 Emily C. Vogler1
Emily C. Vogler1 Matthew Mahavongtrakul1
Matthew Mahavongtrakul1 Kristianna Sarkan1
Kristianna Sarkan1 Ryan C. Bohannan1Silvina Catuara-Solarz1
Ryan C. Bohannan1Silvina Catuara-Solarz1 Jorge Busciglio1,2,3*
Jorge Busciglio1,2,3*- 1Department of Neurobiology and Behavior, University of California, Irvine, Irvine, CA, United States
- 2Institute for Memory Impairments and Neurological Disorders, University of California, Irvine, Irvine, CA, United States
- 3Center for the Neurobiology of Learning and Memory, University of California, Irvine, Irvine, CA, United States
Vesicular Zn2+ (zinc) is released at synapses and has been demonstrated to modulate neuronal responses. However, mechanisms through which dysregulation of zinc homeostasis may potentiate neuronal dysfunction and neurodegeneration are not well-understood. We previously reported that accumulation of soluble amyloid beta oligomers (AβO) at synapses correlates with synaptic loss and that AβO localization at synapses is regulated by synaptic activity and enhanced by the release of vesicular Zn2+ in the hippocampus, a brain region that deteriorates early in Alzheimer's disease (AD). Significantly, drugs regulating zinc homeostasis inhibit AβO accumulation and improve cognition in mouse models of AD. We used both sexes of a transgenic mouse model lacking synaptic Zn2+ (ZnT3KO) that develops AD-like cognitive impairment and neurodegeneration to study the effects of disruption of Zn2+ modulation of neurotransmission in cognition, protein expression and activation, and neuronal excitability. Here we report that the genetic removal of synaptic Zn2+ results in progressive impairment of hippocampal-dependent memory, reduces activity-dependent increase in Erk phosphorylation and BDNF mRNA, alters regulation of Erk activation by NMDAR subunits, increases neuronal spiking, and induces biochemical and morphological alterations consistent with increasing epileptiform activity and neurodegeneration as ZnT3KO mice age. Our study shows that disruption of synaptic Zn2+ triggers neurodegenerative processes and is a potential pathway through which AβO trigger altered expression of neurotrophic proteins, along with reduced hippocampal synaptic density and degenerating neurons, neuronal spiking activity, and cognitive impairment and supports efforts to develop therapeutics to preserve synaptic zinc homeostasis in the brain as potential treatments for AD.
1. Introduction
Zinc is one of the most abundant trace elements in the brain, with its distribution and levels in the central nervous system tightly regulated by various proteins, including ZnT3, which transports Zn2+ into presynaptic vesicles of glutamatergic neurons in the mossy fiber tract of the hippocampus (1, 2). This synaptic pool of Zn2+ is released with glutamate and modulates neurotransmission, influencing different ion channels and receptors (3–6). Dysregulation of zinc homeostasis has been implicated in the pathology of Alzheimer's disease (AD). Drugs targeting zinc in the brain have been shown to inhibit amyloid beta (Aβ) accumulation (7), reduce Aβ oligomer (AβO) levels, and restore cognition (8, 9), dendritic spine density, and synaptic protein levels (10) in mouse models of AD. AβO also bind and sequester Zn2+, resulting in synaptic loss (11) and inhibition of long-term potentiation (12) in rat hippocampus. Zn/AβO inhibit hippocampal long-term potentiation (LTP) and activate hippocampal microglia, while removal of Zn2+ leads to restoration of Zn/AβO to Aβ fibrils (13). In humans, non-demented subjects with AD neuropathology have lower total zinc levels and preserved ZnT3 expression compared to AD patients, implicating dysregulation of synaptic Zn2+ in cognitive impairment (14). Zn2+ and ZnT3 have been implicated in other neurodegenerative disorders, including Parkinson's disease (15) and Huntington's disease (16). Zinc depletion has been found to promote expression of the apoptotic protein Caspase-3 (17), reduce adult hippocampal neurogenesis (18), and negatively affect memory, expression of proteins critical to neuronal health and synaptic function (19).
We have previously reported that the accumulation of AβO in synapses is regulated by synaptic activity and enhanced by the presence of synaptic Zn2+. Furthermore, we found that AβO colocalize with N-methyl-D-aspartate receptor (NMDAR) NR2B subunits and that synaptic accumulation of AβO correlates with synaptic loss in AD brains (20). AβO has also been demonstrated to induce apoptotic cell death through interaction with NR2B-containing NMDARs in hippocampal cultures (21). Zn2+ is a potent inhibitor of NMDAR responses (22–27), which is dependent on NMDAR subunit composition (28–34). Zn2+ has been shown to diffuse beyond the synaptic cleft and inhibit extrasynaptic NMDARs (6) and inhibit postsynaptic NR2A NMDARs (35), strongly suggesting that the interaction between Zn2+ and AβO disrupts NMDAR signaling.
Genetic elimination of ZnT3 results in the absence of Zn2+ in synaptic vesicles and enhanced susceptibility to induced seizures (36, 37). Similarly, Zn2+ chelation induces hyperexcitability in hippocampus (38), and infusing Zn2+ into the hippocampus delays the development of seizures in kindling models of epilepsy (39). In addition to inhibiting NMDARs, Zn2+ activates TrkB receptors (3, 40) and regulates activation of Erk1/2 (17, 41). Consequently, AβO sequestration of Zn2+ at synaptic sites may trigger neuronal hyperactivity and neurodegenerative pathways (42). Disruption of neuromodulation by synaptic Zn2+ is modeled in a transgenic ZnT3 knockout (ZnT3KO) mouse model. Though cognitively normal at a young age, ZnT3KO mice exhibit increased electroencephalogram (EEG) spiking activity after kainate-induced seizures (37) and develop age-dependent cognitive impairment and loss of synaptic and neurotrophic proteins similar to those found in AD (43).
Due to an increase in aging populations and little progress with therapeutics to effectively target this impaired cognition, there is an immediate need for understanding the relationship between neurodegeneration and biometals. To further understand the effects of disrupting Zn2+ neuromodulation, we profiled the progression of impairment in hippocampal-dependent memory as ZnT3KO mice age and we assessed NMDA signaling, Erk phosphorylation, neuronal spiking activity, and markers of epileptiform activity and neurodegeneration. We found a progressive decline in cognition, reduced activity-dependent increases in Erk phosphorylation and brain-derived neurotrophic factor (BDNF), and differential regulation of NMDAR subunits, along with increased neuronal spiking and alterations consistent with increasing epileptiform activity and neurodegeneration. These findings demonstrate that synaptic Zn2+ is critical in normal neuronal function and that disruption of Zn2+ neurotransmission triggers progressive, age-dependent alterations consistent with those triggered by AβO-mediated AD pathology.
2. Materials and methods
2.1. Experimental design and statistical analysis
ZnT3KO mice are reported to initially have normal cognition that becomes impaired with age, indicating that the lack of synaptic zinc is not the direct cause of impaired cognition, but induces neurodegenerative effects leading to impairment. Consequently, we sought to investigate the effects of disruption of Zn2+ modulation of neurotransmission in cognition, protein expression and activation, and neuronal excitability in ZnT3KO mice. Basal biochemical and activity-dependent alterations in neurotrophic NMDAR signaling and BDNF mRNA expression in ZnT3KO mice before the onset of cognitive deficits were assessed in acute hippocampal slice cultures from juvenile WT and ZnT3KO mice. Neuronal hyperactivity before the onset of cognitive deficits in ZnT3KO was assessed using EEG recordings in young WT and ZnT3KO. Progressive age-dependent cognitive decline, neurodegenerative protein expression, and neurodegenerative alterations in both biochemistry and brain cytoarchitecture associated with epileptiform activity were assessed in cohorts of aged WT and ZnT3KO at 3, 6, 12, and 15 months of age. All animal experiments were approved by the University of California, Irvine Institutional Animal Care and Use Committee and conformed to all University and USDA animal care regulations. Homozygous male and female 129Sv wild-type (WT) and ZnT3KO mice (from Richard Palmiter, University of Washington) were housed in the vivarium at the University of California, Irvine, maintained on a 12-h day/night cycle in separate WT and ZnT3KO colonies, and provided food and water ad libitum. After the eighth generation all of the adult mice in the colonies were genotyped, using the Transnetyx genotyping service, to check for genetic drift. For experiments using adult mice, dams and pups from litters born within 5 days of each other were combined in one cage to a maximum of 3 dams and 14 pups until weaning at 21 days of age. The weaned mice were sexed and housed 5 per cage by sex, with equal numbers of males and females. Equal numbers of males and females were used for experiments unless health precluded participation of an individual. For experiments using hippocampal slices from 3 to 4-week-old mice, hippocampal tissue from litters consisting of at least 4 pups were combined as one sample. GraphPad Prism 7 software was used for statistical analysis. Data is presented as mean ± SEM, with raw data from two compared conditions analyzed by two-tailed Student's t-test, reporting t-test statistics (t), degrees of freedom (df), and p-value (p) in the results.
2.2. Object location memory (OLM)
OLM, also referred to as “novel place learning” or “novel object location,” is a memory task that assesses rodents' preference for novelty; in this case, the novelty of the object is placement in a different location (44). Mice were handled for 2 min for 5 consecutive days and habituated to the testing arena for 5 min for 6 consecutive days, with days 4 and 5 of handling overlapping with days 1 and 2 of habituation. On training day, two identical objects were introduced into the training area, spaced equally apart. Animals were allowed to explore the objects for 10 min to become familiarized with the objects and their location. After 24 h, the mice were tested for long-term object location memory by returning the mice to the testing arena with one of the previous objects placed in the same location and the other object placed in a novel location, and allowing the mice to explore the objects. All extra-maze cues in the room were kept constant throughout training and testing. Habituation, training, and testing were all filmed and analyzed in EthoVision software. Exploration of the objects was scored by two investigators blind to subjects' condition. Time of exploration (t) was determined by the subject being within 1 cm of the object and the elongated line from eyes to nose would intersect the object. The discrimination index (DI) was determined by:
2.3. Preparation of hippocampal slices
Three- to four-week-old WT or ZnT3KO mice were used to prepare acute hippocampal slices. Animals were anesthetized with 5% isoflurane and decapitated and their brains were rapidly removed and rinsed with ice-cold artificial cerebrospinal fluid (ACSF). The hippocampus was dissected out and sliced into 350 μm-thick sections with a chilled Stoelting tissue chopper and transferred to individual wells in a 48-well plate containing cold ACSF. The ACSF was replaced with 500 μL of 37°C Neurobasal medium and stabilized at 37°C and 5% CO2 for 1 hour before experimental procedures. Slices from two subjects were pooled for each experiment to result in an equal number of slices for each condition in the experiment. Conditions were no treatment, KCl, and KCl with NMDAR subunit-specific antagonists.
2.4. Chemical modulation of neuronal activity
Hippocampal slices were incubated with KCl (20 mM, Sigma). The following drugs were used to modulate NMDA receptor activity: NR2A antagonist [(R)-[(S)-1-(4-bromo-phenyl)-ethylamino]-(2,3-dioxo-1,2,3,4-tetrahydroquinoxalin-5-yl)-methyl]-phosphonic acid (PEAQX or NVP-AAM077), Sigma; NR2B antagonist 4-[2-[4-(cyclohexylmethyl)-1-piperidinyl]-1-hydroxypropyl]phenol (ifenprodil), Sigma. Drugs were reconstituted with sterile H2O and diluted in Neurobasal media.
