 Yuchao Chen
Yuchao Chen Xiaodong Lu1
Xiaodong Lu1- 1Department of Neurology, The Affiliated Hospital of Hangzhou Normal University, Hangzhou, China
- 2Translational Medicine Center, The Affiliated Hospital of Hangzhou Normal University, Hangzhou, China
- 3Department of Neurology and Research Center of Neurology in Second Affiliated Hospital, Zhejiang University School of Medicine, Hangzhou, China
Mutations in the SACS gene have been linked to autosomal recessive spastic ataxia of Charlevoix Saguenay (ARSACS). It is a clinically and genetically heterogeneous disease characterized by slow progressive ataxia, spasticity, sensorimotor neuropathy, and a combination of other manifestations, such as lack of spasticity, hearing loss, and epileptic seizures. Currently, there have been very few case reports regarding the SACS gene mutation in Chinese patients. Here, we describe a 35-year-old Chinese patient carrying a novel variant in SACS (c.11486C>T) presenting with progressive ataxia and demyelinating peripheral neuropathy. We then reviewed 22 Chinese cases carrying SACS gene mutations, including our patient. All of them had a cerebellar ataxia gait and showed cerebellar atrophy on brain magnetic resonance imaging (MRI). A total of 28 SACS mutations were identified in these patients. Our study further expands the mutation spectrum of the SACS gene and contributes to the evaluation of genotype-phenotype correlations.
Introduction
Autosomal recessive spastic ataxia of Charlevoix-Saguenay (ARSACS) is one of the most common autosomal recessive ataxia caused by biallelic mutations within the SACS (OMIM: 270550) gene (1). The majority of patients with ARSACS present three core typical phenotypes of early-onset cerebellar ataxia, spasticity, peripheral neuropathy, and other atypical manifestations, including cognition disability, lacking spasticity, epileptic seizures, and hearing loss (2, 3). Brain magnetic resonance imaging (MRI) often revealed remarkable findings of cerebellum atrophy and linear T2 hypointensities in the pons. The optical coherence tomography (OCT) presented a remarkable abnormality in the retinal nerve fiber layer (RNFL) hypertrophy. However, in clinical practice, the absence of remarkable finds in brain MRI or retinal OCT were also present in some ARSACS cases (4, 5).
Genetically, over 200 mutations have been described in the SACS gene, most of which have been detected in the gigantic exon 10. The majority of the mutation's types were missense mutation and small deletions subsequently. The identical same mutation leading to different clinical features were described, even in siblings (6). These findings suggested that ARSACS is a clinically and genetically heterogeneous disease and it usually confuses us to make a precise diagnosis. Here, we describe the case of a Chinese patient carrying a novel variant in SACS presented with progressive ataxia and demyelinating peripheral neuropathy.
Case Presentation
The patient is a 35-year-old male from a consanguineous family (Figure 1A). He had delayed developmental motor milestones and began ambulating at 36 months of age. Frequent falls, particularly during running, notably occurred during childhood. He developed a progressive ataxic gait and dysarthria at the age of 28 years. However, with the progression, he needed a mobility aid to protect himself when walking and suffered from dysphagia at the age of 35 years. He did not have a history of seizures, constipation, urinary urgency, or visual problems. His parents did not have any symptoms, but two of his uncles had similar symptoms. The young uncle showed gait problems as a child. These symptoms gradually progressed and resulted in him using a wheelchair at the age of 38. The older uncle died at the age of 59 with similar symptoms. Neurologic examination of cranial nerves revealed significant gaze-evoked nystagmus and moderate dysarthria. Limb examination presented muscular atrophy in lower limbs, and the muscle strength of the distal part of the lower extremities was Medical Research Council (MRC) grade 4. There was decreased tone and tendon reflex in the upper and lower limbs. The sensation examination was symmetric, but the pain sense seemed to be more insensitive in the distal limbs. Extensor plantar reflexes were positive bilaterally. Hammer toes and pes cavus were present (Figures 2A,B). Bilateral finger-to-nose tests, alternate motion tests, and heel-to-shin tests were all awkward. Romberg's sign was positive. The score of the Scale for the Assessment and Rating of Ataxia (SARA) and the International Cooperative Ataxia Rating Scale (ICARS) were 25/40 and 23/100, respectively. The total score of the disease-specific severity index for autosomal recessive spastic ataxia of Charlevoix-Saguenay (DSI-ARSACS) was 23.5, and the clinical Spastic Paraplegia Rating Scale (SPRS) was 22.
