 Sindhu Menon1
Sindhu Menon1 Sabrina Armstrong1Amir Hamzeh1
Sabrina Armstrong1Amir Hamzeh1 Naomi P. Visanji1,2,3
Naomi P. Visanji1,2,3 Sergio Pablo Sardi4
Sergio Pablo Sardi4 Anurag Tandon1,5*
Anurag Tandon1,5*- 1Tanz Centre for Research in Neurodegenerative Diseases, Toronto, ON, Canada
- 2Department of Laboratory Medicine and Pathobiology, University of Toronto, Toronto, ON, Canada
- 3Krembil Research Institute, Toronto, ON, Canada
- 4Sanofi, Framingham, MA, United States
- 5Department of Medicine, University of Toronto, Toronto, ON, Canada
α-Synuclein (asyn) is a key pathogenetic factor in a group of neurodegenerative diseases generically known as synucleinopathies, including Parkinson's disease (PD), dementia with Lewy bodies (DLB) and multiple system atrophy (MSA). Although the initial triggers of pathology and progression are unclear, multiple lines of evidence support therapeutic targeting of asyn in order to limit its prion-like misfolding. Here, we review recent pre-clinical and clinical work that offers promising treatment strategies to sequester, degrade, or silence asyn expression as a means to reduce the levels of seed or substrate. These diverse approaches include removal of aggregated asyn with passive or active immunization or by expression of vectorized antibodies, modulating kinetics of misfolding with small molecule anti-aggregants, lowering asyn gene expression by antisense oligonucleotides or inhibitory RNA, and pharmacological activation of asyn degradation pathways. We also discuss recent technological advances in combining low intensity focused ultrasound with intravenous microbubbles to transiently increase blood-brain barrier permeability for improved brain delivery and target engagement of these large molecule anti-asyn biologics.
Rationale for Targeting Asyn
Pathological accumulation of alpha-synuclein (asyn) is the common distinguishing trait amongst the group of brain disorders known as synucleinopathies, which include Parkinson's disease (PD), Dementia with Lewy bodies (DLB), and Multiple System Atrophy (MSA). These disorders progressively develop neuronal and glial inclusions enriched in misfolded, phosphorylated, and detergent-insoluble asyn (1, 2). Asyn is a 140 amino acid protein predominantly localized to nerve terminals. Although it lacks a transmembrane domain, its amino-terminal domain can reversibly adopt an amphipathic alpha-helical structure that enables it to partially embed into synaptic vesicle membranes (3). Its localization and behavior during neuronal activity is highly suggestive of a role in vesicle trafficking, though its precise function remains incompletely understood (4). While the unbound, freely-diffusible pool of asyn in cytoplasm is generally thought to be intrinsically disordered, poorly characterized pathological triggers induce rigid beta sheet-enriched conformations that can polymerize into oligomers and fibrils (5). These asyn multimers accumulate for a variety of reasons including superior conformational stability, inefficient removal by protein degradation machinery in neurons, and ongoing self-expansion by interaction with native asyn which then acquires the seed conformation (6).
This process of recruitment and misfolding of native asyn, called permissive templating, is not dependent on cofactors and is readily observed in cell-free conditions with purified recombinant protein. However, the kinetics and range of conformations are constrained by mutations, binding partners, and by oxidative or post-translational modifications. The first evidence of this process in human brain was the recognition that host-to-graft transmission of Lewy bodies (LB) may have occurred in PD patients who had received fetal neuron grafts 10–15 years earlier (7, 8). This seminal observation prompted a paradigm shift in our understanding of the underlying pathophysiological mechanisms and their role in the stereotypical patterns of disease progression previously identified by Braak et al. (1, 9, 10). Subsequent characterizations of asyn internalization, mixing with cellular asyn, and export of misfolded asyn have been replicated in multiple cell and animal models (11). For example, the host-to-graft transmission of cerebral asyn pathology was observed in transgenic asyn mice implanted with neuronal grafts, as well as simpler experimental systems such as co-cultured neurons (12, 13). Furthermore, extracellular seeding with recombinant pre-formed fibrils or synucleinopathy-derived asyn aggregates readily triggers intracellular neuronal inclusions (14–18) and corresponding intracerebral inoculations into transgenic mice expressing human asyn replicate pathological and clinical features of spreading synucleinopathies (19–24).
There is also an additional layer of complexity evident in the differential spatio-temporal patterns of the pathological asyn spread between PD, MSA, and DLB, which imply that each disease develops a unique pattern of neuronal vulnerability. Imaging and post-mortem analyses suggest neuronal connectivity between brain regions is an essential but insufficient predictor of the pathological asyn spread (25). Other local factors such as asyn expression, origin of pathology, and susceptibility to asyn conformational strains play an important role (26–29). Structurally distinct asyn isoforms generated from recombinant proteins have been serially propagated in cells and animals (28, 30, 31), providing unequivocal evidence for prion-like intercellular spread of conformational strains (32).
The multitude of cell and animal models demonstrating intercellular exchange of asyn aggregates have provided a wealth of mechanistic information on asyn internalization and trafficking, and prompted the development and testing of new therapeutic approaches (33, 34). These studies support the idea that overlapping mechanisms exist between the synucleinopathies that initiate and sustain the pathological spread, regardless of the brain regions affected by early pathology or the underlying trigger. Those commonalities offer useful starting points for the development of disease-modifying treatments.
Biological Pathways That Enable Asyn Pathology Spread
Mechanisms of Intercellular Asyn Transfer and Consequent Impact on Design of Therapeutics
The possibility that misfolded asyn could be transferred between adjacent neurons and contribute to pathological spread provided the impetus to establish the mechanisms of asyn transfer (35). Extracellular asyn is detectable in saliva, plasma and cerebrospinal fluid suggesting physiological mechanisms to enable its secretion. Intercellular exchange of asyn is complicated by the fact that it is a cytosolic protein, so secretion of asyn or its multimers necessitates passage through one or more lipid bilayers. Asyn has been detected within large intracellular vesicles and a non-traditional calcium-dependent exocytic process has been proposed (36–40). Some asyn may also escape due to neuronal injury or death, though the contribution of bolus asyn release is likely minor with predominantly local effects (12). For example, neuronal injury and degeneration in rats injected with AAV containing human asyn was accompanied by a substantial reduction of caudo-rostral asyn propagation suggesting that transmission is preferentially mediated by an active mechanism (41). Anatomical evaluation of spreading pathology following seeding with asyn fibrils also implicate neuronal connectivity as a key determinant of its spread across distal brain regions (29).
An active mechanism that can enable intercellular asyn transmission is via small extracellular lipid bilayer vesicles known as exosomes, which contain proteins and RNA and are secreted by all cell types (Figure 1A) (42). These vesicles originate from the endosomal pathway and are generated when the late endosomal compartment known as a multivesicular body (MVB) fuses with the plasma membrane, releasing intralumenal vesicles as exosomes as well as cytosolic components (43). Post-mortem analyses of synucleinopathy brains have revealed dysfunction in the autophagy-lysosomal pathway, which is connected to the endosomal pathway and exosome release. Impairments in the clearance of autophagic vesicles contributes to the accumulation and aggregation of asyn in synucleinopathies (44). In this scheme, exosomes may represent a neuroprotective response to remove pathological asyn intermediates from cells that have defective clearance pathways. Numerous studies have demonstrated that pharmacological or genetic inhibition of the autophagy-lysosomal pathway increases the release of exosome-associated α-syn and promotes its intercellular transfer (37, 45–47). The exosome lumen also provides a concentrating milieu to facilitate further asyn self-assembly, and consequently, exosomes can transport these replicating particles to different brain areas (48, 49).
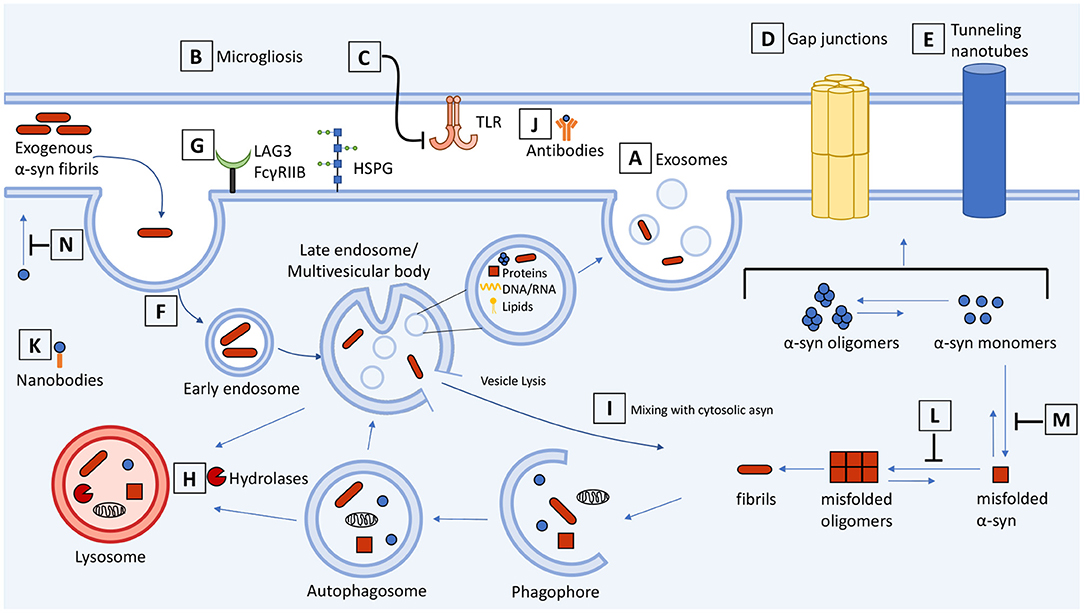
Figure 1. Schematic representation of cell-to-cell transmission, internalization, and trafficking of α-synuclein and therapeutic targets along the pathways. Molecular mechanisms summarized in the context of neuron-neuron and neuron-glial transmission. (A) Secretion and exosome-mediated release of asyn from neurons and (B) activated microglia contributes to the spread of asyn. (C) Inhibitors of microgliosis and toll-like receptors reduce pathological transmission of asyn. Transmission of asyn is also mediated through direct cell-to-cell contact through (D) gap junctions and (E) tunneling nanotubes. Internalization of asyn occurs through (F) endocytosis, (G) macropinocytosis, and receptor-mediated endocytosis which can be targeted to attenuate the transmission of asyn. (H) Once internalized, asyn localizes to the endosomal compartments where it is degraded by the autophagosome-lysosome pathway. Drugs that upregulate autophagy or restore lysosome function can promote the clearance of asyn and prevent its accumulation. (I) Internalized asyn fibrils can rupture endocytic vesicles, enter the cytosolic compartment, and induce templated misfolding of native asyn. (J) Antibodies can bind and sequester extracellular asyn. (K) Antibody fragments can be expressed inside the cell and bind to and enhance turnover of intracellular asyn. (L,M) Compounds can inhibit asyn from misfolding and aggregating as well as (N) inhibit lipid-induced asyn aggregation by displacing it from the membrane. The therapeutic targets and drugs are listed in Table 1.
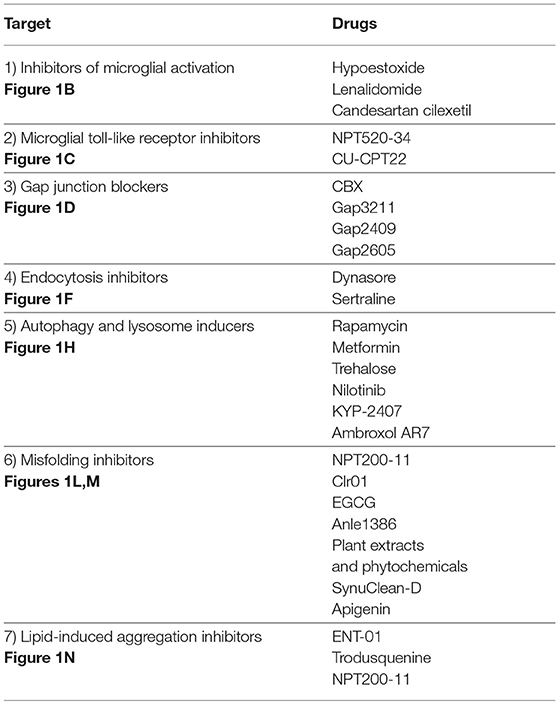
Table 1. Therapeutic targets and drugs from Figure 1.
An important side-benefit of CNS-derived exosomes, if they are indeed bona fide mediators of disease, is their potential for use as biomarkers. Analyses of plasma CNS-derived exosomes from PD patients and healthy controls reveal correlations between the amount of exosomal asyn with PD diagnoses and severity (50–52). In another study, the ratio of multimeric and phosphorylated asyn species to total asyn was higher in PD patients' plasma exosomes when compared to healthy controls (53). Together, these studies suggest that pathological exosomal cargo could serve as an early warning system for diagnosing PD.
Exosomes also play a key role in neuroinflammatory pathways that exacerbate asyn pathology. While microglia provide support in brain by phagocytosing dead cells and clearing asyn aggregates, they also release exosomes that may contribute to the spread of asyn pathology. Exposure of asyn-laden microglia to an inhibitor of exosome synthesis reduced asyn transmission to adjacent neurons and, conversely, treatment of mice with a microglial depleting compound partially suppressed asyn aggregation and transmission (54, 55). There was also less asyn aggregation in these neurons suggesting that microglial exosomes may be a therapeutic target to halt the spread of aberrant α-syn (Figure 1B). In addition, interaction of aggregated asyn with TLR2 and TLR4 on microglia induces microglial activation contributing to neurotoxicity (56–60) (Figure 1C). Microglia activated by oligomeric asyn increased their secretion of proinflammatory cytokines, and microglia-derived exosomes induced asyn pathology and apoptosis in neurons (54, 55, 61). The anti-hypertension drug candesartan cilexetil inhibited the expression of TLR2 and reversed the activated proinflammatory response of primary microglia exposed to oligomeric asyn in vitro (62). Modulation or inhibition of NF-κB signaling with hypoestoxide and lenalidomide, respectively, reduced microgliosis, prevented the loss of dopaminergic neurons, and reduced motor behavioral deficits in mouse models of PD (63, 64). Microglia are clearly involved in propagating asyn pathology and drugs that prevent microglial activation could be a therapeutic approach for synucleinopathies.
Transfer of multimeric asyn in neurons and oligodendrocytes can occur through direct contact with the gap junction protein connexin-32 (Cx32) (Figure 1D). Pharmacological blockade of Cx32 activity with CBX or synthetic peptide mimetics Gap3211, Gap2409, and Gap2605 decreased oligomeric asyn uptake in a concentration dependent manner (65). Likewise, asyn enhances opening of connexin-43 hemichannels in astrocytes and pannexin-1 channels, activating cytokines and inducing cell death (66). While there are conflicting findings reviewed here (67), the evidence that blocking gap junctions is beneficial in neurodegenerative models is exciting for the development of new therapeutics.
Tunneling nanotubes (TNTs) also permit asyn exchange through direct cell-to-cell contact (68–70) (Figure 1E). These membranous channels are composed of F-actin and provide plasma membrane continuity between remote cells to facilitate the transfer of vesicles, organelles and cytoplasmic molecules (71). The addition of exogenous asyn fibrils to cells induces TNT formation and fibrils are directed to the lysosome for degradation. The TNTs are a means by which cells can eliminate damaged lysosomes containing asyn fibrils, though this process can enable further asyn seeding in nearby cells (68). These TNTs are also induced as a “defense mechanism” to actively transfer toxic asyn to healthy astrocytes (70), human neuronal precursors (72), or microglia (73) which can efficiently degrade fibrillar asyn. The Wnt/Ca2+ pathway and βCaMKII are involved in the formation of TNTs and the transfer of vesicles and aggregated asyn (74). Drugs that destabilize TNTs or modulate the Wnt/Ca2+ pathway could impair TNT-mediated propagation of asyn. Although current evidence suggests that TNTs could be a target for therapeutic intervention, the contribution of TNT-mediated transfer in vivo and to synucleinopathies is unclear. Therefore, there is a need to assess TNTs in various cell types and animal models to distinguish contexts where TNTs contribute to asyn pathology vs. when they carry out normal physiological functions.
Internalization of Asyn
Insights into asyn uptake are relevant to understanding the relationship between neuronal function and pathological spread, and for developing therapeutics. Extracellular aggregated asyn is cytotoxic when added to cell culture and is implicated in disease (12, 15). Internalization of asyn fibrils and oligomers occurs through temperature- and dynamin-sensitive endocytosis (75–77) along the entire length of the neuron (soma, dendrites and axons) (15, 78, 79) (Figure 1F). Once internalized, asyn moves through the endosomal compartments before being degraded by the lysosomes. This mechanism of receptor-mediated endocytosis and lysosomal degradation results in the clearance of potentially toxic asyn aggregates from the extracellular space (75). Inside the neuron internalized asyn forms inclusions in recipient cells and induces apoptosis (12, 15).
In vivo studies also demonstrate intercellular transfer of ectopically-expressed human asyn from rat and mouse host brains to dopaminergic neuron grafts, where the transferred asyn localized to endosomes and seeded aggregation of asyn (13, 80). Neuronal internalization of asyn was decreased when endocytosis was blocked by co-injection of the dynamin inhibitor Dynasore (13). Other mechanisms of asyn endocytosis include: macropinocytosis, an actin-dependent process that utilizes heparan sulfate proteoglycans (HSPGs) (18, 81, 82) and N-linked glycan-dependent binding to neurexin 1β mediated by N-acetylation of asyn (83), lymphocyte-activation gene 3 (LAG3) receptors (84, 85), and FcγRIIB receptors (86) (Figure 1G). There is hope that these pathways may be therapeutically targetable to attenuate cell-to-cell transmission of asyn. However, the relative contributions of each pathway to pathogenicity is poorly understood, and their cell type and species specificity remain under investigation. For example, the localization and role of LAG3 in propagating synuclein pathology has been recently questioned (87).
