 Fan Wang1,2†
Fan Wang1,2† Xiangyi Liu
Xiangyi Liu Lu Chen
Lu Chen Lu Tang
Lu Tang Dongsheng Fan
Dongsheng Fan
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Neurol., 11 April 2022
Sec. Dementia and Neurodegenerative Diseases
Volume 13 - 2022 | https://doi.org/10.3389/fneur.2022.865264
ERBB4 is related to amyotrophic lateral sclerosis (ALS) in patients with a family history and is thought to cause ALS-19. We screened 448 ALS patients, including 364 sporadic ALS (sALS) and 84 familial ALS (fALS) patients with ERBB4 variants, in a Chinese cohort. In total, 12 missense variants were identified in this study. Of these, 3 (p.Arg106His, p.Gln164Pro, and p.Val212Leu) were absent from the in-house healthy control cohort and population databases and predicted to be likely pathogenic. Genetic burden analysis did not reveal an increase in damaging variants of the ERBB4 gene. We considered that most of the missense variants in ERBB4 were not pathogenic, but certain variants, such as p.Arg106His, p.Gln164Pro, and p.Val212Leu, were likely pathogenic. The phenotype of these three patients carrying ERBB4 variants revealed the typical clinical manifestations of ALS without cognitive dysfunction. We concluded that ERBB4 likely pathogenic variants account for ~0.67% of ALS patients in China. It is necessary to interpret the relationship between the disease and variants carefully for ALS patients with ERBB4 gene variants.
Amyotrophic lateral sclerosis (ALS) is a progressive and fatal neurodegenerative disease that mainly affects the upper and lower motor neurons of the spinal cord and brain (1). The ALS incidence in European populations has been estimated to be 2.16 cases per 100,000 people per year, and it usually leads to death ~3–4 years after onset (2, 3). In the Chinese population, the ALS incidence is 1.65 per 100,000 people per year (4), and the median survival time is 71 months after symptom onset (5). Furthermore, the clinical characteristics seem to be different between Chinese ALS patients and ALS patients in Caucasian population (6).
Approximately 5–10% of patients have a positive family history (fALS), while the remaining cases are sporadic (sALS). Interestingly, the gene mutations of fALS are also present in sALS. Genetic factors play an important role in the pathogenesis of ALS (7). The genetic cause of ~10% of sALS and 2/3 of fALS in the Caucasian population has been determined (8). Importantly, there is likely to be genetic heterogeneity between ethnic groups with ALS (9). Our previous studies have shown that the frequency of gene variant differences between Chinese and Caucasian ALS patients could be a primary reason underlying the distinct clinical features (10, 11). To date, more than 25 mutant genes have been shown to cause or significantly increase the risk of ALS.
ERBB4 is a transmembrane tyrosine kinase of the epidermal growth factor receptor (EGFR) family that includes ERBB1, ERBB2, ERBB3, and ERBB4 (12). After binding of neuregulin (NRG) to other members of the subfamily, such as ERBB2 or ERBB3, ERBB4 forms homodimers or heterodimers to activate the C-terminal domains and autophosphorylation of its tyrosine kinase, thereby mediating various downstream signaling cascades (13). ERBB4 plays an essential role in neurodevelopment, such as nerve conduction and synaptic plasticity, and it is also related to the occurrence of mental illness and epilepsy (14, 15). The ERBB4 mutation was first found in a family of ALS patients in 2013 and was believed to be a new pathogenic gene of ALS-19 (16). A study conducted a genetic analysis of known ALS/FTD patients that did not carry the C9orf72 expansion mutation and found that the ERBB4 mutation (c.1997T>C, p.Ile666Thr) was the probable pathogenic variant (17). A heterozygous mutation in ERBB4 (c.2136T>G, p.Ile712Met) was also reported in late-onset alS/FTD (18). Moreover, in the pathophysiology of ALS, immunofluorescence staining showed that ERBB4 immunoreactivity was decreased in the spinal cord of sALS patients, which further indicates that ERBB4 is involved in the pathophysiological process of sALS (19). In this study, we screened ERBB4 variants in a large cohort of ALS patients from mainland China to determine their pathogenicity and frequency.
This study included 448 Chinese ALS patients who were admitted to the Department of Neurology, Peking University Third Hospital from January 2015 to July 2021. All patients were diagnosed with definite, probable, or laboratory-supported probable ALS according to the revised El Escorial criteria (20), and patients who had ALS-like syndrome caused by paraneoplastic or autoimmune diseases or with suspected or possible ALS were excluded. A total of 1,812 control subjects without any neurological disease history were included in the in-house control cohort. Written informed consent for genetic analysis was obtained from all participants. This study was approved by the ethics committee at Peking University Third Hospital (IRB No. 00006761).