2.5. Quantification of protein levels after chemical modulation
Hippocampal slices were transferred to RIPA buffer containing 10 μL/mL Halt protease and phosphatase inhibitor cocktail (Thermo Fisher Scientific), homogenized for 20 s with an IKA Ultra-Turrax T8 homogenizer, and centrifuged at 14,000 x g for 1 h. The supernatant was removed and stored at −80°C until immunoblotting. Protein concentration was assayed using the Thermo Fisher Scientific Pierce BCA protein assay kit.
2.6. Western blot
Proteins were prepared for SDS-PAGE using the BioRad Criterion system. Twenty micrograms of proteins were equalized with RIPA buffer, diluted 1:1 with Laemmli sample buffer (BioRad)/β-Mercaptoethanol prepared at a ratio of 95:5, heated to 100°C for 5 min, loaded into wells of Criterion 4-20% Tris/HCl 1.0 mm precast gels, and separated by gel electrophoresis for 2 h at 125 mV in BioRad tris/glycine/SDS running buffer. Proteins were then transferred onto PVDF membranes (Immobilon) by Western blot for 3 h at 4°C in Towbin buffer (BioRad) with Precision Plus Protein All Blue Standards (BioRad) to identify molecular weights. Membranes were blocked overnight at 4°C in 5% non-fat milk or 5% BSA with 0.1% Tween 20 in TBS and probed with primary antibodies overnight at 4°C in 2.5% non-fat milk or BSA with 0.05% Tween 20 in TBS. Membranes were then washed three times for 5 min each with 0.1% Tween 20 in TBS and incubated in goat anti-mouse and goat anti-rabbit IgG (H+L) HRP-conjugated secondary antibodies (Thermo Fisher Scientific) in 2.5% non-fat milk or BSA with 0.05% Tween 20 in TBS for 2 h at room temperature. Membranes were washed three times for 5 min each with 0.1% Tween 20 in TBS and incubated for 5 min in SuperSignal West Femto, Dura, or Pico chemiluminescent substrate and imaged on a BioRad ChemiDoc XRS+ imaging system and analyzed for quantification with ImageLab software (BioRad). All data were normalized to GAPDH (glyceraldehyde 3-phosphate dehydrogenase). Primary antibodies used were mouse anti-GAPDH (Abcam 125247), rabbit anti-GAPDH (Abcam 37168), rabbit anti-BDNF (Abcam 6201), rabbit anti-MAPK (ERK1/2; Cell Signaling Technology 4695), rabbit anti-phospho-p44/42 MAPK (ERK1/2; Cell Signaling Technology 9106), rabbit anti-Akt (pan; Cell Signaling Technology 4691), rabbit anti-phospho-Akt (Cell Signaling Technology 2965), and mouse anti-calbindin (Swant 300). Secondary antibodies were Alexa Fluor 488 goat anti-rabbit, Alexa Fluor 488 goat anti-mouse, Alexa Fluor 594 goat anti-rabbit, and Alexa Fluor 594 goat anti-mouse (Molecular Probes).
2.7. Dot blot
Twenty micrograms of proteins were equalized with RIPA buffer and loaded into adjacent wells of an S&S manifold-I dot-blot apparatus housing a nitrocellulose membrane. The samples were drawn through the apparatus and into the membrane by vacuum and blocked overnight at 4°C in 5% non-fat milk with 0.1% Tween 20 in TBS and probed with primary antibodies overnight at 4°C in 2.5% non-fat milk with 0.1% Tween 20 in TBS. Antibodies used were rabbit anti-NPY (Abcam 10980) and mouse anti-MAP2 (Millipore MAB4318). Membranes were washed three times for 5 min each with 0.1% Tween 20 in TBS and incubated in goat anti-mouse and goat anti-rabbit IgG (H+L) HRP-conjugated secondary antibodies (Thermo Fisher Scientific) for 2 h at room temperature. Membranes were washed three times for 5 min each with 0.1% Tween 20 in TBS and incubated for 5 min in SuperSignal West Dura chemiluminescent substrate and imaged on a BioRad ChemiDoc XRS+ imaging system and analyzed for quantification with QuantityOne 1-D analysis software (BioRad). All data were normalized to MAP2 adjacent samples.
2.8. Extraction of RNA from tissue and purification
Hippocampal slices from 3- to 4-week-old WT or ZnT3KO mice were prepared as previously described for chemical modulation of neuronal activity and incubated in 20 mM KCl or vehicle for 3.5 h at 37°C and 5% CO2, transferred to 1 mL of Trizol (Invitrogen 15596-026) and homogenized for 20 s with an IKA Ultra-Turrax T8 homogenizer. RNA was isolated using the Trizol method (Invitrogen) and stored at −80°C until purification using a Qiagen RNeasy mini-kit (Qiagen 74104) with on-column DNase I digestion according to the manufacturer's protocol. RNA concentrations and purity were measured using a NanoDrop 2000 spectrophotometer and analyzed using NanoDrop software (Thermo Fisher Scientific).
2.9. RT-PCR and qPCR
cDNA was prepared from up to 1.0 μg RNA using the BioRad iScript Reverse Transcription (RT) Supermix system (BioRad 170-8840) and RT-PCR was performed with a PCR Sprint thermal cycler using the BioRad iScript RT protocol. The resulting cDNA was diluted 1:5 in DNAse/RNAse-free H2O and used for qPCR by the SYBR green method (BioRad 170-8880) with a BioRad DNAengine Peltier thermal cycler using the iQ SYBR green protocol. Primer sequences (Eurofins MWG Operon) used were:
TCGTTCCTTTCGAGTTAGCC (mBDNFexS)
TTGGTAAACGGCACAAAAC (mBDNFexAS)
AGGTATCCTGACCCTGAAG (mActinS)
GCTCATTGTAGAAGGTGTGG (mActinAS).
The resulting data was analyzed using MJ Opticon Monitor (BioRad) and mRNA expression was measured with satisfactory reproducibility between triplicates and fold change relative to average ΔCt was calculated as 2−ΔΔCt and normalized to actin.
2.10. Immunohistochemistry and image analysis
Free-floating 40 μm-thick sections of 4% paraformaldehyde (PFA)-fixed mouse brain sections were permeabilized and blocked with 5% BSA/0.3% Triton X-100/PBS for one-to-two hours and incubated overnight at 4°C with primary antibodies in 2.5% BSA/0.015% Triton X-100/PBS. Primary antibodies used were mouse anti-calbindin 1:1,000 (Swant 300), rabbit anti-NPY 1:500 (Abcam 10980), mouse anti-GAP-43 1:1,000 (Calbiochem CP09L), rabbit anti-synaptoporin 1:750 (Synaptic Systems 102002), mouse anti-synaptophysin 1:1,000 (Cell Signaling 7H12) and mouse anti-synaptophysin 1:1000 (Abcam ab8049). The sections were washed three times for 5 min each with PBS and incubated for 1 hour at room temperature with secondary antibodies in 2.5% BSA/0.015% Triton X-100/PBS. Secondary antibodies used were goat anti-rabbit and goat anti-mouse fluorescent conjugated Alexa 488 and Alexa 594 1:5,000 (Invitrogen). The sections were washed again three times for 5 min each with PBS and incubated in Hoescht 33342 solution 1:10,000 (Invitrogen) in PBS for 5 min, washed again as before and mounted on a microscope slide with Vectashield mounting media (Vector Laboratories) with a microscope cover glass. Slides were dried for 1 h at 37°C and stored at 4°C.
Images were captured with an Axiovert 200 inverted microscope and processed using AxioVision software (Zeiss) for visualization of calbindin and NPY immunolabeling. An Apotome device (Zeiss) was used for optical “Z” sectioning of multifluorescence signals, then a maximum intensity projection of all z planes was created for analysis of synaptoporin and GAP-43 immunolabeling. This allowed visualization of individual processes through multiple planes of focus.
Calbindin and NPY immunolabeling in specific hippocampal regions were quantified as percent area in two fields from two samples for each condition. The mean fluorescence was measured in each sample in cell-free areas to determine background autofluorescence and the images adjusted to normalize the level of background across green and red channels. Synaptoporin puncta were counted at 63X magnification with three fields imaged for each subject in the molecular layer of the dentate gyrus of ZnT3KO mice and values were expressed as ratio relative to WT puncta for each subject. Puncta were counted if the size and intensity were above a threshold that was determined to be background fluorescence. GAP-43 levels were analyzed in ten fields from each subject imaged at 63X magnification and the percent area of GAP-43 immunofluorescent labeling in each field was quantified using the AxioVision (Zeiss) automated measurement program and the average percent area was calculated.
2.11. Fluorojade labeling and image analysis
40 μm-thick PFA-fixed mouse brain sections were mounted on microscope slides with 0.5% gelatin (Sigma) mounting solution and dried for 5 min at 45°C. The slides were then immersed in 100% ethanol for 5 min, 70% ethanol for 2 min, and rinsed in deionized water for 2 min. Slides were then immersed in a solution of 0.06% KMnO4 (Sigma) for 10 min with gentle shaking and rinsed in deionized water for 2 min. Slides were then immersed in a solution of 0.0004% Fluorojade B (Millipore) in 0.1% acetic acid for 20 min with gentle shaking in the dark, rinsed in deionized water three times for 1 min each and dried at 45°C for 10 min. Slides were then immersed in xylene for three changes of 1 min each and coverslipped with DPX mountant (Sigma).
Two-to-four images of the CA3 region of the hippocampus were captured at 20X with an Axiovert 200 inverted microscope and processed using AxioVision software (Zeiss).
Background intensity was normalized between samples and Fluorojade puncta were marked if the size and intensity were above a threshold that was determined to be background fluorescence and counted as positive labeling if the Fluorojade puncta was co-localized with Hoescht staining to eliminate non-specific Fluorojade label. The number of Fluorojade- and Hoescht-positive cells counted in the multiple images was averaged to quantify degenerating neurons for each animal.
2.12. EEG detection of epileptiform activity
EEG activity was recorded in 2-month-old freely moving mice using a Neurologger recording device (TSE Systems) and processed with Spike2 v8.07 (Cambridge Electronic Design) analysis software. Surgeries were performed as previously described in Vogler et al. (45). Briefly, mice were anesthetized with 3% isoflurane, the scalp removed, then 0.255 mm diameter electrodes were implanted 1 mm deep into 0.26 mm holes drilled into the skull and fastened with cyanoacrylate glue. A mounting pedestal was fashioned with dental cement over the electrode wires and exposed skull, and the wound sealed with cyanoacrylate glue. Two recording electrodes were placed at −1.34 mm A/P, ± 1.5 mm M/L and two at −5.34 mm A/P, ± 3.00 mm M/L relative to bregma and the reference electrode was placed at −7.00 mm A/P, 0.50 mm L relative to bregma. Mice were intraperitoneally injected with 3 mg/kg ketoprofen prior to surgery, provided with 1.25 mg/ml acetaminophen in their drinking water for 3 days post-surgery, and housed individually in a home cage for the duration of the experiment. Mice were briefly anesthetized with isoflurane at least 4 days post-surgery and the Neurologger was affixed to the pedestal. The mice were then returned to their home cage, which was then placed in a grounded Faraday cage for a recording period of 20–26 h. Recordings with poor signal from any electrode were rejected. EEG data during movement detected by the accelerometer function of the Neurologger device was removed manually and the raw EEG data was bandpass filtered from 1 to 80 Hz. Spike threshold was a peak amplitude 2.5X greater than the average baseline to determine spikes/hour.