Filamentous asyn inclusions have also been described in glial cells of synucleinopathy patients (88). Asyn multimers may be transmitted to glial cells and further contribute to disease progression. Neuronal and astrocyte co-cultures demonstrated cell-to-cell transmission of asyn through endocytosis, forming inclusion bodies, and inducing an inflammatory response (89). These asyn multimers then localize to lysosomes in astrocytes where they can be degraded (90). Neuron-to-oligodendroglia transmission of asyn by dynamin-dependent endocytosis (91) and neuron-to-microglia transmission via GM1 ganglioside and lipid rafts have also been reported (92). Identification of receptors that bind asyn aggregates in neuronal and non-neuronal cells might provide alternative therapeutics that inhibit asyn internalization or promote clearance from the extracellular space to prevent asyn spread.
Post-translational modifications can also affect asyn uptake. Modification with O-linked N-acetylglucosamine (O-GlcNAc) inhibits asyn aggregation and reduces its toxicity to neurons in vitro (93). Inhibitors of glycoside hydrolase O-GlcNAcase (OGA) increase O-GlcNAc modification of amyloid proteins (94). Recently, OGA inhibition with a brain-penetrant inhibitor Thiamet-G increased levels of O-GlcNAc-modified proteins and impaired the uptake of asyn fibrils in mouse primary neurons (95). Pharmacological induction of O-GlcNAc could potentially be useful as a therapeutic to prevent aggregation and reduce spread of asyn.
Intracellular Trafficking and Accumulation
Following internalization, asyn fibrils are trafficked through endosomal compartments from early to late endosomes and finally to lysosomes where they can persist for days (18, 75, 79, 84, 91). A recent study demonstrated that neutral autophagosomes and autolysosomes are located near the center of the cells at the microtubule organizing center (MTOC), and following acidification, the mature autophagosomes and autolysosomes containing asyn are transported to the periphery of the cell. In contrast, lysosomes are generated in the periphery and transported to the MTOC where they fuse with autophagosomes (Figure 1H). Thus, the lumenal pH of these vesicles is believed to regulate their subcellular localization. Asyn aggregates are located at the MTOC where autophagy occurs and once localized to vesicles they are also transported to the periphery (96). The purpose of this subcellular segregation could be to separate lysosome reformation from lysosome fusion and to secrete indigestible asyn aggregates through the plasma membrane as is the case with secretory autophagy (97).
Cells have several routes to degrade asyn, with the autophagosome-lysosome pathway bearing the primary responsibility for asyn degradation. Lysosome inhibition can accelerate intracellular inclusion formation (12, 18, 77, 98). Proteasomal inhibition has comparatively little effect, though variable results between studies could be due to differences in cell lines and inhibitors used (12, 99, 100). Chaperone-mediated autophagy (CMA) and macroautophagy can also process asyn degradation (101–103). For example, CMA-specific activator AR7 [7-Chloro-3-(4-methylphenyl)-2H-1,4-benzoxazine] increases lysosome activity and was found to attenuate asyn oligomer accumulation in vitro (104). Lysosomal processing and integrity are important for the clearance of asyn seeds and autophagy is a potential therapeutic target in the treatment of synucleinopathies.
Mutations in the GBA1 gene, which encodes the lysosomal enzyme glucocerebrosidase (GCase), are a risk factor for sporadic PD and DLB (105). GCase is a lysosomal hydrolase that degrades the glycosphingolipids, glucosylceramide and glucosylsphingosine, into ceramide and sphingosine. The mechanisms by which GBA1 mutations increase the risk of synucleinopathies are not fully understood. A reciprocal association between GCase activity and asyn has been demonstrated. Elevated glucosylceramide levels due to reduced GCase activity, can lead to asyn accumulation and, conversly, increased GCase activity reduces asyn accumulation and prevents nigrostriatal degeneration (106–108). Preclinical evaluation of venglustat, a brain-penetrant inhibitor of glucosylceramide synthase, reduced asyn pathology and the associated cognitive decline in mouse models of GBA1-related synucleinopathy (109). However, a Phase 2 clinical trial (NCT02906020) testing venglustat in GBA-PD patients was terminated after missing its primary and secondary endpoints. Ambroxol, another drug commonly used in cough medicine, can restore GCase activity and lysosomal function and also reduce markers of oxidative stress in GBA1 fibroblasts (110, 111). Recently, a clinical trial with 17 PD patients determined that ambroxol was safe, well tolerated, crosses the blood-brain barrier, and binds the β-glucocerebrosidase enzyme (112). While promising, future placebo-controlled trials are required to determine if ambroxol therapy could be a disease modifying intervention for PD.
Another effect of asyn aggregation is mediated through its actions at the endoplasmic reticulum (ER), which becomes fragmented in PD, impairing proteostasis and the activation of unfolded protein response (UPR), and retention of GCase within the ER (113). Interestingly, the asyn-induced deficit of ER proteostasis was rescued by inhibitors of the ryanodine receptors which modulate ER calcium influx, including with diltiazem, an FDA approved drug for hypertension and angina. Binding of asyn to the cytosolic SNARE protein ypkt6 separately interferes with ER-Golgi trafficking, which can be corrected with farnesyl transferase inhibitors (114). The combined inhibition of the ryanodine receptors and farnesyl transferase was found to correct the GCase trafficking and also lowered asyn levels in PD-derived cells, suggesting that rescue of proteostasis and trafficking in synucleinopathies may be targeted with small molecule drugs (113).
Rapamycin, an FDA approved drug, increases autophagy levels by inhibiting the mTOR pathway and can reduce dopaminergic cell death, decrease levels of phosphorylated asyn, and improve mitochondrial function in cell and animal models of synucleinopathy (115–117). Rapamycin also interferes with other autophagy independent pathways and therefore, the downstream target of mTOR, lysosomal transcription factor EB (TFEB), may be a more appropriate target. TFEB overexpression decreased pathologic asyn oligomers in rats (118), and TFEB overexpression in oligodendroglia rescued nigrostriatal neurodegeneration in an MSA mouse model (119). In contrast to rapamycin, metformin, trehalose, and nilotinib induce AMP-activated protein kinase (AMPK)-dependent autophagy. This can also prevent asyn accumulation by promoting its clearance and is neuroprotective in PD models (120–122). To determine the synergistic effects of mTOR-dependent and -independent autophagy stimulation, trehalose and rapamycin were tested in an MPTP-induced PD mouse model and showed an additive effect in recovery of dopaminergic neurons and neuroprotection (123). However, one concern of inducing autophagy is the potential co-clearance of other proteins with differential turnover rates (124). While upregulation of autophagy is generally beneficial (125, 126), high autophagic flux can also induce cell death (127). Understanding the precise pathways of asyn clearance and homeostasis will be important for the development of safe autophagy-based therapies.
Direct contact between misfolded and native asyn is necessary for permissive templating. How is this interaction mediated if misfolded asyn is lumenal, enclosed within endocytic vesicles, whereas the native asyn substrate is cytosolic? There is evidence that asyn aggregates can rupture endocytic vesicles following internalization (76, 128, 129) (Figure 1I). The ruptured vesicles can also fuse together to form LB inclusions, which are heterogenous in their composition, containing a mixture of asyn filaments, vesicle lipids and fragmented membranes, and other organelles (130, 131). To prevent these pathological structures, the porphyrin phtalocyanine tetrasulfonate was used to bind to and stabilize vesicle-associated asyn to block its misfolding and aggregation (132). Despite these advances, our knowledge of the molecular players and mechanisms involved in the internalization and transmission of asyn remains inadequate and poses a challenge for identifying therapeutics that target asyn transport.
Therapeutics Targeting Asyn Levels
Targeting Extracellular Asyn
The ability of antibodies to sequester misfolded proteins with high-affinity and high-specificity make them attractive as therapeutics for neurodegenerative diseases (Figure 1J). Extensive research over the past two decades has focused on developing active and passive immunity to target extracellular asyn and several anti-asyn vaccines are currently being tested in early phase clinical trials (133). The initial vaccine studies from Masliah and colleagues were encouraging and showed that both active and passive immunization could lower intracellular asyn aggregates and rescue motor dysfunction in transgenic asyn overexpressing mice (134, 135). Subsequent work has targeted carboxyl-terminal asyn epitopes. Reports of active immunization indicate improved immune cell recruitment and high antibody titers that selectively reduce asyn pathology (136, 137). Antibodies generated to asyn oligomeric species recognize asyn aggregates in human PD tissue but were not sufficient to rescue pathology or the motor phenotype in transgenic mice overexpressing human asyn (138). Whether this was due to specificity of the immunizing oligomer conformation or disease vs. overexpression differences is unclear. Active immunization also carries the risk of generating an autoimmune response against asyn, which can be minimized by passive immunization using humanized or engineered antibodies. Preclinical testing of such antibodies showed promise in preventing neurodegeneration and rescuing motor deficits in mouse and primate models of synucleinopathy (135, 139–145). These findings prompted the testing of a humanized IgG1 monoclonal antibody PRX002 (Prasinezumab, Roche/Prothena) that recently completed its Phase 2a clinical trial. Although the study could not meet its primary outcome measure of change in patient MDS-UPDRS score from baseline, some motor scores showed positive trends, and the study has now progressed to Phase 2b of the trial (NCT03100149)(146).
Active immunization with peptides mimicking the asyn carboxyl-terminus effectively reduced pathological aggregate formation and significantly mitigated motor deficits in transgenic human asyn mice (147). These peptides were further developed by Affiris as PD01A and PD03A (AFFITOPE) for active immunization clinical studies. Importantly, Phase I clinical trials using these antibodies in patients with PD were successfully completed in 2016, showing that both antibodies were locally and systemically well tolerated with no serious adverse effects, which resulted in their progress into Phase II clinical trials (148–150). In addition, a synthetic asyn peptide representing oligomeric and fibrillar asyn developed by United Neuroscience (UB312) is currently undergoing a Phase 1 clinical trial for PD (NCT04075318) (151).
Biogen's BIIB054 (Cinpanemab) monoclonal antibody, on the other hand, did not meet its primary and secondary endpoints in a Phase 2 clinical trial, despite promising pre-clinical results (152), and the trial was discontinued in February 2021 (NCT03318523). Several other antibodies targeting asyn are currently in Phase I trials for passive immunotherapy, including MEDI1341 (AstraZeneca), Lu-AF82422 (Lundbeck), ABBV-0805 (AbbVie/BioArctic), ABL301 (Sanofi/ABL Bio) and UCB7853 (UCB Biopharma/Novartis) (clinicaltrials.gov).
Notwithstanding the significant advances in immunotherapy as a disease modifying strategy, achieving adequate BBB penetrance and target engagement remain key challenges for the use of these therapeutics in the clinic. Retrospective evaluations of multiple immunotherapy clinical trials conducted for Alzheimer's disease (AD) have shown limited benefits (153, 154). The low immunoglobulin levels detected in CSF (<0.5% of serum IgG levels), and lower still in the brain parenchyma, may account for the absence of therapeutic benefits (155). Strategies to improve BBB passage are therefore an important and urgent requirement for developing efficient therapeutic antibodies. Another potential challenge remains the structurally distinct asyn strains that have been identified in human synucleinopathy brains (156–159). These conformers represent reproducibly stable and distinct conformations of asyn. It is unclear whether these be preferentially targeted by conformation-specific immune approaches. The possibility that immune reagents could be developed to target pathogenic conformations of asyn is very appealing, particularly if the non-disease asyn conformers were selectively spared.
Reduction of Intracellular Expression of the Asyn Gene SNCA
As a complementary strategy to targeting extracellular asyn, reducing expression of SNCA (the gene encoding asyn) and intracellular asyn levels in recipient neurons could also mitigate disease progression by reducing the native substrate available for templated misfolding. RNA interference (RNAi) has been widely used in cell and animal models to effectively counteract asyn accumulation. Small interfering RNA (siRNA) are 21–23 nucleotide long double stranded RNA molecules that drive degradation of target mRNA upon sequence-specific binding. Direct infusion into mouse hippocampus of naked anti-asyn siRNA successfully downregulated SNCA expression (160). Short hairpin RNA (shRNA), which is processed by the cellular machinery to form active siRNA, has also been used to lower SNCA expression in neuronal cultures (161), in murine models (162–164), and in non-human primates (165). Although these studies have shown that downregulation of asyn expression significantly reduces asyn transmission, reducing asyn expression in vivo has potential disadvantages including possible neuronal dysfunction, neurotoxicity, and loss of dopaminergic neurons. In vivo studies on the viability of this approach for counteracting PD have been controversial; some reports suggest that >90% suppression of asyn could be neurotoxic and cause the loss of nigral tyrosine hydroxylase expression (165–167), whereas other studies have not observed toxicity with up to 60–70% asyn knockdown (164, 168). Cumulatively, the evidence suggests that partial knockdown of asyn levels is safely achievable, and that even a moderate 30–40% asyn reduction is beneficial against spreading asyn pathology (169, 170). However, if asyn knockdown is found to induce some loss-of-function deficits, it is conceivable that these might be circumvented by corresponding expression of a non-aggregation prone homolog, such as β-synuclein (bsyn), which lacks the hydrophobic core of the non-amyloid component (NAC) domain.
Genome-wide association analyses (GWAS) have revealed that expression profiles of certain micro RNAs (miRNAs) including include miRNA-30, miRNA- 485, miRNA-29, miRNA-26, and let-7 are altered in the brains of PD patients (171, 172). MicroRNAs are small non-coding RNAs that post-transcriptionally regulate target mRNAs and their therapeutic potential in the treatment of PD has consequently generated considerable interest. Four different miRNAs including miR-7, miR-153, miR34b/c, and miR-214 were found to directly regulate asyn, suggesting a potential role for these miRNAs in asyn accumulation and disease progression (173–176). Moreover, incorporating an asyn targeting sequence within an miR-30 structure was shown to improve SNCA silencing and provide protection against dopaminergic neuronal loss and motor deficits in a murine model of PD that overexpresses human asyn (177). AAV9-mediated expression of miR-30-SNCA-shRNA in mouse brain also suppressed asyn levels (168). Moreover, AAV1 expression of an artificial miRNA bearing an asyn targeting sequence reduced the spread of asyn pathology and the associated motor deficits in an aggressive mouse model induced by intracerebral inoculation with synucleinopathy brain homogenates (170).
Delivery vectors for nucleic acid-based therapies can be broadly classified into non-viral and viral vectors. Non-viral vectors display low cytotoxicity and immunogenicity, are cost-effective, and can be easily modified for target binding (178). For example, non-viral vectors for applications in PD include modified peptides, polymers, lipids, exosomes and inorganic materials, or hybrid systems comprising combinations of these vectors. A key disadvantage of non-viral therapeutics is their transient action and requirement for periodic re-administration. Transient asyn suppression can be induced with anti-sense oligonucleotides (ASOs), which are single-stranded oligonucleotide sequences that activate endonuclease RNaseH upon binding to the target mRNA and result in the degradation of the DNA-RNA hybrid. Studies employing asyn ASOs have reported reduced asyn protein expression without causing dopaminergic neurodegeneration (169, 179, 180). BIIB101, an anti-asyn ASO developed by Ionis and Biogen is currently in a Phase I clinical trial in MSA patients (NCT04165486). Results from this study, if successful, will undoubtedly pave the way for ASO-based therapeutics for PD. Despite their relatively long half-life, ASOs will require regular intrathecal administration to maintain stable asyn silencing.
Viral vectors have been historically preferred for gene therapy targeting neurodegenerative disorders because of their stable and long-term expression profiles. Adeno-associated viral vectors (AAVs) are currently the most advanced vectors for CNS gene therapy, with AAV serotypes 1,2,5 and 9 being the most frequently used (34, 181). Lentiviral vectors have also been tested for PD therapeutics, as they allow larger DNA inserts and can encode multiple genes simultaneously. Lentivirus-based PD clinical trials have focused primarily on augmenting dopamine levels by expressing multiple enzymes necessary for its synthesis (182). Viral vectors do have some disadvantages, including inherent immunogenicity, species-specific tissue-tropism and challenges with cost-effective manufacture of virion particles, which may restrict their widespread application (183).
Direct gene editing is being developed as an interesting alternative to repeated administration. Although it is still at an early preclinical stage, gene editing bears the potential to fundamentally alter how genetic disorders are treated by enabling fine tuning of disease gene expression or by correcting mutations. For example, zinc-finger nuclease (ZFN) mediated genome editing was used to correct point mutations in the SNCA gene in patient-derived human induced pluripotent stem cell (hiPSCs) (184). Gene editing with the CRISPR/Cas9 system targeted a DNA methylation site to successfully reduce asyn expression in human iPSC-derived neurons from a PD patient with the SNCA triplication (185). Recently, a CRISPR-based approach was used in transgenic AD-related animals to selectively target the mutated amyloid precursor protein gene, while leaving wild-type alleles intact (186). This significantly reduced APP amyloidosis and gliosis. Although much research is required to explore its full potential, genome editing has immense potential for the precision treatment of genetic disorders such as PD.
A central focus of CNS targeted gene therapy is the development of vectors that can efficiently bypass the blood brain barrier (BBB). The BBB is highly selective and restricts the entry of molecules larger than 400 daltons such that it is impermeable to the vast majority of systemically administered small molecule drugs and nearly all biological therapeutics (187) (discussed further in Section Non-invasive Drug Delivery to Brain). Although direct intracerebral injections are commonly used for gene transfer in pre-clinical models, they are impractical for CNS disorders that require broad bilateral gene expression. Invasive needle tracts induce mechanical trauma to the surrounding tissue, exacerbate local neuroinflammatory processes, and increase the risk of hemorrhage. Intranasal delivery is another potential delivery route to circumvent the BBB (179). It is non-invasive and practically feasible, but low dosage volume, susceptibility to degradation by mucosal enzymes, and substantial variabilities in target engagement make this delivery option suboptimal at present (188). Hence, vectors that can efficiently transfer therapeutic payloads across the BBB are important for the success of gene therapy approaches. Recently, novel brain-penetrating AAV9 variants, such as AAV-PHP.B, have been generated by selecting among random modifications of the viral capsid (189–192). Preclinical testing of these novel AAVs have shown efficacy in ameliorating rodent disease models following systemic delivery, but challenges with differential species- and strain-specific tropism remain (193). The gene therapy vectors that have been used to knockdown SNCA in various PD models have been summarized in Table 2.