According to standard procedures, patient and control genomic DNA was extracted from peripheral blood samples (Qiagen, Valencia, California). DNA was analyzed predominantly by targeted next-generation sequencing (NGS), which was carried out at Kangso Medical Inspection Company (Beijing, China) following standard experimental protocols. The panel of ALS related gene tested in this study were list in Supplementary Table 1. Sequencing reactions were designed to include all coding regions, as well as the flanking ~100 bp of intronic DNA for each exon and 3′ and 5′ untranslated regions (UTRs) of the ERBB4 gene (NM_005235). The quality of the sequencing data was assessed using the BWA, Samtools, Picard, and Genome Analysis Toolkit (GATK) available from Babraham Bioinformatics (http://www.bioinformatics.babraham.ac.uk/). Assessing factors included the allele balance, coverage uniformity, the total count of reads, the percentage of reads that matched the sequence of the human genome and were located inside the target sequence of the target gene, and the average sequencing depth. DNASTAR Lasergene v7.1 was used to compare the cDNA sequence with the reference ERBB4 mRNA sequence. The ERBB4 variants were compared with those reported in the Single Nucleotide Polymorphism Database (dbSNP; https://www.ncbi.nlm.nih.gov/snp), ChinaMAP (http://www.mbiobank.com/), gnomAD (East Asian, EA) (https://gnomad.broadinstitute.org) and 1000 Genomes Projects (East Asian, EA) (http://browser.1000genomes.org) databases.
In silico tools were applied to identify the functional effects of the ERBB4 missense variants, including PolyPhen2 (http://genetics.bwh.harvard.edu/pph2/), SIFT (http://sift.jcvi.org), Mutation Taster (http://www.mutationtaster.org), and CADD. Sequence homologs were aligned to analyze the level of evolutionary conservation using the UniProt website (http://www.uniprot.org/). Three methods were used for the burden analysis of the rare variants of the ERBB4 gene, including the C-alpha test, the Rare-Variant Weighted Aggregate Statistic (RWAS) and the Sequence Kernel Association Test (SKAT), using the R package AssotesteR. Two levels of rare variants were evaluated: (1) rare exon variants with minor allele frequency (MAF) <1% in the population database (gnomAD) (the rare variants set); (2) ultrarare missense variants (presented less than twice in the gnomAD database, according to the prevalence of ALS) with Combined Annotation Dependent Depletion (CADD) score>20 (the ultrarare variants set). The p-values of 10,000 resamplings were also reported. Suspected variants were interpreted mainly based on the American College of Medical Genetics and Genomics (ACMG) guidelines. The structures of wild-type and mutated ERBB4 were calculated according to their alignment by SWISS-MODEL Server (Swiss Institute of Bioinformatics, Lausanne, Switzerland) (21). Graphics were generated using Swiss-Pdb viewer 3.7 software after put the structures of wild-type and mutated ERBB4 into it (22). The ERBB4 protein domain was derived from the National Center for Biotechnology Information (NCBI, https://www.ncbi.nlm.nih.gov) after searched with ERBB4 as a keyword in NCBI. All cases with potentially pathogenic variants in ERBB4 were also sequenced for TARDBP, FUS, SOD1, and C9ORF72.
In total, 448 participants with ALS and 1,812 healthy control subjects were screened. The proportions of definite, probable, or laboratory-supported probable ALS patients were 18.38, 39.46, and 42.16%, respectively. Among the patients, 84 patients were unrelated fALS probands, and 364 patients were considered to have sALS. In total, 293 patients were males, and the remaining 155 patients were females. The mean age of onset ± SD was 42 ± 11.31 years old.
In this study, the total count of reads was 8-9 G, and the percentage of reads that matched the human genome sequence was ~99.9%. The percentage of reads that were located inside the target sequence of the ERBB4 gene was above 99.8%, and the average sequencing depth was above 100 × .
Twelve missense variants were identified in 16 unrelated ALS patients, and their allele frequencies in the healthy control cohort and the public population databases are listed in Table 1. Neither small insertion and deletion mutations nor splice site mutations were found. The variants were all heterozygous. All 12 missense variants were rare variants that were defined as heterozygous variants in dominant ALS-causative genes with MAF <0.1% in any of the following databases: ChinaMAP, gnomAD (EA) and 1000 Genomes Projects (EA). We also identified 278 non-synonymous variants in the control cohort (Supplementary Table 2).
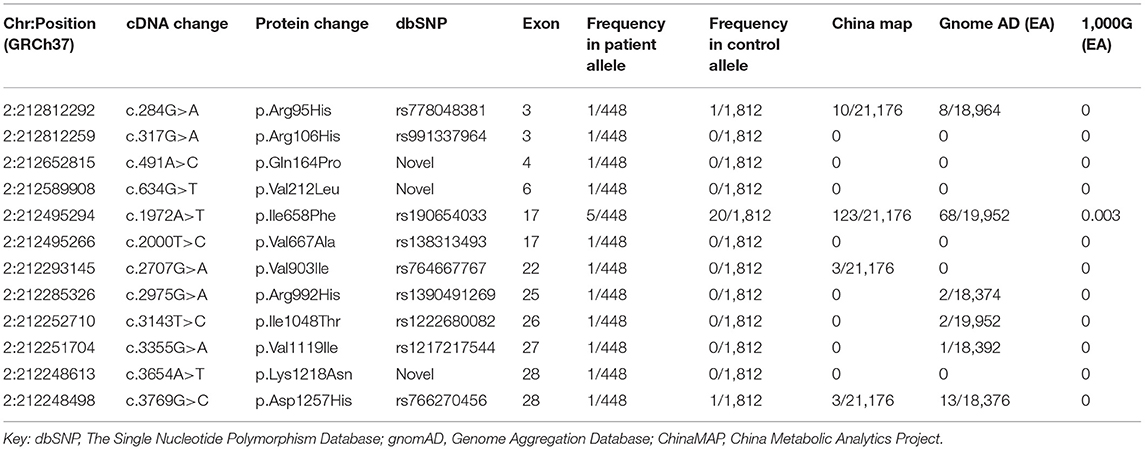
Table 1. ERBB4 missense variants identified both in ALS patients and control subjects with related information in the public database.
