 Yuping Chen
Yuping Chen Xiaoyong Tao1
Xiaoyong Tao1 Jinming Han
Jinming Han
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Neurol. , 09 May 2022
Sec. Neuromuscular Disorders and Peripheral Neuropathies
Volume 13 - 2022 | https://doi.org/10.3389/fneur.2022.873599
This article is part of the Research Topic Phenotypes of Myasthenia Gravis View all 19 articles
Objective: This study aimed to summarize the clinical characteristics and prognosis of patients with anti- acetylcholine receptor (AChR) positive myasthenia gravis (MG) with a combination of anti-LRP4 or Titin antibodies.
Methods: A total of 188 patients with generalized MG before immunotherapy were retrospectively collected and then divided into three groups: single anti-AChR positive-MG (AChR-MG, 101 cases), anti-AChR combined with anti-low-density lipoprotein receptor-related protein four-positive MG (AChR+LRP4-MG, 29 cases), and anti-AChR combined with anti-Titin-positive MG (AChR+Titin-MG, 58 cases). Clinical manifestations, therapeutic responses to immunotherapy, and follow-up information were analyzed.
Results: Of the 188 seropositive MG patients, 29 (15.4%) were positive for both AChR and LRP4 antibodies, and 58 (30.9%) were positive for both AChR and Titin antibodies. The mean disease onset ages in the three groups were 47.41 ± 7.0, 49.81 ± 9.2, and 48.11 ± 6.5 years, respectively. AChR+LRP4-MG showed female predominance (27.6% were males and 72.4% were females), with mild overall clinical symptoms. The AChR+Titin-MG group showed shorter times for conversion to generalized MG (5.14 ± 0.0 months) than the AChR-MG group (11.69 ± 0.0 months) and the AChR+LRP4-MG group (13.08 ± 0.5 months; P < 0.001 in both cases). Furthermore, AChR+Titin-MG group had increased bulbar dysfunction, higher incidences of thymoma (32.8 vs. 19.8% and 3.4%, P=0.035), more severe quantitative MG scores, as assessed by both QMG scores [15.5 (11.75–22.5) vs. 13 (8–19), P = 0.005; and 9 (6–14) P < 0.001], and MG-ADL scores [10 (8–13) vs. 8 (5–13), P = 0.018; and 6 (4–8), P < 0.001]. Treatment for AChR+Titin-MG was largely dependent on corticosteroids and immunosuppressive agents (56.7 vs. 19.2% and 16.7%, p = 0.028). The rates of achieving s(MMS) or better within 2 years following immunotherapy in the three groups were 51.5, 62.1, and 51.7%, respectively (P = 0.581).
Conclusion: Clinical symptoms of anti-AChR positive MG combined with Titin antibody were more severe and progressed faster than those in the AChR + LRP4 and AChR groups. Regardless of antibody status, all patients responded well to immunotherapy and had relatively good prognoses.
Myasthenia gravis (MG) is an autoimmune disease involving antibody-mediated destruction of the neuromuscular junction, which causes fatigable weakness (1). There are three confirmed pathogenic antibodies in MG: acetylcholine receptor antibody (AChR-Ab), muscle-specific tyrosine kinase antibody (MuSK-Ab), and low-density lipoprotein receptor-related protein 4 antibody (LRP4-Ab) (1–3). These main concomitant antibodies target different muscle proteins, including titin, myosin, tropomyosin, and the ryanodine receptor (RyR) (3–6). It is generally believed that pathogenic antibodies are closely related to the occurrence, development, and prognosis of autoimmune diseases (7–10). Although the pathogenicity of these concomitant antibodies is unclear, diagnostic and prognostic values for Titin and RyR antibodies have already been established based on intracellular localization of their target antigens (4, 11, 12). Serum antibody detection also plays an important role in the clinical diagnosis of MG, and more than one autoantibody against extracellular or intracellular targets has been noted in patients with MG (13, 14). However, the clinical value of these antibodies remains unclear. In this study, we retrospectively analyzed the prevalence, clinical features, and prognosis of anti-AChR positive MG combined with anti-LRP4 or anti-Titin antibodies.