3. Results
3.1. While juvenile ZnT3KO mice have normal hippocampal-dependent cognition, there is a progressive cognitive decline as they age
The Morris water maze task (MWM) has been used to assess memory in ZnT3KO mice by several research groups. Those previous findings indicate that 6-10-week-old ZnT3KO mice are unimpaired (46), while 3-month-old ZnT3KO gave variable results in different studies (43, 47, 48), and 6-month-old ZnT3KO mice exhibit significant impairment (43). As ZnT3 and synaptic Zn2+ are heavily concentrated in the hippocampus, we selected a task requiring hippocampal-dependent spatial memory; specifically, the object location memory (OLM) task (44, 49), which has no requirement to learn a task or to introduce a motivating factor. Instead, OLM relies on rodents' inherent preference for novelty over familiar circumstances, including familiar objects placed in novel locations (50, 51), and requires hippocampal function for encoding, consolidation, and retrieval of memory for the location of the objects (52, 53). We profiled hippocampal-dependent memory in ZnT3KO mice at 6 weeks, 3 months, and 6 months of age in both male and females, finding normal cognition at 6 weeks (DI, WT, 26.31 ± 3.714, n = 13; KO, 30.26 ± 2.859, n = 16), impairment at 3 months (DI, WT, 27.22 ± 4.083, n = 9; KO, 12.07 ± 3.93, n = 14; t = 2.567 df = 21, p = 0.018) and profound impairment at 6 months (DI, WT, 28.63 ± 5.193, n = 8; KO, −2.972 ± 6.136, n = 14; t = 3.487 df = 20, p = 0.0023; Figure 1), with no difference between male and females in performance at each age (data not shown). Thus, our results suggest that the lack of synaptic Zn2+ does not directly cause cognitive impairment as young ZnT3KO have intact cognition, but does trigger progressive neurodegenerative processes as ZnT3KO age, leading to impaired cognition.
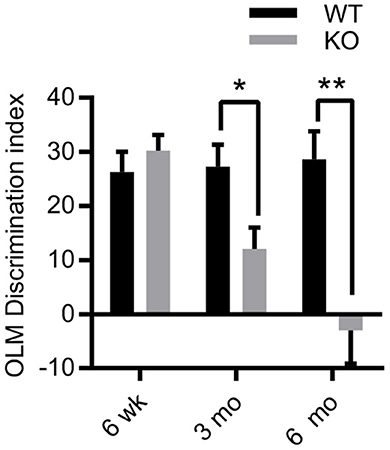
Figure 1. Age-dependent deficits in hippocampal-dependent memory in ZnT3KO mice. Object location memory (OLM) task discrimination index shows age-dependent memory impairment in ZnT3KO mice, becoming significant at 3 months of age and profound by 6 months. WT, wild type, n = 8–13; KO, ZnT3KO, n = 14–16; DI, discrimination index [(novel-familiar)/(novel+familiar)*100]; *p < 0.05, **p < 0.01. Data were analyzed by two-tailed unpaired t-test. Error bars indicate the mean ± SEM.
3.2. Reduced hippocampal expression and phosphorylation of Erk1/2 in ZnT3KO mice
Because Zn2+ activates TrkB receptors and inhibits NMDA receptors (3, 25, 40, 54), we investigated the effects of the removal of synaptic Zn2+ in the expression and activation of two proteins found in both signaling pathways: Erk1/2 and AKT (31, 55–60). Acute hippocampal slices from 3-4-week-old ZnT3KO and WT mice were treated for 10 min with vehicle or 20mM KCl to stimulate vesicle release to assess basal expression and activity-induced phosphorylation levels, followed by protein extraction and analysis by Western blot. Densitometry normalized to GAPDH showed reduced levels of AKT and Erk1/2 in ZnT3KO under basal conditions (Figure 2A and Supplementary Figure 1A). The percent increase of p-AKT after treatment with KCl was similar in both genotypes (data not shown). However, the increase in p-Erk1/2 after treatment with KCl was significant in WT mice (Figure 2B and Supplementary Figure 1B), with a reduced percent increase of p-Erk in ZnT3KO mice (p-Erk1/2 % increase, WT, 72.8 ± 3.9%, n = 4; KO, n = 5, 41.1 ± 3.2%; t = 6.341 df = 7, p = 0.0004). Levels of p-Erk1/2 at basal conditions were also reduced in ZnT3KO mice (p-Erk NT, WT, 0.41 ± 0.02, n = 4; KO, 0.26 ± 0.02, n = 5; t = 4.054 df = 7, p = 0.0048). These results invited further investigation of the role of synaptic Zn2+ in activation of Erk1/2.
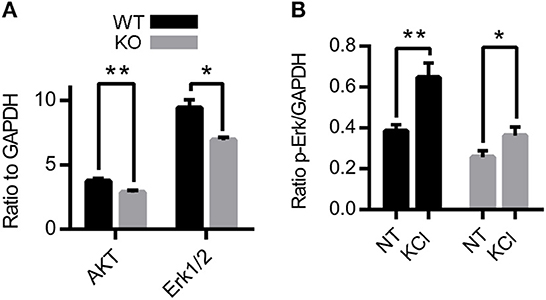
Figure 2. Protein level and activity-dependent phosphorylation of AKT and Erk1/2 are reduced in ZnT3KO hippocampus. (A) Basal levels of AKT and Erk1/2 proteins are reduced in ZnT3KO. (B) Incubation with 20 mM KCl results in a greater increase in p-Erk1/2 in WT than in ZnT3KO hippocampus. WT, wild type; KO, ZnT3KO; AKT, Erk1/2, n = 6; p-Erk1/2, WT, n = 4, KO, n = 5; NT, no treatment (vehicle); KCl, 10-min incubation in 20 mM KCl; *p < 0.05, **p < 0.01; values expressed as normalized densitometry relative to GAPDH. Data were analyzed by two-tailed unpaired t-test. Error bars indicate the mean ± SEM.
3.3. Zn2+ inhibits NR2A-mediated activation of Erk1/2
NMDARs have differential effects on the activation of Erk1/2 depending on NMDAR location and subunit composition, but conflicting results have been reported (31, 33, 57). Consequently, we investigated the effects of removal of synaptic Zn2+ on differential activation of Erk1/2 by NMDAR subunits using antagonists of NR2A (PEAQX) and NR2B (ifenprodil) NMDAR subunits. Acute hippocampal slices were treated with vehicle, 20 mM KCl, or 20 mM KCl+antagonist and the protein samples were prepared for Western blot. Treatment with KCl+ifenprodil reduced levels of p-Erk1/2 in both WT and ZnT3KO, with similar percent decrease in both genotypes (p-Erk1/2 % increase, WT −35.19 ± 1.41%, n = 4; KO, −33.87 ± 3.13%, n = 6, t = 0.3234 df = 8, p = 0.7546; Figure 3A and Supplementary Figure 2A). This indicates that the lack of synaptic Zn2+ does not alter NR2B-mediated activation of Erk1/2. Unexpectedly, treatment with KCl+PEAQX increased levels of p-Erk, with a significantly greater increase in the ZnT3KO (p-Erk1/2 % increase, WT, 10.95 ± 6.21%, n = 5; KO, 78.90 ± 11.55%, n = 5; t = 5.181 df = 8, p = 0.0008; Figure 3B and Supplementary Figure 2B). This observation suggests that synaptic Zn2+ inhibits NR2A-mediated Erk1/2 activation as the removal of Zn2+ resulted in increased p-Erk1/2.
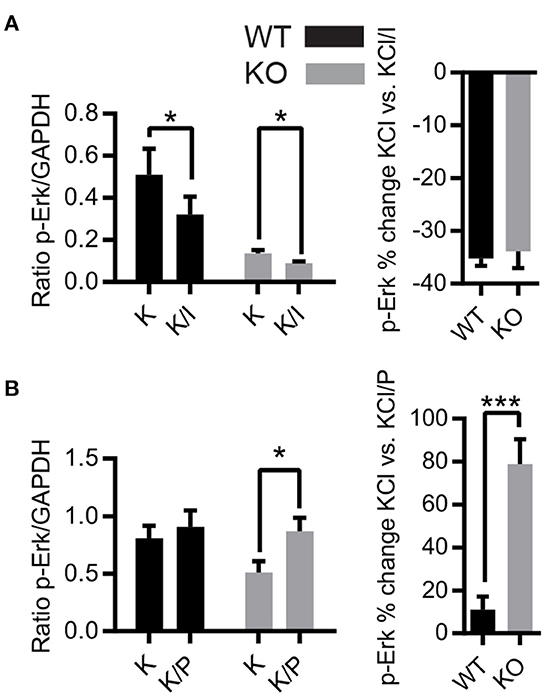
Figure 3. Activity-dependent phosphorylation of Erk1/2 is differentially regulated by NMDAR subunits in ZnT3KO hippocampus. (A) Treatment with ifenprodil, an antagonist of the NMDAR NR2B subunit, showed a decrease in activity-dependent Erk1/2 phosphorylation with a similar percent decrease in both WT and ZnT3KO mice. (B) The activity-dependent phosphorylation of Erk1/2 is increased by incubation with the NR2A antagonist PEAQX, with a significant percent increase in ZnT3KO. WT, wild type; KO, ZnT3KO; ifenprodil, WT, n = 4 KO, n = 6; PEAQX, WT and KO, n = 5; K, 10-min incubation in 20 mM KCl; K/I, 10-min co-incubation with 20 mM KCl and 50 μM ifenprodil: K/P, 30-min incubation with 0.5 μM PEAQX followed by 10-min co-incubation with 0.5 μM PEAQX/20 mM KCl; *p < 0.05, ***p < 0.0005; values expressed as normalized densitometry relative to GAPDH. Data were analyzed by two-tailed unpaired t-test. Error bars indicate the mean ± SEM.
3.4. BDNF expression is downregulated in ZnT3KO hippocampus
Exogenous Zn2+ has differential modulation of NMDAR subunits dependent on membrane voltage and Zn2+ concentration (24, 25), highlighting the complex interactions of endogenous Zn2+ in NMDAR-mediated activation of Erk1/2. To further explore this relationship, we investigated the effects of the removal of synaptic Zn2+ on basal and activity-dependent expression of BDNF, which is linked to NMDAR activation of Erk1/2 signaling (34, 61), and is critical for cognitive function and neuronal health. To determine if the removal of synaptic Zn2+ alters BDNF expression, we assayed BDNF protein levels in age-matched hippocampus tissue from 6-, 12-, and 15-month-old WT and ZnT3KO mice. Proteins extracted from tissue lysates and analyzed by Western blot normalized to GAPDH showed an age-dependent decrease in BDNF levels becoming significant by 15 months of age (BDNF, WT, 0.93 ± 0.09, n = 6; KO, 0.59 ± 0.07, n = 6; t = 2.932 df = 10, p = 0.015; Figure 4A and Supplementary Figure 3). We also investigated whether the increase of BDNF mRNA expression that occurs after depolarization with KCl (62) is altered in ZnT3KO mice using acute hippocampus slices from 3-4-week-old WT and ZnT3KO mice. The slices were incubated for 3.5 h in vehicle or 20 mM KCl, then assayed through RT-PCR and qPCR. The results showed no significant difference in basal levels of BDNF mRNA and a suppressed increase after treatment with KCl in ZnT3KO mice (Ratio BDNF/actin, NT, WT 1.91 x 10−3 ± 5.28 x 10−7, n = 6, KO, 2.31 x 10−3 ± 1.86 x 10−6, n = 6, p < 0.05; ratio BDNF/actin KCl, WT 2.83 ± 0.72, n = 6; KO 1.22 ± 0.32, n = 6; t = 4.93, df = 10, p < 0.0001; Figure 4B). These results suggest that ZnT3KO mice have impaired activity-dependent increases in transcription of BDNF mRNA but may also have compensatory mechanisms to maintain BDNF homeostasis that become less effective as they age.