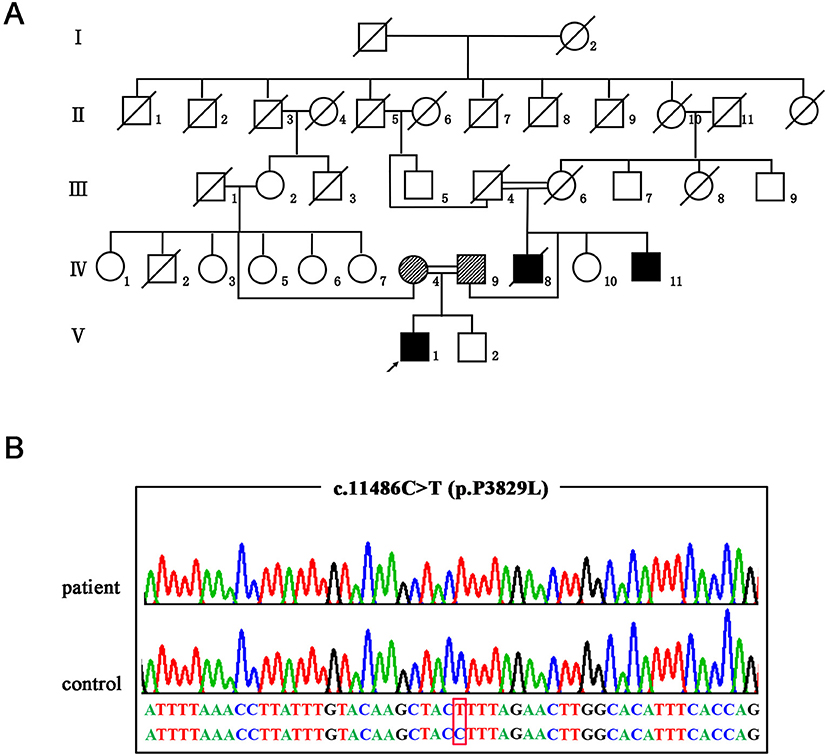
Figure 1. The genetic manifestations of the patient. (A) Pedigree of the family. The arrow indicates the proband. (B) Validation of the SACS mutation (NM_014363: c.11486C>T p.P3829L) by Sanger sequencing.

Figure 2. The clinical and neuroimaging features of the patient. (A,B) Neurological examinations show Hammer toes and pes cavus. (C) Thinning spinal cord in sagittal T2-weighted image (arrow). (D,G) The brain MRI shows the cerebellum atrophy in the sagittal T1-weighted image (arrow), hypointensities in the pons in the axial T2-weighted image (arrow). (E,H) Fundus photographs of the right and left eyes show swollen papilla with a combed aspect of the interpapillomacular region. (F,I) Optical coherence tomography imaging of the right and left eyes shows thickened RNFL (arrow).
Nerve conduction studies (NCS) showed sensorimotor demyelinating polyneuropathy with secondary axonal loss (Supplementary Table 1). A spine MRI revealed thinning spinal cord (Figure 2C). A brain MRI revealed atrophy of the cerebellum on T1-weighted images, and bilateral hypointense stripes in the pons on T2 sequences (Figures 2D,G). Fundus photographs of the eyes demonstrated swollen papilla with a combed aspect of the interpapillomacular region (Figures 2E,H), and OCT depicted hypertrophy in the mild retinal nerve fiber layer(RNFL) (Figures 2F,I).
After genetic counseling, the patient and his parents gave informed consent and the Ethical Committee of the Affiliated Hospital of Hangzhou Normal University in China gave approval. First, we screened causative genes for SCA1, 2, 3, 6, 7, 8, 10, 12, 17, Friedreich's ataxia (FRDA), and Dentatorubral-pallidoluysian atrophy (DRPLA) on the proband. In doing so, we did not identify any pathogenic repeat expansions. We then carried out whole-exome sequencing (WES) and detected a novel homozygous missense variant in the SACS gene (NM_014363.5: c.11486C>T p.P3829L). Afterward, segregation analysis by Sanger sequencing confirmed that the patient's parents were heterozygous carriers and his affected young uncle was also homozygote (Figure 1B). No other known pathogenic variants were identified in the WES study. The variant of c.11486C>T in the SACS gene was absent in databases of dbSNP, gnomAD, and ExAC. SIFT, Polyphen-2, Mutationtaster, and CADD all predicted that the novel missense variant was deleterious. According to the American College of Medical Genetics and Genomics (ACMG), the variant c.11486C>T within SACS is a variant of likely pathogenic (PM2, PP1_Moderate, PP3, PP4).