Medical records and follow up data from 1,109 MG patients who were treated in our hospital between January 2013 and December 2019 were retrospectively reviewed and analyzed. The inclusion criteria included: (1) Patients who had been diagnosed with MG and were over 18 years of age. The MG diagnosis was based on fluctuating weakness symptoms along with supporting pharmacological, serologic, and electrophysiologic tests; (2) Onset symptoms and signs were compatible with generalized MG; (3) Patients were not treated with steroids, immunosuppressive agents, IVIG, or plasma exchange for at least 6 months before antibody detection; (4) Anti-AChR, MuSK, LRP4, and Titin-Ab were measured; (5) Patients were seropositive for anti-AChR antibody.
The following patients were excluded from the study: (1) A total of 171 patients who were younger than 18 years old at the time of admission; (2) Patients who had ocular MG or who only had ocular muscle involvement but for <2 years (180 cases); (3) Patients who had been treated with immunosuppressive agents (tacrolimus in 62 cases, cyclosporine in 78 cases, azathioprine in 20 cases, cyclophosphamide in 40 cases and steroids in 280 cases), had plasma exchanges (PLEX) or had intravenous immunoglobulin (IVIG) treatment (30 cases) within 6 months prior to antibody detection; (4) Patients who were negative for anti-AChR (32 cases); (5) Patients who had incomplete data regarding anti-AChR; or within whom Musk, LRP4, and Titin-Ab were not detected (28 cases). Pregnant individuals were also excluded in this study. We ultimately enrolled 188 patients in our study (Figure 1).
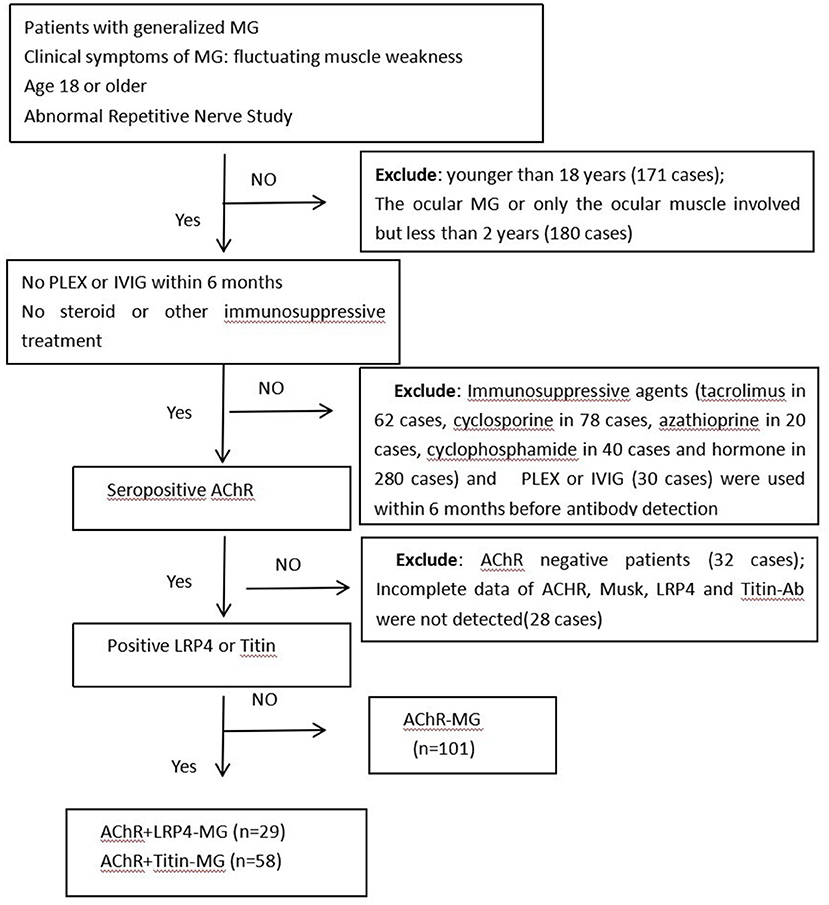
Figure 1. Flowchart of participants included in this study. Abbreviations: MG, Myasthenia gravis; PLEX, plasma exchange; IVIG, intravenous immunoglobulin; AChR, acetylcholine receptor; LRP4, low-density lipoprotein receptor-related protein 4; n, number of patients.
Myasthenia gravis was diagnosed by senior neurologists based on the guidelines of the International Consensus Guidance for Management of Myasthenia Gravis (2, 5). Patients were classified into three groups: AChR-MG, AChR+LRP4-MG, and AChR+Titin-MG. Clinical, diagnostic, therapeutic, and prognosis data, including gender, age of onset, initial symptoms, disease progression, clinical classification, disease severity, the incidence of myasthenia crisis, thymus histopathology, therapeutic options, and prognosis were collected.