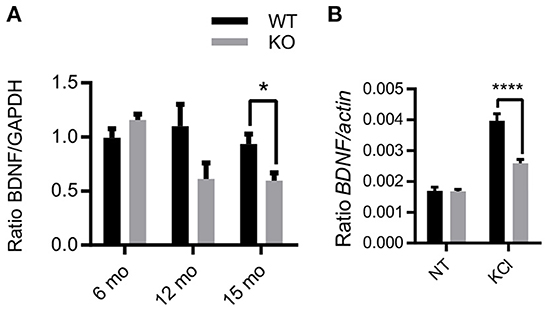
Figure 4. Activity and age-dependent alterations in BDNF expression in ZnT3KO hippocampus. (A) Age-dependent alterations in BDNF protein levels were assessed in hippocampus tissue from age-matched WT and ZnT3KO mice by Western blot analysis, finding that ZnT3KO hippocampus has an age-dependent decrease in BDNF protein levels, becoming significant by 15 months of age. (B) Activity-dependent increase in BDNF mRNA was assessed through RT-PCR and qPCR analysis in acute hippocampal slices from 3-4-week-old WT and ZnT3KO after 3.5-h incubation in 20 mM KCl. WT, wild type; KO, ZnT3KO; BDNF immunoblots, 6 mo, n = 6; 12 mo, n = 5; 15 mo, n = 6; BDNF mRNA, n = 4; NT, no treatment (vehicle); KCl, 3.5 h incubation in 20 mM KCl, n = 6; *p < 0.05, ****p < 0.0001; immunoblot values expressed as normalized densitometry relative to GAPDH; BDNF mRNA values expressed as fold change ratio to actin. Data were analyzed by two-tailed unpaired t-test. Error bars indicate the mean ± SEM.
3.5. Age-dependent increase of markers of epileptiform activity in ZnT3KO hippocampus
ZnT3KO mice exhibit enhanced susceptibility to induced seizures, suggesting that the lack of synaptic Zn2+ causes elevated neuronal activity that reaches seizure threshold with less stimulation, or results in reduced capacity to inhibit excessive activity. Consequently, we investigated the possibility of epileptiform activity in ZnT3KO hippocampus using the biochemical and morphological markers of decreased calbindin expression in the dentate gyrus, increased neuropeptide Y (NPY) expression in the CA3, and aberrant mossy fiber sprouting into the molecular layer of the dentate gyrus. Our results show that ZnT3KO hippocampus has an age-dependent reduction in calbindin protein levels (12 mo., WT, 1.04 ± 0.05, KO, 0.85 ± 0.05, n = 8, t = 2.697 df = 14, p = 0.0173; 15 mo., WT 0.94 ± 0.04, n = 7, KO 0.80 ± 0.04, n = 7, t = 2.314 df = 12, p = 0.0392; Figure 5A and Supplementary Figure 4A), with a larger percent decrease of calbindin immunoreactivity in the dentate gyrus (WT, 19.85 ± 0.75, n = 2, KO 2.62 ± 0.15, n = 2, t = 22.36 df = 2, p = 0.002) compared with CA1 (WT, 31.9 ± 1.5, n = 2, KO, 33.98 ± 1.5, n = 2, t = 0.9843, df = 2; Figures 5B, C). We also found an age-dependent increase in NPY protein levels in ZnT3KO hippocampus (12 mo., WT, 0.97 ± 0.04, n = 5, KO 1.16 ± 0.05, n = 5, t = 3.017, df = 8, p = 0.0166; 15 mo., WT, 0.94 ± 0.14, n = 4, KO, 1.64 ± 0.04, n = 4, t = 4.902, df = 6, p = 0.0027; Figure 5D and Supplementary Figure 4B), particularly in CA3 region (12 mo., WT, 1.66 ± 0.06. n = 2, KO, 4.72 ± 0.16, n = 2, t = 17.86, df = 2, p = 0.0031; Figures 5E, F).
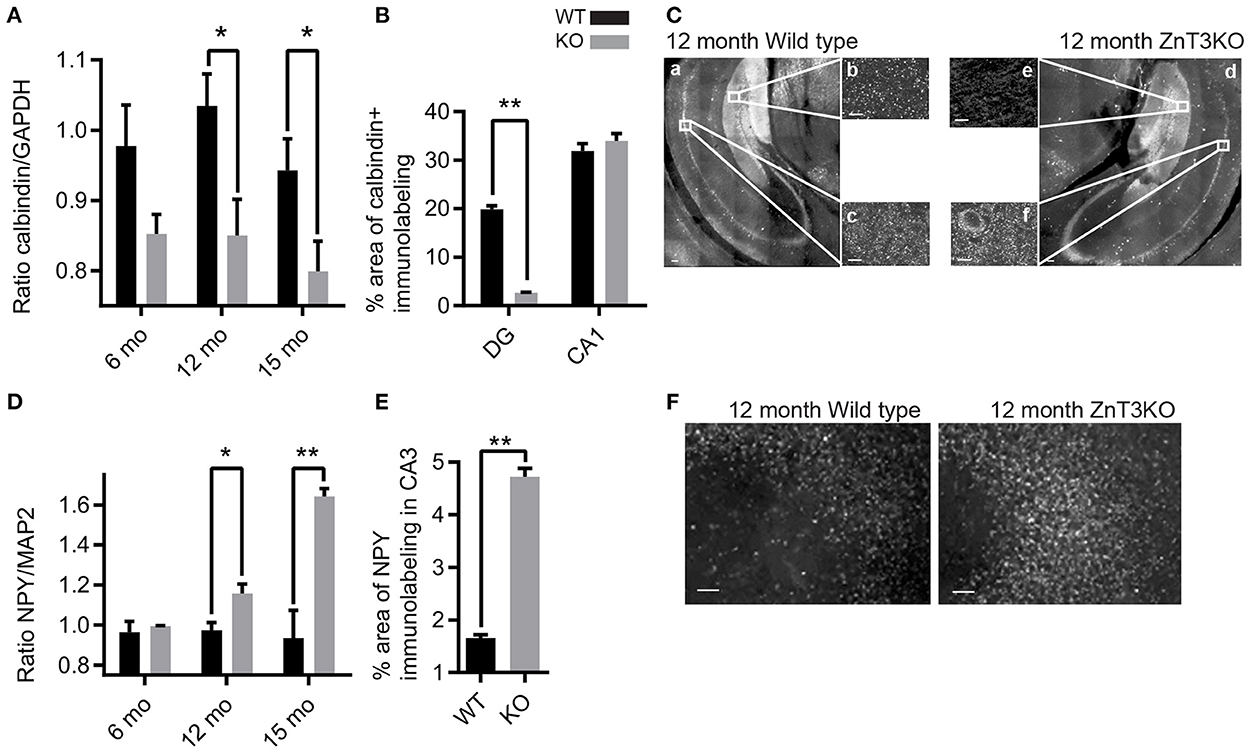
Figure 5. ZnT3KO hippocampus exhibit age-dependent alterations in calbindin and NPY expression that are consistent with alterations resulting from epileptiform activity. (A) Western blot analysis of hippocampal tissue shows an age-dependent decrease of calbindin protein levels in ZnT3KO mice. (B) Quantification of calbindin immunolabeling shows reduced calbindin in the dentate gyrus, but not in the CA1, region of ZnT3KO hippocampus. (C) Representative images of 12-month-old ZnT3KO and WT mice quantified in (B). (D) Dot blot analysis of hippocampal tissue shows an age-dependent increase in NPY protein levels in ZnT3KO mice. (E) Quantification of NPY immunolabeling shows NPY expression is increased in the CA3 region of ZnT3KO hippocampus. (F) Representative images of 12-month old ZnT3KO and WT hippocampus quantified in (E). WT, wild type; KO, ZnT3KO; calbindin immunoblots, 3 and 6 mos. n = 5, 12 mo. n = 8, 15 mo. n = 7; NPY dot blots, 3 and 15 mos. n = 4, 6 mo. n = 3, 12 mo. n = 5; scale bars a and d, 100 μm; b, c, e, and f, 10 μm; *p < 0.05, **p < 0.001; two fields from each subject was imaged at 63x magnification, quantified using the Axiovision (Zeiss) automated measurement program and the percent area calculated. Immunoblot values expressed as normalized densitometry relative to GAPDH or MAP2. Data were analyzed by two-tailed unpaired t-test. Error bars indicate the mean ± SEM.
Mossy fiber sprouting is commonly identified in Zn2+-rich mossy fiber tracts using Timm's staining to label Zn2+. However, ZnT3KO animals have negligible Zn2+ levels in mossy fibers. Thus, we used two different markers to visualize aberrant mossy fiber sprouting: synaptoporin, a synaptic protein preferentially expressed in mossy fibers, and GAP-43, a growth-associated protein found in growth cones of sprouting axons. We found an age-dependent increase in synaptoporin puncta counts expressed as a ratio of ZnT3KO relative to WT (6 mo., WT, 1.00 ± 0.09, n = 8, KO, 1.89 ± 0.29, n = 8, t = 2.975, df = 14, p = 0.01; 13 mo., WT, 1.00 ± 0.11, n = 8, KO, 1.69 ± 0.29, n = 7, t = 2.366, df = 13, p = 0.0342; Figures 6A, B) and GAP-43 percent area (6 mo., WT, 1.19 ± 0.09, n = 5, KO, 2.79 ± 0.23, n = 5, t = 6.441, df = 8, p = 0.0002; 12 mo., WT, 1.17 ± 0.27, n = 5, KO, 3.00 ± 0.56, n = 5, t = 2.956, df = 8, p = 0.0183; Figures 6C, D), in the molecular layer of the dentate gyrus in ZnT3KO relative to WT mice. Together, these results provide biochemical and morphological evidence of age-dependent epileptiform activity which becomes evident beginning at 6 months of age in ZnT3KO mice.
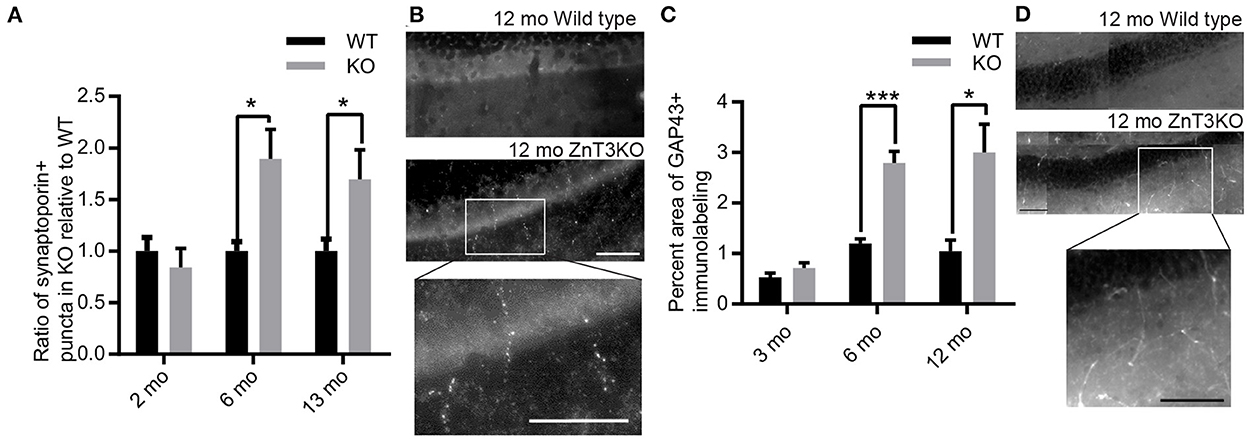
Figure 6. Aberrant, age-dependent mossy fiber sprouting in ZnT3KO hippocampus. (A) Quantification of synaptoporin puncta shows an age-dependent increase in mossy sprouting in the molecular layer of the dentate gyrus in ZnT3KO hippocampus. (B) Representative image of synaptoporin immunolabeling quantified in A and magnification of KO inset box. (C) Quantification of the percent area of GAP43 immunolabeling in the molecular layer of the dentate gyrus shows an age-dependent increase in ZnT3KO hippocampus. (D) Representative image of GAP43 immunolabeling quantified in C and magnification of KO inset box. WT, wild type; KO, ZnT3KO; synaptoporin, 6 mo. n = 10, 13 mo. WT n = 8, KO n = 11; scale bars 100 μm; *p < 0.05, ***p < 0.0002; three fields from each subject were imaged at 63X, puncta counted by an experimenter blinded to subject conditions and values expressed as fold change relative to WT puncta for each subject; GAP43, n = 5; ten fields from each subject were imaged at 63X, quantified using the Axiovision (Zeiss) automated measurement program and the average percent area calculated. Data were analyzed by two-tailed unpaired t-test. Error bars indicate the mean ± SEM.
