
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
CASE REPORT article
Front. Neurol., 22 June 2022
Sec. Dementia and Neurodegenerative Diseases
Volume 13 - 2022 | https://doi.org/10.3389/fneur.2022.909944
This article is part of the Research TopicDementia and Neurodegenerative Diseases – Case Report Collection 2022View all 25 articles
 Adit Friedberg1,2,3
Adit Friedberg1,2,3 Eliana Marisa Ramos4Zhongan Yang5
Eliana Marisa Ramos4Zhongan Yang5 Luke W. Bonham1,6
Luke W. Bonham1,6 Jennifer S. Yokoyama1,2,3
Jennifer S. Yokoyama1,2,3 Peter A. Ljubenkov1
Peter A. Ljubenkov1 Kyan Younes1
Kyan Younes1 Daniel H. Geschwind7,8,9
Daniel H. Geschwind7,8,9 Bruce L. Miller1,2*
Bruce L. Miller1,2*CSF1R-related leukoencephalopathy is an autosomal dominant neurodegenerative disease caused by mutations in the tyrosine kinase domain of the colony stimulating factor 1 receptor (CSF1R). Several studies have found that hematogenic stem cell transplantation is an effective disease modifying therapy however the literature regarding prodromal and early symptoms CSF1R-related leukoencephalopathy is limited. We describe a 63-year-old patient with 4 years of repetitive scratching and skin picking behavior followed by 10 years of progressive behavioral, cognitive, and motor decline in a pattern suggesting behavioral variant of frontotemporal dementia. Brain MRI demonstrated prominent frontal and parietal atrophy accompanied by underlying bilateral patchy white matter hyperintensities sparing the U fibers and cavum septum pellucidum. Whole-exome sequencing revealed a novel, predicted deleterious missense variant in a highly conserved amino acid in the tyrosine kinase domain of CSF1R (p.Gly872Arg). Given this evidence and the characteristic clinical and radiological findings this novel variant was classified as likely pathogenic according to the American College of Medical Genetics standard guidelines. Detailed description of the prodromal scratching and skin picking behavior and possible underlying mechanisms in this case furthers knowledge about early manifestations of CSF1R-related leukoencephalopathy with the hope that early detection and timely administration of disease modifying therapies becomes possible.
CSF1R-related leukoencephalopathy is an autosomal dominant neurodegenerative disease caused by mutations in the tyrosine kinase domain of the colony stimulating factor 1 receptor (CSF1R) (1). It accounts for ~10% of adult-onset leukodystrophies in European cohorts (2). Clinically, CSF1R-related leukoencephalopathy presents with variable combinations of progressive cognitive decline, neuropsychiatric symptoms, pyramidal weakness and spasticity, parkinsonism, gait impairment and seizures. Often the pattern of behavioral and emotional change is similar to behavioral variant frontotemporal dementia (bvFTD) with apathy as a particularly prominent feature (1, 3). Disease onset is typically in the fourth or fifth decade of life and progression is relentless with mean disease duration till death of 6.8 years (4). Consistent radiological features on brain magnetic resonance imaging (MRI) include white matter hyperintensities (WMH) primarily involving the frontal and parietal lobes, with sparing of the U-fibers. Usually, these WMH are bilateral, initially patchy evolving to confluency with restricted diffusion in many cases. Thinning of the corpus callosum is characteristic. Gray matter atrophy is evident over the frontal and parietal lobes. A high incidence of a cavum septum pellucidum and cavum vergae was observed in several case series (5–7). Brain CT demonstrates calcifications distributed in the frontal white matter adjacent to the anterior horns of lateral ventricles and in the parietal subcortical white matter.
The CSF1R gene encodes a transmembrane tyrosine kinase receptor expressed in the brain, predominantly in microglia. Binding of CSF1R to its ligands (CSF1 and interleukin-34) leads to initiation of signal transductions, which contribute to development, maintenance, and activation of microglia (8). In zebrafish models, haploinsufficiency in CSF1R leads to reduced microglial density and in postmortem human brain tissue from patients with CSF1R-related leukoencephalopathy widespread microglia depletion is also evident (9). Moreover, homozygous mutations in CSF1R result in a congenital absence of microglia, abnormal brain development, and pediatric-onset leukoencephalopathy (10). CSF1R-related leukoencephalopathy is increasingly conceptualized as a neurodegenerative disease that occurs in large part due to microglial dysfunction—a primary microgliopathy (11). There are now more than 70 different pathogenic variants in CSF1R, most of which occur in the tyrosine kinase domain resulting in disruption of protein function (1).