All patients were tested for MG-related antibodies in the serum before immunotherapy. If AChR Ab was positive, MuSK, LRP4, Titin, and RyR Ab were further tested in these patients.
A radioimmunoassay for the AChR antibody was performed according to the manufacturer's protocol (RSR Limited, United Kingdom). Patients were defined as antibody positive if antibody titers were ≥0.5 nmol/l (AChR Ab). Blood was tested for MuSK, LRP4, and Titin using enzyme-linked immunoassay (ELISA) as previously described (15). The investigators who performed the ELISA experiments were blinded to clinical diagnoses.
Therapeutic strategies for generalized MG include acetylcholinesterase inhibitors and immunotherapeutic agents. Symptomatic treatment with oral pyridostigmine bromide was used in patients who responded positively to the neostigmine trial. Most patients with generalized MG require induction therapy with glucocorticosteroids. During therapeutic periods, the steroid dosage was gradually increased or decreased according to patients' conditions. Immunosuppressive agents, including azathioprine, cyclosporine A, tacrolimus, or cyclophosphamide, were used in combination with corticosteroids if needed. If the disease was severe (i.e., involved respiratory muscle and bulbar muscle), patients were treated with IVIG and plasma exchanges. These patients were followed up for 2 years. Patients who received either azathioprine, tacrolimus, cyclosporine, or cyclophosphamide were considered to receive immunosuppressive therapy.
Clinical status and disease severity were evaluated based on MGFA classifications, quantitative MG scores (QMGs), and the daily living scale (MG-ADL), respectively. In terms of MGFA post-intervention status (PIS), the classification of “Minimal Manifestation Status (MMS) or better” included minimal manifestation Status (MM0-3), pharmacological remission (PR), and complete stable remission (CSR).
All individuals were followed up and evaluated for 2 years after different treatments. The study was stopped after 2 years. The proportion of patients in the three groups who reached an "MMS or better” state after treatment, and maintained it for more than 6 months were analyzed.
SPSS 26.0 statistical software (IBM, Armonk, New York) was used for statistical analysis. Categorical data were represented as frequencies (%). Continuous data were represented as mean±standard deviation (SD), and ANOVA tests were used for quantitative data. The median (interquartile interval) was used for non-normally distributed statistical descriptions, and nonparametric tests were used for inter-group comparisons. Qualitative statistics were evaluated using two-tailed Fisher's exact tests. A P < 0.05 was considered statistically significant.
Of the 188 seropositive generalized MG patients, 29 patients were positive for both AChR and LRP4 antibodies, while 58 cases were positive for both AChR and Titin. The mean age of disease onset was 47.41 ± 7.0, 49.81 ± 9.2, and 48.11 ± 6.5 years in the AChR-MG, AChR+LRP4-MG, and AChR+Titin-MG groups, respectively. AChR+LRP4-MG showed female predominance (27.6 vs. 72.4%). The proportion of men and women in the AChR-MG and AChR+Titin-MG groups was relatively equal (48.5 vs. 51.5, 58.6 vs. 41.4%; respectively), and there were no significant gender differences between the two groups (Table 1).
