
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
MINI REVIEW article
Front. Neurol., 20 May 2022
Sec. Multiple Sclerosis and Neuroimmunology
Volume 13 - 2022 | https://doi.org/10.3389/fneur.2022.912050
This article is part of the Research TopicImmune-mediated Disorders of the Spinal Cord: Diagnosis, Treatment Strategies, and Outcomes in the 21st CenturyView all 7 articles
 Samir Alkabie1
Samir Alkabie1 Adrian Budhram1,2*
Adrian Budhram1,2*Autoimmune myelopathies are immune-mediated disorders of the spinal cord that can cause significant neurologic disability. Discoveries of antibodies targeting aquaporin-4 (AQP4-IgG) and myelin oligodendrocyte glycoprotein (MOG-IgG) have facilitated the diagnosis of autoimmune myelopathies that were previously considered to be atypical presentations of multiple sclerosis (MS) or idiopathic, and represent major advancements in the field of autoimmune neurology. The detection of these antibodies can substantially impact patient diagnosis and management, and increasing awareness of this has led to a dramatic increase in testing for these antibodies among patients with suspected autoimmune myelopathy. In this review we discuss test methodologies used to detect these antibodies, the role of serum vs. cerebrospinal fluid testing, and the value of antibody titers when interpreting results, with the aim of helping laboratorians and clinicians navigate this testing when ordered as part of the diagnostic evaluation for suspected autoimmune myelopathy.
Autoimmune myelopathies are immune-mediated disorders of the spinal cord that can cause profound weakness, numbness, and bowel/bladder dysfunction. Antibodies targeting aquaporin-4 (AQP4-IgG) and myelin oligodendrocyte glycoprotein (MOG-IgG) define disease processes that can cause immune-mediated spinal cord dysfunction and have clinical features, treatment considerations and prognoses distinct from multiple sclerosis (MS) (1–17). Recognition of their substantial impact on patient diagnosis and management has led to a dramatic increase in testing for these antibodies among patients with suspected autoimmune myelopathy, which parallels an increase in neural antibody testing for suspected neurological autoimmunity more generally (18–20). We herein discuss test methodologies used to detect these antibodies, the role of serum vs. cerebrospinal fluid (CSF) testing, and the value of antibody titers in diagnostic interpretation of results. Key testing considerations for these antibodies are summarized in Table 1. Other aspects of these antibodies separate from their diagnostic utility in the clinical evaluation of suspected autoimmune myelopathy, such as how their titers relate to disease severity, how their persistent positivity informs risk of relapse, or how they interact with the complement system, are not the focus of this review but have been studied and discussed elsewhere (15, 21–26).
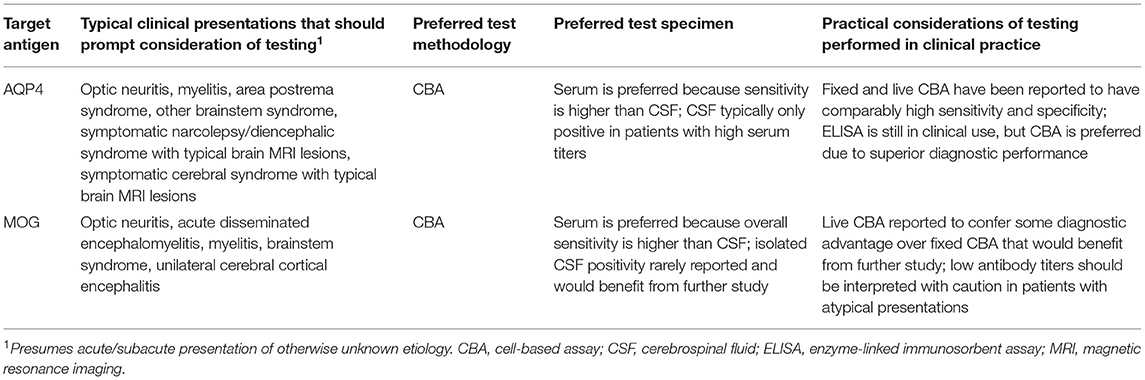
Table 1. Key considerations when testing for antibodies against aquaporin-4 (AQP4) and myelin oligodendrocyte glycoprotein (MOG).
Neuromyelitis optica (NMO), now termed neuromyelitis optica spectrum disorders (NMOSD) and historically known as Devic's syndrome, is a neuro-inflammatory disease that classically presents with relapsing optic neuritis and myelitis (27). In 2004, a serum IgG that characteristically stained mouse brain, termed NMO-IgG, was discovered in patients with this condition and soon determined to target aquaporin-4 (28, 29). While tissue indirect immunofluorescence (TIIF) was initially used to detect AQP4-IgG, evaluations of alternative test methodologies followed rapidly. Immunoprecipitation assays were reported to moderately enhance sensitivity particularly when combined with TIIF, although occasional false-positives were described in patients without characteristic TIIF staining (1, 30). Evaluations of enzyme-linked immunosorbent assay (ELISA) also suggested higher overall sensitivity than TIIF (31). However, the possibility for false-positive results using ELISA was also reported, highlighting the need for more sensitive and specific assays to detect AQP4-IgG (32).