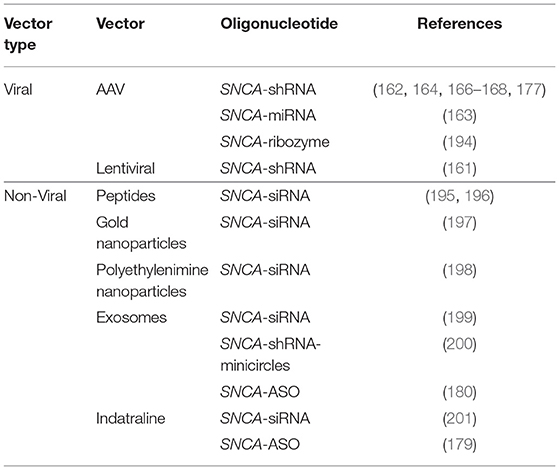
Table 2. Delivery vectors for asyn targeted gene therapy.
Reduction of Intracellular Asyn Protein Expression Using Gene Therapy
The primary drawback of using antibodies is their high molecular weight, which challenges their therapeutic potential. Intrabodies (also called nanobodies) are small antibody fragments containing only the antigen-binding domain, derived from either heavy-chain only antibodies (HCAbs) found in all Camelidae and some shark sera or recombinantly produced from conventional antibodies (202). The two main types of nanobodies are short chain variable fragments (scFv), which consists of the variable light and variable heavy chains linked with a polypeptide sequence, and single-domain (sd) nanobodies (Figure 1K). Among the advantages that nanobodies have over conventional antibodies is their smaller size (typically 15–30 kDa), allowing for greater tissue penetrance, and faster clearance (203–205). Their smaller size (scFv: 250 aa, sdAbs: 120–140 aa) also enables their recombinant DNA to be easily vectorized for gene delivery (206). Given that only a small proportion of peripherally administered antibodies reach the CNS, vectorized intrabodies may overcome this challenge by enabling expression in CNS neurons. The lower immunogenicity, and ability to genetically engineer fusion sequences also enhance the safety and functional potential of intrabodies.
Multiple scFvs targeting different conformations and epitopes of asyn have been selected either using phage display alone or in combination with atomic force microscopy, previously reviewed (207, 208). A recent study produced a scFv from a monoclonal antibody, specific for asyn fibrils (Syn-F2) and found it was able to inhibit seeding of asyn aggregation in vitro. This intrabody was fused with a cell-penetrating peptide, underscoring the ability for genetic engineering to enhance the functionality of intrabodies (209). Despite the ability of anti-asyn scFvs to inhibit formation of toxic oligomeric asyn in vitro, only a few have been further tested in synucleinopathy models to date. NbSyn87 is a camelid-derived nanobody, selected using phage-display with affinity for epitopes in the N-terminal and C-terminal of asyn, allowing it to bind to both monomeric and early fibrillary forms of asyn (210). NbSyn87 inhibited the formation of toxic oligomers in vitro (211) and, following expression in an asyn-overexpression rodent model, efficiently cleared phosphorylated asyn (p-asyn) and partially rescued motor dysfunction (212). The same study also tested a single-domain nanobody, VH14, a human-derived clone with high affinity for the NAC domain of asyn. While both nanobodies were effective, VH14 produced a greater reduction of p-asyn deposits, and unlike NbSyn87, did not generate an inflammatory response (212). One shortcoming of intrabodies is their relatively poor solubility in the cytoplasm, though this can be improved with further engineering. For example, fusion of a protein degradation sequence rich in proline, glutamine, serine and threonine, hence the name PEST, was shown to improve the intracellular solubility of various anti-asyn nanobodies including VH14 and NbSyn87 in vitro by increasing their overall negative charge (213). PEST also functions as a ubiquitin-independent proteasome-degradation signal, thereby enhancing the turnover of cytoplasmic nanobody-asyn conjugates (213, 214). Engineering intrabodies to possess proteolysis targeting sequences represent a promising approach to reducing intracellular asyn concentration, thereby not only inhibiting the misfolding, but also decreasing the amount of substrate to be recruited for aggregation.
Pharmacological Reduction of Asyn Expression and Aggregation
Targeting “druggable” cellular pathways known to modulate asyn transcription may be a useful approach to decreasing asyn levels. Table 3 summarizes pharmacological therapeutics that reduce asyn. For example, agonists of the Beta2 adrenoreceptor (β2AR) downregulate transcription of SNCA gene by decreasing histone H3 lysine 27 acetylation of its promoters and enhancers (215). Three β2AR agonists, metaporterenol, clenbuterol, and salbutamol were found to reduce endogenous SNCA mRNA in a dose- and time-dependent manner following treatment in neuroblastoma and rat primary cortical neurons. Conversely, treatment with β2AR antagonist, propranolol, increased asyn mRNA. Clenbuterol and salmeterol, both BBB-permeable, can protect dopaminergic neurons from MPTP toxicity in a PD mouse model (215, 216). Clenbuterol also significantly reduced asyn mRNA in SNCA triplication iPSC derived neuronal cells. These pre-clinical findings were strengthened by epidemiological data of Norwegian populations prescribed salbutamol (most commonly to treat asthma) and propranolol (hypertension), and salbutamol was associated with reduced risk of developing PD (rate ratio of 0.66) and, conversely, propranolol was associated with increased risk of PD (rate ratio 2.20). Additional epidemiological investigations have reported the same relationship (240) or no association (241). In addition to its SNCA transcription regulation, β2AR agonists also have anti-inflammatory effects (216), making it an especially promising therapeutic target in PD. Although β2AR agonists have shown to be safe and effective in other neurological conditions including multiple sclerosis (242) and ALS (243), no large scale clinical trials in PD have been conducted to date. However, three open label trials observed that salbutamol added to levodopa treatment provided some symptomatic benefit in a small number of PD patients tested (244–246).
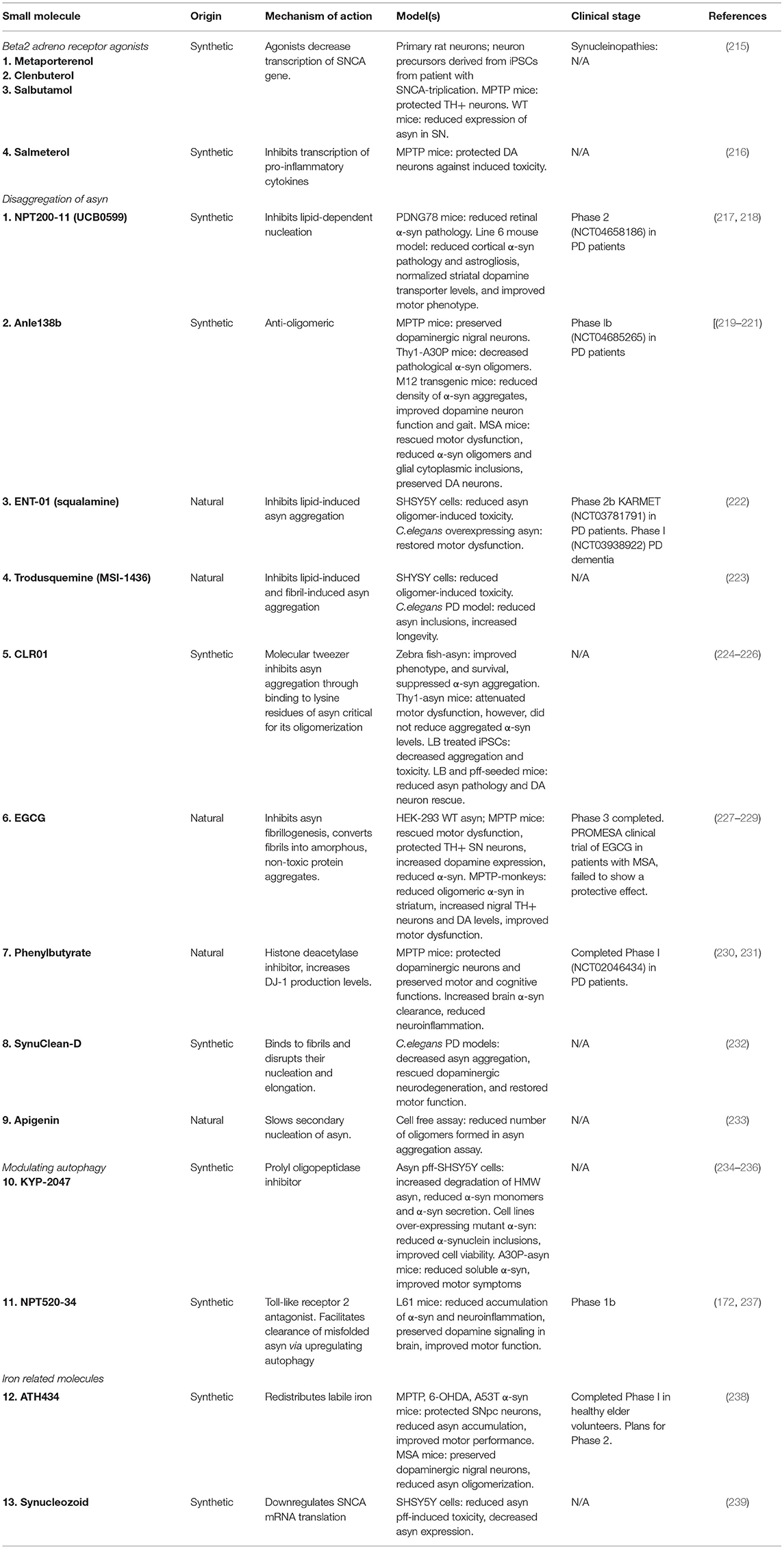
Table 3. Pharmacological approaches to reduce asyn.
Small molecules targeting asyn mRNA to downregulate its expression are being evaluated. A sequence-based design was used to identify molecules that bound to the iron-responsive element (IRE) of SNCA mRNA, a region involved in regulating translation (239). The most promising candidate, synucleozoid, was most effective at inhibiting the translation of asyn, and showed a dose-dependent reduction in asyn fibril-induced cytotoxicity. Mechanistically, synucleozoid lowered asyn protein levels by reducing the amount of SNCA loaded onto polysomes and, importantly, was selective for asyn mRNA (239). This study provides encouraging evidence that pharmacologically modifying the structural elements in asyn mRNA can reduce asyn expression.
Preventing asyn aggregation with small molecule inhibitors is being investigated (Figure 1L), but few candidates have reached human clinical trials. Epigallocatechin-3-gallate (EGCG), a polyphenol, was shown to prevent the conversion of asyn into toxic oligomers (227, 228) and was tested in a Phase III trial (PROMESA, NCT02008721) in patients with MSA although it failed to show efficacy, perhaps due to its low bioavailability and sub-acute toxicity in some patients (247). The lack of specificity for asyn hinders EGCG's viability as it inhibits fibrillation of at least 14 different proteins; however, it does appear to have anti-inflammatory and neuroprotective effects in preclinical and clinical trials (229). Plant extracts and phytochemicals such as isorhynchophylline, paeoniflorin, baicalein, curcumin, cuminaldehyde, gallic acid, ginsenosides, and resveratrol have been proposed as possible drug candidates for PD as they reduce asyn oligomerization and fibril formation (Figure 1M) in vitro and in vivo, reviewed by (248). Recently a small molecule kinetic-based screen was developed to identify inhibitors of asyn aggregation and the compound apigenin significantly reduced the formation of oligomers (233). Another small molecule, SynuClean-D, was able to not only reduce asyn aggregation but also rescue dopaminergic neuron degeneration in worms (232). A promising anti-aggregant from Neuropore Therapies, UCB0599, recently entered a Phase 2 clinical trial (NCT04658186) in 450 early-stage PD patients with an expected completion date in mid-2024. This second-generation compound with improved brain penetration, previously named NPT-200-11, interacts with the asyn carboxyl-terminal domain and reduces asyn pathology, inflammation, and associated motor deficits in multiple mouse models with asyn aggregation (217, 218).
As phospholipid binding may affect the formation of asyn fibrils (249), modulating asyn lipid binding could effectively reduce its aggregation (Figure 1N). Various molecules which compete with asyn for lipid membrane binding have been tested in vitro, although only one has advanced beyond Phase I clinical trials. ENT-01 was demonstrated to compete with asyn for membrane binding and effectively restored peristalsis in mouse models of PD (222). A 50-patient open label Phase 2a study found that orally administered ENT-01 tablets were safe and reduced constipation in over 80% of PD patients, and mildly improved both motor and non-motor symptoms, and a Phase 2b trial is ongoing assessing both constipation and neurological symptoms in 152 PD patients (KARMET, NCT03781791). Although targeting peripheral asyn within the enteric nervous system before it potentially travels to the CNS/brainstem may be beneficial, ENT-01 cannot cross the BBB. A structurally similar BBB-permeable molecule, trodusquemine, has also demonstrated ability to displace asyn and its oligomers from the membrane, inhibiting its lipid-induced aggregation in vitro and in a C. elegans model of PD (223), and is in development for clinical trials.
Anle138b, another BBB-permeable drug, developed by MODAG is proposed to directly inhibit inter-peptide interactions and formation of beta-sheet rich asyn structures (219). Oral administration of anle138b reduced asyn oligomers, and restored striatal dopamine and motor function in both MSA (221) and PD (220) mouse models. Anle138b treatment increased the number of small, monomeric asyn species, while decreasing the inner density of the larger insoluble asyn aggregates, suggesting it interfered with the asyn aggregation process and potentially destabilized existing asyn aggregates (220). Assessment of the tolerability and pharmacodynamics of anle138b is currently underway in a double-blind placebo controlled ascending dose trial in PD patients, with expected completion in June 2022 (NCT04685265).
Artificial Intelligence Approaches for Drug Repurposing
Drug repurposing represents a fast and cost-effective option for developing new treatments for PD [recently reviewed in (250)]. This approach takes advantage of previously approved drugs and re-targets them for a new indication. Repurposing regulatory agency-approved molecules, with an existing safety and pharmacokinetic record in humans, bypasses the high-risk phase of the drug development process and can enter development for a new indication at Phase IIa, providing significant savings in both cost and time. Indeed, drug repurposing can cost as little as $250,000, take as little as 4 years and has a significantly greater chance of reaching patients than a novel drug (251, 252).
A recent review of all clinical trials for PD found that 50 of 145 drugs in phase I-III clinical trial for PD were repurposed, illustrating the strong contribution of repurposing as a strategy to enhance the drug development pipeline for PD (253). Notably, of these 50 repurposed drugs, only two purportedly target asyn; mannitol and memantine. Mannitol is a sugar approved for reducing intracranial pressure, cerebral edema and intraocular pressure. It can also function as a chaperone to stabilize asyn structure in vitro and attenuate synucleinopathy in asyn transgenic mice (254) and is currently being tested in a Phase II clinical trial for effects in PD (NCT03823638). The NMDA receptor antagonist memantine, indicated for moderate and severe AD, is currently being tested in 50 patients in a Phase II clinical trial for effects on the cell-to-cell transmission of asyn (NCT03858270). Since the review of McFarthing and colleagues, the histone deactetylase inhibitor phenylbutyrate, currently indicated for rare pediatric metabolic disorders, has also begun a small Phase 1 study to test the effects on elimination of asyn from the brain to the blood in individuals with PD (NCT02046434), having been previously shown to increase DJ-1 expression and attenuate asyn aggregation in Y39C asyn transgenic mice (230).
Despite the significant advantages over traditional drug development, a major challenge in repurposing remains the efficient identification of drugs with the appropriate efficacy and prioritizing them for development, from the thousands of regulatory approved compounds. To address this challenge, a machine learning approach using artificial intelligence was recently deployed to identify existing drugs that might prevent oligomerization of asyn. The computational approach uses text mining of published abstracts to generate mathematical vectors that can be used to rank a set of candidate drugs based on semantic similarity to compounds with a known effect (prevention of oligomerization of asyn) (255). Based on their predicted ability to inhibit the asyn aggregation, 620 approved drugs were ranked with this methodology. Pharmacoepidemiologic analysis of 15 of the top 50 drugs replicated expected associations between allopurinol and fenofibrate and PD and further identified 3 novel drugs associated with a decreased odds of incident PD; pentoxifylline, theophylline and dexamethasone (256). From a list of 1832 FDA approved drugs, several antihypertensive drugs were identified that might reduce asyn oligomerization. Combinations of angiotensin receptor II blockers and dihydropyridine calcium channel blockers, angiotensin converting enzyme inhibitors and diuretics were found to be inversely associated with time to PD diagnosis and thus be of interest for having disease modifying potential in PD (257). Recently, following an AI-based screen, several compounds were systematically evaluated in cell and animal models of asyn overexpression and revealed rifabutin as a potential drug to reduce nigrostriatal degeneration (258).
Although these data require replication in independent cohorts and/or investigation in laboratory-based assays or animal models of asyn aggregation, they do provide promising real-world data on the potential of using a computational approach to efficiently identify drugs targeting oligomerization of asyn, that are amenable to repurposing for disease modification in PD. To our knowledge there are still no drugs with regulatory approval that were identified using artificial intelligence, however given the pace of discovery and significant investment in this domain, the true promise of this technology should become apparent in the coming years.
Non-invasive Drug Delivery To Brain
A key challenge for asyn therapeutics is their delivery across the BBB, a protective and selectively permeable microvasculature around the CNS composed of tight junctions, endothelial cells, pericytes and astrocytic end foot processes. Receptors, ion channels and transporters on the BBB tightly regulate the transport of molecules into the brain parenchyma, thereby restricting the bioavailability of CNS targeted drugs (187). Indeed, several therapeutics that have shown promise in pre-clinical studies have been unsuccessful in clinical trials, primarily because of their difficulty in crossing the BBB and poor target engagement (259).