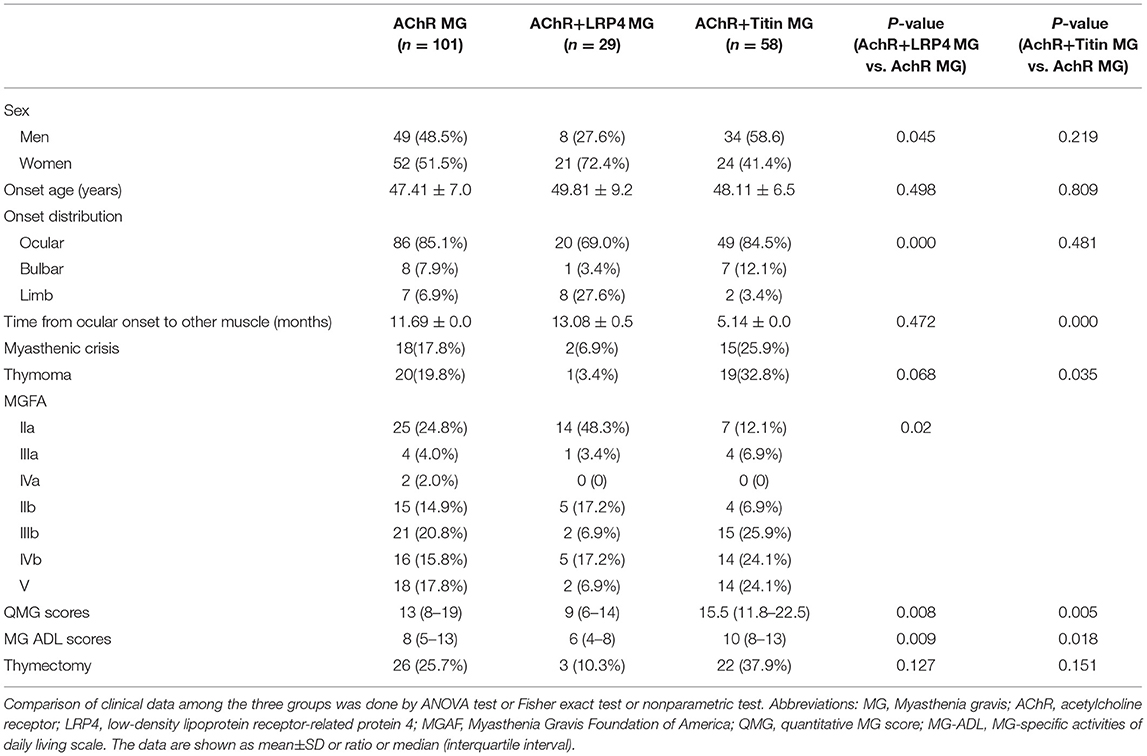
Table 1. Characteristics of patients with AChR-MG, AChR+LRP4-MG and AChR+Titin-MG.
Ocular muscle weakness was the most common onset symptom in all three groups. More patients in the AChR+LRP4-MG suffered from limb weakness onset than in the AChR-MG and AChR+Titin-MG groups (27.6 vs. 6.9% and 11.5%; P < 0.001). Compared to the AChR-MG and AChR+LRP4-MG groups, patients in the AChR+Titin-MG group tended to have shorter conversion times from ocular to generalized MG (5.14 ± 0.0 vs. 11.69 ± 0.0 and 13.08 ± 0.5 months; P < 0.001), Furthermore, AChR+Titin-MG patients had greater bulbar dysfunction, higher incidences of thymoma (32.8 vs. 19.8 and 3.4%; P = 0.006), and more severe QMG scores [15.5 (11.75–22.5) vs. 13 (8–19) in AChR-the MG group (P=0.005), and 9 (6–14) in the AChR+LRP4-MG group (P < 0.001)]. MG-ADL scores were also significantly increased in the AChR+Titin-MG group [10 (8–13) vs. 8 (5–13) in the AChR-MG group, P = 0.018; and 6 (4–8) in the AChR+LRP4-MG group, P< 0.001].
The most common MGFA classification in AChR+LRP4-MG patients was MGFA IIa (48.3%), while 25.9% of AChR+Titin-MG patients were classified as MGFA IIIb. Additionally, more patients were classified as MGFA IVb-V in the AChR+Titin-MG group than in either the AChR-MG and AChR+LRP4-MG groups (48.2 vs. 33.2, and 24.1%, P = 0.02).
Affected muscles in the three groups were analyzed at different time points (6 months, 12 months, and 24 months; see Figure 2). Our results showed that clinical symptoms did not differ significantly among the three groups during different time points.