Cell-based assays (CBAs) using HEK cells transfected with AQP4 were shown to have high sensitivity and specificity, although initially their restriction to specialized centers limited evaluation of their use in a high-thoroughput laboratory setting (1, 30, 33). The advent of commercially available AQP4-IgG ELISA and CBA led to increased test accessibility across clinical service laboratories, and created the need for comparative studies evaluating their diagnostic performance. A multicenter comparison study found that CBAs detecting AQP4-IgG by quantitative flow cytometry (FACS) or visual observation of immunofluorescence (IF) had excellent specificity and the highest sensitivity for NMOSD when compared to other assays (34). AQP4-IgG ELISA with lowered cut-off values was reported to have high sensitivity but at some expense to specificity, suggesting the need for confirmatory testing by CBA in patients with low ELISA values (34). Meanwhile, both TIIF and immunoprecipitation assays were found to have suboptimal sensitivity for AQP4-IgG, limiting their usefulness in screening patients with suspected NMOSD for this antibody (34). These findings indicated that CBAs are the preferred methodology when testing for AQP4-IgG, a conclusion supported by a subsequent systematic review of AQP4-IgG assays (35).
Several studies have investigated potential differences in variations across CBAs, including the choice of transfected AQP4 isoform, use of live or fixed cells, and use of CBA-FACS or CBA-IF. Human AQP4 exists as two major isoforms that differ in their N termini, the M1 isoform and the M23 isoform (36). It has been debated whether CBAs using the shorter M23 isoform might have higher sensitivity (37, 38). However, a study comparing a widely used commercial M1-based CBA to a newly developed M23-based CBA found no significant differences in test performance, similar to the observation by another group that increased signal of an M23-based CBA was offset by an increase in background fluorescence (35, 39). A multicenter comparison of AQP4-IgG assays that included in-house live M23 CBAs suggested these assays had slightly higher sensitivity, but with rare-false positives reported (40). Another study that included comparison of in-house live M1 and M23 CBAs found that the M1 CBA was preferable in a clinical laboratory service setting, due to false-positive results using the M23 CBA (41). Overall, it would seem that M1 CBAs are suitable for routine clinical use; while there may be potential for increased sensitivity by using M23 CBAs, care should be taken to ensure this does not come at the expense of specificity if developed for clinical service implementation.
With regard to live vs. fixed CBAs for AQP4-IgG detection, an early study reported a slight reduction in sensitivity of a commercial AQP4-IgG assay using formaldehyde-fixed HEK cells when compared to live HEK cells in a small cohort of patients, which the authors posited might be related to epitope alteration due to fixation (34, 35). However, a more recent study that incorporated the 2015 international consensus diagnostic criteria for NMOSD into its comparison of AQP4-IgG assays found excellent specificity and a comparably high sensitivity of both fixed and live CBAs, and confirmed superiority of CBAs over ELISA and TIIF (42). The findings of this study indicate that commercial fixed CBA is a viable option for clinical laboratories that are interested in offering AQP4-IgG testing but do not have the capability to perform live CBA, which is relatively time-consuming and labor-intensive. Among specialized laboratories that offer live CBA, both CBA-IF and CBA-FACS are in use. A multicenter study that included both live CBA-IF and CBA-FACS assays found, somewhat surprisingly, that live CBA-FACS assays varied substantially in terms of sensitivity and specificity, highlighting the importance of live CBA-FACS optimization prior to clinical implementation (40). This is exemplified by one center that, after rigorously evaluating live CBA-FACS compared to other AQP4-IgG assays and determining their live M1 CBA-FACS assay to perform optimally, subsequently demonstrated high sensitivity and exceptional specificity of their assay without false-positives in a high throughput setting (41, 43).
While the majority of early investigations into AQP4-IgG were based on serum testing alone, reports emerged of CSF positivity in seronegative patients with NMOSD. One study described three patients with compatible disease phenotypes and NMO-IgG positivity by TIIF that was restricted to CSF; however, the significance of this finding was unclear, given the potential for increased non-specific background staining of serum TIIF that may impede visualization of NMO-IgG staining (44). A study using CBA to detect AQP4-IgG found that only 8/20 patients with serum AQP4-IgG positivity were also positive in CSF, indicating that serum is more sensitive than CSF (45). Another comparison study of serum and CSF AQP4-IgG positivity using CBA found that CSF was positive in only 21/31 patients with AQP4-IgG seropositive NMOSD and none of 14 patients with AQP4-IgG seronegative NMOSD, thereby providing additional evidence that the sensitivity of serum is higher than CSF (46). This was further corroborated by another study evaluating serum vs. CSF testing for AQP4-IgG by CBA as part of clinical service evaluation, in which serum was more sensitive than CSF and no additional cases were detected by CSF testing (47). High serum AQP4-IgG titers have consistently been found to predict CSF positivity, suggesting a relative lack of intrathecal synthesis and need for a critical serum/CSF gradient before AQP4-IgG is detectable in CSF (45–47). Given these findings, serum is recommended over CSF when submitting samples to test for AQP4-IgG by CBA.
Overall, it would seem that any marginal differences in the performance of various AQP4-IgG CBAs reported in the literature, such as those using the M1 vs. M23 isoform or live vs. fixed cells, has less relevance to clinical practice than the overall superior diagnostic performance of CBAs when compared to other test methodologies. Raising awareness of this among both clinicians and laboratorians is critical because assays that are liable to generate false-positive results, such as AQP4-IgG ELISA, are still commonly ordered for patients with suspected NMOSD and can lead to misdiagnosis (48, 49). Such assays should be phased out of clinical practice wherever possible, because accessibility to CBA is now widespread due to commercial assays using fixed cells that perform comparably well to live CBAs offered at specialized centers. Given that AQP4-IgG serostatus now features prominently in diagnostic criteria and clinical trials of drugs for NMOSD, accurate detection of this antibody has become especially important to patient diagnosis and treatment (10, 50).
