB, Kogelnik N, et al. 1-Oleyl-lysophosphatidic acid (LPA) promotes polarization of BV-2 and primary murine microglia towards an M1-like phenotype. J Neuroinflammation. (2016) 13:205. doi: 10.1186/s12974-016-0701-9
23. Nikodemova M, Kimyon RS De I, Small AL, Collier LS, Watters JJ. Microglial numbers attain adult levels after undergoing a rapid decrease in cell number in the third postnatal week. J Neuroimmunol. (2015) 278:280–8. doi: 10.1016/j.jneuroim.2014.11.018
24. Tay TL, Carrier M, Tremblay M. Physiology of microglia. Adv Exp Med Biol. (2019) 1175:129–48. doi: 10.1007/978-981-13-9913-8_6
25. Ginhoux F, Garel S. The mysterious origins of microglia. Nat Neurosci. (2018) 21:897–9. doi: 10.1038/s41593-018-0176-3
26. Bilimoria PM, Stevens B. Microglia function during brain development: new insights from animal models. Brain Res. (2015) 1617:7–17. doi: 10.1016/j.brainres.2014.11.032
27. Casella G, Garzetti L, Gatta AT, Finardi A, Maiorino C, Ruffini F, et al. IL4 induces IL6-producing M2 macrophages associated to inhibition of neuroinflammation in vitro and in vivo. J Neuroinflammation. (2016) 13:139. doi: 10.1186/s12974-016-0596-5
28. Schneider UC, Davids AM, Brandenburg S, Müller A, Elke A, Magrini S, et al. Microglia inflict delayed brain injury after subarachnoid hemorrhage. Acta Neuropathol. (2015) 130:215–31. doi: 10.1007/s00401-015-1440-1
29. You W, Wang Z, Li H, Shen H, Xu X, Jia G, et al. Inhibition of mammalian target of rapamycin attenuates early brain injury through modulating microglial polarization after experimental subarachnoid hemorrhage in rats. J Neurol Sci. (2016) 367:224–31. doi: 10.1016/j.jns.2016.06.021
30. Wang G, Shi Y, Jiang X, Leak RK, Hu X, Wu Y, et al. HDAC inhibition prevents white matter injury by modulating microglia/macrophage polarization through the GSK3β/PTEN/Akt axis. Proc Natl Acad Sci USA. (2015) 112:2853–8. doi: 10.1073/pnas.1501441112
31. Yang S, Wang H, Yang Y, Wang R, Wang Y, Wu C, et al. Baicalein administered in the subacute phase ameliorates ischemia-reperfusion-induced brain injury by reducing neuroinflammation and neuronal damage. Biomed Pharmacother. (2019) 117:109102. doi: 10.1016/j.biopha.2019.109102
32. Weng L, Wu Z, Zheng W, Meng H, Han L, Wang S, et al. Malibatol A enhances alternative activation of microglia by inhibiting phosphorylation of Mammalian Ste20-like kinase1 in OGD-BV-2 cells. Neurol Res. (2016) 38:342–8. doi: 10.1080/01616412.2016.1174423
33. Zheng X, Huang H, Liu J, Li M, Liu M, Luo T. Propofol attenuates inflammatory response in LPS-activated microglia by regulating the miR-155/SOCS1 pathway. Inflammation. (2018) 41:11–9. doi: 10.1007/s10753-017-0658-6
34. Ji J, Xiang P, Li T, Lan L, Xu X, Lu G, et al. NOSH-NBP, a novel nitric oxide and hydrogen sulfide- releasing hybrid, attenuates ischemic stroke-induced neuroinflammatory injury by modulating microglia polarization. Front Cell Neurosci. (2017) 11:154. doi: 10.3389/fncel.2017.00154
35. Chen A-Q, Fang Z, Chen X-L, Yang S, Zhou Y-F, Mao L, et al. Microglia-derived TNF-α mediates endothelial necroptosis aggravating blood brain–barrier disruption after ischemic stroke. Cell Death Dis. (2019) 10:487. doi: 10.1038/s41419-019-1716-9