We summarized the clinical and genetic features of our case and the reported Chinese patients (Table 1) (2–4, 7–17). The majority of the families were from Southeastern China (Figure 3). Among them, 16 were men and 11 were women. The age of disease onset in the cases (77.7%, 21/27) was no more than 6 years. All of the 27 cases had onset with ataxia, and 15 of them had spastic gait, whereas three cases showed an absence of spasticity, 16 cases showed pes cavus, and 21 patients presented peripheral neuropathy. Almost all of them revealed cerebellar atrophy on a brain MRI, except for one case, due to his young age (3 years). In 15 patients, the MRI displayed signal hypointensities within the pons, and eight cases showed thickening RNFL. Genetically, 12 of 27 cases carried homozygous mutations in the SACS gene. A total of 35 SACS gene mutations were identified in the Chinese patients, including 10 missenses, seven non-senses, 16 small deletions, and two gross deletions. Except for two gross deletions, only one mutation was located in exon 8, while almost all mutations were identified in exon 10 of the SACS gene.

Table 1. Clinical data of Chinese autosomal recessive spastic ataxia of Charlevoix Saguenay (ARSACS) cases from the present study and other related published studies.
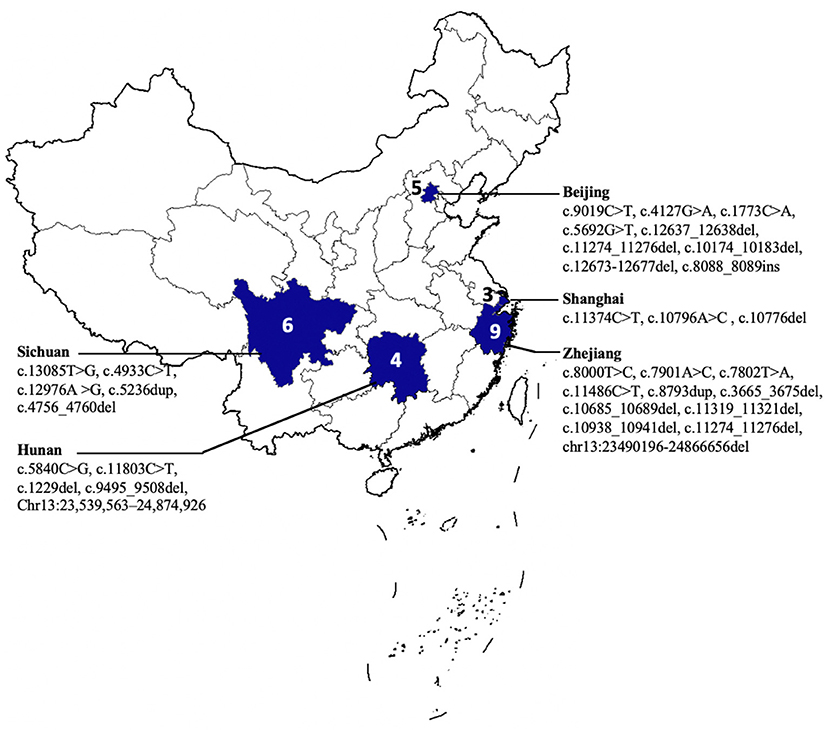
Figure 3. The geographical distribution of patients with autosomal recessive spastic ataxia of Charlevoix Saguenay (ARSACS).
Discussion
Currently, there have been very few case reports regarding SACS gene mutation in Chinese patients. In this study, we presented a Chinese ARSACS patient exhibiting early-onset cerebellar ataxia, pyramidal signs, and peripheral neuropathy. By performing a WES and segregation analysis, we identified a novel variant c.11486C>T mutation at the homozygous state in the SACS gene.
Different from the clinical features of progressive spasticity and preserving tendon reflexes throughout the disease course in Quebec patients (1, 18), our index case lacked the signs of leg spasticity and decreased tendon reflexes. Additionally, a few ARSACS cases without spasticity in the lower limbs were also observed in different racial groups (19, 20). This finding is in line with other previous studies of patients with ARSACS. There is a possibility that the presence of severe neuropathy with demyelinating features could mask any spasticity. However, there was one ARSACS case in the previous reports, which presented neither spasticity nor neuropathy (6).
The lacking-spasticity phenotype may be associated with the localization of the SACS mutation. In our patient, this new variant (c.11486C>T p.P3829L) was located downstream of the UBE3A binding domain (UBD) in the C-terminal of the sacsin protein. The UBD domain may interact with the ubiquitin ligase Ube3A, acting as an important role in hereditary spastic paraplegia (HSP) (21, 22). The patients carrying SACS mutations in the UBD domain usually showed leg spasticity and obvious features of ataxia (21). However, another two patients harboring the homozygous variant (c.11542_11544del) located downstream of the UBD domain presented with ataxia without spasticity (6). Together, the SACS variants located downstream may not affect the UBD domain's function. Additional functional studies are needed in order to confirm the role of those domains at the C-terminus in the SACS gene in protein.