Myelin oligodendrocyte glycoprotein (MOG) is membrane protein specific to the central nervous system that is found predominantly on the surface of myelin sheaths (51). A study of MOG-IgG detected by western blot in patients with clinical isolated syndrome found that antibody positivity was a significant predictor of early conversion to clinically definite MS, but this finding was not reproducible in another study using the same western blot protocol (52, 53). These contradictory data are representative of much of the conflicting early works investigating MOG-IgG detected by immunoblots or ELISAs that use denatured protein as the substrate, which have been summarized previously (11). The lack of consistency across studies using these assays, coupled with their high positivity rate among healthy controls, has been attributed to the detection of antibodies against non-native MOG epitopes (11).
The development of an assay to detect antibodies against natively-folded MOG was a breakthrough that led to the determination of MOG-IgG positivity in approximately 20% of patients with acute disseminated encephalomyelitis (ADEM), compared to only 1% of patients from North America with adult-onset MS (54). Using live CBAs expressing conformational MOG it was subsequently demonstrated that a minority of patients with AQP4-IgG seronegative NMOSD were MOG-IgG positive, expanding the clinical phenotype of this antibody beyond ADEM to include optic neuritis and myelitis (55–57). It has been found that live CBAs expressing full-length human MOG α1 isoform enable detection of MOG-IgG with high sensitivity, while use of secondary antibodies to IgG1 or IgG-Fcγ has been reported to improve specificity (6, 58, 59). Reports of differential IgG binding patterns to MOG isoforms, as well as of reactivities of MOG-IgG subclasses other than IgG1, are of interest and would benefit from further study (59–61). Such future studies could facilitate the refinement of CBAs used to detect MOG-IgG, which have helped to define MOG-antibody-associated disease (MOGAD) as an entity distinct from MS (62–64).
In addition to live CBAs implemented at specialized centers, fixed CBA for MOG-IgG detection has become commercially available. This has spurred comparative studies of live and fixed CBAs for MOG-IgG, which can serve to inform the utility of these assays in clinical laboratories. A multicenter case-control study of three different MOG-IgG CBAs (two live CBAs and one fixed CBA) reported an overall high degree of agreement across assays, although the specificity of fixed CBA (98%) was slightly lower than both live CBAs (>99%) (65). The sensitivity of CBAs in this study, which defined any patient with a compatible MOGAD phenotype as a true-positive, ranged from 23 to 28%, with sensitivity of fixed CBA falling between that of the two live CBAs. A subsequent study compared the reproducibility of 11 antibody assays, including seven live MOG-IgG CBAs and one fixed MOG-IgG CBA (66). The overall concordance of these eight CBAs was 90% when analyzing clearly positive or negative samples, which were classified as such by four testing centers using live CBA. This increased to 96% if the fixed CBA was excluded, which was driven by the exclusion of four false-negatives by fixed CBA (66). This would seem to suggest higher sensitivity of live CBA, although comparison to changes in overall concordance when excluding each live CBA was not reported. Taken together with the findings of the previous study, live CBAs for MOG-IgG likely have slightly higher diagnostic accuracy than fixed CBA, and could thus be useful to reconcile unexpected false-negative or false-positive results by fixed CBA. More striking, however, was that the overall concordance across all 8 CBAs when analyzing low-positive samples was only 33%, which is considerably lower than concordance across CBAs used to detect low levels of AQP4-IgG (40, 66). This greater variability in detection of low levels of MOG-IgG across both live and fixed CBAs calls into question whether these low levels can reliably distinguish MOGAD from other disease processes (discussed later on), and represents a barrier to patient disease classification for the purposes of sensitivity/specificity analyses in diagnostic accuracy studies. The development of international consensus criteria for MOGAD that are independent of the antibody result would be helpful to analyze patients with discordant low-positive MOG-IgG across CBAs, and could be incorporated into future comparative studies of assay performance like has been done with AQP4-IgG testing for NMOSD (42).
Similar to AQP4-IgG, an early investigation of serum vs. CSF testing of MOG-IgG by CBA found a relative lack of intrathecal synthesis and indicated that serum was the preferred sample to test (67). In this study only 12/18 seropositive MOG-IgG patients also had MOG-IgG detected in CSF, although seronegative MOG-IgG patients with a compatible MOGAD phenotype did not undergo CSF MOG-IgG testing to assess for potential identification of additional cases (67). Another study identified three patients with non-MS demyelinating disease that were seronegative for MOG-IgG but found to be positive in CSF using CBA (68). This led the authors to hypothesize that, while overall sensitivity of serum testing for MOG-IgG was higher, CSF testing might identify some additional cases of MOGAD (68). This hypothesis was supported by autopsy findings of a patient with rapidly progressive meningoencephalomyelitis and MOG-IgG positivity in CSF only, which showed similar neuropathological findings to that described in seropositive MOG-IgG cases (69). A recent comparative study determined that 11/38 patients with MOGAD had CSF positivity only compared to 0/36 patients with AQP4-IgG-positive NMOSD, with a significantly higher proportion of patients with MOGAD found to have evidence of intrathecal synthesis of MOG-IgG (70). This was corroborated by another study that identified nine patients with non-MS inflammatory demyelinating diseases compatible with MOGAD and isolated CSF MOG-IgG positivity, eight of whom were found to have intrathecal MOG-IgG synthesis (71). Importantly, however, four patients with MS and isolated CSF MOG-IgG positivity were also reported, suggesting the possibility of false-positive CSF results (71). To better determine the utility of serum vs. CSF MOG-IgG testing, a national laboratory analyzed consecutive samples referred for testing; among 118 patients with positivity in either fluid, isolated CSF MOG-IgG was only found in 4/118 and one had bacterial meningitis rather than MOGAD (72). Overall, these findings suggest that while a minority of patients with disease phenotypes consistent with MOGAD may exhibit isolated CSF MOG-IgG positivity, routine testing of CSF MOG-IgG in addition to serum is low-yield and the possibility of false-positives exists. Additionally, many CBAs used to test for MOG-IgG in serum are not currently clinically validated for detection of this antibody in CSF. For these reasons, it would seem reasonable at this time to limit CSF testing for MOG-IgG to patients in whom there is a high index of clinical suspicion and no more likely alternative diagnosis, pending further investigation of its diagnostic value.