3.6. Increased neuronal spiking activity in the hippocampus of young ZnT3KO mice
Evidence of age-dependent increases in markers of epileptiform activity invited investigation to determine the age of onset of aberrant EEG in ZnT3KO mice. Using a wireless EEG recording device, we analyzed epileptiform spiking activity in freely moving 2-month-old mice in their home cages during ≈24-h recording sessions, finding a 147% increase in the rate of spikes per hour (WT, 0.40 ± 0.06, n = 3; KO, 0.99 ± 0.19, n = 3; t = 2.864, df = 4, p = 0.0457) in ZnT3KO mice (Figures 7A, B). The presence of increased epileptiform spiking in 2-month-old ZnT3KO is possibly the genesis or precursor of more extensive neuronal hyperactivity resulting in markers of epileptiform activity by 6 months of age.
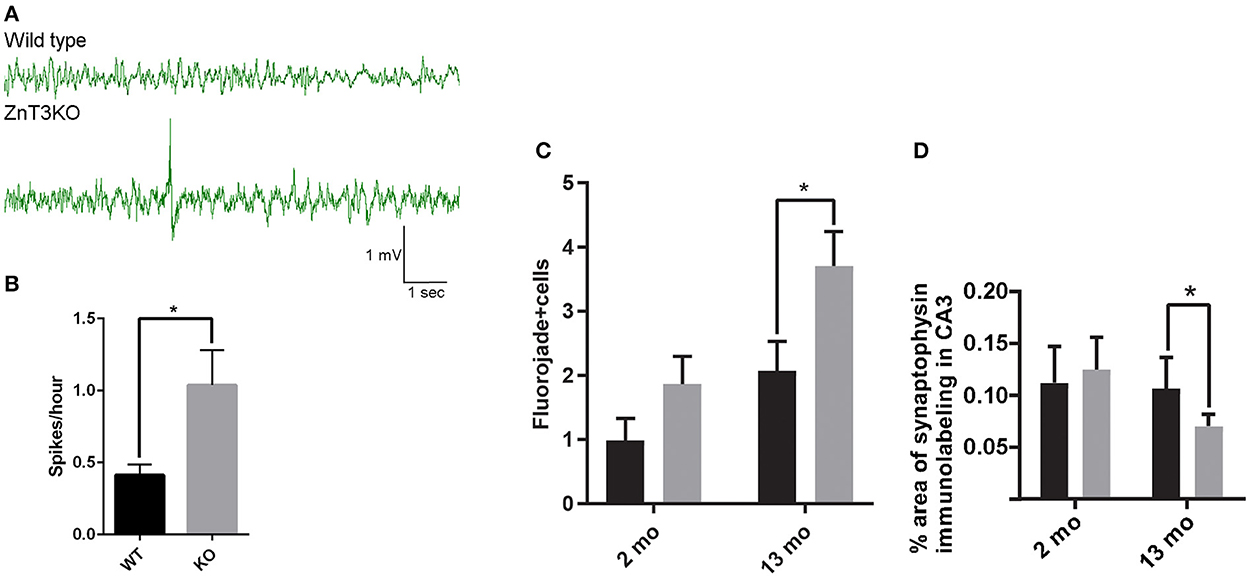
Figure 7. ZnT3KO mice exhibit elevated epileptiform spiking activity, fluorojade+ cells, and reduced synaptophysin expression. (A) Representative EEG of 2-month-old WT and ZnT3KO shows spiking epileptiform activity in ZnT3KO. (B) 2-month-old ZnT3KO have an elevated rate of spikes per hour. n = 3. EEG data during movement detected by implanted Neurologger recording device was manually removed and raw EEG data was bandpass filtered from 1 to 80 Hz. Spike threshold was a peak amplitude 2.5X greater than the average baseline. (C) Image analysis of immunolabeling in hippocampus CA3 region shows an age-dependent decrease in synaptophysin protein levels in ZnT3KO. 2 mo: WT, n = 8, KO, n = 9; 13 mo: n = 6; scale bars 20 μm; one field from each subject was imaged at 63X, quantified using the Axiovision (Zeiss) automated measurement program and the average percent area calculated. (D) Quantification of Fluorojade+ cells counted in the CA3 region of the hippocampus show that there is an increased number in 13-month-old KO. 2 mo: WT, n = 11, KO, n = 9; 13 mo. WT, n = = 9, ZnT3KO n = 11; the CA3 region was imaged at 20X magnification using the Mosaic tool in Axiovision to image and stitch together adjacent fields and the number of Fluorojade positive cells were counted by an experimenter blinded to subject conditions. *p < 0.05. Data were analyzed by two-tailed unpaired t-test. Error bars indicate the mean ± SEM.
3.7. Age-dependent increase in markers of neurodegeneration in the hippocampus of ZnT3KO mice
Altered expression and activation of neurotrophic proteins and epileptiform activity may result in synaptic loss and neurodegeneration. We quantified synaptic density using synaptophysin immunolabeling and degenerating neurons through Fluorojade labeling. Image analysis showed a significant reduction in synaptic density in the CA3 region of 13-month-old ZnT3KO mice determined by percent area of synaptophysin immunolabeling (WT, 0.167 ± 0.034, n = 6; KO, 0.070 ± 0.011; n = 6; t = 2.687, df = 10, p = 0.0228; Figure 7C) and frequent degenerating neurons in the CA3 region of the hippocampus in 13-month old ZnT3KO mice (WT, 2.08 ± 0.46, n = 11; KO, 3.70 ± 0.54; n = 11; t = 2.87, df = 20, p = 0.0095; Figure 7D), with no significant difference found in 2- or 3-month-old ZnT3KO mice, indicating that the lack of synaptic Zn2+ leads to age-dependent synaptic and neuronal loss.
4. Discussion
ZnT3KO mice were initially reported to have normal cognition and sensorimotor responses, suggesting that synaptic zinc has minimal neuromodulatory effects or its effects are easily compensated for. Other studies have found that ZnT3KO mice develop cognitive deficits and neurodegenerative biochemical alterations as they age. These studies indicate that the genetic removal of synaptic Zn2+, while not directly causing cognitive impairment, disrupts the normal role of Zn2+ in neurotransmission, leading to neurodegenerative processes as the mice age. Zn2+ is a modulator of NMDAR responses in both neurotrophic and neurodegenerative pathways (29) and also modulates the development of neuronal hyperactivity and seizures (38, 39). We used acute hippocampal slice cultures from juvenile ZnT3KO mice to assess basal protein expression and protein activation modulated by NMDARs, identifying altered expression and activation of proteins that have been demonstrated to be modulated by the addition or chelation of Zn2+ in cultures of dissociated neurons (3, 19, 25, 40, 54). We used EEG recordings from young ZnT3KO to assess neuronal hyperactivity before the onset of cognitive deficits, finding that 2-month-old ZnT3KO mice have elevated EEG spiking activity, which is consistent with findings of a lower threshold for induced seizures in ZnT3KO (36, 37). We used aged ZnT3KO to assess age-dependent alterations in cognition, protein expression modulated by NMDARs, and biochemical and brain cytoarchitecture consistent with epileptiform activity. We found a profile of progressive cognitive impairment beginning at 3 months of age and becoming profound by 6 months, age dependent decline in BDNF, a neurotrophic protein differentially regulated by NMDARs, and an age dependent increase in markers of epileptiform activity.
Besides structural and synaptic zinc, there is another pool of zinc in the brain: a transient pool referred to as tonic, or “free” zinc, which is unbound Zn2+ present in the cytosol or interstitial fluid yet to be sequestered by Zn2+-binding proteins (1). Zinc homeostasis appears to be disrupted with age (12, 63). Our findings are consistent with recent reports which indicate that both synaptic and tonic Zn2+ can modulate neurotransmission (6, 35). Basal expression of Erk1/2 and AKT after treatment with vehicle, a condition mimicking the effects of tonic Zn2+, are reduced in the ZnT3KO hippocampus, indicating that elimination of synaptic Zn2+ may reduce the pool of tonic Zn2+ available to regulate protein expression. The activity-dependent increase in phosphorylated Erk1/2 and BDNF mRNA levels are also reduced in ZnT3KO mice, demonstrating direct effects of the release of synaptic Zn2+ on protein regulation. The reduced activation of Erk1/2 and no change in activation of AKT in ZnT3KO mice indicates that synaptic Zn2+ released following depolarization modulates the Raf/MEK/Erk signaling cascade, but not the PI3K/AKT pathway, and supports the recent observation that synaptic Zn2+ does not promote TrkB activation in vivo (64, 65).
NMDAR-mediated activation of Erk1/2 has differential regulation of BDNF expression that is dependent on NMDAR location, subunit composition, and intensity of neuronal stimulation (29, 66). To investigate how synaptic Zn2+ influences NMDAR activity, we used antagonists of the NR2A and NR2B subunits, finding that the NR2B antagonist ifenprodil reduces p-Erk1/2 similarly in both genotypes, indicating that synaptic Zn2+ does not modulate NR2B-mediated activation of Erk1/2. Conversely, the NR2A antagonist PEAQX restores p-Erk in ZnT3KO, indicating that Zn2+ modulation of NR2A inhibits activation of Erk1/2. Conflicting roles for the NR2A subunit in Erk1/2 activation have been reported (32, 67), suggesting that NR2A-mediated activation of Erk1/2 is dependent on variable factors. Zinc has biphasic regulation of NMDARs, facilitatory at low levels and repressive at high levels (6, 35, 68), with high affinity for NR2A subunits (27), and tonic inhibition of NR2A (69). Zinc inhibition of NR2A at high levels of synaptic activity indicate a mechanism for zinc to attenuate excessive excitatory neurotransmission.