36. Zhang L, Zhang J, You Z. Switching of the microglial activation phenotype is a possible treatment for depression disorder. Front Cell Neurosci. (2018) 12:306. doi: 10.3389/fncel.2018.00306
37. Zheng J, Sun Z, Liang F, Xu W, Lu J, Shi L, et al. AdipoRon attenuates neuroinflammation after intracerebral hemorrhage through adipoR1-AMPK pathway. Neuroscience. (2019) 412:116–30. doi: 10.1016/j.neuroscience.2019.05.060
38. Lei B, Dawson HN, Roulhac-Wilson B, Wang H, Laskowitz DT, James ML. Tumor necrosis factor α antagonism improves neurological recovery in murine intracerebral hemorrhage. J Neuroinflammation. (2013) 10:103. doi: 10.1186/1742-2094-10-103
39. Liew HK, Pang CY, Hsu CW, Wang MJ Li TY, Peng HF, et al. Systemic administration of urocortin after intracerebral hemorrhage reduces neurological deficits and neuroinflammation in rats. J Neuroinflammation. (2012) 9:13. doi: 10.1186/1742-2094-9-13
40. Liu DL, Zhao LX, Zhang S, Du JR. Peroxiredoxin 1-mediated activation of TLR4/NF-κB pathway contributes to neuroinflammatory injury in intracerebral hemorrhage. Int Immunopharmacol. (2016) 41:82–9. doi: 10.1016/j.intimp.2016.10.025
41. Kolosowska N, Keuters MH, Wojciechowski S, Keksa-Goldsteine V, Laine M, Malm T, et al. Peripheral administration of IL-13 induces anti-inflammatory microglial/macrophage responses and provides neuroprotection in ischemic stroke. Neurotherapeutics. (2019) 16:1304–19. doi: 10.1007/s13311-019-00761-0
42. McGeer PL, McGeer EG. Targeting microglia for the treatment of Alzheimer's disease. Expert Opin Ther Targets. (2015) 19:497–506. doi: 10.1517/14728222.2014.988707
43. Qin C, Zhou LQ, Ma XT, Hu ZW, Yang S, Chen M, et al. Dual functions of microglia in ischemic stroke. Neurosci Bull. (2019) 35:921–33. doi: 10.1007/s12264-019-00388-3
44. Wang J, Xing H, Wan L, Jiang X, Wang C, Wu Y. Treatment targets for M2 microglia polarization in ischemic stroke. Biomed Pharmacother. (2018) 105:518–25. doi: 10.1016/j.biopha.2018.05.143
45. Jurga AM, Paleczna M, Kuter KZ. Overview of general and discriminating markers of differential microglia phenotypes. Front Cell Neurosci. (2020) 14:198. doi: 10.3389/fncel.2020.00198
46. Michell-Robinson MA, Touil H, Healy LM, Owen DR, Durafourt BA, Bar-Or A, et al. Roles of microglia in brain development, tissue maintenance and repair. Brain. (2015) 138:1138–59. doi: 10.1093/brain/awv066
47. Fumagalli S, Perego C, Pischiutta F, Zanier ER, De Simoni MG. The ischemic environment drives microglia and macrophage function. Front Neurol. (2015) 6:81. doi: 10.3389/fneur.2015.00081
48. Kim E, Cho S. Microglia and monocyte-derived macrophages in stroke. Neurotherapeutics. (2016) 13:702–18. doi: 10.1007/s13311-016-0463-1
49. Wang L, Zhang Z, Liang L, Wu Y, Zhong J, Sun X. Anti-high mobility group box-1 antibody attenuated vascular smooth muscle cell phenotypic switching and vascular remodelling after subarachnoid haemorrhage in rats. Neurosci Lett. (2019) 708:134338. doi: 10.1016/j.neulet.2019.134338
50. Liu R, Diao J, He S, Li B, Fei Y, Li Y, et al. XQ-1H protects against ischemic stroke by regulating microglia polarization through PPARgamma pathway in mice. Int Immunopharmacol. (2018) 57:72–81. doi: 10.1016/j.intimp.2018.02.014