c.317G>A (p.Arg106His), c.491A>C (p.Gln164Pro), c.634G>T (p.Val212Leu), and c.2000T>C (p.Val667Ala), c.3654A>T (p.Lys1218Asn) were present neither in the control cohort nor in any of the public population databases, including ChinaMAP, gnomAD (EA), 1000 Genomes Projects (EA) and dbSNP. The functional effects of these 5 missense variants were predicted by PolyPhen2, SIFT, Mutation Taster, and CADD (Table 2). The functional effects of c.2000T>C (p.Val667Ala) and c.3654A>T (p.Lys1218Asn) were benign predicted by in silico tools. The patients who carried other 3 variants (p.Arg106His, p.Gln164Pro and p.Val212Leu) did not carry common pathogenic mutations related to ALS, such as mutations in SOD1, TARDBP, FUS, and C9ORF72 (Supplementary Figure 1). Sequences of the 3 variant sites were validated by Sanger sequencing (Figure 1).
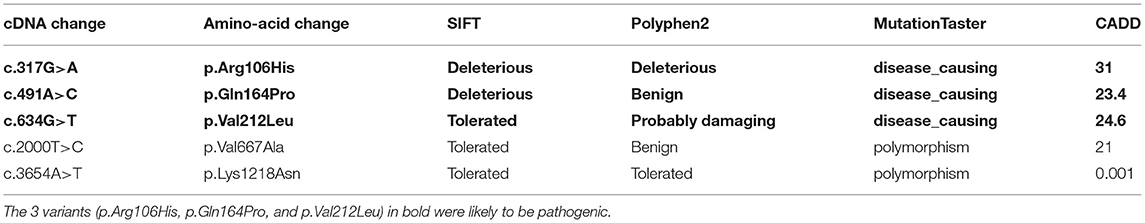
Table 2. Results predicted by in silico tools of the missense variants which were present neither in control cohort nor in any of the population polymorphism databases.
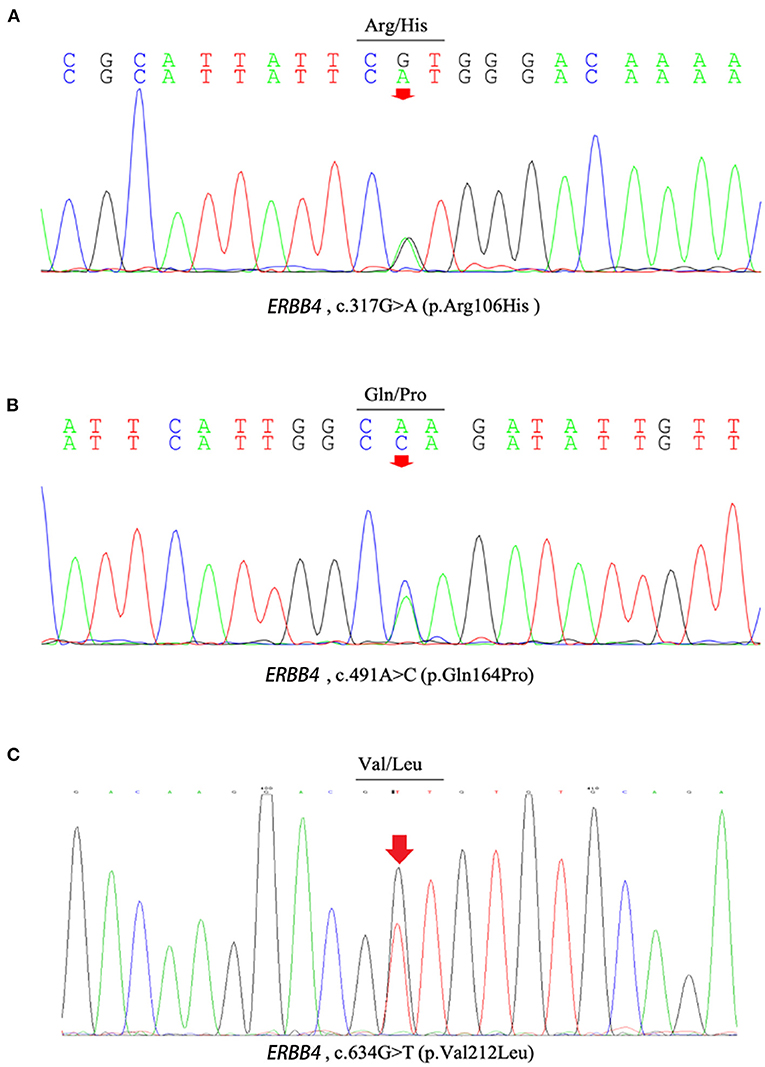
Figure 1. The chromatograph of ERBB4 variants (p.Arg106His, p.Gln164Pro and p.Val212Leu). (A) The chromatograph of ERBB4 p.Arg106His variant. (B) The chromatograph of ERBB4 p.Gln164Pro variant. (C) The chromatograph of ERBB4 p.Val212Leu variant.
