 Keigo Takahashi1
Keigo Takahashi1 Hemanth R. Nelvagal
Hemanth R. Nelvagal Jenny Lange
Jenny Lange Jonathan D. Cooper
Jonathan D. Cooper
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Neurol. , 04 April 2022
Sec. Pediatric Neurology
Volume 13 - 2022 | https://doi.org/10.3389/fneur.2022.886567
This article is part of the Research Topic Neuronal Ceroid Lipofuscinosis: a Multidisciplinary Update View all 12 articles
While significant efforts have been made in developing pre-clinical treatments for the neuronal ceroid lipofuscinoses (NCLs), many challenges still remain to bring children with NCLs a cure. Devising effective therapeutic strategies for the NCLs will require a better understanding of pathophysiology, but little is known about the mechanisms by which loss of lysosomal proteins causes such devastating neurodegeneration. Research into glial cells including astrocytes, microglia, and oligodendrocytes have revealed many of their critical functions in brain homeostasis and potential contributions to neurodegenerative diseases. Genetically modified mouse models have served as a useful platform to define the disease progression in the central nervous system across NCL subtypes, revealing a wide range of glial responses to disease. The emerging evidence of glial dysfunction questions the traditional “neuron-centric” view of NCLs, and would suggest that directly targeting glia in addition to neurons could lead to better therapeutic outcomes. This review summarizes the most up-to-date understanding of glial pathologies and their contribution to the pathogenesis of NCLs, and highlights some of the associated challenges that require further research.
Lysosomal storage disorders (LSDs) are a group of more than 70 monogenetic diseases characterized by defects in lysosomal metabolism and subsequent accumulation of substrates. Most LSDs present with a broad phenotypic spectrum in multiple organs. This is consistent with the fact that nearly all lysosomal enzymes are ubiquitously expressed and their deficiency will therefore affect many tissue types (1). The neuronal ceroid lipofuscinoses (NCLs or Batten disease) are a group of fatal neurodegenerative LSDs affecting children and young adults. In contrast to other non-neuronopathic LSDs, the NCLs primarily affect the central nervous system (CNS), usually including the retina. The NCLs are remarkably heterogeneous diseases, with studies in both humans and animal models showing that each of 13 subtypes is caused by mutations in different individual genes and have different ages of onset, clinical symptoms, and rate of disease progression (2, 3) (Table 1). As comprehensively reviewed elsewhere (2, 3), a mutation (or mutations) in a different NCL gene causes each form of NCL. Some of these mutations are in soluble lysosomal enzymes (e.g., CLN1, CLN2, CLN10, CLN13), others are in transmembrane proteins within the lysosome (e.g., CLN3, CLN7) or elsewhere in the cell (e.g., CLN6, CLN8), or a range of proteins that vary widely in their nature and location (e.g., CLN4, CLN5, CLN11, CLN12, CLN14).
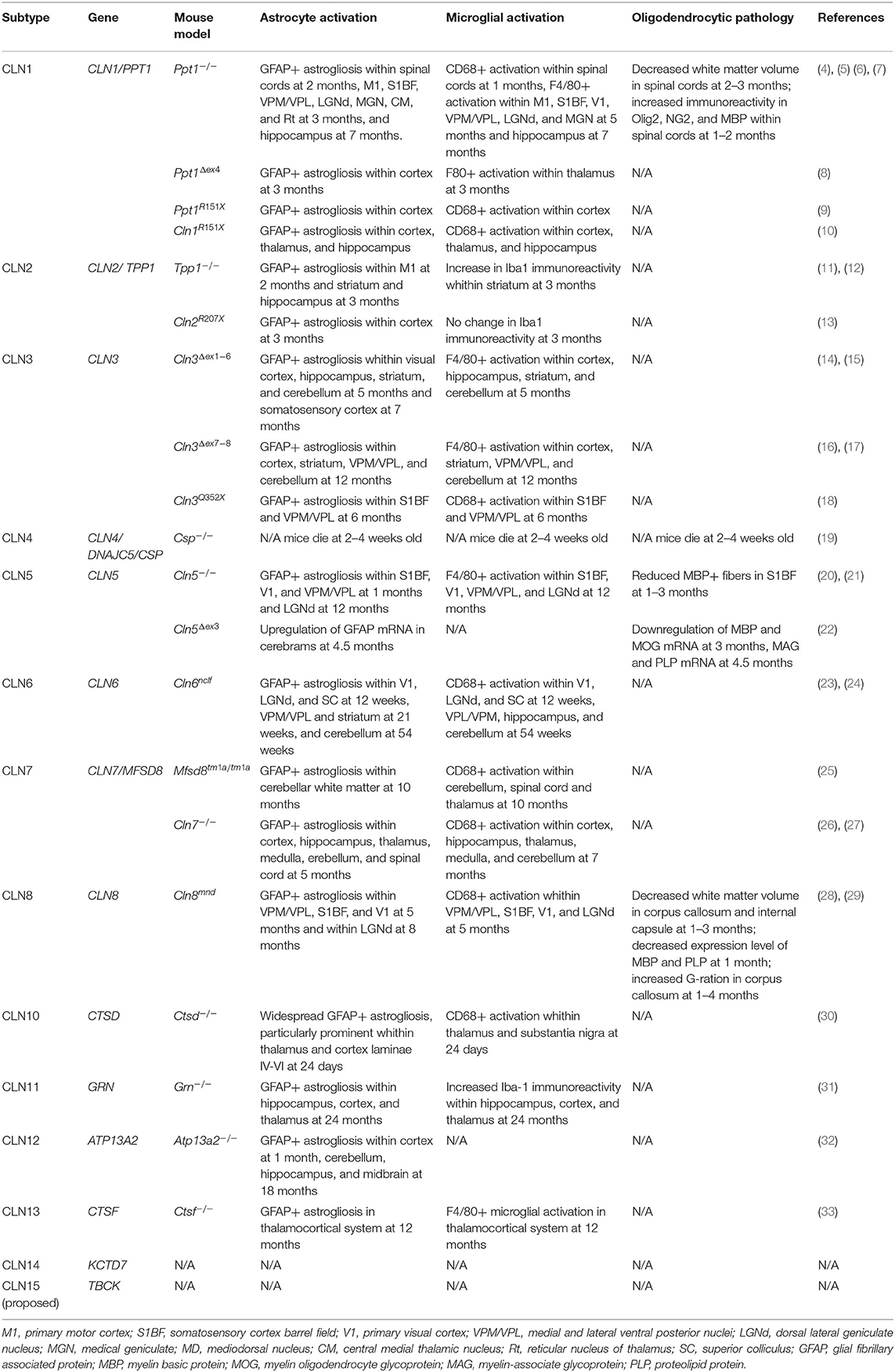
Table 1. Summary of glial changes in mouse models of neuronal ceroids lipofuscinoses.
Research into treatments for most LSDs has primarily focused on the replacement of the missing gene product responsible for each disease. Enzyme replacement therapy (ERT) for several soluble enzyme-deficient forms of NCL including CLN1 and CLN2 diseases has been studied (11, 34–38), which led to the recent FDA approval of cerliponase alfa for CLN2 disease (39). However, ERT is only disease-modifying, and several longer-term challenges regarding whether efficacy will be maintained, the delivery systems used and potential immune responses remain (36, 37, 40). Furthermore, ERT is not an option for those subtypes of NCL caused by defects in transmembrane proteins such as CLN3 disease, which is the most common form of NCL (2). Viral vector-mediated gene therapy has been intensively explored as an alternative therapeutic strategy for the NCLs. This approach theoretically has the advantage that a single one-time administration of viral vector should restore deficient lysosomal proteins to transduced cells (41, 42). Preclinical studies of gene therapy in animal models of CLN1, CLN2, CLN3, CLN6, CLN7, and CLN10 diseases have shown promising results (4, 23, 34, 43–47). However, clinical studies in children with CLN2 disease treated with gene therapy showed considerably less efficacy (48, 49), highlighting the difficulty of translating advances from mice directly into human patients (50). Indeed, none of the therapies that are currently available or being tested clinically are curative. Therefore, devising optimal therapeutic strategies for the NCLs will certainly require a better understanding of pathophysiology in each form of NCL.
Neuropathology in the NCLs was initially characterized in human autopsy studies, revealing marked neuron loss accompanied by intra-lysosomal accumulation of autofluorescent storage material (AFSM), whose major protein component is subunit C of mitochondrial ATP synthase (SCMAS), in addition to astrogliosis, and microglial activation (51, 52). With the limited availability of genetically validated human autopsy samples, many longitudinal studies in animal models have been performed, in order to understand the staging of neuropathological processes from the earliest events to the end-stage of disease. Interestingly, AFSM accumulation, astrogliosis, microglial activation and neuron loss in animal models of NCL are remarkably selective in their early stages, becoming more widespread with disease progression (53). This suggests that despite the ubiquitous expression of these proteins, such selective vulnerability may be due to them playing physiological roles of greater importance in some cell populations than others.