In the past two decades, the use of MRI-guided focused ultrasound (MRIgFUS) has developed as a revolutionary technology that allows for localized and targeted delivery of CNS therapeutics across the BBB without the need for invasive surgery. Although the first demonstrated use of focused ultrasound waves resulted in BBB opening around a lesion site (260), a subsequent landmark study by Hynynen and colleagues combined intravenously injected preformed gas microbubbles with low intensity focused ultrasound (FUS) waves to safely open the BBB in rabbits (261). The coincident application of low-intensity focused ultrasound waves to the target brain areas causes the circulating microbubbles to oscillate and mechanically induce a localized and transient opening of the microvasculature tight junctions. With appropriate calibration, the inflammatory responses, thermal changes, and brain hemorrhage observed with high-intensity ultrasound can be avoided. Importantly, coupling intravenous drug delivery with the transient increase in BBB permeability in specific brain regions has been successfully used to increase the CNS bioavailability of an array of biologics including antibodies, viral vectors and nucleic acids for gene therapy in multiple species from rat to non-human primates (259, 262).
Importantly, BBB opening induced by FUS can be widely adapted to small, large, and multiple brain regions by sonicating single or multiple foci. For example, discrete or multiple foci enabled restricted or broad whole-hemisphere gene transfer and drug delivery in rodents (168, 263, 264). In humans, although the technology is still in clinical trials, brain volumes from 63 mm3 to 25 cm3 have been safely targeted with MRIgFUS for increased BBB permeability (265–269).
Several studies have evaluated MRIgFUS in preclinical models of PD. For example, MRIgFUS was used with intravenous AAV9-SNCA-shRNA to target PD-relevant brain regions as defined by Braak staging in a transgenic mouse model overexpressing human asyn resulting in a 40–60% reduction in asyn levels in the targeted brain regions (168). Another study used FUS to deliver glial cell line-derived neurotrophic factor (GDNF) in 6-hydroxydopamine (6-OHDA) treated rats and rescued dopaminergic neurons and the corresponding motor deficits (270). Liposome-mediated gene delivery of GDNF in combination with FUS was also shown to be neuroprotective in a 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine (MPTP) mouse model of PD (271). Unilateral FUS-mediated opening of mouse BBB coupled with intranasal brain-derived neurotrophic factor (BDNF) significantly enhanced BDNF uptake into the ipsilateral hemisphere of mice (272). Similarly, intranasal delivery of BDNF followed by FUS in MPTP-treated mice rescued dopaminergic neurons and behavioral deficits (273). Recent studies also seem to indicate that FUS mediated opening of the BBB by itself may promote neuroplasticity. For example, treatment with MRIgFUS alone in an AD animal model reduced tau and Aβ protein aggregates (274). The mechanism is not fully understood, but the treatment also increased neurogenesis in the dentate gyrus, possibly by transient upregulation of neurotrophic and pro-inflammatory factors. Whether FUS-mediated BBB opening induces a similar clearance of asyn aggregates remains to be determined, though neuroinflammation is associated with the accumulation of misfolded asyn (275).
With the abundant pre-clinical evidence supporting multiple applications of FUS-mediated BBB opening, its safety and feasibility in humans is now under evaluation for several neurodegenerative diseases. A Phase I clinical trial used the transcranial ExAblate system to repeatedly open the BBB in five patients with AD (NCT02343991) (266). Importantly, the integrity of the BBB was restored within 24 h in all 5 patients. A follow-up Phase IIa trial will assess the safety of targeting multiple brain regions in a larger AD cohort (NCT03739905). Another Phase I study reported the safe and reversible BBB opening in four patients with amyotrophic lateral sclerosis (ALS) (265). An ongoing clinical study (NCT03608553) is also evaluating the safety and feasibility of opening the BBB in patients with mild to moderate Parkinson's disease with dementia (268). These studies represent an important step toward the non-invasive delivery of disease modifying therapeutics for CNS diseases.
Taken together, growing pre-clinical and clinical evidence indicates that FUS mediated BBB opening offers several advantages for therapeutic delivery. First, the FUS mediated increase in BBB permeability is non-invasive and therefore, repeated therapeutic applications, if required, may be a viable strategy. Second, MRIgFUS allows for precise target engagement, either into discreet or broad brain regions, unilaterally or bilaterally as needed. Third, the FUS mediated BBB opening is transient, with a therapeutic window of ~6 h, thus safely returning the BBB to its prior intact state. Fourth, FUS allows for administration of combinations of therapeutics. Several lines of evidence indicate that neurotrophic factors like GDNF can promote dopaminergic regeneration and may be required in addition to therapeutics that aid in targeting and clearing pathological asyn (270, 276). In support of this, FUS-mediated delivery of the neuroprotective agent D3 promoted neuronal survival and rescued memory deficits in a transgenic mouse model of Alzheimer's disease (AD) (277).
Nevertheless, some factors are still unknown and need to be addressed before FUS can be safely employed as a routine therapeutic strategy for brain targeting. For one, more data is required regarding the safety profile of BBB opening, and how it may be affected by age and disease onset. This is especially relevant for neurodegenerative diseases where the BBB integrity may be partially compromised (278). Secondly, more data on the long-term impact of chronic FUS treatment and associated neuroinflammation is warranted, although current evidence seems to suggest that FUS-mediated opening of BBB generates only a transient immune response and repeat administration does not affect BBB integrity or cause haemorrhagic complications (259). Additionally, data on how factors such as microbubble size, exposure parameters and acoustic energy can be used to fine-tune the size and duration of BBB opening may be important to determine ideal and personalized therapeutic approaches.
Final Remarks
New tests to amplify and identify pathologically misfolded asyn in biopsy tissues are increasing in sensitivity (279–284) and may offer earlier detection of disease processes. In combination with prodromal clinical markers (285), these assays offer a critical opportunity to not only segregate synucleinopathy patients into distinct groups, but to also target asyn misfolding and accumulation using the precision tools discussed in this review. Initially, this will involve targeting the asyn assemblies with specific antibodies or by lowering asyn expression in specific brain regions to reduce the asyn substrate available for seeding. Ongoing improvements in viral vectors and gene delivery to the brain may offer less invasive and longer-term treatments to mitigate further pathological spread. Although much remains uncertain regarding age-related changes to other contributing factors such as proteostasis, inflammation, and oxidation, combinations of therapeutics targeting these ancillary pathways that regulate asyn processing could also be included as adjuncts to minimize neurodegeneration.
Author Contributions
All authors listed have made a substantial, direct, and intellectual contribution to the work and approved it for publication.
Funding
AT is supported by grants from the Canadian Institutes of Health Research (CIHR PJT180582) and the Weston Brain Institute (RR193223). SM received a postdoctoral fellowship from the Edmond J. Safra foundation. NPV is supported by the Rossy Foundation and Blidner Family Foundation. AH received a studentship from the Canadian Institutes of Health Research (CIHR-CGS-M).
Conflict of Interest
SPS is a Sanofi employee and stockholder.
The remaining authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher's Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Goedert M, Spillantini MG, Del Tredici K, Braak H. 100 years of Lewy pathology. Nat Rev Neurol. (2013) 9:13–24. doi: 10.1038/nrneurol.2012.242
2. Poewe W, Seppi K, Tanner CM, Halliday GM, Brundin P, Volkmann J, et al. Parkinson disease. Nat Rev Dis Primers. (2017) 3:17013. doi: 10.1038/nrdp.2017.13
3. Silva BA, Breydo L, Uversky VN. Targeting the chameleon: a focused look at alpha-synuclein and its roles in neurodegeneration. Mol Neurobiol. (2013) 47:446–59. doi: 10.1007/s12035-012-8334-1
4. Burre J, Sharma M, Sudhof TC. Cell biology and pathophysiology of alpha-synuclein. Cold Spring Harb Perspect Med. (2018) 8:a024091. doi: 10.1101/cshperspect.a024091
5. Snead D, Eliezer D. Intrinsically disordered proteins in synaptic vesicle trafficking and release. J Biol Chem. (2019) 294:3325–42. doi: 10.1074/jbc.REV118.006493
6. Asher DM, Belay E, Bigio E, Brandner S, Brubaker SA, Caughey B, et al. Risk of transmissibility from neurodegenerative disease-associated proteins: experimental knowns and unknowns. J Neuropathol Exp Neurol. (2020) 79:1141–6. doi: 10.1093/jnen/nlaa109
7. Kordower JH, Chu Y, Hauser RA, Freeman TB, Olanow CW. Lewy body-like pathology in long-term embryonic nigral transplants in Parkinson's disease3. Nat Med. (2008) 14:504–6. doi: 10.1038/nm1747
8. Li JY, Englund E, Holton JL, Soulet D, Hagell P, Lees AJ, et al. Lewy bodies in grafted neurons in subjects with Parkinson's disease suggest host-to-graft disease propagation. Nat Med. (2008) 14:501–3. doi: 10.1038/nm1746
9. Braak H, Del Tredici K, Bratzke H, Hamm-Clement J, Sandmann-Keil D, Rub U. Staging of the intracerebral inclusion body pathology associated with idiopathic Parkinson's disease (preclinical and clinical stages). J.Neurol. (2002) 249:III/1–III/5. doi: 10.1007/s00415-002-1301-4
10. Beach TG, Adler CH, Sue LI, Vedders L, Lue L, White Iii CL, et al. Multi-organ distribution of phosphorylated alpha-synuclein histopathology in subjects with Lewy body disorders. Acta Neuropathol. (2010) 119:689–702. doi: 10.1007/s00401-010-0664-3
11. Rodriguez L, Marano MM, Tandon A. Import and export of misfolded alpha-synuclein. Front Neurosci. (2018) 12:344. doi: 10.3389/fnins.2018.00344
12. Desplats P, Lee HJ, Bae EJ, Patrick C, Rockenstein E, Crews L, et al. Inclusion formation and neuronal cell death through neuron-to-neuron transmission of alpha-synuclein. Proc Natl Acad Sci USA. (2009) 106:13010–5. doi: 10.1073/pnas.0903691106
13. Hansen C, Angot E, Bergstrom AL, Steiner JA, Pieri L, Paul G, et al. alpha-Synuclein propagates from mouse brain to grafted dopaminergic neurons and seeds aggregation in cultured human cells1. J Clin Invest. (2011) 121:715–25. doi: 10.1172/JCI43366
14. Luk KC, Song C, O'Brien P, Stieber A, Branch JR, Brunden KR, et al. Exogenous alpha-synuclein fibrils seed the formation of Lewy body-like intracellular inclusions in cultured cells2. Proc Natl Acad Sci USA. (2009) 106:20051–6. doi: 10.1073/pnas.0908005106
15. Volpicelli-Daley LA, Luk KC, Patel TP, Tanik SA, Riddle DM, Stieber A, et al. Exogenous alpha-synuclein fibrils induce Lewy body pathology leading to synaptic dysfunction and neuron death1. Neuron. (2011) 72:57–71. doi: 10.1016/j.neuron.2011.08.033
16. Reyes JF, Olsson TT, Lamberts JT, Devine MJ, Kunath T, Brundin P. A cell culture model for monitoring alpha-synuclein cell-to-cell transfer. Neurobiol Dis. (2014) 77:266–75. doi: 10.1016/j.nbd.2014.07.003
17. Woerman AL, Stohr J, Aoyagi A, Rampersaud R, Krejciova Z, Watts JC, et al. Propagation of prions causing synucleinopathies in cultured cells. Proc Natl Acad Sci U S A. (2015) 112:E4949–58. doi: 10.1073/pnas.1513426112
18. Karpowicz RJ Jr, Haney CM, Mihaila TS, Sandler RM, Petersson EJ, Lee VM. Selective imaging of internalized proteopathic alpha-synuclein seeds in primary neurons reveals mechanistic insight into transmission of synucleinopathies. J Biol Chem. (2017) 292:13482–97. doi: 10.1074/jbc.M117.780296
19. Rey NL, Petit GH, Bousset L, Melki R, Brundin P. Transfer of human alpha-synuclein from the olfactory bulb to interconnected brain regions in mice. Acta Neuropathol. (2013) 126:555–73. doi: 10.1007/s00401-013-1160-3
20. Rey NL, Steiner JA, Maroof N, Luk KC, Madaj Z, Trojanowski JQ, et al. Widespread transneuronal propagation of alpha-synucleinopathy triggered in olfactory bulb mimics prodromal Parkinson's disease. J Exp Med. (2016) 213:1759–78. doi: 10.1084/jem.20160368
21. Mougenot AL, Nicot S, Bencsik A, Morignat E, Verchere J, Lakhdar L, et al. Prion-like acceleration of a synucleinopathy in a transgenic mouse model. Neurobiol Aging. (2012) 33:2225–8. doi: 10.1016/j.neurobiolaging.2011.06.022
22. Luk KC, Kehm VM, Zhang B, O'Brien P, Trojanowski JQ, Lee VM. Intracerebral inoculation of pathological alpha-synuclein initiates a rapidly progressive neurodegenerative alpha-synucleinopathy in mice. J Exp Med. (2012) 209:975–86. doi: 10.1084/jem.20112457
23. Watts JC, Giles K, Oehler A, Middleton L, Dexter DT, Gentleman SM, et al. Transmission of multiple system atrophy prions to transgenic mice. Proc Natl Acad Sci U S A. (2013) 110:19555–60. doi: 10.1073/pnas.1318268110
24. Sacino AN, Brooks M, McKinney AB, Thomas MA, Shaw G, Golde TE, et al. Brain injection of alpha-synuclein induces multiple proteinopathies, gliosis, and a neuronal injury marker. J Neurosci. (2014) 34:12368–78. doi: 10.1523/JNEUROSCI.2102-14.2014
25. Surmeier DJ, Obeso JA, Halliday GM. Selective neuronal vulnerability in Parkinson disease. Nat Rev Neurosci. (2017) 18:101–13. doi: 10.1038/nrn.2016.178
26. Brundin P, Melki R. Prying into the prion hypothesis for Parkinson's disease. J Neurosci. (2017) 37:9808–18. doi: 10.1523/JNEUROSCI.1788-16.2017
27. Alegre-Abarrategui J, Brimblecombe KR, Roberts RF, Velentza-Almpani E, Tilley BS, Bengoa-Vergniory N, et al. Selective vulnerability in alpha-synucleinopathies. Acta Neuropathol. (2019) 138:681–704. doi: 10.1007/s00401-019-02010-2
28. Lau A, So RWL, Lau HHC, Sang JC, Ruiz-Riquelme A, Fleck SC, et al. alpha-Synuclein strains target distinct brain regions and cell types. Nat Neurosci. (2020) 23:21–31. doi: 10.1038/s41593-019-0541-x
29. Rahayel S, Misic B, Zheng YQ, Liu ZQ, Abdelgawad A, Abbasi N, et al. Differentially targeted seeding reveals unique pathological alpha-synuclein propagation patterns. Brain. (2021). doi: 10.1093/brain/awab440
30. Bousset L, Pieri L, Ruiz-Arlandis G, Gath J, Jensen PH, Habenstein B, et al. Structural and functional characterization of two alpha-synuclein strains. Nat Commun. (2013) 4:2575. doi: 10.1038/ncomms3575
31. Peelaerts W, Bousset L, Van der Perren A, Moskalyuk A, Pulizzi R, Giugliano M, et al. alpha-Synuclein strains cause distinct synucleinopathies after local and systemic administration. Nature. (2015) 522:340–4. doi: 10.1038/nature14547
32. Watts JC. Calling alpha-synuclein a prion is scientifically justifiable. Acta Neuropathol. (2019) 138:505–8. doi: 10.1007/s00401-019-02058-0