51. Choi JY, Kim JY, Kim JY, Park J, Lee WT, Lee JE. M2 phenotype microglia-derived cytokine stimulates proliferation and neuronal differentiation of endogenous stem cells in ischemic brain. Exp Neurobiol. (2017) 26:33–41. doi: 10.5607/en.2017.26.1.33
52. Provencio JJ, Swank V, Lu H, Brunet S, Baltan S, Khapre RV, et al. Neutrophil depletion after subarachnoid hemorrhage improves memory via NMDA receptors. Brain Behav Immun. (2016) 54:233–42. doi: 10.1016/j.bbi.2016.02.007
53. Lyu J, Xie D, Bhatia TN, Leak RK, Hu X, Jiang X. Microglial/macrophage polarization and function in brain injury and repair after stroke. CNS Neurosci Ther. (2021) 27:515–27. doi: 10.1111/cns.13620
54. Ma DC, Zhang NN, Zhang YN, Chen HS. Salvianolic acids for injection alleviates cerebral ischemia/reperfusion injury by switching M1/M2 phenotypes and inhibiting NLRP3 inflammasome/pyroptosis axis in microglia in vivo and in vitro. J Ethnopharmacol. (2021) 270:113776. doi: 10.1016/j.jep.2021.113776
55. Ganbold T, Bao Q, Zandan J, Hasi A, Baigude H. Modulation of microglia polarization through silencing of NF-kappaB p65 by functionalized curdlan nanoparticle-mediated RNAi. ACS Appl Mater Interfaces. (2020) 12:11363–74. doi: 10.1021/acsami.9b23004
56. Hu X, Li P, Guo Y, Wang H, Leak RK, Chen S, et al. Microglia/macrophage polarization dynamics reveal novel mechanism of injury expansion after focal cerebral ischemia. Stroke. (2012) 43:3063–70. doi: 10.1161/STROKEAHA.112.659656
57. Collmann FM, Pijnenburg R, Hamzei-Taj S, Minassian A, Folz-Donahue K, Kukat C, et al. Individual in vivo profiles of microglia polarization after stroke, represented by the genes iNOS and Ym1. Front Immunol. (2019) 10:1236. doi: 10.3389/fimmu.2019.01236
58. Xia CY, Zhang S, Chu SF, Wang ZZ, Song XY, Zuo W, et al. Autophagic flux regulates microglial phenotype according to the time of oxygen-glucose deprivation/reperfusion. Int Immunopharmacol. (2016) 39:140–8. doi: 10.1016/j.intimp.2016.06.030
59. Zhang X, Zhu XL Ji BY, Cao X, Yu LJ, Zhang Y, et al. LncRNA-1810034E14Rik reduces microglia activation in experimental ischemic stroke. J Neuroinflammation. (2019) 16:75. doi: 10.1186/s12974-019-1464-x
60. Taetzsch T, Levesque S, McGraw C, Brookins S, Luqa R, Bonini MG, et al. Redox regulation of NF-kappaB p50 and M1 polarization in microglia. Glia. (2015) 63:423–40. doi: 10.1002/glia.22762
61. Hou L, Che Y, Sun F, Wang Q. Taurine protects noradrenergic locus coeruleus neurons in a mouse Parkinson's disease model by inhibiting microglial M1 polarization. Amino Acids. (2018) 50:547–56. doi: 10.1007/s00726-018-2547-1
62. Su D, Cheng Y, Li S, Dai D, Zhang W, Lv M. Sphk1 mediates neuroinflammation and neuronal injury via TRAF2/NF-κB pathways in activated microglia in cerebral ischemia reperfusion. J Neuroimmunol. (2017) 305:35–41. doi: 10.1016/j.jneuroim.2017.01.015
63. Xiang B, Zhong P, Fang L, Wu X, Song Y, Yuan H. miR-183 inhibits microglia activation and expression of inflammatory factors in rats with cerebral ischemia reperfusion via NF-κB signaling pathway. Exp Ther Med. (2019) 18:2540–6. doi: 10.3892/etm.2019.7827