A significant finding made in multiple mouse models across subtypes of NCL is the profound loss of thalamic neurons, which typically precedes neuron loss in the corresponding region of the cortex to which these thalamic neurons relay (5, 14, 16, 17, 20, 28, 30, 54). Strikingly, these studies in mouse models also revealed that localized astrocytic and microglial activation, which both occur early in disease progression, accurately predict where subsequent selective neuron loss occurs in mouse models of a majority of NCL subtypes. Such findings cast doubt on traditional perspectives of the NCLs as predominantly “neuronal” diseases, and lead to the hypothesis that abnormalities in glial cells may contribute to the neurodegeneration associated with the NCLs.
In the “neuron-centric” past of neuroscience, glial cells were often relegated to being considered as undefined passive structural elements, and in the diseased state glial activation was often considered a secondary response to neuron dysfunction or damage. Over recent decades, this traditional neuron-centric conception of the CNS has been challenged by a large body of research aiming to provide a better understanding of glial function, revealing that glial cells including astrocytes, microglia, and oligodendrocytes have more active roles in both neuronal homeostasis and neurodegeneration (55–57). Notably, recent technological advancements have enabled us to study the heterogeneity of each glial cell type, and have revealed their bimodal or multimodal roles in neurodegenerative diseases (58, 59). This review aims to summarize the recent progress in our understanding of glial pathologies and their contribution to NCL pathogenesis and examines where NCL research currently stands in the field of glial biology. This review focusses primarily upon CLN1, CLN2 and CLN3 diseases as the three most common forms of NCL, in which a consideration of glial dysfunction or the contribution to pathogenesis has been undertaken or is underway. However, where available, the extent of astrogliosis and microglial activation or oligodendrocyte pathology is listed in mouse models of other forms of NCL in (Table 1). As discussed below, these immunohistochemically detectable changes may be due to dysfunction of glial cell types (which is largely unexplored in most NCLs), or reflect their response to ongoing neuronal dysfunction or loss.
Neuroimmune responses mediated by both astrocytes and microglia have crucial roles in all CNS insults including brain injury, infection, and neurodegenerative diseases (60, 61). In response to these insults, astrocytes and microglia become “activated” or “reactive” by altering their morphology, protein expression, and secretion profile. The fact that astrocytes and microglia typically both become activated in concert has made it difficult to distinguish the relative contributions of astrocytes to neurodegeneration, and whether these are distinct from those of microglia. Nonetheless, understanding their distinct patterns of activation in disease states is very important.
Upregulation of intermediate filaments, most notably glial fibrillary acidic protein (GFAP), is a classic marker for astrogliosis in mammalian models, and the expression level of GFAP or immunohistochemical detection of this marker has proved a useful tool to assess the extent of astrogliosis (62). As summarized in Table 1, GFAP-positive astrogliosis has been documented in all characterized mouse models of NCL. Although astrogliosis is observed in multiple CNS regions toward the end stage of disease, typical astrogliosis in the NCLs is characterized by its regional specificity and timing; astrogliosis especially occurs early and is pronounced in thalamocortical pathways where considerable subsequent neuron loss occurs (reviewed in 26, see individual references in Table 1). This is in contrast to many other neuropathic LSDs such as mucopolysaccharidosis (MPS), in which astrogliosis tends to be more generalized across the CNS throughout disease progression (63, 64). Interestingly, the extent of GFAP reactivity and morphological alteration in astrocytes varies across the NCLs. For example, hypertrophy of astrocyte cell bodies and processes and GFAP upregulation in Cln3−/− mice appears to be more subtle or perhaps attenuated compared to astrocytes observed in Ppt1−/− mice (14), implying that CLN1 and CLN3 diseases differ in the extent to which astrocytes are intrinsically dysfunctional and/or respond to extracellular stimuli. These differences in astrogliosis in CLN1 and CLN3 diseases are also recapitulated by in vitro experiments using primary astrocytes derived from the relevant mouse models; Ppt1−/− astrocytes exhibit a more activated morphology and higher expression levels of GFAP, and enhanced secretion of cytokine and chemokine compared with the wild-type astrocytes (65). In contrast, Cln3−/− astrocytes showed attenuated changes in morphology and GFAP expression in response to pharmacological stimulation with reduced secretion of a range of neuroprotective factors, mitogens, cytokines, and chemokines (66). It will therefore be important to further investigate the nature of astrocytic dysfunction using similar tissue culture methods for other forms of NCL such as CLN2 disease.
Recently, it has been demonstrated that GFAP depalmitoylation is regulated by PPT1, and blocking palmitoylation by the unique palmitoylated residue in GFAP attenuates both astrogliosis and the concurrent neurodegenerative pathology in CLN1 mice (67). This is the first evidence suggesting that loss of NCL proteins in astrocytes directly impacts an intrinsic astrocyte response rather than “reactive astrogliosis” occurring solely in response to ongoing neuronal damage. However, these findings appear somewhat contradictory to previous evidence showing that prevention of GFAP upregulation by knocking out both GFAP and Vimentin in Ppt1−/− mice (Gfap−/−; Vimentin−/−; Ppt1−/−) exacerbates disease pathology, which had been interpreted as evidence for a protective role of GFAP upregulation in CLN1 disease (68). Not only do such findings imply multi-dimensional roles of astrogliosis, which will be discussed shortly, but also potentially different pathological impacts depending on NCL subtype, affected brain regions and staging of disease progression.
Recent efforts have focused on gene expression profiling of activated astrocytes both in vitro and in vivo to decipher their functional properties in the context of neurodegeneration. The paradigm of neurotoxic “A1” astrocytes and neuroprotective “A2” astrocytes is now a generally recognized concept (62, 69). Astrocytes resembling “A1” or neurotoxic status have been reported in more common neurodegenerative diseases such as Alzheimer's disease (AD) (70), amyotrophic lateral sclerosis (ALS) (71), and Parkinson's disease (72). Similarly, the pronounced typical A1-specific molecular signature has been recently reported in the forebrains of Ppt1−/− mice (73), suggesting a neurotoxic function of astrocytes in CLN1 disease. However, caution is needed in using the current A1/A2 classifications to interpret pathological roles of astrocytes, because such a binary A1/A2 paradigm may be an oversimplification of potentially more wide-ranging and heterogeneous states of astrogliosis (74). Indeed, the recent RNA sequencing data of Tpp1−/− mice have shown changes in the expression of a restricted subset of A1- or A2-specific genes, which does not match the typical A1/A2 classification (75). A lack of clear A1/A2 signature has also been reported in other diseases including Huntington disease (76), highlighting that astrocyte heterogeneity may convolute A1/A2 boundaries. Nevertheless, there is a potential that these widely accepted A1/A2 markers can still be useful for both investigating the pathological contribution of astrogliosis, comparing astrocyte phenotypes in the NCLs to other neurodegenerative conditions and assessing the efficacy of therapeutic approaches for NCLs.
Astrocytes also exert pathological influences on neuronal health through multiple non-inflammatory functions such as neurotransmitter recycling, ion buffering, and the release of growth factors (77, 78). In addition, the role of phagocytosis by astrocytes in synaptic connectivity is now in the spotlight but has been relatively understudied in neurodegenerative diseases (79). Considering their close relationship with lysosomal calcium signaling and lysosomal exocytosis, it is plausibly speculated that the loss of NCL proteins could affect many of these non-inflammatory functions of astrocytes. Impaired calcium signaling in primary astrocytes derived from Ppt1−/− and Cln3−/− mice has been documented (65, 66). Therefore, it will be important to decipher the molecular bases of possibly more diverse forms of astrocytic dysfunction in the NCLs rather than solely focusing on astrogliosis to better understand the pathological role of astrocytes in NCL pathogenesis.
Microglia, the CNS tissue resident macrophage population, also become “activated” or “reactive” by changing their gene expression, morphology, motility, migration, metabolism, secretome, phagocytosis, proliferation, and death in response to CNS pathology (61). Microglial-astrocyte crosstalk via the release of diverse signaling molecules is particularly thought to mediate neurodegeneration (80), with recent studies suggesting that neurotoxic A1 astrocytes are triggered by fragmented mitochondria released from microglia to propagate and trigger neuronal death (81, 82).
Classically, immunoreactivity of several molecular markers including CD68, MHC antigen class II, F4/80, and Iba1 have been widely used to define the activated state of microglia (83, 84). Longitudinal studies using several of these markers have confirmed that where examined microglial activation is invariably present in the CNS of NCL mouse models, and anatomical distribution and onset of microglial activation largely overlap those of astrogliosis (Table 1). Although comprehensive profiling of multiple microglial markers is still underway, data so far suggest that the nature of microglial activation appears to be different in each NCL. This subtype-dependent nature of microglial activation is buttressed by in vitro primary culture experiments in CLN1 and CLN3 disease; Ppt1−/− microglia are morphologically more activated with increased secretion of IL-1β (65), whereas Cln3−/− microglia exhibit attenuated morphological responses to pharmacological stimulation with reduced secretion of several chemokines (66). Notably, when Ppt1−/− astrocytes and microglia were co-cultured, they appeared to cross-prime one another to exacerbate neuron loss (65), implicating the involvement of astrocyte-microglia crosstalk in CLN1 disease pathophysiology.