There is emerging evidence that hematopoietic stem cell transplantation (HSCT) is a disease modifying therapy for CSF1R-related leukoencephalopathy that stabilizes disease progression, highlighting the clinical importance of early detection of individuals with pathogenic CSF1R variants (12–14). Nevertheless, detailed description of early/prodromal behavioral changes in CSF1R-related leukoencephalopathy is limited.
In this case study we report a patient with a novel likely pathogenic variant in CSF1R who presented with repetitive scratching behavior 4 years prior to the onset of progressive relentless behavioral, cognitive and motor decline accompanied by radiologic features highly consistent with CSF1R-related encephalopathy. Our goals are to enhance the field's knowledge about early phenotypic and genotypic features of CSF1R-related leukoencephalopathy and to further elucidate the heterogenous genetic causes of bvFTD clinical syndrome. We also discuss possible mechanisms underlying the scratching behavior.
Clinical and genetic evaluation was performed at the University of California, San Francisco, Memory and Aging Center as part of a prospective research study: “Frontotemporal Dementia: Genes, Images, and Emotions” (15). For patient anonymity some details have been modified.
Whole-brain structural MRI images were acquired using a 3T (Magnetom Prisma) scanner manufactured by Siemens implementing previously published acquisition protocols (16).
Whole-exome sequencing (WES) was conducted to screen known genes associated with dementia and leukoencephalopathies. Whole-exome regions were captured with the SeqCap EZ Human Exome Kit v3 and sequenced on an Illumina HiSeq4000 at the UCLA Neuroscience Genomics Core (semel.ucla.edu/ungc). Sequence reads were mapped to the GRCh37/hg19 reference genome and variants joint-called according to the Genome Analysis Toolkit best practices recommendations (17). Series of filtering steps were applied to prioritize variants, and candidate variants were confirmed by Sanger sequencing. The hexanucleotide repeat of C9orf72 was screened using both fluorescent and repeat-primed PCR, as previously described (18).
L was a 63-year-old right-handed female who presented to evaluation in the memory clinic due to 10 years of progressive behavioral changes and cognitive decline. Her early developmental history was notable for mild reading and writing difficulties in elementary school. In her youth she suffered mild brain trauma, falling twice from her bike. With both events there was loss consciousness for a short duration and no residual symptoms. After high school L attended college, where she obtained a BA. She married in her twenties. Her baseline personality was described as delightful, kind and social. At the age of 45 she presented with an adult-onset epilepsy with generalized tonic-clonic seizures. The seizures were successfully controlled with valproic acid. At the age of 49 she developed a new behavior, repetitively scratching and picking the skin of her left torso and shoulder. She never explained to her family members the reason for the scratching and did not explicitly complain of pruritus. She did not report an increasing sense of tension prior to scratching or relief during or after the behavior. The scratching increased in frequency and severity. The behavior was not accompanied by any other perseverative, stereotyped or compulsive/ritualistic behaviors. She was examined by a dermatologist who found no causative skin lesion and defined her condition as “neurogenic itch.” Four years later, at the age of 53 years she became socially withdrawn and less communicative. Her husband lost substantial amounts of their money shopping but she took no action to protect herself. At the age of 55 years she could no longer manage her finances or run her own business. She was unable to operate her computer or other technological equipment. She asked her dentist to remove all her teeth and replace them with dentures, without expressing a reason for this request. At the age of 61 she started to purchase recklessly. She got into a physical fight with her husband and subsequently they divorced. L did not express concern about the divorce and seemed to show less empathy for others. She flew balloons in the neighborhood park for multiple hours a day. She also became hyperoral with preference to sweets. At the age of 63 when she presented to evaluation, the family noticed word finding difficulties. She gradually stopped talking but acknowledged people with eye contact and head movement. She undressed in front of her family and collected soda bottle caps. Her scratching behavior worsened, and she scratched her skin to the point of bleeding. Family history was taken for four generations and was unremarkable for either early onset dementia, psychiatric disease epilepsy or motor decline. Both of her parents lived into their eighties with no cognitive decline. Her paternal and maternal families originated from Western Europe. Neurological examination was remarkable for increased axial tone and rigidity and bradykinesia in her upper limbs more so on the right. In her mental status examination L seemed inattentive, abulic and stimulus bound. Her affect was flat. Verbal output was decreased with occasional Yes/No answers, and use of overlearned phrases such as “I don't think so.” She could not follow instructions required for praxis examination. Her Mini-Mental State Examination score was 6/30. General physical examination was unremarkable except for minor excoriations over her torso secondary to scratching.