64. Xin P, Xu X, Deng C, Liu S, Wang Y, Zhou X, et al. The role of JAK/STAT signaling pathway and its inhibitors in diseases. Int Immunopharmacol. (2020) 80:106210. doi: 10.1016/j.intimp.2020.106210
65. Owen KL, Brockwell NK, Parker BS. JAK-STAT signaling: a double-edged sword of immune regulation and cancer progression. Cancers. (2019) 11:2002. doi: 10.3390/cancers11122002
66. Yang X, Xu S, Qian Y, Xiao Q. Resveratrol regulates microglia M1/M2 polarization via PGC-1α in conditions of neuroinflammatory injury. Brain Behav Immun. (2017) 64:162–72. doi: 10.1016/j.bbi.2017.03.003
67. Qu Z, Zheng N, Wei Y, Chen Y, Zhang Y, Zhang M, et al. Effect of cornel iridoid glycoside on microglia activation through suppression of the JAK/STAT signalling pathway. J Neuroimmunol. (2019) 330:96–107. doi: 10.1016/j.jneuroim.2019.01.014
68. Wang H, Zang C, Liu XS, Aster JC. The role of Notch receptors in transcriptional regulation. J Cell Physiol. (2015) 230:982–8. doi: 10.1002/jcp.24872
69. Yao L, Cao Q, Wu C, Kaur C, Hao A, Ling EA. Notch signaling in the central nervous system with special reference to its expression in microglia. CNS Neurol Disord Drug Targets. (2013) 12:807–14. doi: 10.2174/18715273113126660172
70. Wu F, Luo T, Mei Y, Liu H, Dong J, Fang Y, et al. Simvastatin alters M1/M2 polarization of murine BV2 microglia via Notch signaling. J Neuroimmunol. (2018) 316:56–64. doi: 10.1016/j.jneuroim.2017.12.010
71. Zhang Y, Liu J, Wang C, Liu J, Lu W. Toll-like receptors gene polymorphisms in autoimmune disease. Front Immunol. (2021) 12:672346. doi: 10.3389/fimmu.2021.672346
72. Sakate R, Nishiyama M, Fukuda Y, Kitaoka S, Furuyashiki T. The transcription factor Hhex regulates inflammation-related genes in microglia. J Pharmacol Sci. (2022) 149:166–71. doi: 10.1016/j.jphs.2022.04.006
73. Lindner S, Dahlke K, Sontheimer K, Hagn M, Kaltenmeier C, Barth TFE, et al. Interleukin 21–induced granzyme B–expressing B cells infiltrate tumors and regulate T cells. Cancer Res. (2013) 73:2468–79. doi: 10.1158/0008-5472.CAN-12-3450
74. Anttila JE, Whitaker KW, Wires ES, Harvey BK, Airavaara M. Role of microglia in ischemic focal stroke and recovery: focus on Toll-like receptors. Prog Neuropsychopharmacol Biol Psychiatry. (2017) 79:3–14. doi: 10.1016/j.pnpbp.2016.07.003
75. Jiang CT, Wu WF, Deng YH, Ge JW. Modulators of microglia activation and polarization in ischemic stroke (Review). Mol Med Rep. (2020) 21:2006–18. doi: 10.3892/mmr.2020.11003
76. Cai W, Yang T, Liu H, Han L, Zhang K, Hu X, et al. Peroxisome proliferator-activated receptor gamma (PPARgamma): a master gatekeeper in CNS injury and repair. Prog Neurobiol. (2018) 163–164:27–58. doi: 10.1016/j.pneurobio.2017.10.002
77. Zhao X, Wang H, Sun G, Zhang J, Edwards NJ, Aronowski J. Neuronal interleukin-4 as a modulator of microglial pathways and ischemic brain damage. J Neurosci. (2015) 35:11281–91. doi: 10.1523/JNEUROSCI.1685-15.2015
78. Di Lucente J, Nguyen HM, Wulff H, Jin LW, Maezawa I. The voltage-gated potassium channel Kv13 is required for microglial pro-inflammatory activation in vivo. Glia. (2018) 66:1881–95. doi: 10.1002/glia.23457
79. Tian DS Li CY, Qin C, Murugan M, Wu LJ, Liu JL. Deficiency in the voltage-gated proton channel Hv1 increases M2 polarization of microglia and attenuates brain damage from photothrombotic ischemic stroke. J Neurochem. (2016) 139:96–105. doi: 10.1111/jnc.13751