The need to consider antibody titer when interpreting positive MOG-IgG results was highlighted in a single-center study of consecutive patients tested for MOG-IgG by live CBA. This study found that the probability of a true-positive result varied significantly by titer, ranging from 100% with a titer of ≥1:1000 to only 51% in those with a titer of 1:20–1:40 (73). These findings indicate that antibody titer can help to gauge the likelihood of a true-positive result when encountering a positive MOG-IgG result in a patient with an atypical or equivocal phenotype for MOGAD, and should therefore be routinely reviewed as part of test interpretation. Importantly, titration may not be performed in clinical laboratories using fixed CBA for MOG-IgG detection, and cut-off titers for MOG-IgG seropositivity by live CBA can vary depending on factors such as the secondary antibody used (66); for these reasons, reporting of titers should be clarified with the testing laboratory to avoid misinterpretation.
One outstanding question is how to interpret MOG-IgG detected by CBA in a small portion of patients with MS, which is typically low-titer and has generally been classified as false-positive in diagnostic accuracy studies. This classification is supported by a literature review of patients with typical lesions for MS on magnetic resonance imaging (MRI) and MOG-IgG positivity by live or fixed CBA, who were found not to differ from what would be expected of clinically definite MS in terms of attack severity, treatment response, MRI evolution over time, and outcomes (74). However, it has been suggested that low titers of MOG-IgG in a patient with MS may indicate that they lie on a disease spectrum between the two entities, in part because of the high proportion of patients with MS among those classified as false-positives in studies of MOG-IgG (75). While it is true that low-titer MOG-IgG values classified as false-positives are often reported in patients with MS, this is confounded by the fact that MS is likely the most common alternative diagnosis among patients being tested for MOG-IgG in clinical practice; the greatest proportion of false-positives is thus expected to be patients with MS, even if no predilection for MOG-IgG detection in MS exists. To this point, a recent study that indiscriminately performed MOG-IgG testing in consecutive neurologic patients found that the majority with low antibody levels had non-inflammatory diseases such as stroke, rather than MS (76). Even if a higher proportion of low-titer MOG-IgG in MS compared to other diseases was systematically demonstrated, an additional challenge would be discerning whether low levels of MOG-IgG in this context contribute to immunopathogenesis or are an epiphenomenon of MS-related demyelination, i.e., whether the antibody is of clinical relevance or not; this adds a layer of complexity to both diagnostic accuracy studies and clinical practice that would benefit from dedicated evaluation.
The discoveries of AQP4-IgG and MOG-IgG have transformed the evaluation of patients with suspected autoimmune myelopathy. Sensitive and specific CBAs to detect these antibodies have facilitated the diagnosis of AQP4-IgG-positive NMOSD and MOGAD in clinical practice, which are distinct disease entities. Serum is the preferred specimen for routine testing of both antibodies; isolated CSF MOG-IgG positivity in a minority of patients with compatible disease phenotypes, while intriguing, is a finding that requires further study. Fixed CBAs, which are relatively easy to implement in clinical laboratories and are thus now in widespread use, have dramatically improved test accessibility. For AQP4-IgG detection, fixed CBA has been found to perform comparably well to live CBA and should motivate replacement of other commercial assays that are still in use despite demonstrably inferior diagnostic performance, such as ELISA. Live CBAs seem to confer some diagnostic advantage over fixed CBAs for MOG-IgG detection, although future comparative studies would benefit from the development of international consensus diagnostic criteria for MOGAD that could aid in patient classifications for the purposes of sensitivity/specificity analyses. Such criteria could also help clinicians to reconcile possible clinically irrelevant or false-positive results that can occur with live or fixed MOG-IgG CBAs, generally at low titers and both in MS as well as various other diseases. In patients with atypical or equivocal phenotypes for MOGAD and a positive MOG-IgG result, discussion with the testing laboratory surrounding assay cut-offs as well as consideration of titer can help to avoid misdiagnosis.
This manuscript was entirely planned and drafted by SA and AB. Both authors contributed to the article and approved the submitted version.