Recent research has been delineating the complex and heterogeneous state of activated microglia, a topic that is still under debate. The classification of pro-inflammatory “M1” microglia vs. anti-inflammatory “M2” microglia using the expression of particular cell surface markers and cytokines had been long recognized (57, 84), despite the validity of such a classification still being under scrutiny. M1 polarization of microglia with upregulation of CD16/32 and CD86 has been reported in Ppt1−/− and Cln3−/− mice, and knocking out of the inflammation-related cell adhesion molecule sialoadhesin in those mice attenuated numbers of M1-polarized microglia, levels of pro-inflammatory cytokines, and altered disease phenotype (85). However, given criticism that the M1/M2 dichotomy provides an oversimplified perspective (86, 87), a new pathological classification that incorporates the concept of disease-associated microglia (DAM) has recently been put forth (58, 88). DAM are molecularly characterized by the expression of typical microglial genes such as Iba1, Cst3, and Hexb, coincident with downregulation of homeostatic microglial genes including P2ry12, P2ry13, Cx3cr1, CD33, and Tmem119 (89). DAM further display upregulation of genes involved in lysosomal, phagocytic, and lipid metabolism pathways such as Apoe, Ctsd, Lpl, Tyrobp, and Trem2, which perhaps makes the DAM classification particularly pertinent to LSDs. RNA sequencing data has revealed the existence of both TREM2-independent and TREM2-dependent DAM in Tpp1−/− mice, suggesting the pro-inflammatory and neurotoxic role of activated microglia in CLN2 disease (75, 90). However, the pathological role of DAM still remains debatable; several recent studies have shown neuroprotective effects of TREM2-dependent DAM in mouse models of AD and GRN haploinsufficiency-causing frontotemporal lobar degeneration (GRN-FTLD) (91, 92), suggesting the pathological contribution of DAM may well be disease-dependent. Interestingly, complete deficiency of Grn−/− is known to cause CLN11 disease (31), suggesting a similar phenotype may exist in some forms of NCL. Therefore, caution should be exercised in overinterpreting data for the expression of, or staining for, DAM markers and it will be wise not to solely rely on such findings when interpreting pathological roles of activated microglia in NCL pathogenesis in future studies.
The secretion of cytokines and chemokines is of paramount importance for both astrocytes and microglia to exert pro- and anti-inflammatory effects on the process of neurodegeneration (93). The progressive elevation of multiple cytokines and chemokines has been confirmed by whole transcriptomics and/or proteomics in the forebrains and cerebella of Tpp1−/− mice (75, 90) and forebrains and spinal cords of Ppt1−/− mice (68, 94, 95). Such evidence for the region- and subtype-specific nature of neuroinflammatory changes in CLN1 and CLN2 diseases correlates with the previously shown region- and subtype-specific immunoreactivity of astrogliosis and microglial activation markers. Pharmacological modulation of neuroinflammation is an emerging therapeutic strategy for neurodegenerative diseases (96). Until now, only a few anti-inflammatory drugs have been preclinically tested for NCLs: fingolimod, teriflunomide, and MW151 in Ppt1−/− mice (97, 98) and ibuprofen and mycophenolate motefil in Cln3−/− deficient mice (99, 100) and provide only partial phenotypic rescue. While modulation of neuroinflammation may provide additional therapeutic benefit, especially when used in combination with other therapies such as ERT or gene therapy, these preclinical results suggest that alteration of central pro-inflammatory cascades in NCL mice might be a non-specific downstream consequence.
Other non-immune-related properties of microglia also have a significant impact on neuronal health. Microglial-mediated phagocytosis is critical in maintaining CNS homeostasis by pruning synapses or phagocytizing dysfunctional, dying or the debris of deceased neurons and other cell types (57, 101). It has been shown that impaired microglial phagocytic function promotes the development of several neurological diseases such as Rett syndrome (102) and tuberous sclerosis complex (103). Since phagocytosis requires focal exocytosis of lysosomes (104), it is plausible to speculate that lysosomal dysfunction due to NCL protein deficiency could also impair phagocytosis in these cells. While there have been several pieces of evidence from RNA sequencing or proteomics analysis suggesting altered phagocytosis in the brains of Ppt1−/− and Tpp1−/− mice (75, 94), microglial-specific alteration of phagocytosis is yet to be elucidated. A better understanding of the nature of such dysfunctional phagocytosis by microglia and its contribution to NCL pathogenesis may therefore inform us of new therapeutic targets.
Demyelination is another pathological change widely seen in multiple neurodegenerative diseases. Consistent with recent evidence suggesting the regulatory roles of lysosomal exocytosis in myelination, abnormal myelination is commonly seen in many LSDs including Niemann-Pick disease, Gaucher disease, metachromatic leukodystrophy, multiple sulfatase deficiency, and Krabbe disease (105–107). In contrast, pathological evidence of either dysmyelination or demyelination in the NCLs has been investigated only in mouse models of CLN1, CLN5, and CLN8 diseases with limited depth of characterization (Table 1). A key question is whether overt demyelination occurs at all in these disorders, or whether any changes in myelin composition occur secondary to loss of axons, as a result of neuron loss. Certainly, changes in white matter volume are evident in both animal models and human autopsy specimens (6, 21, 29), but its basis is poorly understood. Of course, any consideration of myelin must necessarily include Schwann cells in the peripheral nervous system (PNS), which serve a similar, but not identical role to oligodendrocytes in the CNS. However, the pathological impact of the NCLs upon the PNS is largely underappreciated, but is currently of renewed interest.
A key question that remains to be answered is whether or not the loss of NCL proteins from glial cells confers any direct cell-autonomous effects on these glial cells themselves and/or non-cell-autonomous effects on other cell types including neurons in either a harmful or protective manner. In in vitro studies using primary astrocytes, neuron-glial co-culture experiments showed that both Ppt1−/− and Cln3−/− glia are detrimental to the survival of both wild-type and mutant neurons (65, 66). Such data raise the possibility that mutant astrocytes and microglia may actively trigger the neurodegenerative changes seen in CLN1 and CLN3 diseases. Such in vitro models are a crucial component in unraveling cell-type-specific contributions to disease pathogenesis and lend themselves to high throughput screening to detect novel phenotypes and assess potential therapeutic interventions (108–110). Using this approach has highlighted disease-modifying pathways in a number of neurodegenerative diseases that may provide valuable therapeutic targets. Furthermore, the advent of induced pluripotent stem cell (iPSC) models allows the close physiological representation of disease-affected cells on a species-specific genetic background. iPSC models have only been used to a limited extent in the NCLs to date and have so far not been used to generate glial cells despite the availability of well-established differentiation protocols (111–113). For the NCLs, it will be vital to further investigate glial phenotypes in vitro and to validate those findings by generating cell-type-specific mutant mice to explore these issues in vivo.
Microglial depletion using CSF-1R inhibitors has enabled us to study the direct effect of microglia on the CNS disease process in mammalian models (114). With this technique, it has been shown that microglial depletion in Ppt1−/− mice attenuated optic nerve pathologies and several behavioral abnormalities (115). Although such findings might be confounded by the fact that completely abolishing microglia is likely to negatively impact CNS homeostasis, such studies still provide a degree of mechanistic insight into microglial contributions to CLN1 disease progression. Since the effectiveness and safety of some CSF-1R inhibitors have been proven in humans (114) and as new and more specific CSF-1R inhibitors become available, microglial depletion may be a clinically relevant approach.
The Cre-LoxP system in mice has proved a powerful tool to investigate the effect of cell-type-specific genetic mutation on neurodegeneration and applied to a wide range of diseases including LSDs in vivo. For example, it has been shown that astrocytic-specific deletion of Sulfatase Modifying Factor 1 (SUMF1) (Sumf1flox/flox; GFAP-Cre) was sufficient to induce neuron loss in a mouse model of multiple sulfatase deficiency (MSD) (116). Also, microglial-specific deletion of NPC1 (Npc1flox/−; Cx3cr1-Cre) has been shown to enhance microglial phagocytotic uptake and impaired lipid trafficking, resulting in impaired myelin turnover in a mouse model of Niemann-Pick type C (NPC) disease (117), caused by a deficiency in the NPC1 protein. In contrast, it has also been shown that astrocytic-specific deletion of NPC1 (Npc1flox/−; GFAP-CreER) does not cause neurodegeneration, but neuron-specific knockout (Npc1flox/−; Syn1-Cre) does in the NPC mouse model (118). Such data suggest that the nature of the glial contribution to pathogenesis is likely to differ between LSDs. However, no study has yet investigated the effect of astrocyte-, microglial-, or oligodendrocyte-specific deletion of NCL genes in vivo has been reported, indicating that NCL research regarding glial pathology is admittedly lagging behind other LSDs. Perhaps this is in part because of the sheer body of work this would entail given the number of NCL subtypes, as well as the fact that several of the genes that are deficient in the NCLs are lysosomal enzymes that are normally secreted and can be taken up by neighboring cells via a variety of receptor subtypes (42). This process of “cross-correction” naturally confounds and complicates any attempts to generate cell-type-specific PPT1 or TPP-1 deficient mice. However, recent work in creating chimeric “tethered” versions of enzymes might indeed enable the creation of conditional cell-type-specific models (119).