Clinical information of the 16 unrelated ALS patients with 12 missense variants are list in Table 3. All three patients carrying c.317G>A (p.Arg106His), c.491A>C (p.Gln164Pro), and c.634G>T (p.Val212Leu) were males. The average onset age was 49.33 ± 7.10 years old. Two patients had limb onset, and one patient had bulbar onset. However, none of these patients had cognitive dysfunction. They were all alive, and their survival times were 27, 30, and 60 months. The patient with the c.317G>A (p.Arg106His) variant had limb onset at the age of 57 years old. He felt weakness in his left foot in 2016, and his condition gradually worsened. He had left foot drops and unsteady walking 2 years later. By 2020, he developed muscle atrophy in his left leg and weakness in his right hand. The patient with the c.491A>C (p.Gln164Pro) variant had limb onset at the age of 43 years old. In May 2019, the patient developed left arm weakness, muscle atrophy, and fasciculations, and his condition gradually worsened too. Seven months later, he was diagnosed with probable ALS. The patient with the c.634G>T (p.Val212Leu) variant had bulbar onset at the age of 48 years, and 18 months later, he was diagnosed with probable ALS. Initially, he mainly suffered from slurred speech, choking on drinking water, and difficulty swallowing in February 2019. Later, he developed finger weakness and difficulty lifting his upper limbs, which was accompanied by fasciculations. Unfortunately, he was a familial patient, and his cousin had similar symptoms and the same mutation site as him. His cousin's son also carried the same mutation at this locus but did not show any clinical symptoms. Interestingly, his cousin's twin brother had no similar symptoms. The chromatograph of ERBB4 p.Val212Leu mutation and its normal control is shown in Figure 2A. The pedigree of this family is shown in Figure 2B. The Val212 residue was highly conserved across different species (Figure 2C). The structures of wild-type ERBB4 and mutated ERBB4 were calculated according to their alignment (Figure 2D). As shown in the schematic of the ERBB4 protein domain, the mutation site was located in the Furin-like cysteine-rich region (shown by the red arrow, Figure 2E).
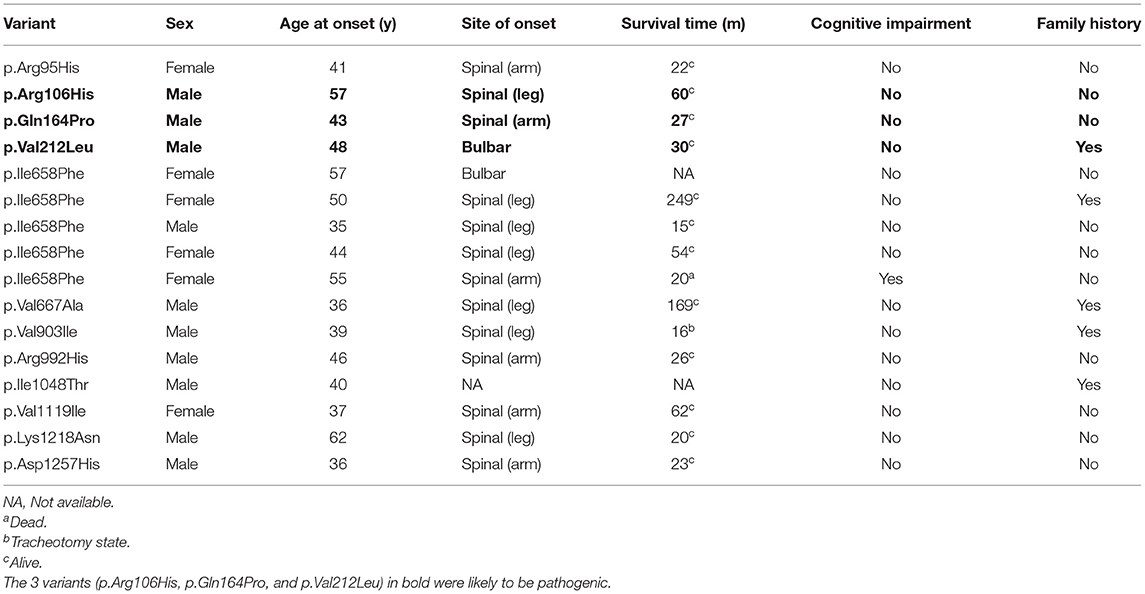
Table 3. Clinical information of the 16 unrelated ALS patients with 12 missense variants.