At the time of presentation, 14 years after the emergence of the scratching behavior, her complete blood count, renal and liver function tests were within normal limits suggesting that systemic causes of pruritus such as a solid or hematologic malignancy, cholestasis or renal insufficiency were not likely. Blood electrolyte levels, thyroid functions, coagulation tests, HBA1C, B12 levels, methylmalonic acid levels and very-long-chain fatty acid (VLCFA), were also all within normal limits.
Brain MRI demonstrated bilateral symmetrical global cortical atrophy (Figure 1). Atrophy was most pronounced in the frontal and parietal lobes, particularly in the medial frontal cortices. Profound thinning of the corpus callosum was evident. T2 FLAIR sequence showed bilateral patchy WMH involving frontal and parietal white matter, sparing the U fibers. Cavum septum pellucidum was noted. Diffusion weighted imaging did not show a pattern of restricted diffusion and T2* sequences did not demonstrate signal abnormalities consistent with calcifications.
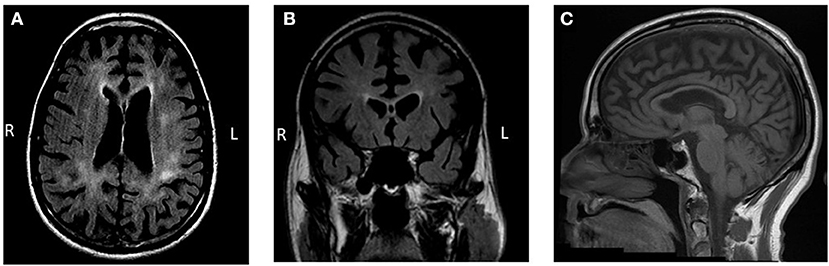
Figure 1. Brain MRI findings. (A,B) Bilateral patchy and predominantly frontoparietal white matter hyperintensities with accompanying marked gray matter atrophy are observed on T2-weighted fluid-attenuated inversion recovery (FLAIR) imaging. Cavum septum pellucidum is also evident. (C) Thinning of the corpus callosum is observed on a T1-weighted sequence.
The clinical syndrome met research criteria for probable bvFTD (19). The combination of gray matter atrophy and WMH led the neurologists to consider underlying neuropathological etiologies associated with white matter signal changes such as corticobasal degeneration and frontotemporal lobar degeneration (FTLD) TDP type A. The pattern of white matter involvement in the setting of protracted clinical course was suggestive of leukodystrophy as a primary etiology, particularly CSF1R-related leukoencephalopathy considering the characteristic brain MRI findings (20) and seizures. To promote the diagnostic process WES was performed to analyze a large panel of genes associated with dementia and leukoencephalopathies.
WES revealed a novel heterozygous missense variant c.2614G>A (p.Gly872Arg) in the CSF1R gene (Figure 2). This variant, not found in the Genome Aggregation Database (gnomAD) (21), was consistently predicted as damaging by multiple in silico tools [PolyPhen-2 (http://genetics.bwh.harvard.edu/pph2), SIFT (http://sift.jcvi.org) and CADD (http://cadd.gs.washington.edu)]. It lies in the tyrosine kinase domain, similarly to many other CSF1R-related leukoencephalopathy causing variants, in a highly conserved amino acid. Segregation analysis of this variant was not feasible due to unreachable relatives of the proband. Given these evidences and the characteristic clinical and radiologic findings (22), this variant was classified as likely pathogenic according to the American College of Medical Genetics standard guidelines (23).