Our relatively poor understanding of the pathomechanisms that operate in the NCLs has certainly hampered the generation of more effective therapeutic strategies. Until recently, glial cells across various neurodegenerative diseases have often been considered as poorly defined passive structural elements. The underappreciated consideration of glial involvement in the NCLs is no exception, which is perhaps reflected by the re-naming of these disorders in the 1960s as “neuronal ceroid lipofuscinoses” (120) to distinguish them from other childhood encephalopathies. The rapidly expanding body of research into normal glial biology and their responses to disease has facilitated a reassessment that glia are not just passive bystanders of pathology in the CNS, but instead are active determinants of neurodegeneration. As summarized in this review, there is substantial evidence suggesting such glial involvement in NCL pathophysiology, and changes in glial activation are frequently used to evaluate therapeutic efficacy in preclinical studies (4, 11, 15, 23, 26, 34, 43–47, 97). Of necessity, this review focusses primarily upon the three most common forms of NCL, CLN1 disease, CLN2 disease and CLN3 disease, in which the issue of glial contribution to pathogenesis has been considered. Nevertheless, as detailed in Table 1, glial activation is present in all forms of NCL and is consistently present before neuron loss occurs. As such, we might anticipate that glia may also be involved in the pathogenesis of these other forms of NCL. However, given the pronounced difference between even CLN1, CLN2 and CLN3 disease that are discussed in this review, it could be expected that the extent and nature of glial involvement may also vary markedly between types of NCL. Nevertheless, although the glial contribution to disease progression has been intensively studied in other neurodegenerative diseases, relatively little is known about whether glia contribute mechanistically to the profoundly neurodegenerative phenotype of most forms of NCL.
There are several remaining issues that still need addressing in order to clarify the contribution of glial pathology in the NCLs. First, all of the many of subcellular alterations known to be associated with NCLs and other LSDs such as impaired autophagy, lysosomal trafficking, and alterations in the mTOR and TFEB signaling pathways have primarily been studied in neurons or fibroblasts, but not specifically in glial cells of any variety (27, 104, 121–126). Indeed, there is considerable potential that studying these pathways in NCL glia will yield valuable mechanistic information about cell-type-specific impacts of disease-causing mutations. Second, while NCL research has predominantly relied on mouse models, recent evidence has suggested species-dependent differences in the functional properties of astrocytes, questioning the translational relevance of information mouse astrocytes (127). As this issue almost certainly applies to microglia and oligodendrocytes as well, the implementation of glia differentiated from human NCL-patient-derived iPSCs is likely to be of considerable benefit (113). Third, as already discussed, studying the cell-autonomous effects of soluble enzyme deficiency in vivo is hampered by “cross-correction,” a phenomenon via which mannose 6-phosphate receptor-mediated endocytosis facilitates extracellularly delivered lysosomal enzymes to be taken up by recipient cells. As a previous example of the way to overcome this challenge, the chimeric GALC enzyme tethered to the lysosomal membrane has been engineered in the Krabbe disease mouse model so the cell-autonomous effect of oligodendrocyte-specific GALC deficiency could be studied (119). It will be important to extend such methodology to PPT1 and TPP1 in order to address the cellular autonomy of CLN1 and CLN2 diseases, respectively.
Modern “omics technologies have greatly contributed to a better understanding of the complex physiological nature of glial pathologies in the NCLs and other LSDs (128, 129). RNA sequencing has been widely used in the field of NCLs now that its cost is substantially reduced, but there are a number of caveats concerning the validity of RNA sequencing results. For example, RNA sequencing of a bulk tissue cannot distinguish molecular events in different cell types. As such distinct molecular changes that occur in specific glial cell populations such as microglia and oligodendrocytes, which comprise a relatively small proportion of the total cells present in these samples, might be masked. The application of the single-cell or single-nucleus RNA sequencing technology can theoretically overcome this issue (101), and is likely to reveal new insights into the broad range of effects upon glia in the NCLs. Another issue, which is perhaps unique in LSD research, is that lysosomal proteins play a crucial role in post-translational modification and intracellular trafficking (104, 130), which transcriptomics analyses cannot address. Proteomics analysis instead is more suitable in this case, but again, proteomic data obtained from bulk tissue cannot distinguish between different cell types. Most recently, single-cell proteomics technologies have been invented (131), and it may be predicted that this approach will be widely used to study glial biology in near future.
Notably, glia also exist outside the CNS in different forms depending on the anatomical region. Schwann cells are the myelinating cells in the peripheral nervous system (PNS) and are involved in maintaining ionic balance and providing support to axons (132). There are also non-myelinating Schwann cells called terminal Schwann cells, residing at the neuromuscular junction (133). Satellite glial cells are found in peripheral ganglia and potentially have similar functions to astrocytes in the CNS (134). There is also a unique population of astrocyte-like cells called enteric glial cells, involved in the regulation of the intestinal epithelial barrier and in regulating the function of neurons within the enteric nervous system (ENS) (135). Given the accumulated evidence for glial abnormalities across multiple forms of NCL, it will be important to investigate the impact of disease upon these “non-CNS glial cells” that are key components of the PNS and ENS. These may represent important cellular targets to obtain better therapeutic outcomes in patients with NCLs.
To conclude, much like the different types of musicians in a band that need to coordinate together with its singer to produce harmonious music, different glial cells provide coordinated support for neuronal health. As in a band it only takes one member to perform sub-optimally for the music to be compromised, and it is very likely that the dysfunction of any one type (or types) of glia similarly contribute to neurodegeneration. With recent technical advances, we are now entering an exciting time for expanding our knowledge of glial dysfunction and its contribution to the pathogenesis of the NCLs. This knowledge will almost certainly help us design more effective and appropriately targeted therapeutic strategies for these disorders.
KT: conceptualization, investigation, and writing the original draft. HN and JL: writing, reviewing, and editing. JC: supervision, conceptualization, reviewing, and editing. All authors contributed to the article and approved the submitted version.
The many studies from the Pediatric Storage Disorders Laboratory (PSDL) cited in this review were funded from a variety of sources: US National Institutes of Health (NS 043205, NS41930, NS44310, NS116574, and NS117635), the Wellcome Trust, UK Medical Research Council, European Union 6th Framework Programme, European Union 7th Framework Programme and Horizon 2020 research and innovation programme under Grant Agreement No. 666918 (BATCure), Sparks Foundation, Batten Disease Support and Research Association (BDSRA), Batten Disease Family Association (BDFA), Beyond Batten Disease Foundation, NCL Stiftung, The Saoirse Foundation and Health Research Board of Ireland, Noah's Hope/Hope for Bridget, The Fore Batten Foundation, Hailey's Heroes, The Children's Brain Diseases Foundation, The Bletsoe Family, and The Natalie Fund. This work was also made possible by institutional support from the Department of Pediatrics, Washington University in St Louis to JC, and a McDonnell International Scholars Academy award to KT.
JC has received research support from BioMarin Pharmaceutical Inc., Abeona Therapeutics Inc., Regenexbio Inc., and Neurogene.
The remaining authors declare that the research was conducted in the absence of any commercial orfinancial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
The continued support of our funders is greatly appreciated, as is the outstanding efforts of members of the laboratory past and present. We would also like to thank Dr. Alison Barnwell for constructive comments on the manuscript.