33. Steiner JA, Quansah E, Brundin P. The concept of alpha-synuclein as a prion-like protein: ten years after. Cell Tissue Res. (2018) 373:161–73. doi: 10.1007/s00441-018-2814-1
34. Savitt D, Jankovic J. Targeting alpha-synuclein in Parkinson's disease: progress towards the development of disease-modifying therapeutics. Drugs. (2019) 79:797–810. doi: 10.1007/s40265-019-01104-1
35. Bras IC, Outeiro TF. Alpha-synuclein: mechanisms of release and pathology progression in synucleinopathies. Cells. (2021) 10:375. doi: 10.3390/cells10020375
36. Lee HJ, Patel S, Lee SJ. Intravesicular localization and exocytosis of alpha-synuclein and its aggregates. J Neurosci. (2005) 25:6016–24. doi: 10.1523/JNEUROSCI.0692-05.2005
37. Emmanouilidou E, Melachroinou K, Roumeliotis T, Garbis SD, Ntzouni M, Margaritis LH, et al. Cell-produced alpha-synuclein is secreted in a calcium-dependent manner by exosomes and impacts neuronal survival1. J Neurosci. (2010) 30:6838–51. doi: 10.1523/JNEUROSCI.5699-09.2010
38. Bae EJ, Ho DH, Park E, Jung JW, Cho K, Hong JH, et al. Lipid peroxidation product 4-hydroxy-2-nonenal promotes seeding-capable oligomer formation and cell-to-cell transfer of alpha-synuclein. Antioxid Redox Signal. (2013) 18:770–83. doi: 10.1089/ars.2011.4429
39. Jang A, Lee HJ, Suk JE, Jung JW, Kim KP, Lee SJ. Non-classical exocytosis of alpha-synuclein is sensitive to folding states and promoted under stress conditions. J Neurochem. (2010) 113:1263–74. doi: 10.1111/j.1471-4159.2010.06695.x
40. Lee HJ, Baek SM, Ho DH, Suk JE, Cho ED, Lee SJ. Dopamine promotes formation and secretion of non-fibrillar alpha-synuclein oligomers. Exp Mol Med. (2011) 43:216–22. doi: 10.3858/emm.2011.43.4.026
41. Ulusoy A, Musgrove RE, Rusconi R, Klinkenberg M, Helwig M, Schneider A, et al. Neuron-to-neuron alpha-synuclein propagation in vivo is independent of neuronal injury. Acta Neuropathol Commun. (2015) 3:13. doi: 10.1186/s40478-015-0198-y
42. Mathivanan S, Ji H, Simpson RJ. Exosomes: extracellular organelles important in intercellular communication. J Proteomics. (2010) 73:1907–20. doi: 10.1016/j.jprot.2010.06.006
43. Hessvik NP, Llorente A. Current knowledge on exosome biogenesis and release. Cell Mol Life Sci. (2018) 75:193–208. doi: 10.1007/s00018-017-2595-9
44. Arotcarena ML, Teil M, Dehay B. Autophagy in synucleinopathy: the overwhelmed and defective machinery. Cells. (2019) 8:565. doi: 10.3390/cells8060565
45. Alvarez-Erviti L, Seow Y, Schapira AH, Gardiner C, Sargent IL, Wood MJ, et al. Lysosomal dysfunction increases exosome-mediated alpha-synuclein release and transmission3. Neurobiol Dis. (2011) 42:360–7. doi: 10.1016/j.nbd.2011.01.029
46. Danzer KM, Kranich LR, Ruf WP, Cagsal-Getkin O, Winslow AR, Zhu L, et al. Exosomal cell-to-cell transmission of alpha synuclein oligomers. Mol Neurodegener. (2012) 7:42. doi: 10.1186/1750-1326-7-42
47. Lee HJ, Cho ED, Lee KW, Kim JH, Cho SG, Lee SJ. Autophagic failure promotes the exocytosis and intercellular transfer of alpha-synuclein. Exp Mol Med. (2013) 45:e22. doi: 10.1038/emm.2013.45
48. Ghidoni R, Benussi L, Binetti G. Exosomes: the Trojan horses of neurodegeneration. Med Hypotheses. (2008) 70:1226–7. doi: 10.1016/j.mehy.2007.12.003
49. Grey M, Dunning CJ, Gaspar R, Grey C, Brundin P, Sparr E, et al. Acceleration of alpha-synuclein aggregation by exosomes. J Biol Chem. (2015) 290:2969–82. doi: 10.1074/jbc.M114.585703
50. Shi M, Liu C, Cook TJ, Bullock KM, Zhao Y, Ginghina C, et al. Plasma exosomal alpha-synuclein is likely CNS-derived and increased in Parkinson's disease. Acta Neuropathol. (2014) 128:639–50. doi: 10.1007/s00401-014-1314-y
51. Niu M, Li Y, Li G, Zhou L, Luo N, Yao M, et al. A longitudinal study on alpha-synuclein in plasma neuronal exosomes as a biomarker for Parkinson's disease development and progression. Eur J Neurol. (2020) 27:967–74. doi: 10.1111/ene.14208
52. Stuendl A, Kraus T, Chatterjee M, Zapke B, Sadowski B, Moebius W, et al. alpha-synuclein in plasma-derived extracellular vesicles is a potential biomarker of Parkinson's disease. Mov Disord. (2021) 36:2508–18. doi: 10.1002/mds.28639
53. Zheng H, Xie Z, Zhang X, Mao J, Wang M, Wei S, et al. Investigation of alpha-synuclein species in plasma exosomes and the oligomeric and phosphorylated alpha-synuclein as potential peripheral biomarker of Parkinson's disease. Neuroscience. (2021) 469:79–90. doi: 10.1016/j.neuroscience.2021.06.033
54. Xia Y, Zhang G, Han C, Ma K, Guo X, Wan F, et al. Microglia as modulators of exosomal alpha-synuclein transmission. Cell Death Dis. (2019) 10:174. doi: 10.1038/s41419-019-1404-9
55. Guo M, Wang J, Zhao Y, Feng Y, Han S, Dong Q, et al. Microglial exosomes facilitate alpha-synuclein transmission in Parkinson's disease. Brain. (2020) 143:1476–97. doi: 10.1093/brain/awaa090
56. Fellner L, Irschick R, Schanda K, Reindl M, Klimaschewski L, Poewe W, et al. Toll-like receptor 4 is required for alpha-synuclein dependent activation of microglia and astroglia. Glia. (2013) 61:349–60. doi: 10.1002/glia.22437
57. Kim C, Ho DH, Suk JE, You S, Michael S, Kang J, et al. Neuron-released oligomeric alpha-synuclein is an endogenous agonist of TLR2 for paracrine activation of microglia. Nat Commun. (2013) 4:1562. doi: 10.1038/ncomms2534
58. Li N, Wu Y, Zhu L, Huang Y, Liu Z, Shi M, et al. Extracellular microvesicles-derived from microglia treated with unaggregated alpha-synuclein attenuate mitochondrial fission and toxicity-induced by Parkinsonian toxin MPP. Biochem Biophys Res Commun. (2019) 517:642–7. doi: 10.1016/j.bbrc.2019.07.084
59. Xia Y, Zhang G, Kou L, Yin S, Han C, Hu J, et al. Reactive microglia enhance the transmission of exosomal alpha-synuclein via toll-like receptor 2. Brain. (2021) 144:2024–37. doi: 10.1093/brain/awab122
60. Choi I, Zhang Y, Seegobin SP, Pruvost M, Wang Q, Purtell K, et al. Microglia clear neuron-released alpha-synuclein via selective autophagy and prevent neurodegeneration. Nat Commun. (2020) 11:1386. doi: 10.1038/s41467-020-15119-w
61. Chang C, Lang H, Geng N, Wang J, Li N, Wang X. Exosomes of BV-2 cells induced by alpha-synuclein: important mediator of neurodegeneration in PD. Neurosci Lett. (2013) 548:190–5. doi: 10.1016/j.neulet.2013.06.009
62. Daniele SG, Beraud D, Davenport C, Cheng K, Yin H, Maguire-Zeiss KA. Activation of MyD88-dependent TLR1/2 signaling by misfolded alpha-synuclein, a protein linked to neurodegenerative disorders. Sci Signal. (2015) 8:ra45. doi: 10.1126/scisignal.2005965
63. Kim C, Ojo-Amaize E, Spencer B, Rockenstein E, Mante M, Desplats P, et al. Hypoestoxide reduces neuroinflammation and alpha-synuclein accumulation in a mouse model of Parkinson's disease. J Neuroinflammation. (2015) 12:236. doi: 10.1186/s12974-015-0455-9
64. Valera E, Mante M, Anderson S, Rockenstein E, Masliah E. Lenalidomide reduces microglial activation and behavioral deficits in a transgenic model of Parkinson's disease. J Neuroinflammation. (2015) 12:93. doi: 10.1186/s12974-015-0320-x
65. Reyes JF, Sackmann C, Hoffmann A, Svenningsson P, Winkler J, Ingelsson M, et al. Binding of alpha-synuclein oligomers to Cx32 facilitates protein uptake and transfer in neurons and oligodendrocytes. Acta Neuropathol. (2019) 138:23–47. doi: 10.1007/s00401-019-02007-x
66. Diaz EF, Labra VC, Alvear TF, Mellado LA, Inostroza CA, Oyarzun JE, et al. Connexin 43 hemichannels and pannexin-1 channels contribute to the alpha-synuclein-induced dysfunction and death of astrocytes. Glia. (2019) 67:1598–619. doi: 10.1002/glia.23631
67. Sanchez OF, Rodriguez AV, Velasco-Espana JM, Murillo LC, Sutachan JJ, Albarracin SL. Role of Connexins 30, 36, and 43 in brain tumors, neurodegenerative diseases, and neuroprotection. Cells. (2020) 9:846. doi: 10.3390/cells9040846
68. Abounit S, Bousset L, Loria F, Zhu S, de Chaumont F, Pieri L, et al. Tunneling nanotubes spread fibrillar alpha-synuclein by intercellular trafficking of lysosomes. EMBO J. (2016) 35:2120–38. doi: 10.15252/embj.201593411
69. Dieriks BV, Park TI, Fourie C, Faull RL, Dragunow M, Curtis MA. alpha-synuclein transfer through tunneling nanotubes occurs in SH-SY5Y cells and primary brain pericytes from Parkinson's disease patients. Sci Rep. (2017) 7:42984. doi: 10.1038/srep42984
70. Rostami J, Holmqvist S, Lindstrom V, Sigvardson J, Westermark GT, Ingelsson M, et al. Human astrocytes transfer aggregated alpha-synuclein via tunneling nanotubes. J Neurosci. (2017) 37:11835–53. doi: 10.1523/JNEUROSCI.0983-17.2017
71. Kimura S, Hase K, Ohno H. The molecular basis of induction and formation of tunneling nanotubes. Cell Tissue Res. (2013) 352:67–76. doi: 10.1007/s00441-012-1518-1
72. Grudina C, Kouroupi G, Nonaka T, Hasegawa M, Matsas R, Zurzolo C. Human NPCs can degrade alpha-syn fibrils and transfer them preferentially in a cell contact-dependent manner possibly through TNT-like structures. Neurobiol Dis. (2019) 132:104609. doi: 10.1016/j.nbd.2019.104609
73. Scheiblich H, Dansokho C, Mercan D, Schmidt SV, Bousset L, Wischhof L, et al. Microglia jointly degrade fibrillar alpha-synuclein cargo by distribution through tunneling nanotubes. Cell. (2021) 184:5089–106.e21. doi: 10.1016/j.cell.2021.09.007
74. Vargas JY, Loria F, Wu YJ, Cordova G, Nonaka T, Bellow S, et al. Hasegawa, van Woerden GM, Trollet C, Zurzolo C. The Wnt/Ca(2+) pathway is involved in interneuronal communication mediated by tunneling nanotubes. EMBO J. (2019) 38:e101230. doi: 10.15252/embj.2018101230
75. Lee HJ, Suk JE, Bae EJ, Lee JH, Paik SR, Lee SJ. Assembly-dependent endocytosis and clearance of extracellular alpha-synuclein. Int J Biochem Cell Biol. (2008) 40:1835–49. doi: 10.1016/j.biocel.2008.01.017
76. Samuel F, Flavin WP, Iqbal S, Pacelli C, Sri Renganathan SD, Trudeau LE, et al. Effects of serine 129 phosphorylation on alpha-synuclein aggregation, membrane association, and internalization. J Biol Chem. (2016) 291:4374–85. doi: 10.1074/jbc.M115.705095
77. Sacino AN, Brooks MM, Chakrabarty P, Saha K, Khoshbouei H, Golde TE, et al. Proteolysis of alpha-synuclein fibrils in the lysosomal pathway limits induction of inclusion pathology. J Neurochem. (2017) 140:662–78. doi: 10.1111/jnc.13743
78. Freundt EC, Maynard N, Clancy EK, Roy S, Bousset L, Sourigues Y, et al. Neuron-to-neuron transmission of alpha-synuclein fibrils through axonal transport. Ann Neurol. (2012) 72:517–24. doi: 10.1002/ana.23747
79. Brahic M, Bousset L, Bieri G, Melki R, Gitler AD. Axonal transport and secretion of fibrillar forms of alpha-synuclein, Abeta42 peptide and HTTExon 1. Acta Neuropathol. (2016) 131:539–48. doi: 10.1007/s00401-016-1538-0
80. Angot E, Steiner JA, Lema Tome CM, Ekstrom P, Mattsson B, Bjorklund A, et al. Alpha-synuclein cell-to-cell transfer and seeding in grafted dopaminergic neurons in vivo. PLoS ONE. (2012) 7:e39465. doi: 10.1371/journal.pone.0039465
81. Holmes BB, DeVos SL, Kfoury N, Li M, Jacks R, Yanamandra K, et al. Heparan sulfate proteoglycans mediate internalization and propagation of specific proteopathic seeds. Proc Natl Acad Sci USA. (2013) 110:E3138–47. doi: 10.1073/pnas.1301440110
82. Hudak A, Kusz E, Domonkos I, Josvay K, Kodamullil AT, Szilak L, et al. Contribution of syndecans to cellular uptake and fibrillation of alpha-synuclein and tau. Sci Rep. (2019) 9:16543. doi: 10.1038/s41598-019-53038-z
83. Birol M, Wojcik SP, Miranker AD, Rhoades E. Identification of N-linked glycans as specific mediators of neuronal uptake of acetylated alpha-Synuclein. PLoS Biol. (2019) 17:e3000318. doi: 10.1371/journal.pbio.3000318
84. Mao X, Ou MT, Karuppagounder SS, Kam TI, Yin X, Xiong Y, et al. Pathological alpha-synuclein transmission initiated by binding lymphocyte-activation gene 3. Science. (2016) 353:aah3374. doi: 10.1126/science.aah3374
85. Angelopoulou E, Paudel YN, Villa C, Shaikh MF, Piperi C. Lymphocyte-activation gene 3 (LAG3) protein as a possible therapeutic target for Parkinson's disease: molecular mechanisms connecting neuroinflammation to alpha-synuclein spreading pathology. Biology. (2020) 9:86. doi: 10.3390/biology9040086
86. Choi YR, Cha SH, Kang SJ, Kim JB, Jou I, Park SM. Prion-like propagation of alpha-synuclein is regulated by the FcgammaRIIB-SHP-1/2 signaling pathway in neurons. Cell Rep. (2018) 22:136–48. doi: 10.1016/j.celrep.2017.12.009
87. Emmenegger M, De Cecco E, Hruska-Plochan M, Eninger T, Schneider MM, Barth M, et al. LAG3 is not expressed in human and murine neurons and does not modulate alpha-synucleinopathies. EMBO Mol Med. (2021) 13:e14745. doi: 10.15252/emmm.202114745
88. Croisier E, Graeber MB. Glial degeneration and reactive gliosis in alpha-synucleinopathies: the emerging concept of primary gliodegeneration. Acta Neuropathol. (2006) 112:517–30. doi: 10.1007/s00401-006-0119-z
89. Lee HJ, Suk JE, Patrick C, Bae EJ, Cho JH, Rho S, et al. Direct transfer of alpha-synuclein from neuron to astroglia causes inflammatory responses in synucleinopathies. J Biol Chem. (2010) 285:9262–72. doi: 10.1074/jbc.M109.081125
90. Loria F, Vargas JY, Bousset L, Syan S, Salles A, Melki R, et al. alpha-Synuclein transfer between neurons and astrocytes indicates that astrocytes play a role in degradation rather than in spreading. Acta Neuropathol. (2017) 134:789–808. doi: 10.1007/s00401-017-1746-2
91. Konno M, Hasegawa T, Baba T, Miura E, Sugeno N, Kikuchi A, et al. Suppression of dynamin GTPase decreases alpha-synuclein uptake by neuronal and oligodendroglial cells: a potent therapeutic target for synucleinopathy. Mol Neurodegener. (2012) 7:38. doi: 10.1186/1750-1326-7-38
92. Park JY, Kim KS, Lee SB Ryu JS, Chung KC, Choo YK, et al. On the mechanism of internalization of alpha-synuclein into microglia: roles of ganglioside GM1 and lipid raft. J Neurochem. (2009) 110:400–11. doi: 10.1111/j.1471-4159.2009.06150.x
93. Levine PM, Galesic A, Balana AT, Mahul-Mellier AL, Navarro MX, De Leon CA, et al. alpha-Synuclein O-GlcNAcylation alters aggregation and toxicity, revealing certain residues as potential inhibitors of Parkinson's disease. Proc Natl Acad Sci U S A. (2019) 116:1511–9. doi: 10.1073/pnas.1808845116
94. Selnick HG, Hess JF, Tang C, Liu K, Schachter JB, Ballard JE, et al. Discovery of MK-8719, a potent O-GlcNAcase inhibitor as a potential treatment for tauopathies. J Med Chem. (2019) 62:10062–97. doi: 10.1021/acs.jmedchem.9b01090
95. Tavassoly O, Yue J, Vocadlo DJ. Pharmacological inhibition and knockdown of O-GlcNAcase reduces cellular internalization of alpha-synuclein preformed fibrils. FEBS J. (2021) 288:452–70. doi: 10.1111/febs.15349
96. Hilverling A, Szego EM, Dinter E, Cozma D, Saridaki T, Falkenburger BH. Maturing autophagosomes are transported towards the cell periphery. Cell Mol Neurobiol. (2021). doi: 10.21203/rs.3.rs-200706/v1
97. Kimura T, Jia J, Kumar S, Choi SW, Gu Y, Mudd M, et al. Dedicated SNAREs and specialized TRIM cargo receptors mediate secretory autophagy. EMBO J. (2017) 36:42–60. doi: 10.15252/embj.201695081
98. Lee HJ, Khoshaghideh F, Patel S, Lee SJ. Clearance of alpha-synuclein oligomeric intermediates via the lysosomal degradation pathway. J Neurosci. (2004) 24:1888–96. doi: 10.1523/JNEUROSCI.3809-03.2004