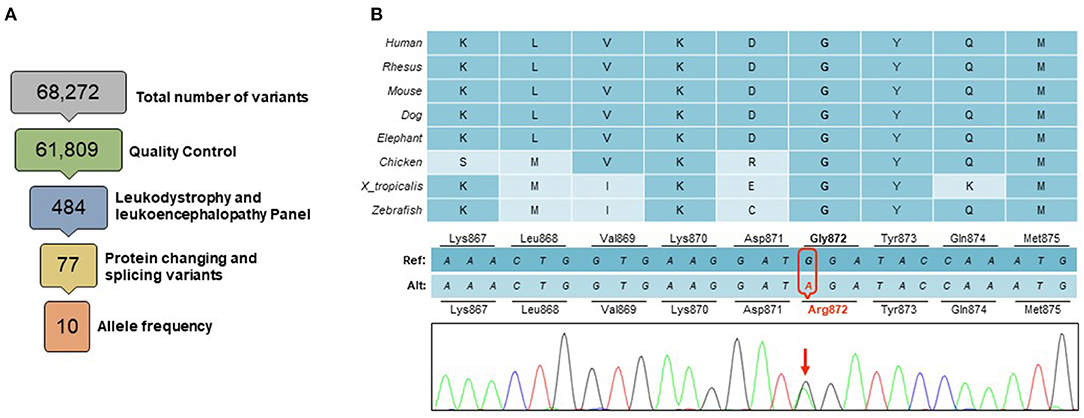
Figure 2. Genetic findings. (A) A total of 68,272 variants were identified within the targeted exome. Variants of low quality were removed through quality control (GQ <20, RD <10, AF <0.25). Among a panel of 165 genes known to be associated with leukodystrophies and leukoencephalopathies, a total of 484 variants were identified, of which 77 were protein changing or splicing variants. Only 10 of these had an allele frequency <0.10 in gnomAD database, including a novel heterozygous missense variant in the CSF1R gene (B) This change in a highly conserved amino acid in the CSF1R tyrosine domain (p.Gly872Arg), was predicted to be deleterious by multiple in silico tools.
At the time L presented to our care, HSCT was not yet considered a therapeutic option and was not offered. L passed away due to her neurogenerative disease and was not enrolled into an autopsy program.
We present a patient with a progressive behavioral cognitive and motor syndrome compatible with bvFTD which was preceded by adult onset generalized epilepsy and 4 years of repetitive scratching and skin picking behavior. The pattern of white and gray matter changes, the profound thinning of the corpus callosum and the occurrence of septum pellucidum cavum were all suggestive of CSF1R-related leukoencephalopathy (22). Genetic analysis revealed a novel likely pathogenic missense variant in the CSF1R gene. Since both parents were not affected clinically this may represent a de novo genetic variant. Other possibilities include incomplete penetrance or genetic mosaicism as was shown in other sporadic cases (1, 24).
Repetitive scratching and skin picking behavior have not yet been described in the literature as an early or prodromal symptom of CSF1R-related leukoencephalopathy. Delineating and highlighting such symptoms may contribute to early detection of patients with pathogenic CSF1R variants, allowing timely treatment with new disease modifying therapies.
We suggest several possible mechanisms underlying the scratching behavior in the context CSF1R-related encephalopathy which are not mutually exclusive and may act synergistically. Scratching is considered a self-grooming behavior aimed to protect the organism of potential external threats and is observed in multiple species. In humans excessive scratching and skin picking, in a pattern like the behavior observed in this case, is observed in skin picking disorder (SPD) (25). A diffuse tensor imaging study in skin picking disorder patients demonstrated reduced white matter integrity in tracts in proximity of the bilateral anterior cingulate cortex (ACC) implicating the importance of the ACC in regulation of this behavior (26). Interestingly L presented with severe atrophy in the ACC bilaterally accompanied by adjacent white matter lesions (Figure 1). This pattern might indicate early regional involvement that accounted for the early onset of repetitive scratching and skin picking behavior. Moreover, a candidate animal model of skin picking disorder is the Hoxb8 knockout mouse. Mice with mutations of the Hoxb8 gene groom excessively, to the point of developing skin lesions (27). In this model Hoxb8 is expressed in specific populations of microglia and neuronal cells (28). The Hoxb8 gene is expressed in the orbital cortex, the anterior cingulate, the striatum, and the limbic system, structures implicated in repetitive and compulsive behaviors. Recent work in this mouse model suggested that the absence of proper maintenance of circuit modulation by Hoxb8 expressing microglia results in the excessive grooming behavior (29). It is possible that early depletion of the Hoxb8 expressing microglia population resulted in the predisposition to repetitive scratching behavior. Interestingly, in mouse models of FTD with progranulin deficiency, excessive grooming behaviors were present (30). This might imply converging neuroanatomical and/or underlying molecular mechanisms with scratching observed in CSF1R-related leukoencephalopathy. Lastly, in the previous decade CSF1R inhibitors were introduced in cancer treatment. Safety assessment of Emactuzumab, a monoclonal antibody that targets and inhibits CSF1R revealed that pruritus was a highly prevalent side effect affecting 56% of treated patients (31). Like other monoclonal antibodies Emactuzumab does not penetrate the blood brain barrier suggesting that CSF1R dysfunction in peripheral tissues might contribute to the scratching behavior.