1. Kingma SDK, Bodamer OA, Wijburg FA. Epidemiology and diagnosis of lysosomal storage disorders; Challenges of screening. Best Pract Res Clin Endocrinol Metab. (2015) 29:145–57. doi: 10.1016/j.beem.2014.08.004
2. Mole SE, Cotman SL. Genetics of the neuronal ceroid lipofuscinoses (Batten disease). Biochim Biophys Acta. (2015) 1852:2237–41. doi: 10.1016/j.bbadis.2015.05.011
3. Mole SE, Anderson G, Band HA, Berkovic SF, Cooper JD, Kleine Holthaus SM, et al. Clinical challenges and future therapeutic approaches for neuronal ceroid lipofuscinosis. Lancet Neurol. (2019) 18:107–16. doi: 10.1016/S1474-4422(18)30368-5
4. Shyng C, Nelvagal HR, Dearborn JT, Tyynelä J, Schmidt RE, Sands MS, et al. Synergistic effects of treating the spinal cord and brain in CLN1 disease. Proc Natl Acad Sci U S A. (2017) 114:E5920–9. doi: 10.1073/pnas.1701832114
5. Bible E, Gupta P, Hofmann SL, Cooper JD. Regional and cellular neuropathology in the palmitoyl protein thioesterase-1 null mutant mouse model of infantile neuronal ceroid lipofuscinosis. Neurobiol Dis. (2004) 16:346–59. doi: 10.1016/j.nbd.2004.02.010
6. Nelvagal HR, Dearborn JT, Ostergaard JR, Sands MS, Cooper JD. Spinal manifestations of CLN1 disease start during the early postnatal period. Neuropathol Appl Neurobiol. (2020) 47:251–67. doi: 10.1111/nan.12658
7. Kielar C, Maddox L, Bible E, Pontikis CC, Macauley SL, Griffey MA, et al. Successive neuron loss in the thalamus and cortex in a mouse model of infantile neuronal ceroid lipofuscinosis. Neurobiol Dis. (2007) 25:150–62. doi: 10.1016/j.nbd.2006.09.001
8. Jalanko A, Vesa J, Manninen T, Von Schantz C, Minye H, Fabritius AL, et al. Mice with Ppt1 Δex4 mutation replicate the INCL phenotype and show an inflammation-associated loss of interneurons. Neurobiol Dis. (2005) 18:226–41. doi: 10.1016/j.nbd.2004.08.013
9. Bouchelion A, Zhang Z, Li Y, Qian H. Mukherjee AB. Mice homozygous for c451C>T mutation in Cln1 gene recapitulate INCL phenotype. Ann Clin Transl Neurol. (2014) 1:1006–23. doi: 10.1002/acn3.144
10. Miller JN, Kovács AD, Pearce DA. The novel Cln1R151X mouse model of infantile neuronal ceroid lipofuscinosis (INCL) for testing nonsense suppression therapy. Hum Mol Genet. (2015) 24:185–96. doi: 10.1093/HMG/DDU428
11. Chang M, Cooper JD, Sleat DE, Cheng SH, Dodge JC, Passini MA, et al. Intraventricular enzyme replacement improves disease phenotypes in a mouse model of late infantile neuronal ceroid lipofuscinosis. Mol Ther. (2008) 16:649–56. doi: 10.1038/mt.2008.9
12. Ghosh A, Rangasamy SB, Modi KK, Pahan K. Gemfibrozil, food and drug administration-approved lipid-lowering drug, increases longevity in mouse model of late infantile neuronal ceroid lipofuscinosis. J Neurochem. (2017) 141:423–35. doi: 10.1111/jnc.13987
13. Geraets RD, Langin LM, Cain JT, Parker CM, Beraldi R, Kovacs AD, et al. tailored mouse model of CLN2 disease: a nonsense mutant for testing personalized therapies. PLoS ONE. (2017) 12:e0176526. doi: 10.1371/journal.pone.0176526
14. Pontikis CC, Cella C V, Parihar N, Lim MJ, Chakrabarti S, Mitchison HM, et al. Late onset neurodegeneration in the Cln3 -/- mouse model of juvenile neuronal ceroid lipofuscinosis is preceded by low level glial activation. Brain Res. (2004) 1023:231–42. doi: 10.1016/j.brainres.2004.07.030
15. Kovács AD, Saje A, Wong A, Ramji S, Cooper JD, Pearce DA. Age-dependent therapeutic effect of memantine in a mouse model of juvenile Batten disease. Neuropharmacology. (2012) 63:769–75. doi: 10.1016/J.NEUROPHARM.2012.05.040
16. Pontikis CC, Cotman SL, MacDonald ME, Cooper JD. Thalamocortical neuron loss and localized astrocytosis in the Cln3Δex7/8 knock-in mouse model of Batten disease. Neurobiol Dis. (2005) 20:823–36. doi: 10.1016/j.nbd.2005.05.018
17. Cotman SL. Cln3 Deltaex7/8 knock-in mice with the common JNCL mutation exhibit progressive neurologic disease that begins before birth. Hum Mol Genet. (2002) 11:2709–21. doi: 10.1093/hmg/11.22.2709
18. Langin L, Johnson TB, Kovács AD, Pearce DA, Weimer JM. A tailored Cln3 Q352X mouse model for testing therapeutic interventions in CLN3 Batten disease. Sci Rep. (2020) 10:1–12. doi: 10.1038/s41598-020-67478-5
19. Alevy J, Burger CA, Albrecht NE, Jiang D, Samuel MA. Progressive myoclonic epilepsy-associated gene Kctd7 regulates retinal neurovascular patterning and function. Neurochem Int. (2019) 129:104486. doi: 10.1016/J.NEUINT.2019.104486
20. von Schantz C, Kielar C, Hansen SN, Pontikis CC, Alexander NA, Kopra O, et al. Progressive thalamocortical neuron loss in Cln5 deficient mice: Distinct effects in Finnish variant late infantile NCL. Neurobiol Dis. (2009) 34:308–19. doi: 10.1016/j.nbd.2009.02.001
21. Schmiedt ML, Blom T, Blom T, Kopra O, Wong A, von Schantz-Fant C, et al. Cln5-deficiency in mice leads to microglial activation, defective myelination and changes in lipid metabolism. Neurobiol Dis. (2012) 46:19–29. doi: 10.1016/j.nbd.2011.12.009
22. Kopra O, Vesa J, von Schantz C, Manninen T, Minye H, Fabritius AL, et al. A mouse model for Finnish variant late infantile neuronal ceroid lipofuscinosis, CLN5, reveals neuropathology associated with early aging. Hum Mol Genet. (2004) 13:2893–906. doi: 10.1093/HMG/DDH312
23. White KA, Nelvagal HR, Poole TA, Lu B, Johnson TB, Davis S, et al. Intracranial delivery of AAV9 gene therapy partially prevents retinal degeneration and visual deficits in CLN6-Batten disease mice. Mol Ther - Methods Clin Dev. (2021) 20:497–507. doi: 10.1016/j.omtm.2020.12.014
24. Thelen M, Daμμe M, Schweizer M, Hagel C, Wong AMS, Cooper JD, et al. Disruption of the autophagy-lysosome pathway is involved in neuropathology of the nclf mouse model of neuronal ceroid lipofuscinosis. PLoS ONE. (2012) 7:e35493. doi: 10.1371/journal.pone.0035493
25. Damme M, Brandenstein L, Fehr S, Jankowiak W, Bartsch U, Schweizer M, et al. Gene disruption of Mfsd8 in mice provides the first animal model for CLN7 disease. Neurobiol Dis. (2014) 65:12–24. doi: 10.1016/J.NBD.2014.01.003
26. Chen X, Dong T, Hu Y, Shaffo FC, Belur NR, Mazzulli JR, Gray SJ. AAV9/MFSD8 gene therapy is effective in preclinical models of neuronal ceroid lipofuscinosis type 7 disease. J Clin Invest. (2022) doi: 10.1172/JCI146286
27. Brandenstein L, Schweizer M, Sedlacik J, Fiehler J, Storch S. Lysosomal dysfunction and impaired autophagy in a novel mouse model deficient for the lysosomal membrane protein Cln7. Hum Mol Genet. (2016) 25:777–91. doi: 10.1093/HMG/DDV615
28. Kuronen M, Lehesjoki AE, Jalanko A, Cooper JD, Kopra O. Selective spatiotemporal patterns of glial activation and neuron loss in the sensory thalamocortical pathways of neuronal ceroid lipofuscinosis 8 mice. Neurobiol Dis. (2012) 47:444–57. doi: 10.1016/j.nbd.2012.04.018
29. Kuronen M, Hermansson M, Manninen O, Zech I, Talvitie M, Laitinen T, et al. Galactolipid deficiency in the early pathogenesis of neuronal ceroid lipofuscinosis model Cln8mnd: implications to delayed myelination and oligodendrocyte maturation. Neuropathol Appl Neurobiol. (2012) 38:471–86. doi: 10.1111/J.1365-2990.2011.01233.X
30. Partanen S, Haapanen A, Kielar C, Pontikis C, Alexander N, Inkinen T, et al. Synaptic changes in the thalamocortical system of cathepsin D-deficient mice. J Neuropathol Exp Neurol. (2008) 67:16–29. doi: 10.1097/nen.0b013e31815f3899
31. Ghoshal N, Dearborn JT, Wozniak DF, Cairns NJ. Core features of frontotemporal dementia recapitulated in progranulin knockout mice. Neurobiol Dis. (2012) 45:395–408. doi: 10.1016/J.NBD.2011.08.029