99. Paxinou E, Chen Q, Weisse M, Giasson BI, Norris EH, Rueter SM, et al. Induction of alpha-synuclein aggregation by intracellular nitrative insult. J Neurosci. (2001) 21:8053–61. doi: 10.1523/JNEUROSCI.21-20-08053.2001
100. Webb JL, Ravikumar B, Atkins J, Skepper JN, Rubinsztein DC. Alpha-Synuclein is degraded by both autophagy and the proteasome. J Biol Chem. (2003) 278:25009–13. doi: 10.1074/jbc.M300227200
101. Alvarez-Erviti L, Rodriguez-Oroz MC, Cooper JM, Caballero C, Ferrer I, Obeso JA, et al. Chaperone-mediated autophagy markers in Parkinson disease brains. Arch Neurol. (2010) 67:1464–72. doi: 10.1001/archneurol.2010.198
102. Cuervo AM, Stefanis L, Fredenburg R, Lansbury PT, Sulzer D. Impaired degradation of mutant alpha-synuclein by chaperone-mediated autophagy. Science. (2004) 305:1292–5. doi: 10.1126/science.1101738
103. Tanik SA, Schultheiss CE, Volpicelli-Daley LA, Brunden KR, Lee VM. Lewy body-like alpha-synuclein aggregates resist degradation and impair macroautophagy. J Biol Chem. (2013) 288:15194–210. doi: 10.1074/jbc.M113.457408
104. Ho PW, Leung CT, Liu H, Pang SY, Lam CS, Xian J, et al. Age-dependent accumulation of oligomeric SNCA/alpha-synuclein from impaired degradation in mutant LRRK2 knockin mouse model of Parkinson disease: role for therapeutic activation of chaperone-mediated autophagy (CMA). Autophagy. (2020) 16:347–70. doi: 10.1080/15548627.2019.1603545
105. Gan-Or Z, Liong C, Alcalay RN. GBA-associated Parkinson's disease and other synucleinopathies. Curr Neurol Neurosci Rep. (2018) 18:44. doi: 10.1007/s11910-018-0860-4
106. Mazzulli JR, Xu YH, Sun Y, Knight AL, McLean PJ, Caldwell GA, et al. Gaucher disease glucocerebrosidase and alpha-synuclein form a bidirectional pathogenic loop in synucleinopathies. Cell. (2011) 146:37–52. doi: 10.1016/j.cell.2011.06.001
107. Rocha EM, Smith GA, Park E, Cao H, Brown E, Hayes MA, et al. Glucocerebrosidase gene therapy prevents alpha-synucleinopathy of midbrain dopamine neurons. Neurobiol Dis. (2015) 82:495–503. doi: 10.1016/j.nbd.2015.09.009
108. Rockenstein E, Clarke J, Viel C, Panarello N, Treleaven CM, Kim C, et al. Glucocerebrosidase modulates cognitive and motor activities in murine models of Parkinson's disease. Hum Mol Genet. (2016) 25:2645–60. doi: 10.1093/hmg/ddw124
109. Viel C, Clarke J, Kayatekin C, Richards AM, Chiang MSR, Park H, et al. Preclinical pharmacology of glucosylceramide synthase inhibitor venglustat in a GBA-related synucleinopathy model. Sci Rep. (2021) 11:20945. doi: 10.1038/s41598-021-00404-5
110. Ambrosi G, Ghezzi C, Zangaglia R, Levandis G, Pacchetti C, Blandini F. Ambroxol-induced rescue of defective glucocerebrosidase is associated with increased LIMP-2 and saposin C levels in GBA1 mutant Parkinson's disease cells. Neurobiol Dis. (2015) 82:235–42. doi: 10.1016/j.nbd.2015.06.008
111. McNeill A, Magalhaes J, Shen C, Chau KY, Hughes D, Mehta A, et al. Ambroxol improves lysosomal biochemistry in glucocerebrosidase mutation-linked Parkinson disease cells. Brain. (2014) 137:1481–95. doi: 10.1093/brain/awu020
112. Mullin S, Smith L, Lee K, D'Souza G, Woodgate P, Elflein J, et al. Ambroxol for the Treatment of Patients With Parkinson Disease With and Without Glucocerebrosidase Gene Mutations: A Nonrandomized, Noncontrolled Trial. JAMA Neurol. (2020) 77:427–34. doi: 10.1001/jamaneurol.2019.4611
113. Stojkovska I, Wani WY, Zunke F, Belur NR, Pavlenko EA, Mwenda N, et al. Rescue of alpha-synuclein aggregation in Parkinson's patient neurons by synergistic enhancement of ER proteostasis and protein trafficking. Neuron. (2022) 110:436–51.e11. doi: 10.1016/j.neuron.2021.10.032
114. Cuddy LK, Wani WY, Morella ML, Pitcairn C, Tsutsumi K, Fredriksen K, et al. Stress-induced cellular clearance is mediated by the SNARE protein ykt6 and disrupted by alpha-synuclein. Neuron. (2019) 104:869–84.e11. doi: 10.1016/j.neuron.2019.09.001
115. Bai X, Wey MC, Fernandez E, Hart MJ, Gelfond J, Bokov AF, et al. Rapamycin improves motor function, reduces 4-hydroxynonenal adducted protein in brain, and attenuates synaptic injury in a mouse model of synucleinopathy. Pathobiol Aging Age Relat Dis. (2015) 5:28743. doi: 10.3402/pba.v5.28743
116. Malagelada C, Jin ZH, Jackson-Lewis V, Przedborski S, Greene LA. Rapamycin protects against neuron death in in vitro and in vivo models of Parkinson's disease. J Neurosci. (2010) 30:1166–75. doi: 10.1523/JNEUROSCI.3944-09.2010
117. Pan T, Rawal P, Wu Y, Xie W, Jankovic J, Le W. Rapamycin protects against rotenone-induced apoptosis through autophagy induction. Neuroscience. (2009) 164:541–51. doi: 10.1016/j.neuroscience.2009.08.014
118. Decressac M, Mattsson B, Weikop P, Lundblad M, Jakobsson J, Bjorklund A. TFEB-mediated autophagy rescues midbrain dopamine neurons from alpha-synuclein toxicity. Proc Natl Acad Sci U S A. (2013) 110:E1817–26. doi: 10.1073/pnas.1305623110
119. Arotcarena ML, Bourdenx M, Dutheil N, Thiolat ML, Doudnikoff E, Dovero S, et al. Transcription factor EB overexpression prevents neurodegeneration in experimental synucleinopathies. JCI Insight. (2019) 4:e129719. doi: 10.1172/jci.insight.129719
120. Dulovic M, Jovanovic M, Xilouri M, Stefanis L, Harhaji-Trajkovic L, Kravic-Stevovic T, et al. The protective role of AMP-activated protein kinase in alpha-synuclein neurotoxicity in vitro. Neurobiol Dis. (2014) 63:1–11. doi: 10.1016/j.nbd.2013.11.002
121. He Q, Koprich JB, Wang Y, Yu WB, Xiao BG, Brotchie JM, et al. Treatment with trehalose prevents behavioral and neurochemical deficits produced in an AAV alpha-synuclein rat model of Parkinson's disease. Mol Neurobiol. (2016) 53:2258–68. doi: 10.1007/s12035-015-9173-7
122. Hebron ML, Lonskaya I, Moussa CE. Nilotinib reverses loss of dopamine neurons and improves motor behavior via autophagic degradation of alpha-synuclein in Parkinson's disease models. Hum Mol Genet. (2013) 22:3315–28. doi: 10.1093/hmg/ddt192
123. Pupyshev AB, Tikhonova MA, Akopyan AA, Tenditnik MV, Dubrovina NI, Korolenko TA. Therapeutic activation of autophagy by combined treatment with rapamycin and trehalose in a mouse MPTP-induced model of Parkinson's disease. Pharmacol Biochem Behav. (2019) 177:1–11. doi: 10.1016/j.pbb.2018.12.005
124. de Wet S, Du Toit A, Loos B. Spermidine and rapamycin reveal distinct autophagy flux response and cargo receptor clearance profile. Cells. (2021) 10:95. doi: 10.3390/cells10010095
125. Lumkwana D, du Toit A, Kinnear C, Loos B. Autophagic flux control in neurodegeneration: Progress and precision targeting-Where do we stand? Prog Neurobiol. (2017) 153:64–85. doi: 10.1016/j.pneurobio.2017.03.006
126. Klionsky DJ, Petroni G, Amaravadi RK, Baehrecke EH, Ballabio A, Boya P, et al. Autophagy in major human diseases. EMBO J. (2021) 40:e108863. doi: 10.15252/embj.2021108863
127. Liu Y, Levine B. Autosis and autophagic cell death: the dark side of autophagy. Cell Death Differ. (2015) 22:367–76. doi: 10.1038/cdd.2014.143
128. Freeman D, Cedillos R, Choyke S, Lukic Z, McGuire K, Marvin S, et al. Alpha-synuclein induces lysosomal rupture and cathepsin dependent reactive oxygen species following endocytosis. PLoS ONE. (2013) 8:e62143. doi: 10.1371/journal.pone.0062143
129. Jiang P, Gan M, Yen SH, McLean PJ, Dickson DW. Impaired endo-lysosomal membrane integrity accelerates the seeding progression of alpha-synuclein aggregates. Sci Rep. (2017) 7:7690. doi: 10.1038/s41598-017-08149-w
130. Flavin WP, Bousset L, Green ZC, Chu Y, Skarpathiotis S, Chaney MJ, et al. Endocytic vesicle rupture is a conserved mechanism of cellular invasion by amyloid proteins. Acta Neuropathol. (2017) 134:629–53. doi: 10.1007/s00401-017-1722-x
131. Shahmoradian SH, Lewis AJ, Genoud C, Hench J, Moors TE, Navarro PP, et al. Lewy pathology in Parkinson's disease consists of crowded organelles and lipid membranes. Nat Neurosci. (2019) 22:1099–109. doi: 10.1038/s41593-019-0423-2
132. Fonseca-Ornelas L, Eisbach SE, Paulat M, Giller K, Fernandez CO, Outeiro TF, et al. Small molecule-mediated stabilization of vesicle-associated helical alpha-synuclein inhibits pathogenic misfolding and aggregation. Nat Commun. (2014) 5:5857. doi: 10.1038/ncomms6857
133. Castonguay AM, Gravel C, Levesque M. Treating Parkinson's Disease with Antibodies: Previous Studies and Future Directions. J Parkinsons Dis. (2021) 11:71–92. doi: 10.3233/JPD-202221
134. Masliah E, Rockenstein E, Adame A, Alford M, Crews L, Hashimoto M, et al. Effects of alpha-synuclein immunization in a mouse model of Parkinson's disease. Neuron. (2005) 46:857–68. doi: 10.1016/j.neuron.2005.05.010
135. Masliah E, Rockenstein E, Mante M, Crews L, Spencer B, Adame A, et al. Passive immunization reduces behavioral and neuropathological deficits in an alpha-synuclein transgenic model of Lewy body disease. PLoS ONE. (2011) 6:e19338. doi: 10.1371/journal.pone.0019338
136. Sanchez-Guajardo V, Annibali A, Jensen PH, Romero-Ramos M. alpha-Synuclein vaccination prevents the accumulation of parkinson disease-like pathologic inclusions in striatum in association with regulatory T cell recruitment in a rat model. J Neuropathol Exp Neurol. (2013) 72:624–45. doi: 10.1097/NEN.0b013e31829768d2
137. Ugen KE, Lin X, Bai G, Liang Z, Cai J, Li K, et al. Evaluation of an alpha synuclein sensitized dendritic cell based vaccine in a transgenic mouse model of Parkinson's disease. Hum Vaccin Immunother. (2015) 11:922–30. doi: 10.1080/21645515.2015.1012033
138. Doucet M, El-Turabi A, Zabel F, Hunn BHM, Bengoa-Vergniory N, Cioroch M, et al. Preclinical development of a vaccine against oligomeric alpha-synuclein based on virus-like particles. PloS ONE. (2017) 12:e0181844. doi: 10.1371/journal.pone.0181844
139. Bae EJ, Lee HJ, Rockenstein E, Ho DH, Park EB, Yang NY, et al. Antibody-aided clearance of extracellular alpha-synuclein prevents cell-to-cell aggregate transmission. J Neurosci. (2012) 32:13454–69. doi: 10.1523/JNEUROSCI.1292-12.2012
140. Games D, Valera E, Spencer B, Rockenstein E, Mante M, Adame A, et al. Reducing C-terminal-truncated alpha-synuclein by immunotherapy attenuates neurodegeneration and propagation in Parkinson's disease-like models. J Neurosci. (2014) 34:9441–54. doi: 10.1523/JNEUROSCI.5314-13.2014
141. Tran HT, Chung CH, Iba M, Zhang B, Trojanowski JQ, Luk KC, et al. Alpha-synuclein immunotherapy blocks uptake and templated propagation of misfolded alpha-synuclein and neurodegeneration. Cell Rep. (2014) 7:2054–65. doi: 10.1016/j.celrep.2014.05.033
142. Spencer B, Valera E, Rockenstein E, Overk C, Mante M, Adame A, et al. Anti-alpha-synuclein immunotherapy reduces alpha-synuclein propagation in the axon and degeneration in a combined viral vector and transgenic model of synucleinopathy. Acta Neuropathol Commun. (2017) 5:7. doi: 10.1186/s40478-016-0410-8
143. El-Agnaf O, Overk C, Rockenstein E, Mante M, Florio J, Adame A, et al. Differential effects of immunotherapy with antibodies targeting alpha-synuclein oligomers and fibrils in a transgenic model of synucleinopathy. Neurobiol Dis. (2017) 104:85–96. doi: 10.1016/j.nbd.2017.05.002
144. Schofield DJ, Irving L, Calo L, Bogstedt A, Rees G, Nuccitelli A, et al. Preclinical development of a high affinity alpha-synuclein antibody, MEDI1341, that can enter the brain, sequester extracellular alpha-synuclein and attenuate alpha-synuclein spreading in vivo. Neurobiol Dis. (2019) 132:104582. doi: 10.1016/j.nbd.2019.104582
145. Henderson MX, Covell DJ, Chung CH, Pitkin RM, Sandler RM, Decker SC, et al. Characterization of novel conformation-selective alpha-synuclein antibodies as potential immunotherapeutic agents for Parkinson's disease. Neurobiol Dis. (2020) 136:104712. doi: 10.1016/j.nbd.2019.104712
146. Jankovic J, Goodman I, Safirstein B, Marmon TK, Schenk DB, Koller M, et al. Safety and tolerability of multiple ascending doses of PRX002/RG7935, an anti-alpha-synuclein monoclonal antibody, in patients with Parkinson disease: a randomized clinical trial. JAMA Neurol. (2018) 75:1206–14. doi: 10.1001/jamaneurol.2018.1487
147. Mandler M, Valera E, Rockenstein E, Weninger H, Patrick C, Adame A, et al. Next-generation active immunization approach for synucleinopathies: implications for Parkinson's disease clinical trials. Acta Neuropathol. (2014) 127:861–79. doi: 10.1007/s00401-014-1256-4
148. Volc D, Poewe W, Kutzelnigg A, Luhrs P, Thun-Hohenstein C, Schneeberger A, et al. Safety and immunogenicity of the alpha-synuclein active immunotherapeutic PD01A in patients with Parkinson's disease: a randomised, single-blinded, phase 1 trial. Lancet Neurol. (2020) 19:591–600. doi: 10.1016/S1474-4422(20)30136-8
149. Meissner WG, Traon AP, Foubert-Samier A, Galabova G, Galitzky M, Kutzelnigg A, et al. Rascol, and AInvestigators FFS A Phase 1 randomized trial of specific active alpha-synuclein immunotherapies PD01A and PD03A in multiple system atrophy. Mov Disord. (2020) 35:1957–65. doi: 10.1002/mds.28218
150. Poewe W, Volc D, Seppi K, Medori R, Luhrs P, Kutzelnigg A, et al. Safety and tolerability of active immunotherapy targeting alpha-synuclein with PD03A in patients with early Parkinson's disease: a randomized, placebo-controlled, phase 1. Study J Parkinsons Dis. (2021) 11:1079–89. doi: 10.3233/JPD-212594
151. Nimmo JT, Verma A, Dodart JC, Wang CY, Savistchenko J, Melki R, et al. Novel antibodies detect additional alpha-synuclein pathology in synucleinopathies: potential development for immunotherapy. Alzheimers Res Ther. (2020) 12:159. doi: 10.1186/s13195-020-00727-x
152. Weihofen A, Liu Y, Arndt JW, Huy C, Quan C, Smith BA, et al. Development of an aggregate-selective, human-derived alpha-synuclein antibody BIIB054 that ameliorates disease phenotypes in Parkinson's disease models. Neurobiol Dis. (2019) 124:276–88. doi: 10.1016/j.nbd.2018.10.016
153. Nimmo JT, Kelly L, Verma A, Carare RO, Nicoll JAR, Dodart JC. Amyloid-beta and alpha-synuclein immunotherapy: from experimental studies to clinical trials. Front Neurosci. (2021) 15:733857. doi: 10.3389/fnins.2021.733857
154. Bittar A, Bhatt N, Kayed R. Advances and considerations in AD tau-targeted immunotherapy. Neurobiol Dis. (2020) 134:104707. doi: 10.1016/j.nbd.2019.104707
155. Zella MAS, Metzdorf J, Ostendorf F, Maass F, Muhlack S, Gold R, et al. Novel immunotherapeutic approaches to target alpha-synuclein and related neuroinflammation in Parkinson's disease. Cells. (2019) 8:105. doi: 10.3390/cells8020105
156. Li B, Ge P, Murray KA, Sheth P, Zhang M, Nair G, et al. Cryo-EM of full-length alpha-synuclein reveals fibril polymorphs with a common structural kernel. Nat Commun. (2018) 9:3609. doi: 10.1038/s41467-018-05971-2
157. Guerrero-Ferreira R, Taylor NM, Arteni AA, Kumari P, Mona D, Ringler P, et al. Two new polymorphic structures of human full-length alpha-synuclein fibrils solved by cryo-electron microscopy. Elife. (2019) 8:e48907. doi: 10.7554/eLife.48907
158. Schweighauser M, Shi Y, Tarutani A, Kametani F, Murzin AG, Ghetti B, et al. Structures of alpha-synuclein filaments from multiple system atrophy. Nature. (2020) 585:464–9. doi: 10.1038/s41586-020-2317-6
159. Martinez-Valbuena I, Visanji NP, Kim A, Lau HHC, So RWL, Alshimemeri S, et al. Alpha-synuclein seeding shows a wide heterogeneity in multiple system atrophy. Transl Neurodegener. (2022) 11:7. doi: 10.1186/s40035-022-00283-4