Zn2+ has been demonstrated to reduce susceptibility to hyperexcitability and seizure (36–39) and potentiate inhibitory glycine receptors at mossy fiber axon terminals (70, 71). Removal of Zn2+ inhibition would tip the balance of neurotransmission toward excessive excitation. Markers of epileptiform activity were assessed in hippocampal tissue from age-matched cohorts of WT and ZnT3KO mice. We found an increase in mossy fiber sprouting at 6 months of age in ZnT3KO, when cognitive impairment becomes profound in this genotype, and biochemical markers of epileptiform activity apparent at 12 months of age in ZnT3KO mice. Synaptic Zn2+ is released in the CA3 and the dentate gyrus (DG) of the hippocampus under both normal and hyperactive conditions, so the removal of Zn2+ would impact those regions significantly. Zn2+-enriched mossy fibers arise from granule cells of the DG and normally project to the CA3 region, where they terminate to form synapses with CA3 pyramidal cells and GABAergic interneurons, and also form small terminals that synapse with GABAergic cells in the hilus of the DG (72). Neuronal hyperactivity in the hippocampus triggers mossy fibers to sprout, forming aberrant excitatory collaterals terminating in the inner molecular layer of the DG, causing excessive excitation of adjacent granule cells that project into the hilus of the DG (73), and to form recurrent excitatory collaterals that generate a feed-forward circuit, increasing neuronal excitability and resulting in increased hyperactivity (74–76). Hyperactivity also stimulates newborn granule cells from adult neurogenesis to develop in abnormal locations in the hilus and receive mossy fiber input (77). Thus, the lack of Zn2+ inhibition in the hippocampus can trigger a cascade of neuronal hyperexcitability leading to epileptiform activity and seizures. NPY suppresses epileptiform activity and its expression is upregulated in response to seizures (78, 79). Consequently, our observation of age-dependent increased NPY expression in the CA3 is accordant with the removal of Zn2+ enhancement of inhibitory response, as is our observation of age-dependent decrease of calbindin, a cytoplasmic protein that buffers intraneuronal calcium and is downregulated by seizures in the DG region but not in CA1 (80). Neurodegenerative markers resulting from the cumulative effects of epileptiform activity and altered neuronal signaling were assessed in hippocampal tissue from age-matched cohorts of WT and ZnT3KO mice. We found a reduction in the synaptic protein synaptophysin and an increase in Fluorojade labeling, two common markers of neurodegeneration, to be most pronounced in the CA3 region of aged ZnT3KO mice, indicating that the lack of synaptic Zn2+ has a profound impact in that hippocampal region.
Aberrant neuronal hyperactivity and hippocampal circuit reorganization has also been found in transgenic mice overexpressing hAPP [(81), reviewed in (82)], and treatment with an anti-seizure drug reverses hippocampal remodeling, synaptic dysfunction and cognitive deficits in hAPP mice (83). In humans, patients with amnestic mild cognitive impairment (aMCI, often a precursor to AD) who also have epilepsy develop symptoms of cognitive decline earlier than aMCI patients without epilepsy, and AD patients with subclinical epileptiform activity also have an earlier onset of cognitive decline (84). Recent research has detected clinically silent seizures and epileptiform spiking in the hippocampi of AD patients (85) and patients with some forms of familial AD are at greater risk of suffering seizures (86). Also, treatment with an anti-seizure drug improves cognition in aMCI patients (87–89). These observations, along with our findings, indicate that clinically silent seizures and epileptiform activity are present early in and contribute to the progression of cognitive decline, and also suggest the possibility that AβO interference with Zn2+ signaling promotes cognitive decline through increasing neuronal hyperactivity.
Our findings provide insight into why ZnT3KO mice begin life phenotypically normal but develop cognitive impairment and neurodegeneration as they age. We found alterations in protein expression, neuronal signaling, and EEG spiking in ZnT3KO mice before the onset of cognitive impairment, which indicates that while these alterations are not sufficient to impair cognition, they can contribute to neurodegeneration. Zn2+ has been shown to have biphasic regulation of NMDARs that is dependent on variable factors (29, 66), as has the expression of BDNF mediated by Erk1/2 activation through NMDARs (34, 61). Using KCl application to hippocampal slice cultures to stimulate neurotransmission, we observed that activity-dependent activation of Erk1/2 and expression of BDNF mRNA is greater in WT than in ZnT3KO, suggesting that Zn2+ is a modulator of Erk1/2-mediated BDNF expression. We also observed that Zn2+ inhibits NR2A-containing NMDARs, indicating that Zn2+ inhibition of NR2A in WT may increase BDNF mRNA expression. These observations invite future investigation of activity-dependent expression of BDNF mRNA in the presence of NR2A antagonists in ZnT3KO to clarify the role of Zn2+ inhibition of NR2A-containing NMDARs in BDNF expression. We also found markers of epileptiform activity beginning with the onset of profound cognitive impairment and subsequent decreased BDNF expression and synaptic density and increased neurodegeneration in aged ZnT3KO mice, advocating for compensatory mechanisms that are eventually overcome by increasing neurodegeneration.
These observations lead us to propose that the cumulative effects of interference with synaptic Zn2+ modulation of neurotransmission triggers neurodegenerative pathways and accelerates neuronal excitotoxicity that ultimately disrupt synaptic signaling and induce significant neurodegeneration in an age dependent manner. ZnT3 levels decline with age in mice, rats and humans, and decline even further in AD (12, 43, 90), suggesting that disruption of zinc homeostasis contributes to AD pathology. The source of cognitive impairment in AD is not well-established. Cognitive decline correlates with soluble Aβ (91) as well as with tangle density, but that does not demonstrate the causative role of tau or Aβ in AD. Multiple studies show the neurotoxic effects of Aβ (92, 93), providing a pathway for cognitive decline. More recently, positive clinical trial results show a delay in cognitive decline in patients with immunotherapy targeting Aβ (Lecanemab), not tau. In addition, multiple evidences show that tau pathology is likely initiated by Aβ meaning that targeting Aβ may still affect tau pathology (94–97). Together these findings support further research into targeted interventions to reduce AβO formation and activity and to preserve synaptic zinc homeostasis in the brain of affected patients.
Data availability statement
The raw data supporting the conclusions of this article will be made available by the authors, without undue reservation.
Ethics statement
The animal study was reviewed and approved by IACUC, University of California-Irvine.
Author contributions
EV, MM, KS, RB, and SC-S performed experiments, analyzed results, and prepared figures. EV and JB designed experiments and interpreted results. EV, MM, and JB wrote and edited manuscript. All authors contributed to the article and approved the submitted version.
Funding
This study was supported by a grant from the California Department of Public Health Alzheimer's Disease Program and NIH/NIA grant AG056850.
Acknowledgments
The authors would like to thank Brenton Phung and Manisha Seelam for their assistance in preparing this manuscript.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher's note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fneur.2022.882635/full#supplementary-material
References
1. Frederickson CJ. Neurobiology of zinc and zinc-containing neurons. Int Rev Neurobiol. (1989) 31:145–238. doi: 10.1016/s0074-7742(08)60279-2
2. Palmiter RD, Cole TB, Quaife CJ, Findley SD. ZnT-3, a putative transporter of zinc into synaptic vesicles. Proc Natl Acad Sci U S A. (1996) 93:14934–9. doi: 10.1073/pnas.93.25.14934
3. Hwang JJ, Park MH, Choi SY, Koh JY. Activation of the Trk signaling pathway by extracellular zinc. Role of metalloproteinases J Biol Chem. (2005) 280:11995–2001. doi: 10.1074/jbc.M403172200
4. Besser L, Chorin E, Sekler I, Silverman WF, Atkin S, Russell JT, et al. Synaptically released zinc triggers metabotropic signaling via a zinc-sensing receptor in the hippocampus. J Neurosci. (2009) 29:2890–901. doi: 10.1523/jneurosci.5093-08.2009
5. Paoletti P, Vergnano AM, Barbour B, Casado M. Zinc at glutamatergic synapses Neuroscience. (2008) 158:126–136. doi: 10.1016/jneuroscience01
6. Anderson CT, Radford RJ, Zastrow ML, Zhang DY, Apfel UP, Lippard SJ, et al. Modulation of extrasynaptic NMDA receptors by synaptic and tonic zinc. Proc Natl Acad Sci U S A. (2015) 112:E2705–2714. doi: 10.1073/pnas.1503348112
7. Cherny RA, Atwood CS, Xilinas ME, Gray DN, Jones WD, McLean CA, et al. Treatment with a copper-zinc chelator markedly and rapidly inhibits beta-amyloid accumulation in Alzheimer's disease transgenic mice. Neuron. (2001) 30:665–76. doi: 10.1016/s0896-6273(01)00317-8
8. Adlard PA, Cherny RA, Finkelstein DI, Gautier E, Robb E, Cortes M, et al. Rapid restoration of cognition in Alzheimer's transgenic mice with 8-hydroxy quinoline analogs is associated with decreased interstitial. Abeta Neuron. (2008) 59:43–55 doi: 10.1016/jneuron06
9. Grossi C, Francese S, Casini A, Rosi MC, Luccarini I, Fiorentini A, et al. Clioquinol decreases amyloid-beta burden and reduces working memory impairment in a transgenic mouse model of Alzheimer's disease. J Alzheimer's Dis JAD. (2009) 17:423–40. doi: 10.3233/JAD-2009-1063
10. Adlard PA, Bica L, White AR, Nurjono M, Filiz G, Crouch PJ, et al. Metal ionophore treatment restores dendritic spine density and synaptic protein levels in a mouse model of Alzheimer's disease. PLoS ONE. (2011) 6:e17669. doi: 10.1371/journal.pone.0017669