32. Kett LR, Stiller B, Bernath MM, Tasset I, Blesa J, Jackson-Lewis V, et al. α-Synuclein-independent histopathological and motor deficits in mice lacking the endolysosomal parkinsonism protein Atp13a2. J Neurosci. (2015) 35:5724. doi: 10.1523/JNEUROSCI.0632-14.2015
33. Smith KR, Dahl HHM, Canafoglia L, Andermann E, Damiano J, Morbin M, et al. Cathepsin F mutations cause Type B Kufs disease, an adult-onset neuronal ceroid lipofuscinosis. Hum Mol Genet. (2013) 22:1417–23. doi: 10.1093/HMG/DDS558
34. Passini MA, Dodge JC, Bu J, Yang W, Zhao Q, Sondhi D, et al. Intracranial delivery of CLN2 reduces brain pathology in a mouse model of classical late infantile neuronal ceroid lipofuscinosis. J Neurosci. (2006) 26:1334–42. doi: 10.1523/JNEUROSCI.2676-05.2006
35. Vuillemenot BR, Katz ML, Coates JR, Kennedy D, Tiger P, Kanazono S, et al. Intrathecal tripeptidyl-peptidase 1 reduces lysosomal storage in a canine model of late infantile neuronal ceroid lipofuscinosis. Mol Genet Metab. (2011) 104:325–37. doi: 10.1016/j.ymgme.2011.06.018
36. Vuillemenot BR, Kennedy D, Cooper JD, Wong AMS, Sri S, Doeleman T, et al. Nonclinical evaluation of CNS-administered TPP1 enzyme replacement in canine CLN2 neuronal ceroid lipofuscinosis. Mol Genet Metab. (2015) 114:281–93. doi: 10.1016/j.ymgme.2014.09.004
37. Schulz A, Ajayi T, Specchio N, de Los Reyes E, Gissen P, Ballon D, et al. Study of intraventricular cerliponase alfa for CLN2 disease. N Engl J Med. (2018) 378:1898–907. doi: 10.1056/NEJMoa1712649
38. Cooper JD, Puhl AC, Wang SH, Eultgen EM, Takahashi K, Le SQ, et al. Devising effective enzyme replacement therapy for infantile onset neuronal ceroid lipofuscinosis (CLN1 disease). Mol Genet Metab. (2021) 132:S28. doi: 10.1016/J.YMGME.2020.12.048
39. Markham A. Cerliponase alfa: first global approval. Drugs. (2017) 77:1247–9. doi: 10.1007/s40265-017-0771-8
40. Lenders M, Stypmann J, Duning T, Schmitz B, Brand SM, Brand E. Serum-mediated inhibition of enzyme replacement therapy in fabry disease. J Am Soc Nephrol. (2016) 27:256–64. doi: 10.1681/ASN.2014121226
41. Rastall DPW, Amalfitano A. Recent advances in gene therapy for lysosomal storage disorders. Appl Clin Genet. (2015) 8:157. doi: 10.2147/TACG.S57682
42. Biffi A. Gene therapy for lysosomal storage disorders: a good start. Hum Mol Genet. (2016) 25:R65–75. doi: 10.1093/hmg/ddv457
43. Sondhi D, Hackett NR, Peterson DA, Stratton J, Baad M, Travis KM, et al. Enhanced survival of the LINCL mouse following CLN2 gene transfer using the rh10 rhesus Macaque-derived Adeno-associated virus vector. Mol Ther. (2007) 15:481–91. doi: 10.1038/sj.mt.6300049
44. Cabrera-Salazar MA, Roskelley EM, Bu J, Hodges BL, Yew N, Dodge JC, et al. Timing of therapeutic intervention determines functional and survival outcomes in a mouse model of late infantile batten disease. Mol Ther. (2007) 15:1782–8. doi: 10.1038/sj.mt.6300249
45. Katz ML, Tecedor L, Chen Y, Williamson BG, Lysenko E, Wininger FA, et al. AAV gene transfer delays disease onset in a TPP1-deficient canine model of the late infantile form of Batten disease. Sci Transl Med. (2015) 7:313ra180. doi: 10.1126/scitranslmed.aac6191
46. Kleine Holthaus SM, Aristorena M, Maswood R, Semenyuk O, Hoke J, Hare A, et al. Gene therapy targeting the inner retina rescues the retinal phenotype in a mouse model of CLN3 batten disease. Hum Gene Ther. (2020) 31:709. doi: 10.1089/HUM.2020.038
47. Pike LS, Tannous BA, Deliolanis NC, Hsich G, Morse D, Tung CH, et al. Imaging gene delivery in a mouse model of congenital neuronal ceroid lipofuscinosis. Gene Ther. (2011) 18:1173–8. doi: 10.1038/gt.2011.118
48. Worgall S, Sondhi D, Hackett NR, Kosofsky B, Kekatpure M V, Neyzi N, et al. Treatment of late infantile neuronal ceroid lipofuscinosis by CNS administration of a serotype 2 adeno-associated virus expressing CLN2 cDNA. Hum Gene Ther. (2008) 19:463–74. doi: 10.1089/hum.2008.022
49. Sondhi D, Kaminsky SM, Hackett NR, Pagovich OE, Rosenberg JB, De BP, et al. Slowing late infantile Batten disease by direct brain parenchymal administration of a rh10 adeno-associated virus expressing CLN2. Sci Transl Med. (2020) 12:5413. doi: 10.1126/SCITRANSLMED.ABB5413
50. Johnson TB, Cain JT, White KA, Ramirez-Montealegre D, Pearce DA, Weimer JM. Therapeutic landscape for Batten disease: current treatments and future prospects. Nat Rev Neurol. (2019) 15:161–78. doi: 10.1038/s41582-019-0138-8
51. Tyynelä J, Cooper JD, Khan MN, Shemilt SJ, Haltia M. Hippocampal pathology in the human neuronal ceroid-lipofuscinoses: distinct patterns of storage deposition, neurodegeneration and glial activation. Brain Pathol. (2006) 14:349–57. doi: 10.1111/j.1750-3639.2004.tb00077.x
52. Anderson GW, Goebel HH, Simonati A. Human pathology in NCL. Biochim Biophys Acta - Mol Basis Dis. (2013) 1832:1807–26. doi: 10.1016/j.bbadis.2012.11.014
53. Nelvagal HR, Lange J, Takahashi K, Tarczyluk-Wells MA, Cooper JD. Pathomechanisms in the neuronal ceroid lipofuscinoses. Biochim Biophys Acta - Mol Basis Dis. (2020) 1866:165570. doi: 10.1016/j.bbadis.2019.165570
54. Morgan JP, Magee H, Wong A, Nelson T, Koch B, Cooper JD, et al. Murine model of variant late infantile ceroid lipofuscinosis recapitulates behavioral and pathological phenotypes of human disease. PLoS ONE. (2013) 8:e78694. doi: 10.1371/journal.pone.0078694
55. Bélanger M, Magistretti PJ. The role of astroglia in neuroprotection. Dialogues Clin Neurosci. (2009) 11:281–95. doi: 10.31887/DCNS.2009.11.3/mbelanger
56. Chung W-S, Allen NJ, Eroglu C. Astrocytes control synapse formation, function, and elimination. Cold Spring Harb Perspect Biol. (2015) 7:a020370. doi: 10.1101/cshperspect.a020370
57. Chen Z, Trapp BD. Microglia and neuroprotection. J Neurochem. (2016) 136:10–7. doi: 10.1111/jnc.13062
58. Bennett ML, Viaene AN. What are activated and reactive glia and what is their role in neurodegeneration? Neurobiol Dis. (2021) 148:105172. doi: 10.1016/J.NBD.2020.105172
59. Sheeler C, Rosa JG, Ferro A, McAdams B, Borgenheimer E, Cvetanovic M. Glia in neurodegeneration: the housekeeper, the defender and the perpetrator. Int J Mol Sci. (2020) 21:1–16. doi: 10.3390/IJMS21239188
60. Sofroniew M V. Astrocyte barriers to neurotoxic inflammation. Nat Rev Neurosci. (2015) 16:249–63. doi: 10.1038/nrn3898
61. Prinz M, Jung S, Priller J. Microglia biology: one century of evolving concepts. Cell. (2019) 179:292–311. doi: 10.1016/j.cell.2019.08.053
62. Liddelow SA, Barres BA. Reactive Astrocytes: Production, Function, and Therapeutic Potential. Immunity. (2017) 46:957–67. doi: 10.1016/j.immuni.2017.06.006
63. Wilkinson FL, Holley RJ, Langford-Smith KJ, Badrinath S, Liao A, Langford-Smith A, et al. Neuropathology in mouse models of mucopolysaccharidosis type I, IIIA and IIIB. PLoS ONE. (2012) 7:e35787. doi: 10.1371/JOURNAL.PONE.0035787
64. Takahashi K, Le SQ, Kan S, Jansen MJ, Dickson PI, Cooper JD. Neuropathology of murine Sanfilippo D syndrome. Mol Genet Metab. (2021) 134:323–9. doi: 10.1016/J.YMGME.2021.11.010
65. Lange J, Haslett LJ, Lloyd-Evans E, Pocock JM, Sands MS, Williams BP, et al. Compromised astrocyte function and survival negatively impact neurons in infantile neuronal ceroid lipofuscinosis. Acta Neuropathol Commun. (2018) 6:74. doi: 10.1186/s40478-018-0575-4
66. Parviainen L, Dihanich S, Anderson GW, Wong AM, Brooks HR, Abeti R, et al. Glial cells are functionally impaired in juvenile neuronal ceroid lipofuscinosis and detrimental to neurons. Acta Neuropathol Commun. (2017) 5:74. doi: 10.1186/s40478-017-0476-y
67. Yuan W, Lu L, Rao M, Huang Y, Liu CE, Liu S, et al. GFAP hyperpalmitoylation exacerbates astrogliosis and neurodegenerative pathology in PPT1-deficient mice. Proc Natl Acad Sci U S A. (2021) 118:e2022261118. doi: 10.1073/PNAS.2022261118/-/DCSUPPLEMENTAL