80. Hamzei Taj S, Kho W, Riou A, Wiedermann D, Hoehn M. MiRNA-124 induces neuroprotection and functional improvement after focal cerebral ischemia. Biomaterials. (2016) 91:151–65. doi: 10.1016/j.biomaterials.2016.03.025
81. Jiang M, Wang H, Jin M, Yang X, Ji H, Jiang Y, et al. Exosomes from MiR-30d-5p-ADSCs reverse acute ischemic stroke-induced, autophagy-mediated brain injury by promoting M2 microglial/macrophage polarization. Cell Physiol Biochem. (2018) 47:864–78. doi: 10.1159/000490078
82. Hamzei Taj S, Kho W, Aswendt M, Collmann FM, Green C, Adamczak J, et al. Dynamic modulation of microglia/macrophage polarization by miR-124 after focal cerebral ischemia. J Neuroimmune Pharmacol. (2016) 11:733–48. doi: 10.1007/s11481-016-9700-y
83. Chen C, Chu S-F, Ai Q-D, Zhang Z, Guan F-F, Wang S-S, et al. CKLF1 Aggravates focal cerebral ischemia injury at early stage partly by modulating microglia/macrophage toward M1 polarization through CCR4. Cell Mol Neurobiol. (2019) 39:651–69. doi: 10.1007/s10571-019-00669-5
84. Liu X, Liu J, Zhao S, Zhang H, Cai W, Cai M, et al. Interleukin-4 is essential for microglia/macrophage M2 polarization and long-term recovery after cerebral ischemia. Stroke. (2016) 47:498–504. doi: 10.1161/STROKEAHA.115.012079
85. Romero-Miguel D, Lamanna-Rama N, Casquero-Veiga M, Gomez-Rangel V, Desco M, Soto-Montenegro ML. Minocycline in neurodegenerative and psychiatric diseases: an update. Eur J Neurol. (2021) 28:1056–81. doi: 10.1111/ene.14642
86. Kohler E, Prentice DA, Bates TR, Hankey GJ, Claxton A, van Heerden J, et al. Intravenous minocycline in acute stroke: a randomized, controlled pilot study and meta-analysis. Stroke. (2013) 44:2493–9. doi: 10.1161/STROKEAHA.113.000780
87. Garrido-Mesa N, Zarzuelo A, Galvez J. Minocycline: far beyond an antibiotic. Br J Pharmacol. (2013) 169:337–52. doi: 10.1111/bph.12139
88. Hickman S, Izzy S, Sen P, Morsett L, El Khoury J. Microglia in neurodegeneration. Nat Neurosci. (2018) 21:1359–69. doi: 10.1038/s41593-018-0242-x
89. Liao B, Zhao W, Beers DR, Henkel JS, Appel SH. Transformation from a neuroprotective to a neurotoxic microglial phenotype in a mouse model of ALS. Exp Neurol. (2012) 237:147–52. doi: 10.1016/j.expneurol.2012.06.011
90. Ghosh R, Tabrizi SJ. Huntington disease. Handb Clin Neurol. (2018) 147:255–78. doi: 10.1016/B978-0-444-63233-3.00017-8
91. Yamasaki R, Lu H, Butovsky O, Ohno N, Rietsch AM, Cialic R, et al. Differential roles of microglia and monocytes in the inflamed central nervous system. J Exp Med. (2014) 211:1533–49. doi: 10.1084/jem.20132477
92. Kuhlmann T, Ludwin S, Prat A, Antel J, Brück W, Lassmann H. An updated histological classification system for multiple sclerosis lesions. Acta Neuropathol. (2016) 133:13–24. doi: 10.1007/s00401-016-1653-y
93. Bemiller SM, McCray TJ, Allan K, Formica SV, Xu G, Wilson G, et al. TREM2 deficiency exacerbates tau pathology through dysregulated kinase signaling in a mouse model of tauopathy. Mol Neurodegener. (2017) 12:74. doi: 10.1186/s13024-017-0216-6