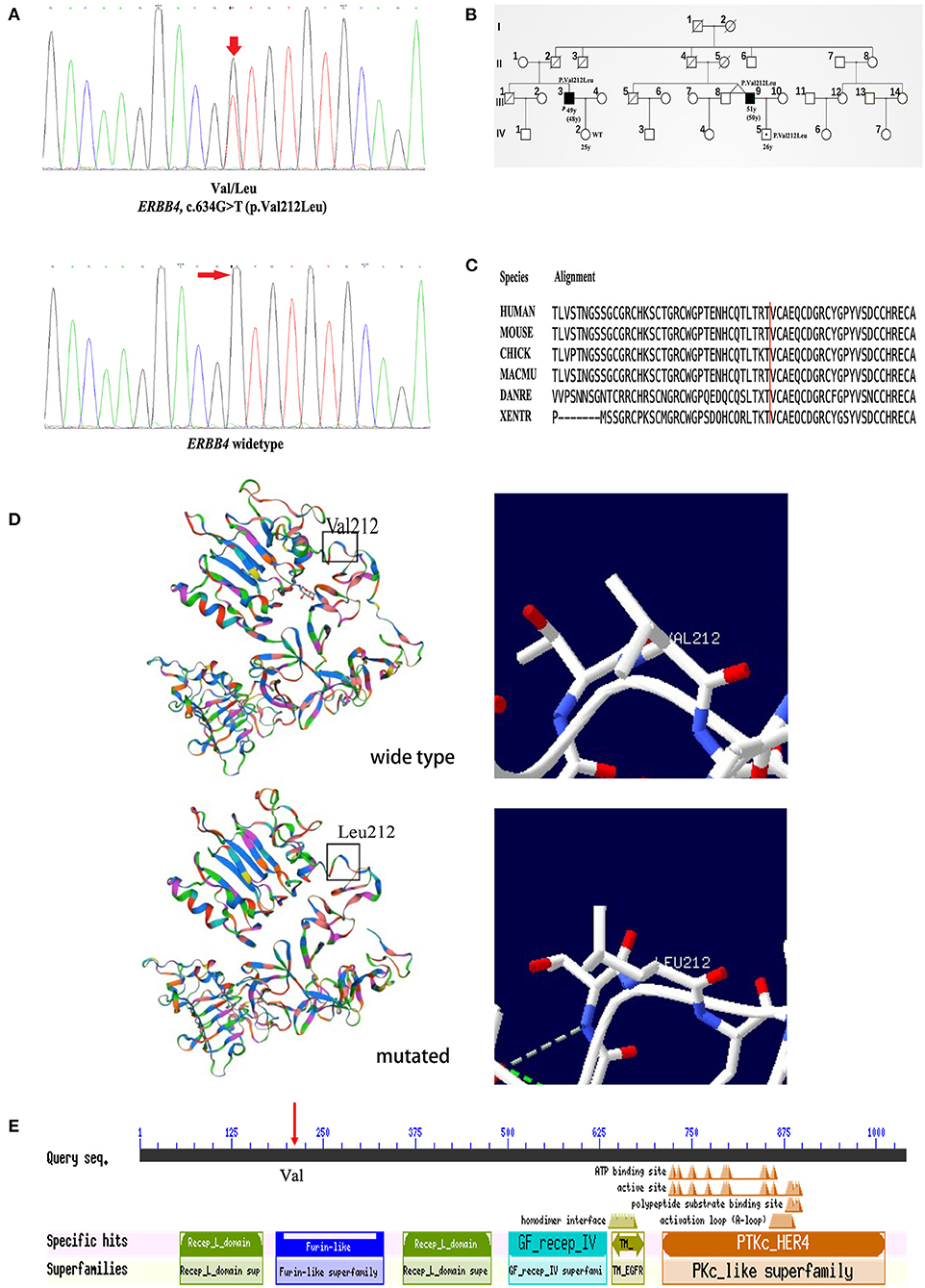
Figure 2. Genetic analysis of the pedigree carrying the ERBB4 p.Val212Leu mutation. (A) The chromatograph of ERBB4 p.Val212Leu mutation and its normal control. (B) Pedigree structure of family carrying the ERBB4 p.Val212Leu mutation (III3 and III9: Affected person carrying mutant gene. IV5: Unaffected person carrying mutant gene). (C) The ERBB4 p.Val212Leu residue is highly conserved across multiple species. (D) The structures of wild-type ERBB4 and mutated ERBB4. (E) The ERBB4 protein domain and the mutation site were located in the Furin-like cysteine-rich region (red arrow).
The results of burden analysis performed by three methods (C-alpha, RWAS, SKAT) are listed in Table 4. A total of 448 ALS patients and 1,812 neurologically normal control individuals were included in the analysis. There was no significant difference between the two variant sets.

Table 4. Results of burden tests in ALS patients and controls.
Here, 448 participants with ALS and 1,812 healthy control subjects were screened. Among them, 84 patients were unrelated fALS probands, and 364 patients were considered to have sALS. We identified 12 missense variants in 16 unrelated ALS patients, and this was the first report of these variants in ALS patients, except for c.284G>A (p.Arg95His) (23). There were 3 variants (p.Arg106His, p.Gln164Pro, and p.Val212Leu) that were likely to be pathogenic, especially the p.Val212Leu variant. The incidence of likely pathogenic variants in our ALS cohort was 0.67% (3/448), and that in the fALS cohort and sALS cohort were 1.19% (1/84) and 0.55% (2/364), respectively. However, the incidence of ERBB4 mutations in sALS was 0.29% (2/691) in central South China (24). This was lower than that in the present study and might be due to the different screening criteria used. In other studies of the genetic spectrum and variability in ALS patients, researchers found several ERBB4 variants, such as p.Met322Lys (25), Glu69Val, Arg103His (26, 27), p.Gly1272Arg, p.His374Gln and p.Met1059Thr, and the p.Gly1272Arg variant was a probable pathogenic variant (28).