68. Macauley SL, Pekny M, Sands MS. The role of attenuated astrocyte activation in infantile neuronal ceroid lipofuscinosis. (2011). doi: 10.1523/JNEUROSCI.3579-11.2011
69. Moulson AJ, Squair JW, Franklin RJM, Tetzlaff W, Assinck P. Diversity of reactive astrogliosis in CNS pathology: heterogeneity or plasticity? Front Cell Neurosci. (2021) 15:703810. doi: 10.3389/fncel.2021.703810
70. Wu T, Dejanovic B, Gandham VD, Gogineni A, Edmonds R, Schauer S, et al. Complement C3 is activated in human AD brain and is required for neurodegeneration in mouse models of amyloidosis and tauopathy. Cell Rep. (2019) 28:2111–23.e6. doi: 10.1016/j.celrep.2019.07.060
71. Guttenplan KA, Weigel MK, Adler DI, Couthouis J, Liddelow SA, Gitler AD, et al. Knockout of reactive astrocyte activating factors slows disease progression in an ALS mouse model. Nat Commun. (2020) 11:1–9. doi: 10.1038/s41467-020-17514-9
72. Yun SP, Kam TI, Panicker N, Kim S, Oh Y, Park JS, et al. Block of A1 astrocyte conversion by microglia is neuroprotective in models of Parkinson's disease. Nat Med. (2018) 24:931–8. doi: 10.1038/s41591-018-0051-5
73. Sadhukhan T, Bagh MB, Appu AP, Mondal A, Zhang W, Liu A, et al. In a mouse model of INCL reduced S-palmitoylation of cytosolic thioesterase APT1 contributes to microglia proliferation and neuroinflammation. J Inherit Metab Dis. (2021) 44:1051–69. doi: 10.1002/jimd.12379
74. Escartin C, Galea E, Lakatos A, O'Callaghan JP, Petzold GC, Serrano-Pozo A, et al. Reactive astrocyte nomenclature, definitions, and future directions. Nat Neurosci. (2021) 24:312. doi: 10.1038/S41593-020-00783-4
75. Domowicz MS, Chan W-C, Claudio-Vázquez P, Gonzalez T, Schwartz NB. Brain transcriptome analysis of a CLN2 mouse model as a function of disease progression. J Neuroinflammation. (2021) 18:1–18. doi: 10.1186/S12974-021-02302-Z
76. Diaz-Castro B, Gangwani MR, Yu X, Coppola G, Khakh BS. Astrocyte molecular signatures in Huntington's disease. Sci Transl Med. (2019) 11:8546. doi: 10.1126/scitranslmed.aaw8546
77. Khakh BS, Sofroniew M V. Diversity of astrocyte functions and phenotypes in neural circuits. Nat Neurosci. (2015) 18:942–52. doi: 10.1038/nn.4043
78. Han RT, Kim RD, Molofsky AV, Liddelow SA. Astrocyte-immune cell interactions in physiology and pathology. Immunity. (2021) 54:211–24. doi: 10.1016/j.immuni.2021.01.013
79. Galloway DA, Phillips AEM, Owen DRJ, Moore CS. Phagocytosis in the brain: Homeostasis and disease. Front Immunol. (2019) 10:790. doi: 10.3389/FIMMU.2019.00790/BIBTEX
80. Jha MK, Jo M, Kim J-H, Suk K. Microglia-astrocyte crosstalk: an intimate molecular conversation. Neurosci. (2019) 25:227–40. doi: 10.1177/1073858418783959
81. Liddelow SA, Guttenplan KA, Clarke LE, Bennett FC, Bohlen CJ, Schirmer L, et al. Neurotoxic reactive astrocytes are induced by activated microglia. Nature. (2017) 541:481–7. doi: 10.1038/nature21029
82. Joshi AU, Minhas PS, Liddelow SA, Haileselassie B, Andreasson KI, Dorn GW, et al. Fragmented mitochondria released from microglia trigger A1 astrocytic response and propagate inflammatory neurodegeneration. Nat Neurosci. (2019) 22:1635–48. doi: 10.1038/s41593-019-0486-0
83. Aguzzi A, Barres BA, Bennett ML. Microglia: scapegoat, saboteur, or something else? Science. (2013) 339:156–61. doi: 10.1126/SCIENCE.1227901
84. Butovsky O, Weiner HL. Microglial signatures and their role in health and disease. Nat Rev Neurosci. (2018) 19:622–35. doi: 10.1038/s41583-018-0057-5
85. Groh J, Ribechini E, Stadler D, Schilling T, Lutz MB, Martini R. Sialoadhesin promotes neuroinflammation-related disease progression in two mouse models of CLN disease. Glia. (2016) 64:792–809. doi: 10.1002/glia.22962
86. Ransohoff RM. A polarizing question: do M1 and M2 microglia exist? Nat Neurosci. (2016) 19:987–991. doi: 10.1038/nn.4338
87. Jurga AM, Paleczna M, Kuter KZ. Overview of general and discriminating markers of differential microglia phenotypes. Front Cell Neurosci. (2020) 0:198. doi: 10.3389/FNCEL.2020.00198
88. Deczkowska A, Keren-Shaul H, Weiner A, Colonna M, Schwartz M, Amit I. Disease-associated microglia: a universal immune sensor of neurodegeneration. Cell. (2018) 173:1073–81. doi: 10.1016/J.CELL.2018.05.003
89. Butovsky O, Jedrychowski MP, Moore CS, Cialic R, Lanser AJ, Gabriely G, et al. Identification of a unique TGF-β-dependent molecular and functional signature in microglia. Nat Neurosci. (2013) 17:131–143. doi: 10.1038/nn.3599
90. Domowicz MS, Chan WC, Claudio-Vázquez P, Henry JG, Ware CB, Andrade J, et al. Global brain transcriptome analysis of a Tpp1 neuronal ceroid lipofuscinoses mouse model. ASN Neuro. (2019) 11:175909141984339. doi: 10.1177/1759091419843393
91. Lee SH, Meilandt WJ, Xie L, Gandham VD, Ngu H, Barck KH, et al. Trem2 restrains the enhancement of tau accumulation and neurodegeneration by β-amyloid pathology. Neuron. (2021) 109:1283–301.e6. doi: 10.1016/J.NEURON.2021.02.010
92. Reifschneider A, Robinson S, Van Lengerich B, Gnörich J, Logan T, Heindl S, et al. Loss of TREM2 rescues hyperactivation of microglia, but not lysosomal deficits and neurotoxicity in models of progranulin deficiency. EMBO J. (2022) 14:e109108. doi: 10.15252/EMBJ.2021109108
93. Salvador AF, de Lima KA, Kipnis J. Neuromodulation by the immune system: a focus on cytokines. Nat Rev Immunol. (2021) 21:526–41. doi: 10.1038/s41577-021-00508-z
94. Nelvagal HR, Hurtado ML, Eaton SL, Kline RA, Lamont DJ, Sands MS, et al. Comparative proteomic profiling reveals mechanisms for early spinal cord vulnerability in CLN1 disease. Sci Rep. (2020) 10:1–16. doi: 10.1038/s41598-020-72075-7
95. Qiao X, Lu J-Y, Hofmann SL. Gene expression profiling in a mouse model of infantile neuronal ceroid lipofuscinosis reveals upregulation of immediate early genes and mediators of the inflammatory response. BMC Neurosci. (2007) 8:95. doi: 10.1186/1471-2202-8-95
96. Guzman-Martinez L, Maccioni RB, Andrade V, Navarrete LP, Pastor MG, Ramos-Escobar N. Neuroinflammation as a common feature of neurodegenerative disorders. Front Pharmacol. (2019) 10:1008. doi: 10.3389/fphar.2019.01008
97. Groh J, Berve K, Martini R. Immune modulation attenuates infantile neuronal ceroid lipofuscinosis in mice before and after disease onset. Brain Commun. (2021) 3: fcab047. doi: 10.1093/BRAINCOMMS/FCAB047
98. Macauley SL, Wong AMS, Shyng C, Augner DP, Dearborn JT, Pearse Y, et al. An anti-neuroinflammatory that targets dysregulated glia enhances the efficacy of CNS-directed gene therapy in murine infantile neuronal ceroid lipofuscinosis. J Neurosci. (2014) 34:13077–82. doi: 10.1523/JNEUROSCI.2518-14.2014
99. Tarczyluk-Wells MA, Salzlechner C, Najafi AR, Lim MJ, Smith D, Platt FM, et al. Combined anti-inflammatory and neuroprotective treatments have the potential to impact disease phenotypes in Cln3–/– mice. Front Neurol. (2019) 10:963. doi: 10.3389/FNEUR.2019.00963/BIBTEX
100. Seehafer SS, Ramirez-Montealegre D, Wong AMS, Chan CH, Castaneda J, Horak M, et al. Immunosuppression alters disease severity in juvenile Batten disease mice. J Neuroimmunol. (2011) 230:169. doi: 10.1016/J.JNEUROIM.2010.08.024
101. Li Q, Barres BA. Microglia and macrophages in brain homeostasis and disease. Nat Rev Immunol. (2018) 18:225–42. doi: 10.1038/nri.2017.125
102. Derecki NC, Cronk JC, Lu Z, Xu E, Abbott SBG, Guyenet PG, et al. Wild-type microglia arrest pathology in a mouse model of Rett syndrome. Nature. (2012) 484:105–9. doi: 10.1038/nature10907