Definite causal evidence with regards to the pathogenicity of this variant was not obtained due to the unavailability of neuropathological data and unreachable family members for segregation analysis. However, building on the knowledge accumulated from multiple studies about the underlying biology and the clinical and radiological presentation of CSF1R-related encephalopathy there is high likelihood that this variant is disease causing. Future work is needed to establish definite causal relationship between this novel genetic variant and CSF1R-related encephalopathy and to delineate the pattern of early behavioral manifestations in this condition.
The datasets presented in this article are not readily available because of ethical and privacy restrictions. Requests to access the datasets should be directed to the corresponding author.
The studies involving human participants were reviewed and approved by University of California, San Francisco Human Research Protection Program Institutional Review Board. The patients/participants provided their written informed consent to participate in this study. Written informed consent was obtained from the individual(s) for the publication of any potentially identifiable images or data included in this article.
AF, ER, and BM conceptualized the study. AF conducted the literature review and wrote the manuscript. BM, KY, and PL examined the patient. ER, ZY, DG, LB, and JY were involved in genetic data generation, analysis, and interpretation. All authors reviewed and revised the final manuscript.
This work was supported by NIH grants P01 AG019724 and P30 AG062422, by grant 2021-A-023-FEL from the Larry L. Hillblom Foundation (AF), by the Global Brain Health Institute, Alzheimer's Association, and Alzheimer's Society Pilot Awards for Global Brain Health Leaders: GBHI ALZ UK-21-721419 (AF) and NIH-NIA grant R01 AG062588 (JY). Samples from the National Cell Repository for Alzheimer's Disease (NCRAD), which receives government support under a cooperative agreement grant (U24 AG021886) awarded by the National Institute on Aging (NIA), were used in this study.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
We thank L and her family for participating in dementia research. We also thank contributors who collected samples.
1. Konno T, Kasanuki K, Ikeuchi T, Dickson DW, Wszolek ZK. CSF1R-related leukoencephalopathy: a major player in primary microgliopathies. Neurology. (2018) 91:1092–104. doi: 10.1212/WNL.0000000000006642
2. Lynch DS, Jaunmuktane Z, Sheerin UM, Phadke R, Brandner S, Milonas I, et al. Hereditary leukoencephalopathy with axonal spheroids: a spectrum of phenotypes from CNS vasculitis to Parkinsonism in an adult onset leukodystrophy series. J Neurol Neurosurg Psychiatry. (2016) 87:512–9. doi: 10.1136/jnnp-2015-310788
3. Ikeuchi T, Mezaki N, Miura T. Cognitive dysfunction and symptoms of movement disorders in adult-onset leukoencephalopathy with axonal spheroids and pigmented glia. Parkinsonism Relat Disord. (2018) 46(Suppl. 1):S39–41. doi: 10.1016/j.parkreldis.2017.08.018
4. Konno T, Yoshida K, Mizuno T, Kawarai T, Tada M, Nozaki H, et al. Clinical and genetic characterization of adult-onset leukoencephalopathy with axonal spheroids and pigmented glia associated with CSF1R mutation. Eur J Neurol. (2017) 24:37–45. doi: 10.1111/ene.13125
5. Konno T, Broderick DF, Mezaki N, Isami A, Kaneda D, Tashiro Y, et al. Diagnostic value of brain calcifications in adult-onset leukoencephalopathy with axonal spheroids and pigmented glia. AJNR Am J Neuroradiol. (2017) 38:77–83. doi: 10.3174/ajnr.A4938