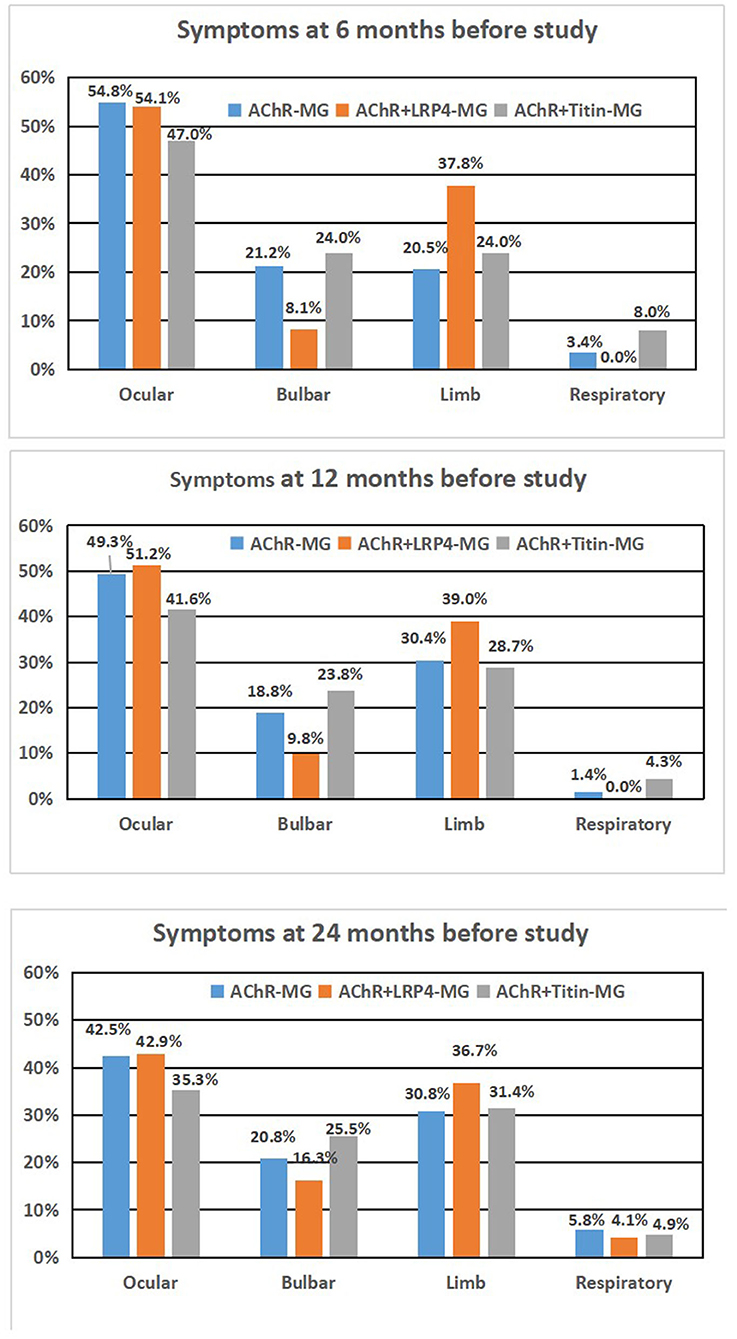
Figure 2. Syptoms of AChR-MG, AChR+LRP4-MG, and AChR+Titan-MG patients during different time period. (A) 6 months before study, (B) 12 months before study, and (C) 24 months before study.
Patients were treated with standard therapies for MG. The rates of achieving MMS or better in the three groups within 2 years after immunosuppressive treatment were 51.5, 62.1, and 51.7%, respectively (Table 2). AChR+Titin-MG treatment was highly dependent on steroids combined with immunosuppressive agents (Table 3).
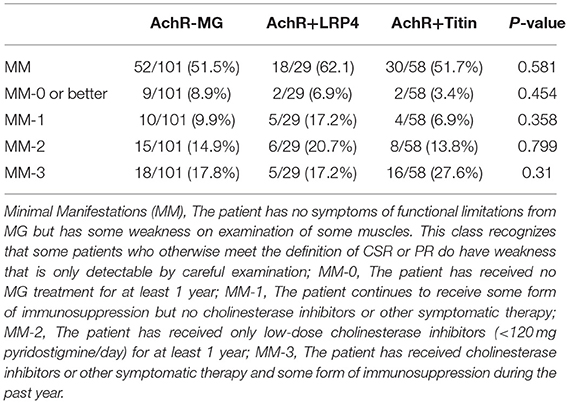
Table 2. Comparison of the prognosis after immunosuppressive therapy among AchR-MG, AchR+LRP4 and AchR+Titin MG.
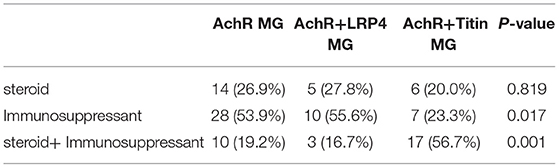
Table 3. Therapeutic strategy for MMS or better among three groups.