AB reports that he holds the London Health Sciences Centre and London Health Sciences Foundation Chair in Neural Antibody Testing for Neuro-Inflammatory Diseases.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
1. Waters P, Jarius S, Littleton E, Leite MI, Jacob S, Gray B, et al. Aquaporin-4 antibodies in neuromyelitis optica and longitudinally extensive transverse myelitis. Arch Neurol. (2008) 65:913–9. doi: 10.1001/archneur.65.7.913
2. Flanagan EP, Weinshenker BG, Krecke KN, Lennon VA, Lucchinetti CF, McKeon A, et al. Short myelitis lesions in aquaporin-4-IgG-positive neuromyelitis optica spectrum disorders. JAMA Neurol. (2015) 72:81–7. doi: 10.1001/jamaneurol.2014.2137
3. Chang KH, Lyu RK, Chen CM, Wu YR, Chang HS, Huang CC, et al. Distinct features between longitudinally extensive transverse myelitis presenting with and without anti-aquaporin 4 antibodies. Mult Scler. (2013) 19:299–307. doi: 10.1177/1352458512451659
4. Zekeridou A, Lennon VA. Aquaporin-4 autoimmunity. Neurol Neuroimmunol Neuroinflamm. (2015) 2:e110. doi: 10.1212/NXI.0000000000000110
5. Dubey D, Pittock SJ, Krecke KN, Morris PP, Sechi E, Zalewski NL, et al. Clinical, radiologic, and prognostic features of myelitis associated with myelin oligodendrocyte glycoprotein autoantibody. JAMA Neurol. (2019) 76:301–9. doi: 10.1001/jamaneurol.2018.4053
6. Marignier R, Hacohen Y, Cobo-Calvo A, Pröbstel AK, Aktas O, Alexopoulos H, et al. Myelin-oligodendrocyte glycoprotein antibody-associated disease. Lancet Neurol. (2021) 20:762–72. doi: 10.1016/S1474-4422(21)00218-0
7. Lopez Chiriboga S, Flanagan EP. Myelitis and other autoimmune myelopathies. Continuum. (2021) 27:62–92. doi: 10.1212/CON.0000000000000900
8. Takahashi T, Miyazawa I, Misu T, Takano R, Nakashima I, Fujihara K, et al. Intractable hiccup and nausea in neuromyelitis optica with anti-aquaporin-4 antibody: a herald of acute exacerbations. J Neurol Neurosurg Psychiatry. (2008) 79:1075–8. doi: 10.1136/jnnp.2008.145391
9. Sato D, Fujihara K. Atypical presentations of neuromyelitis optica. Arq Neuropsiquiatr. (2011) 69:824–8. doi: 10.1590/S0004-282X2011000600019
10. Wingerchuk DM, Banwell B, Bennett JL, Cabre P, Carroll W, Chitnis T, et al. International consensus diagnostic criteria for neuromyelitis optica spectrum disorders. Neurology. (2015) 85:177–89. doi: 10.1212/WNL.0000000000001729
11. Reindl M, Waters P. Myelin oligodendrocyte glycoprotein antibodies in neurological disease. Nat Rev Neurol. (2019) 15:89–102. doi: 10.1038/s41582-018-0112-x
12. Budhram A, Mirian A, Le C, Hosseini-Moghaddam SM, Sharma M, Nicolle MW. Unilateral cortical FLAIR-hyperintense lesions in anti-MOG-associated encephalitis with Seizures (FLAMES): characterization of a distinct clinico-radiographic syndrome. J Neurol. (2019) 266:2481–7. doi: 10.1007/s00415-019-09440-8
13. Ogawa R, Nakashima I, Takahashi T, Kaneko K, Akaishi T, Takai Y, et al. MOG antibody-positive, benign, unilateral, cerebral cortical encephalitis with epilepsy. Neurol Neuroimmunol Neuroinflamm. (2017) 4:e322. doi: 10.1212/NXI.0000000000000322
14. Jarius S, Paul F, Aktas O, Asgari N, Dale RC, de Seze J, et al. MOG encephalomyelitis: international recommendations on diagnosis and antibody testing. J Neuroinflammation. (2018) 15:134. doi: 10.1186/s12974-018-1144-2
15. López-Chiriboga AS, Majed M, Fryer J, Dubey D, McKeon A, Flanagan EP, et al. Association of MOG-IgG serostatus with relapse after acute disseminated encephalomyelitis and proposed diagnostic criteria for MOG-IgG-associated disorders. JAMA Neurol. (2018) 75:1355–63. doi: 10.1001/jamaneurol.2018.1814
16. Fadda G, Alves CA, O'Mahony J, Castro DA, Yeh EA, Marrie RA, et al. Comparison of spinal cord magnetic resonance imaging features among children with acquired demyelinating syndromes. JAMA Netw Open. (2021) 4:e2128871. doi: 10.1001/jamanetworkopen.2021.28871
17. Fadda G, Armangue T, Hacohen Y, Chitnis T, Banwell B. Paediatric multiple sclerosis and antibody-associated demyelination: clinical, imaging, and biological considerations for diagnosis and care. Lancet Neurol. (2021) 20:136–49. doi: 10.1016/S1474-4422(20)30432-4
18. Budhram A, Yang L, Bhayana V, Mills JR, Dubey D. Clinical sensitivity, specificity, and predictive value of neural antibody testing for autoimmune encephalitis. J Appl Lab Med. (2022) 7:350–6. doi: 10.1093/jalm/jfab127
19. Budhram A, Dubey D, Sechi E, Flanagan EP, Yang L, Bhayana V, et al. Neural antibody testing in patients with suspected autoimmune encephalitis. Clin Chem. (2020) 66:1496–509. doi: 10.1093/clinchem/hvaa254
20. Dubey D, Pittock SJ, Kelly CR, McKeon A, Lopez-Chiriboga AS, Lennon VA, et al. Autoimmune encephalitis epidemiology and a comparison to infectious encephalitis. Ann Neurol. (2018) 83:166–77. doi: 10.1002/ana.25131