The p.Val212Leu variant was found in fALS. The Val212 residue is highly conserved across different species, and high evolutionary conservation always suggests the functional importance of a position. Several lines of evidence support the pathogenicity of the p.Val212Leu variant. First, the variant was located in the well-studied functional domain without benign variation (PM1). Second, the variant was not reported in any of the public databases, such as dbSNP, ChinaMAP, gnomAD (EA) and 1000 Genomes Projects (EA) (PM2). Third, the variant cosegregated with the phenotype of ALS in the affected family members (PP1). Furthermore, this variant was classified as disease-causing by several in silico predictions, and the Val212 residue was highly conserved across species (PP3). Last, the patients' clinical phenotype in this family was highly consistent with the disease caused by the ERBB4 variant (PP4). Therefore, according to the ACMG variant classification criteria, the p.Val212Leu variant may be classified as “likely pathogenic” (PM1+PM2+PP1+PP3+PP4) (29). Combining the functional prediction results of the in silico tools with a CADD score>20, we also inferred that the c.317G>A (p.Arg106His), c.491A>C (p.Gln164Pro), and c.634G>T (p.Val212Leu) variants were likely to be pathogenic.
The mutation region was in the Furin-like cysteine-rich region of the ERBB4 protein. Studies have proven that the Furin-like cysteine-rich region mainly mediates the Wnt/β-catenin signaling pathway, which plays a potential role in the pathogenesis of ALS (30–33). The abnormal activation of this pathway is related to neuronal degeneration and glial cell proliferation (33). Two previous studies found that the pathogenesis of ERBB4 mutations in ALS was related to decreased autophosphorylation (16, 18). According to the structure prediction, there was no obvious differences between mutant and wildtype protein. Previous study has shown that the RMSD/RMSF plot is helpful in determining the effect of amino acid changes on protein structure (34), other methods including mRNA expression level and animal models are also needed to verify the pathogenicity of p.Val212Leu in the future.
Although we considered the p.Arg106His, p.Gln164Pro, and p.Val212Leu variants were likely pathogenic, the burden analysis showed that the rare variants in our study were not significantly different from the controls. It was indicated that most of the missense variants in ERBB4 were not pathogenic, but certain variants, such as p.Arg106His, p.Gln164Pro, and p.Val212Leu, were likely pathogenic. Therefore, it is necessary to interpret the relationship between the disease and variants carefully for ALS patients with ERBB4 gene variants.
In our study, clinical information of the three likely pathogenic variants (p.Arg106His, p.Gln164Pro, and p.Val212Leu) revealed that all three patients were males, and the average age at onset was 49.33 ± 7.10 years old. The ratio of bulbar onset to limb onset was 1:2, and none of these patients had cognitive dysfunction. A study of 11 ALS patients with ERBB4 variants in southern China demonstrated similar clinical characteristics to our present study. The male/female ratio was 9:2, and the average onset age was 49.5 ± 12.6 years old. The limb-onset/bulbar-onset ratio was 10:1, and none of these patients had cognitive dysfunction (23). In our previous study on the clinical characteristics of ALS patients from 2015 to 2018, it was shown that the male/female ratio was 1.85:1, the average onset age was 53.0 ± 11 years old, and the limb-onset/bulbar-onset ratio was 3.71:1 (6). Similar to our study, most previous reports have shown that ALS patients with ERBB4 variants did not have cognitive dysfunction, except for two heterozygous variants of ERBB4 (c.2136T>G, p.Ile712Met; c.1997T>C, p.Ile666Thr) (17, 18).
This study has certain limitations. Due to some special reasons, such as the impact of the new coronavirus epidemic, our collection of the data on ALS families was not comprehensive, which affected our judgment on the pathogenicity of the mutation. Long-term follow-up observation of the families is needed. In addition, because ALS is both clinically and genetically a highly heterogeneous disease (35), it may reduce the power to detect some effects. Furthermore, all patients who participated in the research were recruited from China. Due to the long history of intermarriage among the population, the patient population in this study was heterogeneous, which may further limit our analysis power.
In this Chinese ALS cohort, 12 missense variants were identified, and 3 of these variants (p.Arg106His, p.Gln164Pro, and p.Val212Leu) were likely pathogenic. We considered that most of the missense variants in ERBB4 were not pathogenic, but certain variants, such as p.Arg106His, p.Gln164Pro, and p.Val212Leu, were likely pathogenic, especially the p.Val212Leu variant. We concluded that the incidence of likely pathogenic variants in our ALS cohort was 0.67% (3/448), and that of the fALS cohort and sALS cohort were 1.19% (1/84) and 0.55% (2/364), respectively. Therefore, it is necessary to interpret the relationship between the disease and variants carefully for ALS patients with ERBB4 gene variants.
The data presented in the study are deposited in the NCBI SRA repository, accession number PRJNA814828, the project information is accessible with the following link: http://www.ncbi.nlm.nih.gov/bioproject/814828.
The studies involving human participants were reviewed and approved by Ethics Committee at Peking University Third Hospital. The patients/participants provided their written informed consent to participate in this study. Written informed consent was obtained from the individual(s) for the publication of any potentially identifiable images or data included in this article.
FW, XL, and DF conceived the study. FW and XL performed the experiments and data analyses. JH, NZ, LT, and LC conducted the patient enrolment and follow-up. FW wrote the manuscript. XL and DF revised the manuscript. All authors contributed to the article and approved the submitted version.