Our study found that clinical symptoms of anti-AChR MG combined with anti-LRP4 or anti-Titin antibody were more severe and progressed faster than anti-AChR positive MG. Regardless of antibody status, all patients responded well to immunotherapy and had relatively good prognoses.
About 85% of MG patients have autoantibodies against AChR, whereas 5%-26% of MG have autoantibodies against MuSK (1–3, 15–17). We focused on AChR positive patients in this study. We only found one patient who was both AChR and MuSK antibody-positive, which was lower than the proportion of patients who had seronegative MG (and who were not excluded in our study).
Most previous studies on LRP4 have largely focused on the seronegative MG population. However, there have been some reports of anti-AChR patients with double-positive LRP4 antibodies in clinical practice (20). Among 1,109 patients diagnosed with MG at our center, we found 29 cases with AChR combined with anti-LRP4 antibodies. The proportion of patients with anti-LRP4-antibodies was 2.61%, which coincided with the proportion in double-negative patients (14) and in other studies (18–20). Titin auto-antibodies were found in 30.9% of seropositive patients [compared with the 20–40% that has previously been reported in the literature (11, 13, 21). We found that the AChR+LRP4-MG phenotype showed a strong female predominance (72.4%), which was consistent with previous studies (22, 23). The mean age of onset in the three groups was 47.41 ± 7.0, 49.81 ± 9.2, and 48.11 ± 6.5 years, respectively. We focused on adults with generalized MG and excluded relatively young ocular patients.
Most MG patients with ocular symptoms at onset may progress to the generalized form of the disease within 2 years (1, 3). Our results confirmed that most MG patients had an ocular-only onset. However, the AChR+LRP4-MG group had significantly higher numbers of patients with limb weakness during disease onset than the AChR-MG or AChR+Titin-MG group. Therefore, AChR+LRP4-MG patients were much more likely to have generalized MG at the time of disease onset. Three of the AChR+LRP4-MG patients presented with MGFA class V in our study (20). It is unknown if other antibodies, such as agrin, were positive because that testing was not done (14).
Compared to the AChR-MG and AChR+LRP4-MG groups, AChR+Titin-MG patients showed shorter progression times from ocular to generalized MG (within 5.1 months). Rapid disease progression following symptom onset maybe because of the involvement of titin antibodies. Additionally, our data on MGFA classifications showed that 25.9% of AChR+Titin-MG patients were classified as MGFA IIIb, while 48.3% of AChR+LRP4-MG patients were classified as MGFA IIa. Moreover, there were more patients with MGFA IVb -V. Our results indicated that AChR+Titin-MG was associated with more severe disease status. Titin antibodies are usually considered to be accompanying antibodies and can only be found in patients with MG and anti-AChR antibodies. It is highly likely that the presence of thymoma in AChR+Titin-MG patients is related to their disease pathology.
Muscles that were involved at different time points (i.e., 6, 12, and 24 months before our study) did not differ significantly among the three groups. Thus, affected muscle groups appear to be similar at different stages of the disease, although disease severity differs.
Current common treatments for MG include AChE inhibitors, immunosuppressive drugs, thymectomy, IVIG, and plasmapheresis (24, 25). In our study, the proportions of patients who have achieved MM-3 or better for more than 6 months in the three groups were 51.5, 62.1, and 51.7%, respectively. These percentages are higher than what was reported in a study conducted by Utsugisawa K (26) but are consistent with other previous studies (27, 28).
All patients were treated with pyridostigmine. Monotherapy with an immunosuppressive agent was used in 53.9 and 55.6% of AChR-MG and AChR+LRP4-MG patients, and immunosuppressive therapy was used in combination therapy with azathioprine or tacrolimus corticosteroids in 56.7% of patients with AChR+Titin-MG.
The proportion of steroids combined with immunosuppressive agents in the AChR+Titin MG group was much higher than in the other two groups, suggesting that AChR+Titin MG needs stronger immunotherapy to achieve the same outcomes and is thus also associated with severe immune dysfunction.
In summary, anti-AChR positive MG can coexist with anti-LRP4 or anti-Titin antibodies. AChR+LRP4-MG has a female predominance and presents with milder symptoms. Furthermore, AChR+Titin-MG shows a shorter conversion time from ocular to generalized MG, a higher incidence of thymoma, and has a more severe presentation than AChR+LRP4-MG. Regardless of antibody status, all patients responded well to immunotherapy.