6. Sundal C, Van Gerpen JA, Nicholson AM, Wider C, Shuster EA, Aasly J, et al. MRI characteristics and scoring in HDLS due to CSF1R gene mutations. Neurology. (2012) 79:566–74. doi: 10.1212/WNL.0b013e318263575a
7. Konno T, Tada M, Tada M, Koyama A, Nozaki H, Harigaya Y, et al. Haploinsufficiency of CSF-1R and clinicopathologic characterization in patients with HDLS. Neurology. (2014) 82:139–48. doi: 10.1212/WNL.0000000000000046
8. Chitu V, Gokhan S, Nandi S, Mehler MF, Stanley ER. Emerging roles for CSF-1 receptor and its ligands in the nervous system. Trends Neurosci. (2016) 39:378–93. doi: 10.1016/j.tins.2016.03.005
9. Oosterhof N, Kuil LE, van der Linde HC, Burm SM, Berdowski W, van Ijcken WFJ, et al. Colony-stimulating factor 1 receptor (CSF1R) regulates microglia density and distribution, but not microglia differentiation in vivo. Cell Rep. (2018) 24:1203–17.e6. doi: 10.1016/j.celrep.2018.06.113
10. Oosterhof N, Chang IJ, Karimiani EG, Kuil LE, Jensen DM, Daza R, et al. Homozygous mutations in CSF1R cause a pediatric-onset leukoencephalopathy and can result in congenital absence of microglia. Am J Hum Genet. (2019) 104:936–47. doi: 10.1016/j.ajhg.2019.03.010
11. Sirkis DW, Bonham LW, Yokoyama JS. The role of microglia in inherited white-matter disorders and connections to frontotemporal dementia. Appl Clin Genet. (2021) 14:195–207. doi: 10.2147/TACG.S245029
12. Eichler FS, Li J, Guo Y, Caruso PA, Bjonnes AC, Pan J, et al. CSF1R mosaicism in a family with hereditary diffuse leukoencephalopathy with spheroids. Brain. (2016) 139:1666–72. doi: 10.1093/brain/aww066
13. Gelfand JM, Greenfield AL, Barkovich M, Mendelsohn BA, Van Haren K, Hess CP, et al. Allogeneic HSCT for adult-onset leukoencephalopathy with spheroids and pigmented glia. Brain. (2020) 143:503–11. doi: 10.1093/brain/awz390
14. Tipton PW, Kenney-Jung D, Rush BK, Middlebrooks EH, Nascene D, Singh B, et al. Treatment of CSF1R-related leukoencephalopathy: breaking new ground. Mov Disord. (2021) 36:2901–9. doi: 10.1002/mds.28734
15. Rankin KP, Kramer JH, Miller BL. Patterns of cognitive and emotional empathy in frontotemporal lobar degeneration. Cogn Behav Neurol. (2005) 18:28–36. doi: 10.1097/01.wnn.0000152225.05377.ab
16. Illan-Gala I, Casaletto KB, Borrego-Ecija S, Arenaza-Urquijo EM, Wolf A, Cobigo Y, et al. Sex differences in the behavioral variant of frontotemporal dementia: A new window to executive and behavioral reserve. Alzheimers Dement. (2021) 17:1329–41. doi: 10.1002/alz.12299
17. McKenna A, Hanna M, Banks E, Sivachenko A, Cibulskis K, Kernytsky A, et al. The genome analysis toolkit: a mapreduce framework for analyzing next-generation DNA sequencing data. Genome Res. (2010) 20:1297–303. doi: 10.1101/gr.107524.110
18. DeJesus-Hernandez M, Mackenzie IR, Boeve BF, Boxer AL, Baker M, Rutherford NJ, et al. Expanded GGGGCC hexanucleotide repeat in noncoding region of C9ORF72 causes chromosome 9p-linked FTD and ALS. Neuron. (2011) 72:245–56. doi: 10.1016/j.neuron.2011.09.011
19. Rascovsky K, Hodges JR, Knopman D, Mendez MF, Kramer JH, Neuhaus J, et al. Sensitivity of revised diagnostic criteria for the behavioural variant of frontotemporal dementia. Brain. (2011) 134:2456–77. doi: 10.1093/brain/awr179
20. Resende LL, de Paiva ARB, Kok F, da Costa Leite C, Lucato LT. Adult leukodystrophies: a step-by-step diagnostic approach. Radiographics. (2019) 39:153–68. doi: 10.1148/rg.2019180081