21. Dinoto A, Sechi E, Flanagan EP, Ferrari S, Solla P, Mariotto S, et al. Serum and cerebrospinal fluid biomarkers in neuromyelitis optica spectrum disorder and myelin oligodendrocyte glycoprotein associated disease. Front Neurol. (2022) 13:866824. doi: 10.3389/fneur.2022.866824
22. Liu J, Tan G, Li B, Zhang J, Gao Y, Cao Y, et al. Serum aquaporin 4-Immunoglobulin G titer and neuromyelitis optica spectrum disorder activity and severity: a systematic review and meta-analysis. Front Neurol. (2021) 12:746959. doi: 10.3389/fneur.2021.746959
23. Asavapanumas N, Tradtrantip L, Verkman AS. Targeting the complement system in neuromyelitis optica spectrum disorder. Expert Opin Biol Ther. (2021) 21:1073–86. doi: 10.1080/14712598.2021.1884223
24. Tradtrantip L, Yeaman MR, Verkman AS. Cytoprotective IgG antibodies in sera from a subset of patients with AQP4-IgG seropositive neuromyelitis optica spectrum disorder. Sci Rep. (2021) 11:21962. doi: 10.1038/s41598-021-01294-3
25. Oliveira LM, Apóstolos-Pereira SL, Pitombeira MS, Bruel Torretta PH, Callegaro D, Sato DK. Persistent MOG-IgG positivity is a predictor of recurrence in MOG-IgG-associated optic neuritis, encephalitis and myelitis. Mult Scler. (2019) 25:1907–14. doi: 10.1177/1352458518811597
26. Keller CW, Lopez JA, Wendel EM, Ramanathan S, Gross CC, Klotz L, et al. Complement activation is a prominent feature of MOGAD. Ann Neurol. (2021) 90:976–82. doi: 10.1002/ana.26226
27. Wingerchuk DM, Hogancamp WF, O'Brien PC, Weinshenker BG. The clinical course of neuromyelitis optica (Devic's syndrome). Neurology. (1999) 53:1107–14. doi: 10.1212/WNL.53.5.1107
28. Lennon VA, Wingerchuk DM, Kryzer TJ, Pittock SJ, Lucchinetti CF, Fujihara K, et al. A serum autoantibody marker of neuromyelitis optica: distinction from multiple sclerosis. Lancet. (2004) 364:2106–12. doi: 10.1016/S0140-6736(04)17551-X
29. Lennon VA, Kryzer TJ, Pittock SJ, Verkman AS, Hinson SR. IgG marker of optic-spinal multiple sclerosis binds to the aquaporin-4 water channel. J Exp Med. (2005) 202:473–7. doi: 10.1084/jem.20050304
30. McKeon A, Fryer JP, Apiwattanakul M, Lennon VA, Hinson SR, Kryzer TJ, et al. Diagnosis of neuromyelitis spectrum disorders: comparative sensitivities and specificities of immunohistochemical and immunoprecipitation assays. Arch Neurol. (2009) 66:1134–8. doi: 10.1001/archneurol.2009.178
31. Jarius S, Franciotta D, Paul F, Bergamaschi R, Rommer PS, Ruprecht K, et al. Testing for antibodies to human aquaporin-4 by ELISA: sensitivity, specificity, and direct comparison with immunohistochemistry. J Neurol Sci. (2012) 320:32–7. doi: 10.1016/j.jns.2012.06.002
32. Kim YJ, Jung SW, Kim Y, Park YJ, Han K, Oh EJ. Detection of anti-aquaporin-4 antibodies in neuromyelitis optica: comparison of tissue-based and cell-based indirect immunofluorescence assays and ELISA. J Clin Lab Anal. (2012) 26:184–9. doi: 10.1002/jcla.21508
33. Takahashi T, Fujihara K, Nakashima I, Misu T, Miyazawa I, Nakamura M, et al. Establishment of a new sensitive assay for anti-human aquaporin-4 antibody in neuromyelitis optica. Tohoku J Exp Med. (2006) 210:307–13. doi: 10.1620/tjem.210.307
34. Waters PJ, McKeon A, Leite MI, Rajasekharan S, Lennon VA, Villalobos A, et al. Serologic diagnosis of NMO: a multicenter comparison of aquaporin-4-IgG assays. Neurology. (2012) 78:665–71. doi: 10.1212/WNL.0b013e318248dec1
35. Waters PJ, Pittock SJ, Bennett JL, Jarius S, Weinshenker BG, Wingerchuk DM. Evaluation of aquaporin-4 antibody assays. Clin Exp Neuroimmunol. (2014) 5:290–303. doi: 10.1111/cen3.12107
36. Furman CS, Gorelick-Feldman DA, Davidson KG, Yasumura T, Neely JD, Agre P, et al. Aquaporin-4 square array assembly: opposing actions of M1 and M23 isoforms. Proc Natl Acad Sci U S A. (2003) 100:13609–14. doi: 10.1073/pnas.2235843100
37. Nicchia GP, Mastrototaro M, Rossi A, Pisani F, Tortorella C, Ruggieri M, et al. Aquaporin-4 orthogonal arrays of particles are the target for neuromyelitis optica autoantibodies. Glia. (2009) 57:1363–73. doi: 10.1002/glia.20855
38. Crane JM, Lam C, Rossi A, Gupta T, Bennett JL, Verkman AS. Binding affinity and specificity of neuromyelitis optica autoantibodies to aquaporin-4 M1/M23 isoforms and orthogonal arrays. J Biol Chem. (2011) 286:16516–24. doi: 10.1074/jbc.M111.227298
39. Jarius S, Paul F, Fechner K, Ruprecht K, Kleiter I, Franciotta D, et al. Aquaporin-4 antibody testing: direct comparison of M1-AQP4-DNA-transfected cells with leaky scanning versus M23-AQP4-DNA-transfected cells as antigenic substrate. J Neuroinflammation. (2014) 11:129. doi: 10.1186/1742-2094-11-129