94. Kim HS, Suh YH. Minocycline and neurodegenerative diseases. Behav Brain Res. (2009) 196:168–79. doi: 10.1016/j.bbr.2008.09.040
95. Plane JM, Shen Y, Pleasure DE, Deng W. Prospects for minocycline neuroprotection. Arch Neurol. (2010) 67:1442–8. doi: 10.1001/archneurol.2010.191
96. Kobayashi K, Imagama S, Ohgomori T, Hirano K, Uchimura K, Sakamoto K, et al. Minocycline selectively inhibits M1 polarization of microglia. Cell Death Dis. (2013) 4:e525. doi: 10.1038/cddis.2013.54
97. Lu Y, Zhou M, Li Y, Li Y, Hua Y, Fan Y. Minocycline promotes functional recovery in ischemic stroke by modulating microglia polarization through STAT1/STAT6 pathways. Biochem Pharmacol. (2021) 186:114464. doi: 10.1016/j.bcp.2021.114464
98. Zhao F, Hua Y, He Y, Keep RF Xi G. Minocycline-induced attenuation of iron overload and brain injury after experimental intracerebral hemorrhage. Stroke. (2011) 42:3587–93. doi: 10.1161/STROKEAHA.111.623926
99. Jin Q, Cheng J, Liu Y, Wu J, Wang X, Wei S, et al. Improvement of functional recovery by chronic metformin treatment is associated with enhanced alternative activation of microglia/macrophages and increased angiogenesis and neurogenesis following experimental stroke. Brain Behav Immun. (2014) 40:131–42. doi: 10.1016/j.bbi.2014.03.003
100. Zhu XC, Jiang T, Zhang QQ, Cao L, Tan MS, Wang HF, et al. Chronic Metformin preconditioning provides neuroprotection via suppression of NF-kappaB-mediated inflammatory pathway in rats with permanent cerebral ischemia. Mol Neurobiol. (2015) 52:375–85. doi: 10.1007/s12035-014-8866-7
101. Han L, Cai W, Mao L, Liu J, Li P, Leak RK, et al. Rosiglitazone promotes white matter integrity and long-term functional recovery after focal cerebral ischemia. Stroke. (2015) 46:2628–36. doi: 10.1161/STROKEAHA.115.010091
102. Cai Y, Xu H, Yan J, Zhang L, Lu Y. Molecular targets and mechanism of action of dexmedetomidine in treatment of ischemia/reperfusion injury. Mol Med Rep. (2014) 9:1542–50. doi: 10.3892/mmr.2014.2034
103. Yeh CH, Hsieh LP, Lin MC, Wei TS, Lin HC, Chang CC, et al. Dexmedetomidine reduces lipopolysaccharide induced neuroinflammation, sickness behavior, and anhedonia. PLoS ONE. (2018) 13:e0191070. doi: 10.1371/journal.pone.0191070
104. Park J, Min JS, Kim B, Chae UB, Yun JW, Choi MS, et al. Mitochondrial ROS govern the LPS-induced pro-inflammatory response in microglia cells by regulating MAPK and NF-kappaB pathways. Neurosci Lett. (2015) 584:191–6. doi: 10.1016/j.neulet.2014.10.016
105. Simon-O'Brien E, Gauthier D, Riban V, Verleye M. Etifoxine improves sensorimotor deficits and reduces glial activation, neuronal degeneration, and neuroinflammation in a rat model of traumatic brain injury. J Neuroinflammation. (2016) 13:203. doi: 10.1186/s12974-016-0687-3
106. Bonsack FT, Alleyne CH Jr., Sukumari-Ramesh S. Augmented expression of TSPO after intracerebral hemorrhage: a role in inflammation? J Neuroinflammation. (2016) 13:151. doi: 10.1186/s12974-016-0619-2
107. Li HD Li M, Shi E, Jin WN, Wood K, Gonzales R, et al. A translocator protein 18 kDa agonist protects against cerebral ischemia/reperfusion injury. J Neuroinflammation. (2017) 14:151. doi: 10.1186/s12974-017-0921-7