11. Grabrucker AM, Schmeisser MJ, Udvardi PT, Arons M, Schoen M, Woodling NS, et al. Amyloid beta protein-induced zinc sequestration leads to synaptic loss via dysregulation of the ProSAP2/Shank3 scaffold. Mol Neurodegener. (2011) 6:65. doi: 10.1186/1750-1326-6-65
12. Datki Z, Galik-Olah Z, Janosi-Mozes E, Szegedi V, Kalman J, Hunya ÁG, et al. Alzheimer risk factors age and female sex induce cortical Aβ aggregation by raising extracellular zinc. Mol Psychiatry. (2020) 25:2728–41. doi: 10.1038/s41380-020-0800-y
13. Lee MC, Yu WC, Shih YH, Chen CY, Guo ZH, Huang SJ, et al. Zinc ion rapidly induces toxic, off-pathway amyloid-beta oligomers distinct from amyloid-beta derived diffusible ligands in Alzheimer's disease. Sci Rep. (2018) 8:4772. doi: 10.1038/s41598-018-23122-x
14. Bjorklund NL, Reese LC, Sadagoparamanujam VM, Ghirardi V, Woltjer RL, Taglialatela G, et al. Absence of amyloid beta oligomers at the postsynapse and regulated synaptic Zn2+ in cognitively intact aged individuals with Alzheimer's disease neuropathology. Mol Neurodegener. (2012) 7:23. doi: 10.1186/1750-1326-7-23
15. Sikora J, Kieffer BL, Paoletti P, Ouagazzal A-M. Synaptic zinc contributes to motor and cognitive deficits in 6-hydroxydopamine mouse models of Parkinson's disease. Neurobiol Dis. (2020) 134:104681. doi: 10.1016/j.nbd.2019.104681
16. Niu L, Li L, Yang S, Wang W, Ye C, Li H, et al. Disruption of zinc transporter ZnT3 transcriptional activity and synaptic vesicular zinc in the brain of Huntington's disease transgenic mouse. Cell Biosci. (2020) 10:106. doi: 10.1186/s13578-020-00459-3
17. Pang W, Leng X, Lu H, Yang H, Song N, Tan L, et al. Depletion of intracellular zinc induces apoptosis of cultured hippocampal neurons through suppression of ERK signaling pathway and activation of caspase-3. Neurosci Lett. (2013) 552:140–145. doi: 10.1016/jneulet07
18. Choi BY, Hong DK, Jeong JH, Lee BE, Koh JY, Suh SW, et al. Zinc transporter 3 modulates cell proliferation and neuronal differentiation in the adult hippocampus. Stem Cells. (2020) 38:994–1006. doi: 10.1002/stem.3194
19. Frazzini V, Granzotto A, Bomba M, Massetti N, Castelli V, d'Aurora M, et al. The pharmacological perturbation of brain zinc impairs BDNF-related signaling and the cognitive performances of young mice. Sci Rep. (2018) 8:9768. doi: 10.1038/s41598-018-28083-9
20. Deshpande A, Kawai H, Metherate R, Glabe CG, Busciglio J. A role for synaptic zinc in activity-dependent Abeta oligomer formation and accumulation at excitatory synapses. J Neurosci. (2009) 29:4004–15. doi: 10.1523/JNEUROSCI.5980-08.2009
21. Costa RO, Lacor PN, Ferreira IL, Resende R, Auberson YP, Klein WL, et al. Endoplasmic reticulum stress occurs downstream of GluN2B subunit of N-methyl-d-aspartate receptor in mature hippocampal cultures treated with amyloid-beta oligomers. Aging Cell. (2012) 11:823–33. doi: 10.1111/j.1474-9726.2012.00848.x
22. Morabito A, Zerlaut Y, Serraz B, Sala R, Paoletti P, Rebola N, et al. Activity-dependent modulation of NMDA receptors by endogenous zinc shapes dendritic function in cortical neurons. Cell Rep. (2022) 38:110415. doi: 10.1016/j.celrep.2022.110415
23. Westbrook GL, Mayer ML. Micromolar concentrations of Zn2+ antagonize NMDA and GABA responses of hippocampal neurons. Nature. (1987) 328:640–3. doi: 10.1038/328640a0
24. Paoletti P, Ascher P, Neyton J. High-affinity zinc inhibition of NMDA NR1-NR2A receptors. J Neurosci. (1997) 17:5711–25. doi: 10.1523/JNEUROSCI.17-15-05711.1997
25. Izumi Y, Auberson YP, Zorumski CF. Zinc modulates bidirectional hippocampal plasticity by effects on NMDA receptors. J Neurosci. (2006) 26:7181–8. doi: 10.1523/JNEUROSCI.1258-06.2006
26. Erreger K, Traynelis SF. Zinc inhibition of rat NR1/NR2A N-methyl-D-aspartate receptors. J Physiol. (2008) 586:763–78. doi: 10.1113/jphysiol.2007.143941
27. Amico-Ruvio SA, Murthy SE, Smith TP, Popescu GK. Zinc effects on NMDA receptor gating kinetics. Biophys J. (2011) 100:1910–8. doi: 10.1016/jbpj02
28. Hardingham GE, Fukunaga Y, Bading H. Extrasynaptic NMDARs oppose synaptic NMDARs by triggering CREB shut-off and cell death pathways. Nat Neurosci. (2002) 5:405–14. doi: 10.1038/nn835
29. Vanhoutte P, Bading H. Opposing roles of synaptic and extrasynaptic NMDA receptors in neuronal calcium signalling and BDNF gene regulation. Curr Opin Neurobiol. (2003) 13:366–71. doi: 10.1016/S0959-4388(03)00073-4
30. Massey PV, Johnson BE, Moult PR, Auberson YP, Brown MW, Molnar E, et al. Differential roles of NR2A and NR2B-containing NMDA receptors in cortical long-term potentiation and long-term depression. J Neurosci. (2004) 24:7821–8. doi: 10.1523/JNEUROSCI.1697-04.2004
31. Kim MJ, Dunah AW, Wang YT, Sheng M. Differential roles of NR2A- and NR2B-containing NMDA receptors in Ras-ERK signaling and AMPA receptor trafficking. Neuron. (2005) 46:745–60. doi: 10.1016/jneuron04
32. Chen Q, He S, Hu XL, Yu J, Zhou Y, Zheng J, et al. Differential roles of NR2A- and NR2B-containing NMDA receptors in activity-dependent brain-derived neurotrophic factor gene regulation and limbic epileptogenesis. J Neurosci. (2007) 27:542–52. doi: 10.1523/JNEUROSCI.3607-06.2007
33. Choo AM, Geddes-Klein DM, Hockenberry A, Scarsella D, Mesfin MN, Singh P, et al. NR2A and NR2B subunits differentially mediate MAP kinase signaling and mitochondrial morphology following excitotoxic insult. Neurochem Int. (2012) 60:506–16. doi: 10.1016/jneuint02
34. Zhou X, Ding Q, Chen Z, Yun H, Wang H. Involvement of the GluN2A and GluN2B subunits in synaptic and extrasynaptic N-methyl-D-aspartate receptor function and neuronal excitotoxicity. J Biol Chem. (2013) 288:24151–9. doi: 10.1074/jbc.M113.482000
35. Vergnano AM, Rebola N, Savtchenko LP, Pinheiro PS, Casado M, Kieffer BL, et al. Zinc dynamics and action at excitatory synapses. Neuron. (2014) 82:1101–14. doi: 10.1016/j.neuron.2014.04.034
36. Cole TB, Robbins CA, Wenzel HJ, Schwartzkroin PA, Palmiter RD. Seizures and neuronal damage in mice lacking vesicular zinc. Epilepsy Res. (2000) 39:153–69. doi: 10.1016/S0920-1211(99)00121-7
37. Qian J, Xu K, Yoo J, Chen TT, Andrews G, Noebels JL, et al. Knockout of Zn transporters Zip-1 and Zip-3 attenuates seizure-induced CA1 neurodegeneration. J Neurosci. (2011) 31:97–104. doi: 10.1523/JNEUROSCI.5162-10.2011
38. Blasco-Ibanez JM, Poza-Aznar J, Crespo C, Marques-Mari AI, Gracia-Llanes FJ, Martinez-Guijarro FJ, et al. Chelation of synaptic zinc induces overexcitation in the hilar mossy cells of the rat hippocampus. Neuroscience letters. (2003) 355:101–4. doi: 10.1016/jneulet10
39. Elsas SM, Hazany S, Gregory WL, Mody I. Hippocampal zinc infusion delays the development of afterdischarges and seizures in a kindling model of epilepsy. Epilepsia. (2009) 50:870–9. doi: 10.1111/j.1528-2008
40. Huang YZ, Pan E, Xiong ZQ, McNamara JO. Zinc-mediated transactivation of TrkB potentiates the hippocampal mossy fiber-CA3 pyramid synapse. Neuron. (2007) 57:546–558 doi: 10.1016/jneuron11
41. Sindreu C, Palmiter RD, Storm DR. Zinc transporter ZnT-3 regulates presynaptic Erk1/2 signaling and hippocampus-dependent memory. Proc Natl Acad Sci U S A. (2011) 108:3366–70. doi: 10.1073/pnas.1019166108
42. Vogler EC, Busciglio J. Disruption of zinc neuromodulation by Aβ oligomers: therapeutic implications. Curr Pharm Des. (2014) 20:2520–4. doi: 10.2174/13816128113199990510
43. Adlard PA, Parncutt JM, Finkelstein DI, Bush AI. Cognitive loss in zinc transporter-3 knock-out mice: a phenocopy for the synaptic and memory deficits of Alzheimer's disease? J Neurosci. (2010) 30:1631–6. doi: 10.1523/JNEUROSCI.5255-09.2010
44. Vogel-Ciernia A, Wood MA. Examining object location and object recognition memory in mice. Curr Protoc Neurosci. (2014) 69:8. doi: 10.1002/0471142301.ns0831s69
45. Vogler EC, Flynn DT, Busciglio F, Bohannan RC, Tran A, Mahavongtrakul M, et al. Low cost electrode assembly for EEG recordings in mice. Front Neurosci. (2017) 11:629. doi: 10.3389/fnins.2017.00629
46. Cole TB, Martyanova A, Palmiter RD. Removing zinc from synaptic vesicles does not impair spatial learning, memory, or sensorimotor functions in the mouse. Brain Res. (2001) 891:253–65. doi: 10.1016/s0006-8993(00)03220-0
47. Martel G, Hevi C, Kane-Goldsmith N, Shumyatsky GP. Zinc transporter ZnT3 is involved in memory dependent on the hippocampus and perirhinal cortex Behav Brain Res. (2011) 223:233–8. doi: 10.1016/jbbr04
48. Thackray SE, McAllister BB, Dyck RH. Behavioral characterization of female zinc transporter 3 (ZnT3) knockout mice. Behav Brain Res. (2017) 321:36–49. doi: 10.1016/j.bbr.2016.12.028
49. Balderas I, Rodriguez-Ortiz CJ, Salgado-Tonda P, Chavez-Hurtado J, McGaugh JL, Bermudez-Rattoni F, et al. The consolidation of object and context recognition memory involve different regions of the temporal lobe. Learn Mem. (2008) 15:618–24. doi: 10.1101/lm.1028008
50. Dix SL, Aggleton JP. Extending the spontaneous preference test of recognition: evidence of object-location and object-context recognition. Behav Brain Res. (1999) 99:191–200. doi: 10.1016/s0166-4328(98)00079-5
51. Murai T, Okuda S, Tanaka T, Ohta H. Characteristics of object location memory in mice: Behavioral and pharmacological studies. Physiol Behav. (2006) 90:116–24. doi: 10.1016/jphysbeh09
52. Mumby DG, Gaskin S, Glenn MJ, Schramek TE, Lehmann H. Hippocampal damage and exploratory preferences in rats: memory for objects, places, and contexts. Learn Mem. (2002) 9:49–57. doi: 10.1101/lm.41302
53. Haettig J, Stefanko DP, Multani ML, Figueroa DX, McQuown SC, Wood MA, et al. HDAC inhibition modulates hippocampus-dependent long-term memory for object location in a CBP-dependent manner. Learn Mem. (2011) 18:71–9. doi: 10.1101/lm.1986911
54. Legendre P, Westbrook GL. The inhibition of single N-methyl-D-aspartate-activated channels by zinc ions on cultured rat neurones. J Physiol. (1990) 429:429–49. doi: 10.1113/jphysiol.1990.sp018266
55. Perkinton MS, Ip JK, Wood GL, Crossthwaite AJ, Williams RJ. Phosphatidylinositol 3-kinase is a central mediator of NMDA receptor signalling to MAP kinase (Erk1/2), Akt/PKB and CREB in striatal neurones. J Neurochem. (2002) 80:239–54. doi: 10.1046/j.0022-3042.2001.00699.x