40. Waters P, Reindl M, Saiz A, Schanda K, Tuller F, Kral V, et al. Multicentre comparison of a diagnostic assay: aquaporin-4 antibodies in neuromyelitis optica. J Neurol Neurosurg Psychiatry. (2016) 87:1005–15. doi: 10.1136/jnnp-2015-312601
41. Fryer JP, Lennon VA, Pittock SJ, Jenkins SM, Fallier-Becker P, Clardy SL, et al. AQP4 autoantibody assay performance in clinical laboratory service. Neurol Neuroimmunol Neuroinflamm. (2014) 1:e11. doi: 10.1212/NXI.0000000000000011
42. Prain K, Woodhall M, Vincent A, Ramanathan S, Barnett MH, Bundell CS, et al. AQP4 antibody assay sensitivity comparison in the era of the 2015 diagnostic criteria for NMOSD. Front Neurol. (2019) 10:1028. doi: 10.3389/fneur.2019.01028
43. Redenbaugh V, Montalvo M, Sechi E, Buciuc M, Fryer JP, McKeon A, et al. Diagnostic value of aquaporin-4-IgG live cell based assay in neuromyelitis optica spectrum disorders. Mult Scler J Exp Transl Clin. (2021) 7:20552173211052656. doi: 10.1177/20552173211052656
44. Klawiter EC, Alvarez E, Xu J, Paciorkowski AR, Zhu L, Parks BJ, et al. NMO-IgG detected in CSF in seronegative neuromyelitis optica. Neurology. (2009) 72:1101–3. doi: 10.1212/01.wnl.0000345066.57745.50
45. Takahashi T, Fujihara K, Nakashima I, Misu T, Miyazawa I, Nakamura M, et al. Anti-aquaporin-4 antibody is involved in the pathogenesis of NMO: a study on antibody titre. Brain. (2007) 130:1235–43. doi: 10.1093/brain/awm062
46. Jarius S, Franciotta D, Paul F, Ruprecht K, Bergamaschi R, Rommer PS, et al. Cerebrospinal fluid antibodies to aquaporin-4 in neuromyelitis optica and related disorders: frequency, origin, and diagnostic relevance. J Neuroinflammation. (2010) 7:52. doi: 10.1186/1742-2094-7-52
47. Majed M, Fryer JP, McKeon A, Lennon VA, Pittock SJ. Clinical utility of testing AQP4-IgG in CSF: guidance for physicians. Neurol Neuroimmunol Neuroinflamm. (2016) 3:e231. doi: 10.1212/NXI.0000000000000231
48. Williams JP, Abbatemarco JR, Galli JJ, Rodenbeck SJ, Peterson LK, Haven TR, et al. Aquaporin-4 autoantibody detection by ELISA: a retrospective characterization of a commonly used assay. Mult Scler Int. (2021) 2021:8692328. doi: 10.1155/2021/8692328
49. Jolliffe EA, Keegan BM, Flanagan EP. Trident sign trumps aquaporin-4-IgG ELISA in diagnostic value in a case of longitudinally extensive transverse myelitis. Mult Scler Relat Disord. (2018) 23:7–8. doi: 10.1016/j.msard.2018.04.012
50. Pittock SJ, Berthele A, Fujihara K, Kim HJ, Levy M, Palace J, et al. Eculizumab in aquaporin-4-positive neuromyelitis optica spectrum disorder. N Engl J Med. (2019) 381:614–25. doi: 10.1056/NEJMoa1900866
51. Brunner C, Lassmann H, Waehneldt TV, Matthieu JM, Linington C. Differential ultrastructural localization of myelin basic protein, myelin/oligodendroglial glycoprotein, and 2',3'-cyclic nucleotide 3'-phosphodiesterase in the CNS of adult rats. J Neurochem. (1989) 52:296–304. doi: 10.1111/j.1471-4159.1989.tb10930.x
52. Berger T, Rubner P, Schautzer F, Egg R, Ulmer H, Mayringer I, et al. Antimyelin antibodies as a predictor of clinically definite multiple sclerosis after a first demyelinating event. N Engl J Med. (2003) 349:139–45. doi: 10.1056/NEJMoa022328
53. Kuhle J, Pohl C, Mehling M, Edan G, Freedman MS, Hartung HP, et al. Lack of association between antimyelin antibodies and progression to multiple sclerosis. N Engl J Med. (2007) 356:371–8. doi: 10.1056/NEJMoa063602
54. O'Connor KC, McLaughlin KA, De Jager PL, Chitnis T, Bettelli E, Xu C, et al. Self-antigen tetramers discriminate between myelin autoantibodies to native or denatured protein. Nat Med. (2007) 13:211–7. doi: 10.1038/nm1488
55. Kitley J, Woodhall M, Waters P, Leite MI, Devenney E, Craig J, et al. Myelin-oligodendrocyte glycoprotein antibodies in adults with a neuromyelitis optica phenotype. Neurology. (2012) 79:1273–7. doi: 10.1212/WNL.0b013e31826aac4e
56. Kitley J, Waters P, Woodhall M, Leite MI, Murchison A, George J, et al. Neuromyelitis optica spectrum disorders with aquaporin-4 and myelin-oligodendrocyte glycoprotein antibodies: a comparative study. JAMA Neurol. (2014) 71:276–83. doi: 10.1001/jamaneurol.2013.5857
57. Mader S, Gredler V, Schanda K, Rostasy K, Dujmovic I, Pfaller K, et al. Complement activating antibodies to myelin oligodendrocyte glycoprotein in neuromyelitis optica and related disorders. J Neuroinflammation. (2011) 8:184. doi: 10.1186/1742-2094-8-184
58. Waters P, Woodhall M, O'Connor KC, Reindl M, Lang B, Sato DK, et al. MOG cell-based assay detects non-MS patients with inflammatory neurologic disease. Neurol Neuroimmunol Neuroinflamm. (2015) 2:e89. doi: 10.1212/NXI.0000000000000089
59. Gastaldi M, Scaranzin S, Jarius S, Wildeman B, Zardini E, Mallucci G, et al. Cell-based assays for the detection of MOG antibodies: a comparative study. J Neurol. (2020) 267:3555–64. doi: 10.1007/s00415-020-10024-0