This study was supported by Grants from the National Natural Science Foundation of China (Grant Numbers 81873784, 82071426, 82001361, and 81974197), Clinical Cohort Construction Program of Peking University Third Hospital (Grant Number BYSYDL2019002).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
The authors thank all the patients for their cooperation in this study.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fneur.2022.865264/full#supplementary-material
1. Brown RH, Al-Chalabi A. Amyotrophic lateral sclerosis. N Engl J Med. (2017) 377:162–72. doi: 10.1056/NEJMra1603471
2. van Es MA, Hardiman O, Chio A, Al-Chalabi A, Pasterkamp RJ, Veldink JH, et al. Amyotrophic lateral sclerosis. Lancet. (2017) 390:2084–98. doi: 10.1016/S0140-6736(17)31287-4
3. Kiernan MC, Vucic S, Cheah BC, Turner MR, Eisen A, Hardiman O, et al. Amyotrophic lateral sclerosis. Lancet. (2011) 377:942–55. doi: 10.1016/S0140-6736(10)61156-7
4. Xu L, Chen L, Wang S, Feng J, Liu L, Liu G, et al. Incidence and prevalence of amyotrophic lateral sclerosis in urban china: a national population-based study. J Neurol Neurosurg Psychiatry. (2020) 91:520–5. doi: 10.1136/jnnp-2019-322317
5. Lu Chen, Zhang B, Chen R, Tang L, Liu R, Yang Y, et al. Natural history and clinical features of sporadic amyotrophic lateral sclerosis in China. J Neurol Neurosurg Psychiatry. (2015) 86:1075–81. doi: 10.1136/jnnp-2015-310471
6. Chen L, Xu L, Tang L, Xia K, Tian D, Zhang G, et al. Trends in the clinical features of amyotrophic lateral sclerosis: a 14-year Chinese cohort study. Eur J Neurol. (2021) 28:2893–900. doi: 10.1111/ene.14943
7. Taylor JP, Brown RH Jr, Cleveland DW. Decoding als: from genes to mechanism. Nature. (2016) 539:197–206. doi: 10.1038/nature20413
8. Marangi G, Traynor BJ. Genetic causes of amyotrophic lateral sclerosis: new genetic analysis methodologies entailing new opportunities and challenges. Brain Res. (2015) 1607:75–93. doi: 10.1016/j.brainres.2014.10.009
9. He J, Mangelsdorf M, Fan D, Bartlett P, Brown MA. Amyotrophic lateral sclerosis genetic studies: from genome-wide association mapping to genome sequencing. Neuroscientist. (2015) 21:599–615. doi: 10.1177/1073858414555404
10. Liu X, Wu C, He J, Zhang N, Fan D. Two rare variants of the anxa11 gene identified in chinese patients with amyotrophic lateral sclerosis. Neurobiol Aging. (2019) 74:235.e9–.e12. doi: 10.1016/j.neurobiolaging.2018.09.020
11. Liu X, He J, Gao FB, Gitler AD, Fan D. The epidemiology and genetics of amyotrophic lateral sclerosis in China. Brain Res. (2018) 1693:121–6. doi: 10.1016/j.brainres.2018.02.035
12. Gregory D, Green JM, Culouscou JM, Carlton GW, Rothwell VM, Buckley S. Heregulin induces tyrosine phosphorylation of Her4/P180erbb4. Nature. (1993) 366:473–5. doi: 10.1038/366473a0
13. Burgess AW. Egfr family: structure physiology signalling and therapeutic targets. Growth Factors. (2008) 26:263–74. doi: 10.1080/08977190802312844
14. Mei L, Nave KA. Neuregulin-erbb signaling in the nervous system and neuropsychiatric diseases. Neuron. (2014) 83:27–49. doi: 10.1016/j.neuron.2014.06.007
15. Zhu JM, Li KX, Cao SX, Chen XJ, Shen CJ, Zhang Y, et al. Increased Nrg1-Erbb4 signaling in human symptomatic epilepsy. Sci Rep. (2017) 7:141. doi: 10.1038/s41598-017-00207-7
16. Takahashi Y, Fukuda Y, Yoshimura J, Toyoda A, Kurppa K, Moritoyo H, et al. Erbb4 mutations that disrupt the neuregulin-Erbb4 pathway cause amyotrophic lateral sclerosis type 19. Am J Hum Genet. (2013) 93:900–5. doi: 10.1016/j.ajhg.2013.09.008
17. Dols-Icardo O, Garcia-Redondo A, Rojas-Garcia R, Borrego-Hernandez D, Illan-Gala I, Munoz-Blanco JL, et al. Analysis of known amyotrophic lateral sclerosis and frontotemporal dementia genes reveals a substantial genetic burden in patients manifesting both diseases not carrying the C9orf72 expansion mutation. J Neurol Neurosurg Psychiatry. (2018) 89:162–8. doi: 10.1136/jnnp-2017-316820
18. Sun L, Cheng B, Zhou Y, Fan Y, Li W, Qiu Q, et al. Erbb4 mutation that decreased Nrg1-Erbb4 signaling involved in the pathogenesis of amyotrophic lateral sclerosis/frontotemporal dementia. J Alzheimers Dis. (2020) 74:535–44. doi: 10.3233/JAD-191230
19. Takahashi Y, Uchino A, Shioya A, Sano T, Matsumoto C, Numata-Uematsu Y, et al. Altered immunoreactivity of Erbb4, a causative gene product for Als19, in the spinal cord of patients with sporadic als. Neuropathology. (2019) 39:268–78. doi: 10.1111/neup.12558