160. Lewis J, Melrose H, Bumcrot D, Hope A, Zehr C, Lincoln S, et al. In vivo silencing of alpha-synuclein using naked siRNA. Mol Neurodegener. (2008) 3:19. doi: 10.1186/1750-1326-3-19
161. Sapru MK, Yates JW, Hogan S, Jiang L, Halter J, Bohn MC. Silencing of human alpha-synuclein in vitro and in rat brain using lentiviral-mediated RNAi. ExpNeurol. (2006) 198:382–90. doi: 10.1016/j.expneurol.2005.12.024
162. Khodr CE, Sapru MK, Pedapati J, Han Y, West NC, Kells AP, et al. An alpha-synuclein AAV gene silencing vector ameliorates a behavioral deficit in a rat model of Parkinson's disease, but displays toxicity in dopamine neurons. Brain Res. (2011) 1395:94–107. doi: 10.1016/j.brainres.2011.04.036
163. Khodr CE, Becerra A, Han Y, Bohn MC. Targeting alpha-synuclein with a microRNA-embedded silencing vector in the rat substantia nigra: positive and negative effects. Brain Res. (2014) 1550:47–60. doi: 10.1016/j.brainres.2014.01.010
164. Zharikov A, Bai Q, De Miranda BR, Van Laar A, Greenamyre JT, Burton EA. Long-term RNAi knockdown of alpha-synuclein in the adult rat substantia nigra without neurodegeneration. Neurobiol Dis. (2019) 125:146–53. doi: 10.1016/j.nbd.2019.01.004
165. Collier TJ, Redmond DE Jr, Steece-Collier K, Lipton JW, Manfredsson FP. Is alpha-synuclein loss-of-function a contributor to parkinsonian pathology? evidence from non-human primates. Front Neurosci. (2016) 10:12. doi: 10.3389/fnins.2016.00012
166. Gorbatyuk OS, Li S, Nash K, Gorbatyuk M, Lewin AS, Sullivan LF, et al. In vivo RNAi-mediated alpha-synuclein silencing induces nigrostriatal degeneration2. MolTher. (2010) 18:1450–7. doi: 10.1038/mt.2010.115
167. Benskey MJ, Sellnow RC, Sandoval IM, Sortwell CE, Lipton JW, Manfredsson FP. Silencing alpha synuclein in mature nigral neurons results in rapid neuroinflammation and subsequent toxicity. Front Mol Neurosci. (2018) 11:36. doi: 10.3389/fnmol.2018.00036
168. Xhima K, Nabbouh F, Hynynen K, Aubert I, Tandon A. Noninvasive delivery of an alpha-synuclein gene silencing vector with magnetic resonance-guided focused ultrasound. Mov Disord. (2018) 33:1567–79. doi: 10.1002/mds.101
169. Cole TA, Zhao H, Collier TJ, Sandoval I, Sortwell CE, Steece-Collier K, et al. Alpha-synuclein antisense oligonucleotides as a disease-modifying therapy for Parkinson's disease. JCI Insight. (2021) 6:e135633. doi: 10.1172/jci.insight.135633
170. Menon S, Kofoed RH, Nabbouh F, Xhima K, Al-Fahoum Y, Langman T, et al. Viral alpha-synuclein knockdown prevents spreading synucleinopathy. Brain Commun. (2021) 3:fcab247. doi: 10.1093/braincomms/fcab247
171. Gui Y, Liu H, Zhang L, Lv W, Hu X. Altered microRNA profiles in cerebrospinal fluid exosome in Parkinson disease and Alzheimer disease. Oncotarget. (2015) 6:37043–53. doi: 10.18632/oncotarget.6158
172. Khan I, Preeti K, Fernandes V, Khatri DK, Singh SB. Role of MicroRNAs, aptamers in neuroinflammation and neurodegenerative disorders. Cell Mol Neurobiol. (2021). doi: 10.1007/s10571-021-01093-4
173. Junn E, Lee KW, Jeong BS, Chan TW, Im JY, Mouradian MM. Repression of alpha-synuclein expression and toxicity by microRNA-7. Proc Natl Acad Sci U S A. (2009) 106:13052–7. doi: 10.1073/pnas.0906277106
174. Doxakis E. Post-transcriptional regulation of alpha-synuclein expression by mir-7 and mir-153. J Biol Chem. (2010) 285:12726–34. doi: 10.1074/jbc.M109.086827
175. Wang ZH, Zhang JL, Duan YL, Zhang QS, Li GF, Zheng DL. MicroRNA-214 participates in the neuroprotective effect of Resveratrol via inhibiting alpha-synuclein expression in MPTP-induced Parkinson's disease mouse. Biomed Pharmacother. (2015) 74:252–6. doi: 10.1016/j.biopha.2015.08.025
176. Zhang Z, Cheng Y. miR-16-1 promotes the aberrant alpha-synuclein accumulation in parkinson disease via targeting heat shock protein 70. ScientificWorldJournal. (2014) 2014:938348. doi: 10.1155/2014/938348
177. Han Y, Khodr CE, Sapru MK, Pedapati J, Bohn MC. A microRNA embedded AAV alpha-synuclein gene silencing vector for dopaminergic neurons. Brain Res. (2011) 1386:15–24. doi: 10.1016/j.brainres.2011.02.041
178. Puhl DL, D'Amato AR, Gilbert RJ. Challenges of gene delivery to the central nervous system and the growing use of biomaterial vectors. Brain Res Bull. (2019) 150:216–30. doi: 10.1016/j.brainresbull.2019.05.024
179. Alarcon-Aris D, Pavia-Collado R, Miquel-Rio L, Coppola-Segovia V, Ferres-Coy A, Ruiz-Bronchal E, et al. Anti-alpha-synuclein ASO delivered to monoamine neurons prevents alpha-synuclein accumulation in a Parkinson's disease-like mouse model and in monkeys. EBioMedicine. (2020) 59:102944. doi: 10.1016/j.ebiom.2020.102944
180. Yang J, Luo S, Zhang J, Yu T, Fu Z, Zheng Y, et al. Exosome-mediated delivery of antisense oligonucleotides targeting alpha-synuclein ameliorates the pathology in a mouse model of Parkinson's disease. Neurobiol Dis. (2021) 148:105218. doi: 10.1016/j.nbd.2020.105218
181. Kimura S, Harashima H. Current Status and Challenges Associated with CNS-Targeted Gene Delivery across the BBB. Pharmaceutics. (2020) 12:1216. doi: 10.3390/pharmaceutics12121216
182. Kakoty V, K CS, Dubey SK, Yang CH, Kesharwani P, Taliyan R. Lentiviral mediated gene delivery as an effective therapeutic approach for Parkinson disease. Neurosci Lett. (2021) 750:135769. doi: 10.1016/j.neulet.2021.135769
183. Zu H, Gao D. Non-viral vectors in gene therapy: recent development, challenges, and prospects. AAPS J. (2021) 23:78. doi: 10.1208/s12248-021-00608-7
184. Soldner F, Laganiere J, Cheng AW, Hockemeyer D, Gao Q, Alagappan R, et al. Generation of isogenic pluripotent stem cells differing exclusively at two early onset Parkinson point mutations. Cell. (2011) 146:318–31. doi: 10.1016/j.cell.2011.06.019
185. Kantor B, Tagliafierro L, Gu J, Zamora ME, Ilich E, Grenier C, et al. Downregulation of SNCA expression by targeted editing of DNA methylation: a potential strategy for precision therapy in PD. Mol Ther. (2018) 26:2638–49. doi: 10.1016/j.ymthe.2018.08.019
186. Duan Y, Ye T, Qu Z, Chen Y, Miranda A, Zhou X, et al. Brain-wide Cas9-mediated cleavage of a gene causing familial Alzheimer's disease alleviates amyloid-related pathologies in mice. Nat Biomed Eng. (2021) 6:168–180. doi: 10.1038/s41551-021-00759-0
187. Pardridge WM. Blood-brain barrier delivery. Drug Discov Today. (2007) 12:54–61. doi: 10.1016/j.drudis.2006.10.013
188. Pardridge WM. Blood-brain barrier and delivery of protein and gene therapeutics to brain. Front Aging Neurosci. (2019) 11:373. doi: 10.3389/fnagi.2019.00373
189. Chan KY, Jang MJ, Yoo BB, Greenbaum A, Ravi N, Wu WL, et al. Engineered AAVs for efficient noninvasive gene delivery to the central and peripheral nervous systems. Nat Neurosci. (2017) 20:1172–9. doi: 10.1038/nn.4593
190. Reynaud-Dulaurier R, Decressac M. PHPB/eB vectors bring new successes to gene therapy for brain diseases. Front Bioeng Biotechnol. (2020) 8:582979. doi: 10.3389/fbioe.2020.582979
191. Goertsen D, Flytzanis NC, Goeden N, Chuapoco MR, Cummins A, Chen Y, et al. AAV capsid variants with brain-wide transgene expression and decreased liver targeting after intravenous delivery in mouse and marmoset. Nat Neurosci. (2021) 25:106–15. doi: 10.1038/s41593-021-00969-4
192. Finneran DJ, Njoku IP, Flores-Pazarin D, Ranabothu MR, Nash KR, Morgan D, et al. Toward development of neuron specific transduction after systemic delivery of viral vectors. Front Neurol. (2021) 12:685802. doi: 10.3389/fneur.2021.685802
193. Huang Q, Chan KY, Tobey IG, Chan YA, Poterba T, Boutros CL, et al. Delivering genes across the blood-brain barrier: LY6A, a novel cellular receptor for AAV-PHPB capsids. PloS ONE. (2019) 14:e0225206. doi: 10.1371/journal.pone.0225206
194. Hayashita-Kinoh H, Yamada M, Yokota T, Mizuno Y, Mochizuki H. Down-regulation of alpha-synuclein expression can rescue dopaminergic cells from cell death in the substantia nigra of Parkinson's disease rat model. Biochem Biophys Res Commun. (2006) 341:1088–95. doi: 10.1016/j.bbrc.2006.01.057
195. Javed H, Menon SA, Al-Mansoori KM, Al-Wandi A, Majbour NK, Ardah MT, et al. Development of nonviral vectors targeting the brain as a therapeutic approach for parkinson's disease and other brain disorders. Mol Ther. (2016) 24:746–58. doi: 10.1038/mt.2015.232
196. Spencer B, Trinh I, Rockenstein E, Mante M, Florio J, Adame A, et al. Systemic peptide mediated delivery of an siRNA targeting alpha-syn in the CNSameliorates the neurodegenerative process in a transgenic model of Lewy body disease. Neurobiol Dis. (2019) 127:163–177. doi: 10.1016/j.nbd.2019.03.001
197. Hu K, Chen X, Chen W, Zhang L, Li J, Ye J, et al. Neuroprotective effect of gold nanoparticles composites in Parkinson's disease model. Nanomedicine. (2018) 14:1123–36. doi: 10.1016/j.nano.2018.01.020
198. Helmschrodt C, Hobel S, Schoniger S, Bauer A, Bonicelli J, Gringmuth M, et al. Polyethylenimine nanoparticle-mediated sirna delivery to reduce alpha-synuclein expression in a model of parkinson's disease. Mol Ther Nucleic Acids. (2017) 9:57–68. doi: 10.1016/j.omtn.2017.08.013
199. Cooper JM, Wiklander PB, Nordin JZ, Al-Shawi R, Wood MJ, Vithlani M, et al. Systemic exosomal siRNA delivery reduced alpha-synuclein aggregates in brains of transgenic mice. Mov Disord. (2014) 29:1476–85. doi: 10.1002/mds.25978
200. Izco M, Blesa J, Schleef M, Schmeer M, Porcari R, Al-Shawi R, et al. Systemic exosomal delivery of shrna minicircles prevents parkinsonian pathology. Mol Ther. (2019) 27:2111–22. doi: 10.1016/j.ymthe.2019.08.010
201. Alarcon-Aris D, Recasens A, Galofre M, Carballo-Carbajal I, Zacchi N, Ruiz-Bronchal E, et al. Selective alpha-synuclein knockdown in monoamine neurons by intranasal oligonucleotide delivery: Potential therapy for parkinson's disease. Mol Ther. (2018) 26:550–67. doi: 10.1016/j.ymthe.2017.11.015
202. Muyldermans S. Nanobodies: natural single-domain antibodies. Annu Rev Biochem. (2013) 82:775–97. doi: 10.1146/annurev-biochem-063011-092449
203. Abulrob A, Sprong H, Van Bergen en Henegouwen P, Stanimirovic D. The blood-brain barrier transmigrating single domain antibody: mechanisms of transport and antigenic epitopes in human brain endothelial cells. J Neurochem. (2005) 95:1201–14. doi: 10.1111/j.1471-4159.2005.03463.x
204. Rutgers KS, Nabuurs RJ, van den Berg SA, Schenk GJ, Verrips CT, van Duinen SG, et al. Transmigration of beta amyloid specific heavy chain antibody fragments across the in vitro blood-brain barrier. Neuroscience. (2011) 190:37–42. doi: 10.1016/j.neuroscience.2011.05.076
205. Li T, Vandesquille M, Koukouli F, Dudeffant C, Youssef I, Lenormand P, et al. Camelid single-domain antibodies: A versatile tool for in vivo imaging of extracellular and intracellular brain targets. J Control Release. (2016) 243:1–10. doi: 10.1016/j.jconrel.2016.09.019
206. Messer A, Butler DC. Optimizing intracellular antibodies (intrabodies/nanobodies) to treat neurodegenerative disorders. Neurobiol Dis. (2020) 134:104619. doi: 10.1016/j.nbd.2019.104619
207. Messer A, Joshi SN. Intrabodies as neuroprotective therapeutics. Neurotherapeutics. (2013) 10:447–58. doi: 10.1007/s13311-013-0193-6
208. De Genst E, Messer A, Dobson CM. Antibodies and protein misfolding: From structural research tools to therapeutic strategies. Biochim Biophys Acta. (2014) 1844:1907–19. doi: 10.1016/j.bbapap.2014.08.016
209. Gupta V, Salim S, Hmila I, Vaikath NN, Sudhakaran IP, Ghanem SS, et al. Fibrillar form of alpha-synuclein-specific scFv antibody inhibits alpha-synuclein seeds induced aggregation and toxicity. Sci Rep. (2020) 10:8137. doi: 10.1038/s41598-020-65035-8
210. Guilliams T, El-Turk F, Buell AK, O'Day EM, Aprile FA, Esbjorner EK, et al. De Genst, Nanobodies raised against monomeric alpha-synuclein distinguish between fibrils at different maturation stages. J Mol Biol. (2013) 425:2397–411. doi: 10.1016/j.jmb.2013.01.040
211. Iljina M, Hong L, Horrocks MH, Ludtmann MH, Choi ML, Hughes CD, et al. Nanobodies raised against monomeric a-synuclein inhibit fibril formation and destabilize toxic oligomeric species. BMC Biol. (2017) 15:57. doi: 10.1186/s12915-017-0390-6
212. Chatterjee D, Bhatt M, Butler D, De Genst E, Dobson CM, Messer A, et al. Proteasome-targeted nanobodies alleviate pathology and functional decline in an alpha-synuclein-based Parkinson's disease model. NPJ Parkinsons Dis. (2018) 4:25. doi: 10.1038/s41531-018-0062-4
213. Butler DC, Joshi SN, Genst E, Baghel AS, Dobson CM, Messer A. Bifunctional anti-non-amyloid component alpha-synuclein nanobodies are protective in situ. PLoS ONE. (2016) 11:e0165964. doi: 10.1371/journal.pone.0165964
214. Joshi SN, Butler DC, Messer A. Fusion to a highly charged proteasomal retargeting sequence increases soluble cytoplasmic expression and efficacy of diverse anti-synuclein intrabodies. MAbs. (2012) 4:686–93. doi: 10.4161/mabs.21696
215. Mittal S, Bjornevik K, Im DS, Flierl A, Dong X, Locascio JJ, et al. beta2-Adrenoreceptor is a regulator of the alpha-synuclein gene driving risk of Parkinson's disease. Science. (2017) 357:891–8. doi: 10.1126/science.aaf3934
216. Qian L, Wu HM, Chen SH, Zhang D, Ali SF, Peterson L, et al. beta2-adrenergic receptor activation prevents rodent dopaminergic neurotoxicity by inhibiting microglia via a novel signaling pathway. J Immunol. (2011) 186:4443–54. doi: 10.4049/jimmunol.1002449
217. Wrasidlo W, Tsigelny IF, Price DL, Dutta G, Rockenstein E, Schwarz TC, et al. A de novo compound targeting alpha-synuclein improves deficits in models of Parkinson's disease. Brain. (2016) 139:3217–36. doi: 10.1093/brain/aww238
218. Price DL, Koike MA, Khan A, Wrasidlo W, Rockenstein E, Masliah E, et al. The small molecule alpha-synuclein misfolding inhibitor, NPT200-11, produces multiple benefits in an animal model of Parkinson's disease. Sci Rep. (2018) 8:16165. doi: 10.1038/s41598-018-34490-9
219. Wagner J, Ryazanov S, Leonov A, Levin J, Shi S, Schmidt F, et al. Anle138b: a novel oligomer modulator for disease-modifying therapy of neurodegenerative diseases such as prion and Parkinson's disease. Acta Neuropathol. (2013) 125:795–813. doi: 10.1007/s00401-013-1114-9
220. Wegrzynowicz M, Bar-On Bar-On D, Calo L, Anichtchik O, Iovino M, Xia J, et al. Depopulation of dense alpha-synuclein aggregates is associated with rescue of dopamine neuron dysfunction and death in a new Parkinson's disease model. Acta Neuropathol. (2019) 138:575–95. doi: 10.1007/s00401-019-02023-x
221. Heras-Garvin A, Weckbecker D, Ryazanov S, Leonov A, Griesinger C, Giese A, et al. Anle138b modulates alpha-synuclein oligomerization and prevents motor decline and neurodegeneration in a mouse model of multiple system atrophy. Mov Disord. (2019) 34:255–63. doi: 10.1002/mds.27562
222. Perni M, Galvagnion C, Maltsev A, Meisl G, Muller MB, Challa PK, et al. A natural product inhibits the initiation of alpha-synuclein aggregation and suppresses its toxicity. Proc Natl Acad Sci U S A. (2017) 114:E1009–17. doi: 10.1073/pnas.1610586114
223. Perni M, Flagmeier P, Limbocker R, Cascella R, Aprile FA, Galvagnion C, et al. Multistep Inhibition of alpha-Synuclein Aggregation and Toxicity in Vitro and in Vivo by Trodusquemine. ACS Chem Biol. (2018) 13:2308–19. doi: 10.1021/acschembio.8b00466