Conclusion
Collectively, we reported a Chinese ARSACS case carrying a novel variant in SACS. Our study further expands the mutation spectrum of SACS and contributes to the evaluation of genotype-phenotype correlations.
Data Availability Statement
The datasets presented in this article are not readily available due to ethical and privacy restrictions. Requests to access the datasets should be directed to the corresponding author.
Ethics Statement
The studies involving human participants were reviewed and approved by the Ethical Committee of the Affiliated Hospital of Hangzhou Normal University. The patients/participants provided their written informed consent to participate in this study. Written informed consent was obtained from the individual(s) for the publication of any potentially identifiable images or data included in this article.
Author Contributions
YC, XL, YJ, DL, XY, CT, MZ, HJ, and HY initiated the project and collected and analyzed the data. YC wrote the manuscript. HY commented on and revised the manuscript and supervised all aspects of the project. All the authors read and approved the final manuscript.
Funding
This study was supported by the Medical and Health Science and Technology Project of Zhejiang Province to YC (2021KY898).
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher's Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Acknowledgments
We gratefully acknowledge all participants for their help and willingness to participate in this study.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fneur.2022.845318/full#supplementary-material
References
1. Bouchard JP, Barbeau A, Bouchard R, Bouchard RW. Autosomal recessive spastic ataxia of Charlevoix-Saguenay. Can J Neurol Sci. (1978) 5:61–9. doi: 10.1017/S0317167100024793
2. Zhang Q, Li H, Chen C, Luan Z, Xu X, Tang S. [Analysis of SACS mutation in a family affected with autosomal recessive spastic ataxia of Charlevoix-Saguenay]. Zhonghua Yi Xue Yi Chuan Xue Za Zhi. (2019) 36:217–20. doi: 10.3760/cma.j.issn.1003-9406.2019.03.006
3. Cheng HL, Shao YR, Dong Y, Dong HL, Yang L, Ma Y, et al. Genetic spectrum and clinical features in a cohort of Chinese patients with autosomal recessive cerebellar ataxias. Transl Neurodegener. (2021) 10:40. doi: 10.1186/s40035-021-00264-z
4. Sun W, Meng Y, Zhuo Y, Wang Z, Yuan Y. Novel spastic ataxia of Charlevoix-Saguenay gene compound heterozygous mutations in late onset autosomal recessive spastic ataxia of Charlevoix-Saguenay. Chin J Neurol. (2017) 50:831–6. doi: 10.3760/cma.j.issn.1006-7876.2017.11.007
5. Xiromerisiou G, Dadouli K, Marogianni C, Provatas A, Ntellas P, Rikos D, et al. A novel homozygous SACS mutation identified by whole exome sequencing-genotype phenotype correlations of all published cases. J Mol Neurosci. (2020) 70:131–41. doi: 10.1007/s12031-019-01410-z
6. Synofzik M, Soehn AS, Gburek-Augustat J, Schicks J, Karle KN, Schüle R, et al. Autosomal recessive spastic ataxia of Charlevoix Saguenay (ARSACS): expanding the genetic, clinical and imaging spectrum. Orphanet J Rare Dis. (2013) 8:41. doi: 10.1186/1750-1172-8-41
7. Chen Z, Wang JL, Tang BS, Sun ZF, Shi YT, Shen L, et al. Using next-generation sequencing as a genetic diagnostic tool in rare autosomal recessive neurologic Mendelian disorders. Neurobiol Aging. (2013) 34:2442.e11–7. doi: 10.1016/j.neurobiolaging.2013.04.029
8. Liu L, Li XB, Zi XH, Shen L, Hu ZhM, Huang ShX, et al. A novel hemizygous SACS mutation identified by whole exome sequencing and SNP array analysis in a Chinese ARSACS patient. J Neurol Sci. (2016) 362:111–4. doi: 10.1016/j.jns.2016.01.026
9. Zeng H, Tang JG, Yang YF, Tan ZP, Tan JQ. A novel homozygous SACS mutation identified by whole-exome sequencing in a consanguineous family with autosomal recessive spastic ataxia of Charlevoix-Saguenay. Cytogenet. Genome Res. (2017) 152:16–21. doi: 10.1159/000477428
10. Li S, Chen Y, Yuan X, Wei Q, Ou R, Gu X, et al. [Identification of compound heterozygous mutations of SACS gene in two patients from a pedigree with spastic ataxia of Charlevoix-Saguenay]. Zhonghua Yi Xue Yi Chuan Xue Za Zhi. (2018) 35:507–10. doi: 10.3760/cma.j.issn.1003-9406.2018.04.010
11. Guan RY, Sun YM, Sun GY, Wang J, Wu JJ. One Chinese autosomal recessive spastic ataxia of charlevoix-saguenay pedigree identified by whole exome sequencing: clinical features and literature review. (2019) 027:644–51.