21. Karczewski KJ, Francioli LC, Tiao G, Cummings BB, Alfoldi J, Wang Q, et al. The mutational constraint spectrum quantified from variation in 141,456 humans. Nature. (2020) 581:434–43. doi: 10.1038/s41586-020-2308-7
22. Konno T, Yoshida K, Mizuta I, Mizuno T, Kawarai T, Tada M, et al. Diagnostic criteria for adult-onset leukoencephalopathy with axonal spheroids and pigmented glia due to CSF1R mutation. Eur J Neurol. (2018) 25:142–7. doi: 10.1111/ene.13464
23. Richards S, Aziz N, Bale S, Bick D, Das S, Gastier-Foster J, et al. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American college of medical genetics and genomics and the association for molecular pathology. Genet Med. (2015) 17:405–24. doi: 10.1038/gim.2015.30
24. Saitoh BY, Yamasaki R, Hayashi S, Yoshimura S, Tateishi T, Ohyagi Y, et al. A case of hereditary diffuse leukoencephalopathy with axonal spheroids caused by a de novo mutation in CSF1R masquerading as primary progressive multiple sclerosis. Mult Scler. (2013) 19:1367–70. doi: 10.1177/1352458513489854
25. Grant JE, Odlaug BL, Chamberlain SR, Keuthen NJ, Lochner C, Stein DJ. Skin picking disorder. Am J Psychiatry. (2012) 169:1143–9. doi: 10.1176/appi.ajp.2012.12040508
26. Grant JE, Odlaug BL, Hampshire A, Schreiber LR, Chamberlain SR. White matter abnormalities in skin picking disorder: a diffusion tensor imaging study. Neuropsychopharmacology. (2013) 38:763–9. doi: 10.1038/npp.2012.241
27. Greer JM. Capecchi MR. Hoxb8 is required for normal grooming behavior in mice. Neuron. (2002) 33:23–34. doi: 10.1016/S0896-6273(01)00564-5
28. De S, Van Deren D, Peden E, Hockin M, Boulet A, Titen S, et al. Two distinct ontogenies confer heterogeneity to mouse brain microglia. Development. (2018) 145:dev175901. doi: 10.1242/dev.152306
29. Nagarajan N, Jones BW, West PJ, Marc RE, Capecchi MR. Corticostriatal circuit defects in Hoxb8 mutant mice. Mol Psychiatry. (2018) 23:1868–77. doi: 10.1038/mp.2017.180
30. Krabbe G, Minami SS, Etchegaray JI, Taneja P, Djukic B, Davalos D, et al. Microglial NFkappaB-TNFalpha hyperactivation induces obsessive-compulsive behavior in mouse models of progranulin-deficient frontotemporal dementia. Proc Natl Acad Sci U S A. (2017) 114:5029–34. doi: 10.1073/pnas.1700477114
31. Cassier PA, Italiano A, Gomez-Roca CA, Le Tourneau C, Toulmonde M, Cannarile MA, et al. CSF1R inhibition with emactuzumab in locally advanced diffuse-type tenosynovial giant cell tumours of the soft tissue: a dose-escalation and dose-expansion phase 1 study. Lancet Oncol. (2015) 16:949–56. doi: 10.1016/S1470-2045(15)00132-1
Keywords: CSF1R-related leukoencephalopathy, frontotemporal dementia, repetitive behaviors, early symptoms, scratching
Citation: Friedberg A, Ramos EM, Yang Z, Bonham LW, Yokoyama JS, Ljubenkov PA, Younes K, Geschwind DH and Miller BL (2022) Case Report: Novel CSF1R Variant in a Patient With Behavioral Variant Frontotemporal Dementia Syndrome With Prodromal Repetitive Scratching Behavior. Front. Neurol. 13:909944. doi: 10.3389/fneur.2022.909944
Received: 31 March 2022; Accepted: 02 June 2022;
Published: 22 June 2022.
Edited by:
Sudeshna Das, Massachusetts General Hospital, United StatesReviewed by:
Sergey Illarioshkin, Research Center of Neurology, RussiaCopyright © 2022 Friedberg, Ramos, Yang, Bonham, Yokoyama, Ljubenkov, Younes, Geschwind and Miller. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Bruce L. Miller, YnJ1Y2UubWlsbGVyQHVjc2YuZWR1
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.