108. Pan J, Jin JL, Ge HM, Yin KL, Chen X, Han LJ, et al. Malibatol A regulates microglia M1/M2 polarization in experimental stroke in a PPARgamma-dependent manner. J Neuroinflammation. (2015) 12:51. doi: 10.1186/s12974-015-0270-3
109. Ma L, Pan X, Zhou F, Liu K, Wang L. Hyperforin protects against acute cerebral ischemic injury through inhibition of interleukin-17A-mediated microglial activation. Brain Res. (2018) 1678:254–61. doi: 10.1016/j.brainres.2017.08.023
110. Zhao Y, Han P, Liu L, Wang X, Xu P, Wang H, et al. Indirubin modulates CD4(+) T-cell homeostasis via PD1/PTEN/AKT signalling pathway in immune thrombocytopenia. J Cell Mol Med. (2019) 23:1885–98. doi: 10.1111/jcmm.14089
111. Walsh JG, Muruve DA, Power C. Inflammasomes in the CNS. Nat Rev Neurosci. (2014) 15:84–97. doi: 10.1038/nrn3638
112. Lepore F, D'Alessandro G, Antonangeli F, Santoro A, Esposito V, Limatola C, et al. CXCL16/CXCR6 axis drives microglia/macrophages phenotype in physiological conditions and plays a crucial role in glioma. Front Immunol. (2018) 9:2750. doi: 10.3389/fimmu.2018.02750
113. Ye Y, Jin T, Zhang X, Zeng Z, Ye B, Wang J, et al. Meisoindigo protects against focal cerebral ischemia-reperfusion injury by inhibiting NLRP3 inflammasome activation and regulating microglia/macrophage polarization via TLR4/NF-kappaB Signaling pathway. Front Cell Neurosci. (2019) 13:553. doi: 10.3389/fncel.2019.00553
114. Bian HJ, Xu SY Li HQ, Jia JQ, Ye L, Shu S, et al. JLX001 ameliorates cerebral ischemia injury by modulating microglial polarization and compromising NLRP3 inflammasome activation via the NF-kappaB signaling pathway. Int Immunopharmacol. (2021) 101:108325. doi: 10.1016/j.intimp.2021.108325
115. Jia H, Qi X, Fu L, Wu H, Shang J, Qu M, et al. NLRP3 inflammasome inhibitor ameliorates ischemic stroke by reprogramming the phenotype of microglia/macrophage in a murine model of distal middle cerebral artery occlusion. Neuropathology. (2022) 42:181–9. doi: 10.1111/neup.12802
116. Bellut M, Papp L, Bieber M, Kraft P, Stoll G, Schuhmann MK. NLPR3 inflammasome inhibition alleviates hypoxic endothelial cell death in vitro and protects blood-brain barrier integrity in murine stroke. Cell Death Dis. (2021) 13:20. doi: 10.1038/s41419-021-04379-z
117. Alexaki VI, Fodelianaki G, Neuwirth A, Mund C, Kourgiantaki A, Ieronimaki E, et al. DHEA inhibits acute microglia-mediated inflammation through activation of the TrkA-Akt1/2-CREB-Jmjd3 pathway. Mol Psychiatry. (2017) 23:1410–20. doi: 10.1038/mp.2017.167
118. Tang Y, Li T, Li J, Yang J, Liu H, Zhang XJ, et al. Jmjd3 is essential for the epigenetic modulation of microglia phenotypes in the immune pathogenesis of Parkinson's disease. Cell Death Differ. (2014) 21:369–80. doi: 10.1038/cdd.2013.159
119. Bottcher C, Schlickeiser S, Sneeboer MAM, Kunkel D, Knop A, Paza E, et al. Human microglia regional heterogeneity and phenotypes determined by multiplexed single-cell mass cytometry. Nat Neurosci. (2019) 22:78–90. doi: 10.1038/s41593-018-0290-2
120. Galatro TF, Holtman IR, Lerario AM, Vainchtein ID, Brouwer N, Sola PR, et al. Transcriptomic analysis of purified human cortical microglia reveals age-associated changes. Nat Neurosci. (2017) 20:1162–71. doi: 10.1038/nn.4597