224. Prabhudesai S, Sinha S, Attar A, Kotagiri A, Fitzmaurice AG, Lakshmanan R, et al. A novel “molecular tweezer” inhibitor of alpha-synuclein neurotoxicity in vitro and in vivo. Neurotherapeutics. (2012) 9:464–76. doi: 10.1007/s13311-012-0105-1
225. Richter F, Subramaniam SR, Magen I, Lee P, Hayes J, Attar A, et al. A molecular tweezer ameliorates motor deficits in mice overexpressing alpha-synuclein. Neurotherapeutics. (2017) 14:1107–19. doi: 10.1007/s13311-017-0544-9
226. Bengoa-Vergniory N, Faggiani E, Ramos-Gonzalez P, Kirkiz E, Connor-Robson N, Brown LV, et al. CLR01 protects dopaminergic neurons in vitro and in mouse models of Parkinson's disease. Nat Commun. (2020) 11:4885. doi: 10.1038/s41467-020-18689-x
227. Ehrnhoefer DE, Bieschke J, Boeddrich A, Herbst M, Masino L, Lurz R, et al. EGCG redirects amyloidogenic polypeptides into unstructured, off-pathway oligomers. Nat Struct Mol Biol. (2008) 15:558–66. doi: 10.1038/nsmb.1437
228. Xu Q, Langley M, Kanthasamy AG, Reddy MB. Epigallocatechin gallate has a neurorescue effect in a mouse model of Parkinson disease. J Nutr. (2017) 147:1926–31. doi: 10.3945/jn.117.255034
229. Chen M, Wang T, Yue F, Li X, Wang P, Li Y, et al. Tea polyphenols alleviate motor impairments, dopaminergic neuronal injury, and cerebral alpha-synuclein aggregation in MPTP-intoxicated parkinsonian monkeys. Neuroscience. (2015) 286:383–92. doi: 10.1016/j.neuroscience.2014.12.003
230. Zhou W, Bercury K, Cummiskey J, Luong N, Lebin J, Freed CR. Phenylbutyrate up-regulates the DJ-1 protein and protects neurons in cell culture and in animal models of Parkinson disease. J Biol Chem. (2011) 286:14941–51. doi: 10.1074/jbc.M110.211029
231. Roy A, Ghosh A, Jana A, Liu X, Brahmachari S, Gendelman HE, et al. Sodium phenylbutyrate controls neuroinflammatory and antioxidant activities and protects dopaminergic neurons in mouse models of Parkinson's disease. PLoS ONE. (2012) 7:e38113. doi: 10.1371/journal.pone.0038113
232. Pujols J, Pena-Diaz S, Lazaro DF, Peccati F, Pinheiro F, Gonzalez D, et al. Small molecule inhibits alpha-synuclein aggregation, disrupts amyloid fibrils, and prevents degeneration of dopaminergic neurons. Proc Natl Acad Sci U S A. (2018) 115:10481–6. doi: 10.1073/pnas.1804198115
233. Staats R, Michaels TCT, Flagmeier P, Chia S, Horne RI, Habchi J, et al. Screening of small molecules using the inhibition of oligomer formation in α-synuclein aggregation as a selection parameter. Communications Chemistry. (2020) 3:191. doi: 10.1038/s42004-020-00412-y
234. Myohanen TT, Hannula MJ, Van Elzen R, Gerard M, Van Der Veken P, Garcia-Horsman JA, et al. A prolyl oligopeptidase inhibitor, KYP-2047, reduces alpha-synuclein protein levels and aggregates in cellular and animal models of Parkinson's disease. Br J Pharmacol. (2012) 166:1097–113. doi: 10.1111/j.1476-5381.2012.01846.x
235. Savolainen MH, Richie CT, Harvey BK, Mannisto PT, Maguire-Zeiss KA, Myohanen TT. The beneficial effect of a prolyl oligopeptidase inhibitor, KYP-2047, on alpha-synuclein clearance and autophagy in A30P transgenic mouse. Neurobiol Dis. (2014) 68:1–15. doi: 10.1016/j.nbd.2014.04.003
236. Rostami J, Jantti M, Cui H, Rinne MK, Kukkonen JP, Falk A, et al. Prolyl oligopeptidase inhibition by KYP-2407 increases alpha-synuclein fibril degradation in neuron-like cells. Biomed Pharmacother. (2020) 131:110788. doi: 10.1016/j.biopha.2020.110788
237. Kwon S, Iba M, Masliah E, Kim C. Targeting microglial and neuronal toll-like receptor 2 in synucleinopathies. Exp Neurobiol. (2019) 28:547–53. doi: 10.5607/en.2019.28.5.547
238. Finkelstein DI, Shukla JJ, Cherny RA, Billings JL, Saleh E, Stefanova N, et al. The compound ATH434 prevents alpha-synuclein toxicity in a murine model of multiple system atrophy. J Parkinsons Dis. (2022) 12:105–115. doi: 10.3233/JPD-212877
239. Zhang P, Park HJ, Zhang J, Junn E, Andrews RJ, Velagapudi SP, et al. Translation of the intrinsically disordered protein alpha-synuclein is inhibited by a small molecule targeting its structured mRNA. Proc Natl Acad Sci U S A. (2020) 117:1457–67. doi: 10.1073/pnas.1905057117
240. Gronich N, Abernethy DR, Auriel E, Lavi I, Rennert G, Saliba W. beta2-adrenoceptor agonists and antagonists and risk of Parkinson's disease. Mov Disord. (2018) 33:1465–71. doi: 10.1002/mds.108
241. Searles Nielsen S, Gross A, Camacho-Soto A, Willis AW, Racette BA. Beta2-adrenoreceptor medications and risk of Parkinson disease. Ann Neurol. (2018) 84:683–93. doi: 10.1002/ana.25341
242. Cosentino M, Marino F. Adrenergic and dopaminergic modulation of immunity in multiple sclerosis: teaching old drugs new tricks? J Neuroimmune Pharmacol. (2013) 8:163–79. doi: 10.1007/s11481-012-9410-z
243. Kinali M, Mercuri E, Main M, De Biasia F, Karatza A, Higgins R, et al. Pilot trial of albuterol in spinal muscular atrophy. Neurology. (2002) 59:609–10. doi: 10.1212/WNL.59.4.609
244. Hishida R, Kurahashi K, Narita S, Baba T, Matsunaga M. “Wearing-off” and beta 2-adrenoceptor agonist in Parkinson's disease. Lancet. (1992) 339:870. doi: 10.1016/0140-6736(92)90313-R
245. Alexander GM, Schwartzman RJ, Nukes TA, Grothusen JR, Hooker MD. Beta 2-adrenergic agonist as adjunct therapy to levodopa in Parkinson's disease. Neurology. (1994) 44:1511–3. doi: 10.1212/WNL.44.8.1511
246. Uc EY, Lambert CP, Harik SI, Rodnitzky RL, Evans WJ. Albuterol improves response to levodopa and increases skeletal muscle mass in patients with fluctuating Parkinson disease. Clin Neuropharmacol. (2003) 26:207–12. doi: 10.1097/00002826-200307000-00011
247. Levin J, Maass S, Schuberth M, Giese A, Oertel WH, Poewe W, et al. Safety and efficacy of epigallocatechin gallate in multiple system atrophy (PROMESA): a randomised, double-blind, placebo-controlled trial. Lancet Neurol. (2019) 18:724–35. doi: 10.1016/S1474-4422(19)30141-3
248. Javed H, Nagoor Meeran MF, Azimullah S, Adem A, Sadek B, Ojha SK. Plant extracts and phytochemicals targeting alpha-synuclein aggregation in Parkinson's disease models. Front Pharmacol. (2018) 9:1555. doi: 10.3389/fphar.2018.01555
249. Fink AL. The aggregation and fibrillation of alpha-synuclein. Acc Chem Res. (2006) 39:628–34. doi: 10.1021/ar050073t
250. Johnston TH, Lacoste AMB, Visanji NP, Lang AE, Fox SH, Brotchie JM. Repurposing drugs to treat l-DOPA-induced dyskinesia in Parkinson's disease. Neuropharmacology. (2019) 147:11–27. doi: 10.1016/j.neuropharm.2018.05.035
251. Braun MM, Farag-El-Massah S, Xu K, Cote TR. Emergence of orphan drugs in the United States: a quantitative assessment of the first 25 years. Nat Rev Drug Discov. (2010) 9:519–22. doi: 10.1038/nrd3160
252. Medicine IO. Drug Repurposing and Repositioning: Workshop Summary. Washington, DC: The National Academies Press (2014).
253. McFarthing K, Buff S, Rafaloff G, Dominey T, Wyse RK, Stott SRW. Parkinson's disease drug therapies in the clinical trial pipeline: 2020. J Parkinsons Dis. (2020) 10:757–74. doi: 10.3233/JPD-202128
254. Shaltiel-Karyo R, Frenkel-Pinter M, Rockenstein E, Patrick C, Levy-Sakin M, Schiller A, et al. A blood-brain barrier (BBB) disrupter is also a potent alpha-synuclein (alpha-syn) aggregation inhibitor: a novel dual mechanism of mannitol for the treatment of Parkinson disease (PD). J Biol Chem. (2013) 288:17579–88. doi: 10.1074/jbc.M112.434787
255. Bakkar N, Kovalik T, Lorenzini I, Spangler S, Lacoste A, Sponaugle K, et al. Artificial intelligence in neurodegenerative disease research: use of IBM Watson to identify additional RNA-binding proteins altered in amyotrophic lateral sclerosis. Acta Neuropathol. (2018) 135:227–47. doi: 10.1007/s00401-017-1785-8
256. Maclagan LC, Visanji NP, Cheng Y, Tadrous M, Lacoste AMB, Kalia LV, et al. Identifying drugs with disease-modifying potential in Parkinson's disease using artificial intelligence and pharmacoepidemiology. Pharmacoepidemiol Drug Saf. (2020) 29:864–72. doi: 10.1002/pds.5015
257. Visanji NP, Madan P, Lacoste AMB, Buleje I, Han Y, Spangler S, et al. Using artificial intelligence to identify anti-hypertensives as possible disease modifying agents in Parkinson's disease. Pharmacoepidemiol Drug Saf. (2021) 30:201–9. doi: 10.1002/pds.5176
258. Chen KS, Menezes K, Rodgers JB, O'Hara DM, Tran N, Fujisawa K, et al. Small molecule inhibitors of alpha-synuclein oligomers identified by targeting early dopamine-mediated motor impairment in C. elegans. Mol Neurodegener. (2021) 16:77. doi: 10.1186/s13024-021-00497-6
259. Meng Y, Hynynen K, Lipsman N. Applications of focused ultrasound in the brain: from thermoablation to drug delivery. Nat Rev Neurol. (2021) 17:7–22. doi: 10.1038/s41582-020-00418-z
260. Bakay L, Ballantine HT Jr, Hueter TF, Sosa D. Ultrasonically produced changes in the blood-brain barrier. AMA Arch Neurol Psychiatry. (1956) 76:457–67. doi: 10.1001/archneurpsyc.1956.02330290001001
261. Hynynen K, McDannold N, Vykhodtseva N, Jolesz FA. Noninvasive MR imaging-guided focal opening of the blood-brain barrier in rabbits. Radiology. (2001) 220:640–6. doi: 10.1148/radiol.2202001804
262. McMahon D, O'Reilly MA, Hynynen K. Therapeutic Agent Delivery Across the Blood-Brain Barrier Using Focused Ultrasound. Annu Rev Biomed Eng. (2021) 23:89–113. doi: 10.1146/annurev-bioeng-062117-121238
263. Thevenot E, Jordao JF, O'Reilly MA, Markham K, Ying-Qi L, Foust K, et al. Targeted delivery of scAAV9 to the brain using MRI-guided focused ultrasound. Hum Gene Ther. (2012) 11:1144–55. doi: 10.1089/hum.2012.013
264. Xhima K, Markham-Coultes K, Kofoed RH, Saragovi HU, Hynynen K, Aubert I. Ultrasound delivery of a TrkA agonist confers neuroprotection to Alzheimer-associated pathologies. Brain. (2021). doi: 10.1093/brain/awab460
265. Abrahao A, Meng Y, Llinas M, Huang Y, Hamani C, Mainprize T, et al. First-in-human trial of blood-brain barrier opening in amyotrophic lateral sclerosis using MR-guided focused ultrasound. Nat Commun. (2019) 10:4373. doi: 10.1038/s41467-019-12426-9
266. Lipsman N, Meng Y, Bethune AJ, Huang Y, Lam B, Masellis M, et al. Blood-brain barrier opening in Alzheimer's disease using MR-guided focused ultrasound. Nat Commun. (2018) 9:2336. doi: 10.1038/s41467-018-04529-6
267. Rezai AR, Ranjan M, D'Haese PF, Haut MW, Carpenter J, Najib U, et al. Noninvasive hippocampal blood-brain barrier opening in Alzheimer's disease with focused ultrasound. Proc Natl Acad Sci U S A. (2020) 117:9180–2. doi: 10.1073/pnas.2002571117
268. Gasca-Salas C, Fernandez-Rodriguez B, Pineda-Pardo JA, Rodriguez-Rojas R, Obeso I, Hernandez-Fernandez F, et al. Blood-brain barrier opening with focused ultrasound in Parkinson's disease dementia. Nat Commun. (2021) 12:779. doi: 10.1038/s41467-021-21022-9
269. Park SH, Baik K, Jeon S, Chang WS, Ye BS, Chang JW. Extensive frontal focused ultrasound mediated blood-brain barrier opening for the treatment of Alzheimer's disease: a proof-of-concept study. Transl Neurodegener. (2021) 10:44. doi: 10.1186/s40035-021-00269-8
270. Fan CH, Ting CY, Lin CY, Chan HL, Chang YC, Chen YY, et al. Targeted, and Non-Viral Ultrasound-Mediated GDNF-Plasmid Delivery for Treatment of Parkinson's Disease. Sci Rep. (2016) 6:19579. doi: 10.1038/srep19579
271. Lin CY, Hsieh HY, Chen CM, Wu SR, Tsai CH, Huang CY, et al. Non-invasive, neuron-specific gene therapy by focused ultrasound-induced blood-brain barrier opening in Parkinson's disease mouse model. J Control Release. (2016) 235:72–81. doi: 10.1016/j.jconrel.2016.05.052
272. Chen H, Yang GZ, Getachew H, Acosta C, Sierra Sanchez C, Konofagou EE. Focused ultrasound-enhanced intranasal brain delivery of brain-derived neurotrophic factor. Sci Rep. (2016) 6:28599. doi: 10.1038/srep28599
273. Ji R, Smith M, Niimi Y, Karakatsani ME, Murillo MF, Jackson-Lewis V, et al. Focused ultrasound enhanced intranasal delivery of brain derived neurotrophic factor produces neurorestorative effects in a Parkinson's disease mouse model. Sci Rep. (2019) 9:19402. doi: 10.1038/s41598-019-55294-5
274. Jordao JF, Thevenot E, Markham-Coultes K, Scarcelli T, Weng YQ, Xhima K, et al. Amyloid-beta plaque reduction, endogenous antibody delivery and glial activation by brain-targeted, transcranial focused ultrasound. Exp Neurol. (2013) 248:16–29. doi: 10.1016/j.expneurol.2013.05.008
275. Gelders G, Baekelandt V, Van der Perren A. Linking neuroinflammation and neurodegeneration in Parkinson's disease. J Immunol Res. (2018) 2018:4784268. doi: 10.1155/2018/4784268
276. Tomac A, Lindqvist E, Lin LF, Ogren SO, Young D, Hoffer BJ, et al. Protection and repair of the nigrostriatal dopaminergic system by GDNF in vivo. Nature. (1995) 373:335–9. doi: 10.1038/373335a0
277. Xhima K, Markham-Coultes K, Nedev H, Heinen S, Saragovi HU, Hynynen K, et al. Focused ultrasound delivery of a selective TrkA agonist rescues cholinergic function in a mouse model of Alzheimer's disease. Sci Adv. (2020) 6:eaax6646. doi: 10.1126/sciadv.aax6646
278. Zlokovic BV. The blood-brain barrier in health and chronic neurodegenerative disorders. Neuron. (2008) 57:178–201. doi: 10.1016/j.neuron.2008.01.003
279. Groveman BR, Orru CD, Hughson AG, Raymond LD, Zanusso G, Ghetti B, et al. Rapid and ultra-sensitive quantitation of disease-associated alpha-synuclein seeds in brain and cerebrospinal fluid by alphaSyn RT-QuIC. Acta Neuropathol Commun. (2018) 6:7. doi: 10.1186/s40478-018-0508-2
280. Fenyi A, Leclair-Visonneau L, Clairembault T, Coron E, Neunlist M, Melki R, et al. Detection of alpha-synuclein aggregates in gastrointestinal biopsies by protein misfolding cyclic amplification. Neurobiol Dis. (2019) 129:38–43. doi: 10.1016/j.nbd.2019.05.002
281. Saijo E, Groveman BR, Kraus A, Metrick M, Orru CD, Hughson AG, et al. Ultrasensitive RT-QuIC seed amplification assays for disease-associated tau, alpha-synuclein, prion aggregates. Methods Mol Biol. (2019) 1873:19–37. doi: 10.1007/978-1-4939-8820-4_2
282. Shahnawaz M, Mukherjee A, Pritzkow S, Mendez N, Rabadia P, Liu X, et al. Discriminating alpha-synuclein strains in Parkinson's disease and multiple system atrophy. Nature. (2020) 578:273–7. doi: 10.1038/s41586-020-1984-7
283. Wang Z, Becker K, Donadio V, Siedlak S, Yuan J, Rezaee M, et al. Skin alpha-synuclein aggregation seeding activity as a novel biomarker for Parkinson disease. JAMA Neurol. (2020) 78:30–40. doi: 10.1001/jamaneurol.2020.3311
284. Manne S, Kondru N, Jin H, Serrano GE, Anantharam V, Kanthasamy A, et al. Blinded RT-QuIC analysis of alpha-synuclein biomarker in skin tissue from Parkinson's disease patients. Mov Disord. (2020) 35:2230–9. doi: 10.1002/mds.28242
Keywords: Parkinson's disease, prion, gene therapy, anti-aggregation, brain delivery of drugs, immunization
Citation: Menon S, Armstrong S, Hamzeh A, Visanji NP, Sardi SP and Tandon A (2022) Alpha-Synuclein Targeting Therapeutics for Parkinson's Disease and Related Synucleinopathies. Front. Neurol. 13:852003. doi: 10.3389/fneur.2022.852003
Received: 10 January 2022; Accepted: 01 April 2022;
Published: 09 May 2022.
Edited by:
Lisa Fellner, University of Innsbruck, AustriaReviewed by:
Kelvin C. Luk, University of Pennsylvania, United StatesGaia Faustini, University of Brescia, Italy
Copyright © 2022 Menon, Armstrong, Hamzeh, Visanji, Sardi and Tandon. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Anurag Tandon, YS50YW5kb25AdXRvcm9udG8uY2E=