The original contributions presented in the study are included in the article/supplementary material, further inquiries can be directed to the corresponding author/s.
The studies involving human participants were reviewed and approved by The First Medical Center of PLA General Hospital. The patients/participants provided their written informed consent to participate in this study. Written informed consent was obtained from the individual(s) for the publication of any potentially identifiable images or data included in this article.
YC and XT acquired the clinical data, reviewed the literature, and drafted the article. JH and FQ designed the study, supervised the initial drafting, and critically revised the article. YW, SX and YY collected and analyzed the clinical data. All authors contributed to the article and approved the submitted version.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
2. Berrih-Aknin S, Frenkian-Cuvelier M, Eymard B. Diagnostic and clinical classification of autoimmune myasthenia gravis. J Autoimmun. (2014) 48-9:143–8. doi: 10.1016/j.jaut.2014.01.003
3. Lazaridis K, Tzartos SJ. Myasthenia Gravis: autoantibody specificities and their role in MG management. Front Neurol. (2020) 11:596981. doi: 10.3389/fneur.2020.596981
4. Skeie GO, Aarli JA, Gilhus NE. Titin and ryanodine receptor antibodies in myasthenia gravis. Acta Neurol Scand Suppl. (2006) 183:19–23. doi: 10.1111/j.1600-0404.2006.00608.x
5. Dresser L, Wlodarski R, Rezania K, Soliven B. Myasthenia gravis: epidemiology, pathophysiology and clinical manifestations. J. Clin. Med. (2021) 10:2235. doi.org/10.3390/jcm10112235. doi: 10.3390/jcm10112235
6. Yamamoto T, Sato T, Sugita H. Antifilamin, antivinculin, and antitropomyosin antibodies in myasthenia gravis. Neurology. (1987) 37:1329–33. doi: 10.1212/WNL.37.8.1329
7. Cordts I, Bodart N, Hartmann K, Karagiorgou K, Tzartos JS, Mei L, et al. Screening for lipoprotein receptor-related protein 4-, agrin-, and titin-antibodies and exploring the autoimmune spectrum in myasthenia gravis. J Neurol. (2017) 264:1193–203. doi: 10.1007/s00415-017-8514-z
8. Illa I, Cortes-Vicente E, Martinez MA, Gallardo E. Diagnostic utility of cortactin antibodies in myasthenia gravis. Ann N Y Acad Sci. (2018) 1412:90–4. doi: 10.1111/nyas.13502
9. Gasperi C, Melms A, Schoser B, Zhang Y, Meltoranta J, Risson V, et al. Anti-agrin autoantibodies in myasthenia gravis. Neurology. (2014) 82:1976–83. doi: 10.1212/WNL.0000000000000478
10. Fichtner ML, Jiang R, Bourke A, Nowak RJ. and O'Connor KC. Autoimmune pathology in myasthenia gravis disease subtypes Is governed by divergent mechanisms of immunopathology. Front Immunol. (2020) 11:776. doi: 10.3389/fimmu.2020.00776
11. Stergiou C, Lazaridis K, Zouvelou V, Tzartos J, Mantegazza R, Antozzi C, et al. Titin antibodies in “seronegative” myasthenia gravis–a new role for an old antigen. J Neuroimmunol. (2016) 292:108–15. doi: 10.1016/j.jneuroim.2016.01.018
12. Romi F, Aarli JA, Gilhus NE. Myasthenia gravis patients with ryanodine receptor antibodies have distinctive clinical features. Eur J Neurol. (2007) 14:617–20. doi: 10.1111/j.1468-1331.2007.01785.x
13. Somnier FE, Engel PJ. The occurrence of anti-titin antibodies and thymomas: a population survey of MG 1970-1999. Neurology. (2002) 59:92–8. doi: 10.1212/WNL.59.1.92
14. Rivner MH, Quarles BM, Pan JX, Yu Z, Corse A, Dimachkie MM, et al. Clinical features of LRP4/agrin-antibody-positive myasthenia gravis: A multicenter study. Muscle Nerve. (2020) 62:333–43. doi: 10.1002/mus.26985