12. Lu Q, Shang L, Tian WT, Cao L, Zhang X, Liu Q. Complicated paroxysmal kinesigenic dyskinesia associated with mutations. Ann Transl Med. (2020) 8:8. doi: 10.21037/atm.2019.11.31
13. Jiao B, Zhou Z, Hu Z, Du J, Liao X, Luo Y, et al. Homozygosity mapping and next generation sequencing for the genetic diagnosis of hereditary ataxia and spastic paraplegia in consanguineous families. Parkinsonism Relat Disord. (2020) 80:65–72. doi: 10.1016/j.parkreldis.2020.09.013
14. Wang Z, Song Y, Wang X, Li X, Xu F, Si L, et al. Autosomal recessive spastic ataxia of Charlevoix-Saguenay caused by novel mutations in SACS gene: a report of two Chinese families. Neurosci Lett. (2021) 752:135831. doi: 10.1016/j.neulet.2021.135831
15. Chen Y, Cen Z, Zheng X, Chen S, Xie F, Luo W. Novel compound heterozygous SACS mutations in a case with a spasticity-lacking phenotype of sacsin-related ataxia. Neurol India. (2021) 69:219–21. doi: 10.4103/0028-3886.310115
16. Chen X, Yuan X, Wei QQ, Ou R, Cao B, Hou Y, et al. Identifying gene mutations in autosomal recessive cerebellar ataxias and extending the mutational spectrum in China. Res Sq. (2021). doi: 10.21203/rs.3.rs-577033/v1
17. Zhou Y, Yang W, Liu X, Dong S, Yu H, Wang C, et al. Novel compound heterozygous SACS mutations in a case with a autosomal recessive spastic ataxia of Charlevoix–Saguenay. Chinese J Nervous Ment Dis. (2021) 47:160–5. doi: 10.3969/j.issn.1002-0152.2021.03.007
18. El Euch-Fayache G, Lalani I, Amouri R, Turki I, Ouahchi K, Hung WY, et al. Phenotypic features and genetic findings in sacsin-related autosomal recessive ataxia in Tunisia. Arch Neurol. (2003) 60:982-8. doi: 10.1001/archneur.60.7.982
19. Shimazaki H, Takiyama Y, Sakoe K, Ando Y, Nakano I. A phenotype without spasticity in sacsin-related ataxia. Neurology. (2005) 64:2129-31. doi: 10.1212/01.WNL.0000166031.91514.B3
20. Shimazaki H, Sakoe K, Niijima K, Nakano I, Takiyama Y. An unusual case of a spasticity-lacking phenotype with a novel SACS mutation. J Neurol Sci. (2007) 255:87-9. doi: 10.1016/j.jns.2007.02.002
21. Gregianin E, Vazza G, Scaramel E, Boaretto F, Vettori A, Leonardi E, et al. A novel SACS mutation results in non-ataxic spastic paraplegia and peripheral neuropathy. Eur J Neurol. (2013) 20:1486-91. doi: 10.1111/ene.12220
Keywords: autosomal recessive spastic ataxia of Charlevoix-Saguenay, whole-exome sequencing, novel variant, SACS, spastic
Citation: Chen Y, Lu X, Jin Y, Li D, Ye X, Tao C, Zhou M, Jiang H and Yu H (2022) A Novel SACS Variant Identified in a Chinese Patient: Case Report and Review of the Literature. Front. Neurol. 13:845318. doi: 10.3389/fneur.2022.845318
Received: 29 December 2021; Accepted: 22 February 2022;
Published: 21 March 2022.
Edited by:
Matthew James Farrer, University of Florida, United StatesReviewed by:
Filippo M. Santorelli, Stella Maris Foundation (IRCCS), ItalyMalco Rossi, Fundación Para la Lucha Contra las Enfermedades Neurológicas de la Infancia (FLENI), Argentina
Copyright © 2022 Chen, Lu, Jin, Li, Ye, Tao, Zhou, Jiang and Yu. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Hao Yu, aGFveXV6anVAemp1LmVkdS5jbg==