121. Masuda T, Sankowski R, Staszewski O, Prinz M. Microglia heterogeneity in the single-cell era. Cell Rep. (2020) 30:1271–81. doi: 10.1016/j.celrep.2020.01.010
122. Masuda T, Sankowski R, Staszewski O, Bottcher C, Amann L. Sagar, et al. Spatial and temporal heterogeneity of mouse and human microglia at single-cell resolution. Nature. (2019) 566:388–92. doi: 10.1038/s41586-019-0924-x
123. Ling Y, Jin L, Ma Q, Huang Y, Yang Q, Chen M, et al. Salvianolic acid A alleviated inflammatory response mediated by microglia through inhibiting the activation of TLR2/4 in acute cerebral ischemia-reperfusion. Phytomedicine. (2021) 87:153569. doi: 10.1016/j.phymed.2021.153569
124. Deng W, Mandeville E, Terasaki Y, Li W, Holder J, Chuang AT, et al. Transcriptomic characterization of microglia activation in a rat model of ischemic stroke. J Cereb Blood Flow Metab. (2020) 40:S34–48. doi: 10.1177/0271678X20932870
125. Wu R, Li X, Xu P, Huang L, Cheng J, Huang X, et al. TREM2 protects against cerebral ischemia/reperfusion injury. Mol Brain. (2017) 10:20. doi: 10.1186/s13041-017-0296-9
126. Backes FN, de Souza A, Bianchin MM. Biomarkers in the prognostic evaluation of ischemic stroke: is there benefit in the measurements of TREM-1 and TREM-2 in the acute phase? Clin Biochem. (2021) 98:10–6. doi: 10.1016/j.clinbiochem.2021.07.016
127. Kawabori M, Kacimi R, Kauppinen T, Calosing C, Kim JY, Hsieh CL, et al. Triggering receptor expressed on myeloid cells 2 (TREM2) deficiency attenuates phagocytic activities of microglia and exacerbates ischemic damage in experimental stroke. J Neurosci. (2015) 35:3384–96. doi: 10.1523/JNEUROSCI.2620-14.2015
128. Belayev L, Hong SH, Freitas RS, Menghani H, Marcell SJ, Khoutorova L, et al. DHA modulates MANF and TREM2 abundance, enhances neurogenesis, reduces infarct size, and improves neurological function after experimental ischemic stroke. CNS Neurosci Ther. (2020) 26:1155–67. doi: 10.1111/cns.13444
129. Ma WY, Wang SS, Wu QL, Zhou X, Chu SF, Chen NH. The versatile role of TREM2 in regulating of microglia fate in the ischemic stroke. Int Immunopharmacol. (2022) 109:108733. doi: 10.1016/j.intimp.2022.108733
130. Ransohoff RM. A polarizing question: do M1 and M2 microglia exist? Nat Neurosci. (2016) 19:987–91. doi: 10.1038/nn.4338
Keywords: ischemic stroke, treatment target, microglia, polarization, dual function
Citation: Mo Y, Xu W, Fu K, Chen H, Wen J, Huang Q, Guo F, Mo L and Yan J (2022) The dual function of microglial polarization and its treatment targets in ischemic stroke. Front. Neurol. 13:921705. doi: 10.3389/fneur.2022.921705
Received: 16 April 2022; Accepted: 14 July 2022;
Published: 23 September 2022.
Edited by:
Keith Pennypacker, University of Kentucky, United StatesReviewed by:
Markus Aswendt, University of Cologne, GermanyJiancheng Zhang, Huazhong University of Science and Technology, China
Ma Yuanyuan, Fudan University, China
Liang Liu, Northern Theater General Hospital, China
Copyright © 2022 Mo, Xu, Fu, Chen, Wen, Huang, Guo, Mo and Yan. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Jun Yan, eWFuanVuQGd4bXUuZWR1LmNu; Ligen Mo, bGlnZW5tb0AxNjMuY29t
†These authors have contributed equally to this work
‡ORCID: Jun Yan orcid.org/0000-0001-6847-2985