15. Zhang Z, Guan Y, Han J, Li M, Shi M, Deng H. Regional features of MuSK antibody-positive myasthenia gravis in northeast China. Front Neurol. (2020) 11:516211. doi: 10.3389/fneur.2020.516211
16. Gilhus NE, Skeie GO, Romi F, Lazaridis K, Zisimopoulou P, Tzartos S. Myasthenia gravis-autoantibody characteristics and their implications for therapy. Nat Rev Neurol. (2016) 12:259–68 doi: 10.1038/nrneurol.2016.44
17. Ohta K, Shigemoto K, Fujinami A, Maruyama N, Konishi T, Ohta M. Clinical and experimental features of MuSK antibody positive MG in Japan. Eur J Neurol. (2007) 14:1029–34. doi: 10.1111/j.1468-1331.2007.01870.x
18. Pevzner A, Schoser B, Peters K, Cosma NC, Karakatsani A, Schalke B, et al. Anti-LRP4 autoantibodies in AChR- and MuSK-antibody-negative myasthenia gravis. J Neurol. (2012) 259:427–35. doi: 10.1007/s00415-011-6194-7
19. Li Y, Zhang Y, Cai G, He D, Dai Q. Anti-LRP4 autoantibodies in Chinese patients with myasthenia gravis. Muscle Nerve. (2017) 56:938–42. doi: 10.1002/mus.25591
20. Zisimopoulou P, Evangelakou P, Tzartos J, Lazaridis K, Zouvelou V, Mantegazza R, et al. A comprehensive analysis of the epidemiology and clinical characteristics of anti-LRP4 in myasthenia gravis. J Autoimmun. (2014) 52:139–45. doi: 10.1016/j.jaut.2013.12.004
21. Szczudlik P, Szyluk B, Lipowska M, Ryniewicz B, Kubiszewska J, Dutkiewicz M, et al. Antititin antibody in early- and late-onset myasthenia gravis. Acta Neurol Scand. (2014) 130:229–33. doi: 10.1111/ane.12271
22. Tsivgoulis G, Dervenoulas G, Kokotis P, Zompola C, Tzartos JS, Tzartos SJ, et al. Double seronegative myasthenia gravis with low density lipoprotein-4 (LRP4) antibodies presenting with isolated ocular symptoms. J Neurol Sci. (2014) 346:328–30. doi: 10.1016/j.jns.2014.09.013
23. Higuchi O, Hamuro J, Motomura M, Yamanashi Y. Autoantibodies to low-density lipoprotein receptor-related protein 4 in myasthenia gravis. Ann Neurol. (2011) 69:418–22. doi: 10.1002/ana.22312
24. Morren J, Li Y. Maintenance immunosuppression in myasthenia gravis, an update. J Neurol Sci. (2020) 410:116648. doi: 10.1016/j.jns.2019.116648
25. Lee JI, Jander S. Myasthenia gravis: recent advances in immunopathology and therapy. Expert Rev Neurother. (2017) 17:287–99. doi: 10.1080/14737175.2017.1241144
26. Utsugisawa K, Nagane Y, Akaishi T, Suzuki Y, Imai T, Tsuda E, et al. Early fast-acting treatment strategy against generalized myasthenia gravis. Muscle Nerve. (2017) 55: 794–801. doi: 10.1002/mus.25397
27. Zhang C, Bu B, Yang H, Wang L., Liu w, Duan RS, et al. Immunotherapy choice and maintenance for generalized myasthenia gravis in China. CNS Neurosci Ther. (2020) 26:1241–54. doi: 10.1111/cns.13468
Keywords: acetylcholine receptor, Myasthenia Gravis, low-density lipoprotein receptor-related protein 4, Titin antibody, minimal manifestations status (MMS)
Citation: Chen Y, Tao X, Wang Y, Xu S, Yang Y, Han J and Qiu F (2022) Clinical Characteristics and Prognosis of Anti-AChR Positive Myasthenia Gravis Combined With Anti-LRP4 or Anti-Titin Antibody. Front. Neurol. 13:873599. doi: 10.3389/fneur.2022.873599
Received: 11 February 2022; Accepted: 06 April 2022;
Published: 09 May 2022.
Edited by:
Xiangjun Chen, Fudan University, ChinaReviewed by:
Hongjun Hao, Peking University First Hospital, ChinaCopyright © 2022 Chen, Tao, Wang, Xu, Yang, Han and Qiu. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Feng Qiu, cWl1ZmVuZ25ldEBob3RtYWlsLmNvbQ==; Jinming Han, aGFuamlubWluZzEyMDJAMTI2LmNvbQ==
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.