103. Zhao X, Liao Y, Morgan S, Mathur R, Feustel P, Mazurkiewicz J, et al. Noninflammatory changes of microglia are sufficient to cause epilepsy. Cell Rep. (2018) 22:2080–93. doi: 10.1016/j.celrep.2018.02.004
104. Lie PPY, Nixon RA. Lysosome trafficking and signaling in health and neurodegenerative diseases. Neurobiol Dis. (2019) 122:94–105. doi: 10.1016/j.nbd.2018.05.015
105. Faust PL, Kaye EM, Powers JM. Myelin lesions associated with lysosomal and peroxisomal disorders. Expert Rev Neurother. (2014) 10:1449–1466. doi: 10.1586/ERN.10.127
106. Chen G. The roles of lysosomal exocytosis in regulated myelination. J Neurol Neuromedicine. (2016) 1:4–8. doi: 10.29245/2572.942X/2016/5.1047
107. Graziano ACE, Cardile V. History, genetic, and recent advances on Krabbe disease. Gene. (2015) 555:2–13. doi: 10.1016/J.GENE.2014.09.046
108. Little D, Ketteler R, Gissen P, Devine MJ. Using stem cell–derived neurons in drug screening for neurological diseases. Neurobiol Aging. (2019) 78:130–41. doi: 10.1016/j.neurobiolaging.2019.02.008
109. Lange J, Wood-Kaczmar A, Ali A, Farag S, Ghosh R, Parker J, et al. Mislocalization of nucleocytoplasmic transport proteins in human Huntington's disease PSC-derived striatal neurons. Front Cell Neurosci. (2021) 15:393. doi: 10.3389/FNCEL.2021.742763/BIBTEX
110. Sherman SP, Bang AG. High-throughput screen for compounds that modulate neurite growth of human induced pluripotent stem cell-derived neurons. Dis Model Mech. (2018) 11:dmm031906 doi: 10.1242/DMM.031906
111. Sima N, Li R, Huang W, Xu M, Beers J, Zou J, et al. Neural stem cells for disease modeling and evaluation of therapeutics for infantile (CLN1/PPT1) and late infantile (CLN2/TPP1) neuronal ceroid lipofuscinoses. Orphanet J Rare Dis. (2018) 13:1–14. doi: 10.1186/S13023-018-0798-2/FIGURES/8
112. Uusi-Rauva K, Blom T, Von Schantz-Fant C, Blom T, Jalanko A, Kyttälä A. Induced pluripotent stem cells derived from a CLN5 patient manifest phenotypic characteristics of neuronal ceroid lipofuscinoses. Int J Mol Sci. (2017) 18:955. doi: 10.3390/IJMS18050955
113. Lojewski X, Staropoli JF, Biswas-legrand S, Simas AM, Haliw L, Selig MK, et al. Human iPSC models of neuronal ceroid lipofuscinosis capture distinct effects of TPP1 and CLN3 mutations on the endocytic pathway. Hum Mol Genet. (2014) 23:2005–22. doi: 10.1093/HMG/DDT596
114. Green KN, Crapser JD, Hohsfield LA. To kill microglia: a case for CSF1R inhibitors. Trends Immunol. (2020) 41:771. doi: 10.1016/J.IT.2020.07.001
115. Berve K, West BL, Martini R, Groh J. Sex- and region-biased depletion of microglia/macrophages attenuates CLN1 disease in mice. J Neuroinflammation. (2020) 17:323. doi: 10.1186/s12974-020-01996-x
116. Di Malta C, Fryer JD, Settembre C, Ballabio A. Astrocyte dysfunction triggers neurodegeneration in a lysosomal storage disorder. Proc Natl Acad Sci U S A. (2012) 109:E2334–42. doi: 10.1073/pnas.1209577109
117. Colombo A, Dinkel L, Müller SA, Sebastian Monasor L, Schifferer M, Cantuti-Castelvetri L, et al. Loss of NPC1 enhances phagocytic uptake and impairs lipid trafficking in microglia. Nat Commun. (2021) 12:1–20. doi: 10.1038/s41467-021-21428-5
118. Yu T, Shakkottai VG, Chung C, Lieberman AP. Temporal and cell-specific deletion establishes that neuronal Npc1 deficiency is sufficient to mediate neurodegeneration. Hum Mol Genet. (2011) 20:4440–51. doi: 10.1093/hmg/ddr372
119. Mikulka CR, Dearborn JT, Benitez BA, Strickland A, Liu L, Milbrandt J, et al. Cell-autonomous expression of the acid hydrolase galactocerebrosidase. Proc Natl Acad Sci. (2020)201917675. doi: 10.1073/pnas.1917675117
120. Haltia M, Goebel HH. The neuronal ceroid-lipofuscinoses: a historical introduction. Biochim Biophys Acta - Mol Basis Dis. (2013) 1832:1795–800. doi: 10.1016/j.bbadis.2012.08.012
121. Seranova E, Connolly KJ, Zatyka M, Rosenstock TR, Barrett T, Tuxworth RI, et al. Dysregulation of autophagy as a common mechanism in lysosomal storage diseases. Essays Biochem. (2017) 61:733–49. doi: 10.1042/EBC20170055
122. Chandrachud U, Walker MW, Simas AM, Heetveld S, Petcherski A, Klein M, et al. Identification of autophagy modifiers in Batten disease Unbiased cell-based screening in a neuronal cell model of Batten disease highlights an interaction between Ca 2+ homeostasis, autophagy, and CLN3 function. J Biol Chem. (2015) 290:14361–80. doi: 10.1074/jbc.M114.621706
123. Palmieri M, Pal R, Nelvagal HR, Lotfi P, Stinnett GR, Seymour ML, et al. mTORC1-independent TFEB activation via Akt inhibition promotes cellular clearance in neurodegenerative storage diseases. Nat Commun. (2017) 8:14338. doi: 10.1038/ncomms14338
124. Bajaj L, Sharma J, di Ronza A, Zhang P, Eblimit A, Pal R, et al. A CLN6-CLN8 complex recruits lysosomal enzymes at the ER for Golgi transfer. J Clin Invest. (2020) 140:4118–32. doi: 10.1172/JCI130955
125. Shlevkov E, Basu H, Bray M-A, Sun Z, Wei W, Apaydin K, et al. A high-content screen identifies TPP1 and Aurora B as regulators of axonal mitochondrial transport. Cell Rep. (2019) 28:3224. doi: 10.1016/J.CELREP.2019.08.035
126. Danyukova T, Ariunbat K, Thelen M, Brocke-Ahmadinejad N, Mole SE, Storch S. Loss of CLN7 results in depletion of soluble lysosomal proteins and impaired mTOR reactivation. Hum Mol Genet. (2018) 27:1711–22. doi: 10.1093/HMG/DDY076
127. Li J, Pan L, Pembroke WG, Rexach JE, Godoy MI, Condro MC, et al. Conservation and divergence of vulnerability and responses to stressors between human and mouse astrocytes. Nat Commun. (2021) 12:1–20. doi: 10.1038/s41467-021-24232-3
128. Parenti G, Medina DL, Ballabio A. The rapidly evolving view of lysosomal storage diseases. EMBO Mol Med. (2021) 13:e12836. doi: 10.15252/EMMM.202012836
129. Rintz E, Gaffke L, Podlacha M, Brokowska J, Cyske Z, Wegrzyn G, et al. Transcriptomic changes related to cellular processes with particular emphasis on cell activation in lysosomal storage diseases from the group of mucopolysaccharidoses. Int J Mol Sci. (2020) 21:3194. doi: 10.3390/IJMS21093194
130. Mukherjee AB, Appu AP, Sadhukhan T, Casey S, Mondal A, Zhang Z, et al. Emerging new roles of the lysosome and neuronal ceroid lipofuscinoses. Mol Neurodegener. (2019) 14:4. doi: 10.1186/s13024-018-0300-6
131. Petelski AA, Emmott E, Leduc A, Huffman RG, Specht H, Perlman DH, et al. Multiplexed single-cell proteomics using SCoPE2. Nat Protoc. (2021) 16:5398–425. doi: 10.1038/s41596-021-00616-z
132. Kidd GJ, Ohno N, Trapp BD. Biology of schwann cells. Handb Clin Neurol. (2013) 115:55–79. doi: 10.1016/B978-0-444-52902-2.00005-9
133. Santosa KB, Keane AM, Jablonka-Shariff A, Vannucci B, Snyder-Warwick AK. Clinical relevance of terminal schwann cells: an overlooked component of the neuromuscular junction. J Neurosci Res. (2018) 96:1125. doi: 10.1002/JNR.24231
134. Hanani M, Spray DC. Emerging importance of satellite glia in nervous system function and dysfunction. Nat Rev Neurosci. (2020) 21:485. doi: 10.1038/S41583-020-0333-Z
Keywords: Batten disease, neuronal ceroid lipofuscinosis, astrocyte, microglia, oligodendrocyte
Citation: Takahashi K, Nelvagal HR, Lange J and Cooper JD (2022) Glial Dysfunction and Its Contribution to the Pathogenesis of the Neuronal Ceroid Lipofuscinoses. Front. Neurol. 13:886567. doi: 10.3389/fneur.2022.886567
Received: 28 February 2022; Accepted: 16 March 2022;
Published: 04 April 2022.
Edited by:
Ruth Elizabeth Williams, Evelina London Children's Hospital, United KingdomReviewed by:
Alessandro Simonati, University of Verona, ItalyCopyright © 2022 Takahashi, Nelvagal, Lange and Cooper. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Jonathan D. Cooper, Y29vcGVyamRAd3VzdGwuZWR1
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.