60. Mariotto S, Ferrari S, Monaco S, Benedetti MD, Schanda K, Alberti D, et al. Clinical spectrum and IgG subclass analysis of anti-myelin oligodendrocyte glycoprotein antibody-associated syndromes: a multicenter study. J Neurol. (2017) 264:2420–30. doi: 10.1007/s00415-017-8635-4
61. Schanda K, Peschl P, Lerch M, Seebacher B, Mindorf S, Ritter N, et al. Differential binding of autoantibodies to MOG isoforms in inflammatory demyelinating diseases. Neurol Neuroimmunol Neuroinflamm. (2021) 8:e1027. doi: 10.1212/NXI.0000000000001027
62. Jurynczyk M, Messina S, Woodhall MR, Raza N, Everett R, Roca-Fernandez A, et al. Clinical presentation and prognosis in MOG-antibody disease: a UK study. Brain. (2017) 140:3128–38. doi: 10.1093/brain/awx276
63. Ramanathan S, Mohammad S, Tantsis E, Nguyen TK, Merheb V, Fung VSC, et al. Clinical course, therapeutic responses and outcomes in relapsing MOG antibody-associated demyelination. J Neurol Neurosurg Psychiatry. (2018) 89:127–37. doi: 10.1136/jnnp-2017-316880
64. Cobo-Calvo A, Ruiz A, Rollot F, Arrambide G, Deschamps R, Maillart E, et al. Clinical features and risk of relapse in children and adults with myelin oligodendrocyte glycoprotein antibody-associated disease. Ann Neurol. (2021) 89:30–41. doi: 10.1002/ana.25909
65. Waters PJ, Komorowski L, Woodhall M, Lederer S, Majed M, Fryer J, et al. A multicenter comparison of MOG-IgG cell-based assays. Neurology. (2019) 92:e1250–5. doi: 10.1212/WNL.0000000000007096
66. Reindl M, Schanda K, Woodhall M, Tea F, Ramanathan S, Sagen J, et al. International multicenter examination of MOG antibody assays. Neurol Neuroimmunol Neuroinflamm. (2020) 7:e674. doi: 10.1212/NXI.0000000000000674
67. Jarius S, Ruprecht K, Kleiter I, Borisow N, Asgari N, Pitarokoili K, et al. MOG-IgG in NMO and related disorders: a multicenter study of 50 patients. Part 1: Frequency, syndrome specificity, influence of disease activity, long-term course, association with AQP4-IgG, and origin. J Neuroinflammation. (2016) 13:279. doi: 10.1186/s12974-016-0717-1
68. Mariotto S, Gajofatto A, Batzu L, Delogu R, Sechi G, Leoni S, et al. Relevance of antibodies to myelin oligodendrocyte glycoprotein in CSF of seronegative cases. Neurology. (2019) 93:e1867–e72. doi: 10.1212/WNL.0000000000008479
69. Carta S, Höftberger R, Bolzan A, Bozzetti S, Bonetti B, Scarpelli M, et al. Antibodies to MOG in CSF only: pathological findings support the diagnostic value. Acta Neuropathol. (2021) 141:801–4. doi: 10.1007/s00401-021-02286-3
70. Akaishi T, Takahashi T, Misu T, Kaneko K, Takai Y, Nishiyama S, et al. Difference in the source of Anti-AQP4-IgG and anti-MOG-IgG antibodies in CSF in patients with neuromyelitis optica spectrum disorder. Neurology. (2021) 97:e1–e12. doi: 10.1212/WNL.0000000000012175
71. Kwon YN, Kim B, Kim JS, Mo H, Choi K, Oh SI, et al. Myelin oligodendrocyte glycoprotein-immunoglobulin G in the CSF: clinical implication of testing and association with disability. Neurol Neuroimmunol Neuroinflamm. (2022) 9:e1095. doi: 10.1212/NXI.0000000000001095
72. Pace S, Orrell M, Woodhall M, Palace J, Leite MI, Irani SR, et al. Frequency of MOG-IgG in cerebrospinal fluid versus serum. J Neurol Neurosurg Psychiatry. (2022) 93:334–5. doi: 10.1136/jnnp-2021-326779
73. Sechi E, Buciuc M, Pittock SJ, Chen JJ, Fryer JP, Jenkins SM, et al. Positive predictive value of myelin oligodendrocyte glycoprotein autoantibody testing. JAMA Neurol. (2021) 78:741–6. doi: 10.1001/jamaneurol.2021.0912
74. Zara P, Floris V, Flanagan EP, Lopez-Chiriboga AS, Weinshenker BG, Solla P, et al. Clinical significance of myelin oligodendrocyte glycoprotein autoantibodies in patients with typical MS lesions on MRI. Mult Scler J Exp Transl Clin. (2021) 7:20552173211048761. doi: 10.1177/20552173211048761
75. Levy M, Yeh EA, Hawkes CH, Lechner-Scott J, Giovannoni G. Implications of low-titer MOG antibodies. Mult Scler Relat Disord. (2022) 59:103746. doi: 10.1016/j.msard.2022.103746
Keywords: myelitis, autoimmune neurology, neuroimmunology, neuroinflammation, autoantibody
Citation: Alkabie S and Budhram A (2022) Testing for Antibodies Against Aquaporin-4 and Myelin Oligodendrocyte Glycoprotein in the Diagnosis of Patients With Suspected Autoimmune Myelopathy. Front. Neurol. 13:912050. doi: 10.3389/fneur.2022.912050
Received: 03 April 2022; Accepted: 26 April 2022;
Published: 20 May 2022.
Edited by:
Elia Sechi, University of Sassari, ItalyReviewed by:
Shuhei Nishiyama, Massachusetts General Hospital and Harvard Medical School, United StatesCopyright © 2022 Alkabie and Budhram. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Adrian Budhram, YWRyaWFuLmJ1ZGhyYW1AbGhzYy5vbi5jYQ==
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.