56. Sutton G, Chandler LJ. Activity-dependent NMDA receptor-mediated activation of protein kinase B/Akt in cortical neuronal cultures. J Neurochem. (2002) 82:1097–105. doi: 10.1046/j.1471-4159.2002.01031.x
57. Krapivinsky G, Krapivinsky L, Manasian Y, Ivanov A, Tyzio R, Pellegrino C, et al. The NMDA receptor is coupled to the ERK pathway by a direct interaction between NR2B and RasGRF1. Neuron. (2003) 40:775–784. doi: 10.1016/s0896-6273(03)00645-7
58. Klann E, Dever TE. Biochemical mechanisms for translational regulation in synaptic plasticity. Nat Rev Neurosci. (2004) 5:931–42. doi: 10.1038/nrn1557
59. Mao L, Tang Q, Samdani S, Liu Z, Wang JQ. Regulation of MAPK/ERK phosphorylation via ionotropic glutamate receptors in cultured rat striatal neurons. Eur J Neurosci. (2004) 19:1207–16. doi: 10.1111/j.1460-9568.2004.03223.x
60. Minichiello L. TrkB signalling pathways in LTP and learning. Nature reviews Neuroscience. (2009) 10:850–60. doi: 10.1038/nrn2738
61. Zheng F, Zhou X, Luo Y, Xiao H, Wayman G, Wang H, et al. Regulation of brain-derived neurotrophic factor exon IV transcription through calcium responsive elements in cortical neurons. PLoS One. (2011) 6:e28441. doi: 10.1371/journal.pone.0028441
62. Cosi C, Spoerri PE, Comelli MC, Guidolin D, Skaper SD. Glucocorticoids depress activity-dependent expression of BDNF mRNA in hippocampal neurones. Neuroreport. (1993) 4:527–30. doi: 10.1097/00001756-199305000-00016
63. Ashraf A, Michaelides C, Walker TA, Ekonomou A, Suessmilch M, Sriskanthanathan A, et al. Regional distributions of iron, copper and zinc and their relationships with glia in a normal aging mouse model. Front Aging Neurosci. (2019) 11:351. doi: 10.3389/fnagi.2019.00351
64. Helgager J, Huang YZ, McNamara JO. Brain-derived neurotrophic factor but not vesicular zinc promotes TrkB activation within mossy fibers of mouse hippocampus in vivo. J Comp Neurol. (2014) 3:23647. doi: 10.1002./cne.23647
65. McAllister BB, Bihelek N, Mychasiuk R, Dyck RH. Brain-derived neurotrophic factor and TrkB levels in mice that lack vesicular zinc: effects of age and sex. Neuroscience. (2019) 425:90–100. doi: 10.1016/jneuroscience11
66. Hardingham GE, Arnold FJ, Bading H. A calcium microdomain near NMDA receptors: on switch for ERK-dependent synapse-to-nucleus communication. Nat Neurosci. (2001) 4:565–6. doi: 10.1038/88380
67. Xiao L, Feng C, Chen Y. Glucocorticoid rapidly enhances NMDA-evoked neurotoxicity by attenuating the NR2A-containing NMDA receptor-mediated ERK1/2 activation. Mol Endocrinol. (2010) 24:497–510. doi: 10.1210/me.2009-0422
68. Shen Z, Haragopal H, Li YV. Zinc modulates synaptic transmission by differentially regulating synaptic glutamate homeostasis in hippocampus. Eur J Neurosci. (2020) 52:3710–22. doi: 10.1111/ejn.14749
69. Rachline J, Perin-Dureau F, Le Goff A, Neyton J, Paoletti P. The micromolar zinc-binding domain on the NMDA receptor subunit NR2B. J Neurosci. (2005) 25:308–17. doi: 10.1523/JNEUROSCI.3967-04.2005
70. Miller PS, Da Silva HM, Smart TG. Molecular basis for zinc potentiation at strychnine-sensitive glycine receptors. J Biol Chem. (2005) 280:37877–84. doi: 10.1074/jbc.M508303200
71. Park EJ, Choi IS, Cho JH, Nakamura M, Lee JJ, Lee MG, et al. Zinc modulation of glycine receptors in acutely isolated rat CA3 neurons. Life Sci. (2008) 83:149–54. doi: 10.1016/jlfs04
72. Acsady L, Kamondi A, Sik A, Freund T, Buzsaki G. GABAergic cells are the major postsynaptic targets of mossy fibers in the rat hippocampus. J Neurosci. (1998) 18:3386–403. doi: 10.1523/JNEUROSCI.18-09-03386.1998
73. Ghosal K, Pimplikar SW. Aging and excitotoxic stress exacerbate neural circuit reorganization in amyloid precursor protein intracellular domain transgenic mice. Neurobiol Aging. (2011) 32:2320.e1–9. doi: 10.1016/j.neurobiolaging.2010.04.020
74. Cronin J, Dudek FE. Chronic seizures and collateral sprouting of dentate mossy fibers after kainic acid treatment in rats. Brain Res. (1988) 474:181–4. doi: 10.1016/0006-8993(88)90681-6
75. Scharfman HE, Sollas AL, Berger RE, Goodman JH. Electrophysiological evidence of monosynaptic excitatory transmission between granule cells after seizure-induced mossy fiber sprouting. J Neurophysiol. (2003) 90:2536–47. doi: 10.1152/jn.00251.2003
76. Ito S, Ishizuka T, Yawo H. Remodeling of hippocampal network in pilocarpine-treated mice expressing synaptopHluorin in the mossy fiber terminals. Neuroscience Res. (2012) 74:25–31. doi: 10.1016/jneures07
77. Pierce JP, Melton J, Punsoni M, McCloskey DP, Scharfman HE. Mossy fibers are the primary source of afferent input to ectopic granule cells that are born after pilocarpine-induced seizures. Exp Neurol. (2005) 196:316–31. doi: 10.1016/jexpneurol08
78. Klapstein GJ, Colmers WF. Neuropeptide Y suppresses epileptiform activity in rat hippocampus in vitro. J Neurophysiol. (1997) 78:1651–61. doi: 10.1152/jn.783.1651
79. Wickham J, Ledri M, Bengzon J, Jespersen B, Pinborg LH, Englund E, et al. Inhibition of epileptiform activity by neuropeptide Y in brain tissue from drug-resistant temporal lobe epilepsy patients. Sci Rep. (2019) 9:19393. doi: 10.1038/s41598-019-56062-1
80. Kohr G, Lambert CE, Mody I. Calbindin-D28K (CaBP) levels and calcium currents in acutely dissociated epileptic neurons. Exp Brain Res. (1991) 85:543–51. doi: 10.1007/BF00231738
81. Palop JJ, Chin J, Roberson ED, Wang J, Thwin MT, Bien-Ly N, et al. Aberrant excitatory neuronal activity and compensatory remodeling of inhibitory hippocampal circuits in mouse models of Alzheimer's disease. Neuron. (2007) 55:697–711. doi: 10.1016/jneuron07
82. Hector A, Brouillette J. Hyperactivity induced by soluble amyloid-β oligomers in the early stages of Alzheimer's disease. Front Mol Neurosci. (2021) 13:600084. doi: 10.3389/fnmol.2020.600084
83. Sanchez PE, Zhu L, Verret L, Vossel KA, Orr AG, Cirrito JR, et al. Levetiracetam suppresses neuronal network dysfunction and reverses synaptic and cognitive deficits in an Alzheimer's disease model. Proc Natl Acad Sci U S A. (2012) 109:E2895–2903. doi: 10.1073/pnas.1121081109
84. Vossel KA, Beagle AJ, Rabinovici GD, Shu H, Lee SE, Naasan G, et al. Seizures and epileptiform activity in the early stages of Alzheimer disease. JAMA Neurol. (2013) 70:1158–66. doi: 10.1001/jamaneurol.2013.136
85. Lam AD, Deck G, Goldman A, Eskandar EN, Noebels J, Cole AJ, et al. Silent hippocampal seizures and spikes identified by foramen ovale electrodes in Alzheimer's disease. Nat Med. (2017) 3:4330. doi: 10.1038./nm.4330
86. Zarea A, Charbonnier C, Rovelet-Lecrux A, Nicolas G, Rousseau S, Borden A, et al. Seizures in dominantly inherited Alzheimer disease. Neurology. (2016) 87:912–9. doi: 10.1212/WNL.0000000000003048
87. Bakker A, Krauss GL, Albert MS, Speck CL, Jones LR, Stark CE, et al. Reduction of hippocampal hyperactivity improves cognition in amnestic mild cognitive impairment. Neuron. (2012) 74:467–74. doi: 10.1016/jneuron03
88. Bakker A, Albert MS, Krauss G, Speck CL, Gallagher M. Response of the medial temporal lobe network in amnestic mild cognitive impairment to therapeutic intervention assessed by fMRI and memory task performance. NeuroImage Clin. (2015) 7:688–98. doi: 10.1016/jnicl02
89. Shiozaki K, Kajihara S. Anti-epileptic drugs improved serial 7s scores on the Mini-Mental State Examination in elderly with cognitive impairment and epileptiform discharge on electroencephalography. Psychogeriatrics. (2018) 19:38–45. doi: 10.1111/psyg.12362
90. Beyer N, Coulson DT, Heggarty S, Ravid R, Irvine GB, Hellemans J, et al. ZnT3 mRNA levels are reduced in Alzheimer's disease post-mortem brain. Mol Neurodegener. (2009) 4:53. doi: 10.1186/1750-1326-4-53
91. Wang J, Dickson, DW, Trojanowski JQ, Lee VM. The levels of soluble versus insoluble brain Abeta distinguish Alzheimer's disease from normal and pathologic aging. Exp Neurol. (1999) 158:328–37.
92. Yankner BA, Duffy, LK, Kirschner, DA. Neurotrophic and neurotoxic effects of amyloid beta protein: reversal by tachykinin neuropeptides. Science. (1990) 250:279–82.
93. Deshpande A, Mina E, Glabe C, Busciglio JA. Different conformations of amyloid β induce neurotoxicity by distinct mechanisms in human cortical neurons. J Neurosci. (2006) 26:6011–8. doi: 10.1523/JNEUROSCI.1189-06.2006
94. Oddo S, Caccamo A, Kitazawa M, Tseng BP, LaFerla FM. Amyloid deposition precedes tangle formation in a triple transgenic model of Alzheimer's disease. Neurobiol Aging. (2003) 24:1063–70. doi: 10.1016/j.neurobiolaging.2003.08.012
95. Vasconcelos B, Stancu IC, Buist A. Heterotypic seeding of Tau fibrillization by pre-aggregated Abeta provides potent seeds for prion-like seeding and propagation of Tau-pathology in vivo. Acta Neuropathol. (2016) 131:549–69. doi: 10.1007/s00401-015-1525-x
96. Welikovitch LA, Do Carmo S, Maglozky Z, Szocsics P, Loke J, Freund T, et al. Evidence of intraneuronal, A.β, accumulation preceding tau pathology in the entorhinal cortex. Acta Neuropathol. (2008) 3:1922. doi: 10.1007./s00401-018-1922-z
97. Wilcock DM, Gharkholonarehe N, Van Nostrand WE, Davis J, Vitek MP, Colton CA, et al. Amyloid reduction by amyloid-β vaccination also reduces mouse tau pathology and protects from neuron loss in two mouse models of Alzheimer's disease. J Neurosci. (2009) 29:7957–65. doi: 10.1523/JNEUROSCI.1339-09.2009
Keywords: Alzheimer's disease (AD), synaptic zinc, neurodegeneration, neuronal hyperactivity, hippocampus, neurotrophic signaling, ZnT3, zinc transporter
Citation: Vogler EC, Mahavongtrakul M, Sarkan K, Bohannan RC, Catuara-Solarz S and Busciglio J (2023) Genetic removal of synaptic Zn2+ impairs cognition, alters neurotrophic signaling and induces neuronal hyperactivity. Front. Neurol. 13:882635. doi: 10.3389/fneur.2022.882635
Received: 24 February 2022; Accepted: 08 December 2022;
Published: 20 January 2023.
Edited by:
Emel Koseoglu, Erciyes University, TurkeyReviewed by:
Lilian Calderon-Garciduenas, University of Montana, United StatesRichard H. Dyck, University of Calgary, Canada
Copyright © 2023 Vogler, Mahavongtrakul, Sarkan, Bohannan, Catuara-Solarz and Busciglio. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Jorge Busciglio,  amJ1c2NpZ2xAdWNpLmVkdQ==
amJ1c2NpZ2xAdWNpLmVkdQ==