20. Brooks BR, Miller RG, Swash M, Munsat TL, World World Federation of Neurology Research Group on Motor Neuron D. El escorial revisited: revised criteria for the diagnosis of amyotrophic lateral sclerosis. Amyotroph Lateral Scler Other Motor Neuron Disord. (2000) 1:293–9. doi: 10.1080/146608200300079536
21. Biasini M, Bienert S, Waterhouse A, Arnold K, Studer G, Schmidt T, et al. Swiss-model: modelling protein tertiary and quaternary structure using evolutionary information. Nucleic Acids Res. (2014) 42:W252–8. doi: 10.1093/nar/gku340
22. Guex N, Peitsch MC. Swiss-model and the swiss-pdbviewer: an environment for comparative protein modeling. Electrophoresis. (1997) 18:2714–23. doi: 10.1002/elps.1150181505
23. Chen W, Xie Y, Zheng M, Lin J, Huang P, Pei Z, et al. Clinical and genetic features of patients with amyotrophic lateral sclerosis in Southern China. Eur J Neurol. (2020) 27:1017–22. doi: 10.1111/ene.14213
24. Liu Z, Yuan Y, Wang M, Ni J, Li W, Huang L, et al. Mutation spectrum of amyotrophic lateral sclerosis in central South China. Neurobiol Aging. (2021) 107:181–8. doi: 10.1016/j.neurobiolaging.2021.06.008
25. Liu ZJ, Lin HX, Wei Q, Zhang QJ, Chen CX, Tao QQ, et al. Genetic spectrum and variability in chinese patients with amyotrophic lateral sclerosis. Aging Dis. (2019) 10:1199–206. doi: 10.14336/AD.2019.0215
26. Narain P, Padhi AK, Dave U, Mishra D, Bhatia R, Vivekanandan P, et al. Identification and characterization of novel and rare susceptible variants in indian amyotrophic lateral sclerosis patients. Neurogenetics. (2019) 20:197–208. doi: 10.1007/s10048-019-00584-3
27. Narain P, Pandey A, Gupta S, Gomes J, Bhatia R, Vivekanandan P. Targeted next-generation sequencing reveals novel and rare variants in indian patients with amyotrophic lateral sclerosis. Neurobiol Aging. (2018) 71:265.e9–.e14. doi: 10.1016/j.neurobiolaging.2018.05.012
28. Borg R, Farrugia Wismayer M, Bonavia K, Farrugia Wismayer A, Vella M, van Vugt J, et al. Genetic analysis of als cases in the isolated island population of Malta. Eur J Hum Genet. (2021) 29:604–14. doi: 10.1038/s41431-020-00767-9
29. Richards S, Aziz N, Bale S, Bick D, Das S, Gastier-Foster J, et al. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the american college of medical genetics and genomics and the association for molecular pathology. Genet Med. (2015) 17:405–24. doi: 10.1038/gim.2015.30
30. Jiang X, Guan Y, Zhao Z, Meng F, Wang X, Gao X, et al. Potential roles of the wnt signaling pathway in amyotrophic lateral sclerosis. Cells. (2021) 10:839. doi: 10.3390/cells10040839
31. Vallée A. Aerobic glycolysis activation through canonical wnt/?-catenin pathway in als. Med Sci. (2018) 34:326–30. doi: 10.1051/medsci/20183404013
32. Lecarpentier Y, Vallée A. Opposite interplay between Ppar gamma and canonical Wnt/Beta-catenin pathway in amyotrophic lateral sclerosis. Front Neurol. (2016) 7:100. doi: 10.3389/fneur.2016.00100
33. Pinto C, Cárdenas P, Osses N, Henríquez JP. Characterization of Wnt/?-Catenin and Bmp/Smad signaling pathways in an in vitro model of amyotrophic lateral sclerosis. Front Cell Neurosci. (2013) 7:239. doi: 10.3389/fncel.2013.00239
34. Vats A, Gourie-Devi M, Verma M, Ramachandran S, Taneja B, Kukreti R, et al. Identification of L84f mutation with a novel nucleotide change C255g > T in the superoxide dismutase gene in a North Indian family with amyotrophic lateral sclerosis. Amyotroph Lateral Scler Frontotemporal Degener. (2016) 17:253–9. doi: 10.3109/21678421.2015.1111906
Keywords: China, amyotrophic lateral sclerosis, ERBB4, variant, clinical features
Citation: Wang F, Liu X, He J, Zhang N, Chen L, Tang L and Fan D (2022) Analysis of ERBB4 Variants in Amyotrophic Lateral Sclerosis Within a Chinese Cohort. Front. Neurol. 13:865264. doi: 10.3389/fneur.2022.865264
Received: 29 January 2022; Accepted: 04 March 2022;
Published: 11 April 2022.
Edited by:
Sudeshna Das, Massachusetts General Hospital and Harvard Medical School, United StatesReviewed by:
Abhishek Vats, University of Pittsburgh, United StatesCopyright © 2022 Wang, Liu, He, Zhang, Chen, Tang and Fan. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Dongsheng Fan, ZHNmYW4yMDEwQGFsaXl1bi5jb20=
†These authors have contributed equally to this work
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.