 Alec Aeby
Alec Aeby Berten Ceulemans
Berten Ceulemans Lieven Lagae
Lieven Lagae- 1Pediatric Neurology, Queen Fabiola Children's University Hospital, Université Libre de Bruxelles (ULB), Brussels, Belgium
- 2Department of Pediatric Neurology, Antwerp University Hospital, University of Antwerp, Antwerp, Belgium
- 3Reference Center for Refractory Epilepsy, Pediatric Neurology, Department of Development and Regeneration, University Hospitals Leuven, Leuven, Belgium
To accelerate the process of licensing antiseizure medication (ASM) in children, extrapolation of efficacy data for focal-onset seizures from adults to children ≥2 or ≥4 years of age is now accepted. We summarized the efficacy evidence from randomized, controlled trials that was used to grant approval for the pediatric indication of focal-onset seizures for the different ASMs available in Europe. Data from high-quality randomized, controlled trials in young children are limited, especially on the use of ASMs in monotherapy. Licensure trials are typically focused on seizure type irrespective of etiology or epilepsy syndrome. We elaborate on the importance of etiology- or syndrome-driven research and treatment, illustrating this with examples of childhood epilepsy syndromes characterized by predominantly focal-onset seizures. Some of these syndromes respond well to standard ASMs used for focal-onset seizures, but others would benefit from a more etiology- or syndrome-driven approach. Advances in molecular genetics and neuroimaging have made it possible to reveal the underlying cause of a child's epilepsy and tailor research and treatment. More high-quality randomized, controlled trials based on etiology or syndrome type are needed, including those assessing effects on cognition and behavior. In addition, study designs such as “N-of-1 trials” could elucidate possible new treatment options in rare epilepsies. Broadening incentives currently in place to stimulate the development and marketing of drugs for rare diseases (applicable to some epilepsy syndromes) to more common pediatric epilepsy types and syndromes might be a means to enable high-quality trials, and ultimately allow more evidence-based treatment in children.
Introduction
Epilepsy, a chronic neurological disorder characterized by the recurrence of unprovoked seizures, affects nearly 50 million people worldwide (1). The epilepsy prevalence is highest in young children and older adults (1–3) and was estimated to be 6.2/1,000 people globally in 2016 (1). Epileptic seizures can initiate in one region of one hemisphere of the brain—focal-onset seizures—or in both hemispheres—generalized-onset seizures (4). Epilepsy types and syndromes can be characterized by either exclusively focal or generalized seizures or by a combination of both (5).
Causes of epilepsy include genetic mutations, infections, metabolic disorders, immune disorders and structural abnormalities (5). Determining the etiology of epilepsy may allow optimizing treatment; for instance, certain focal brain malformations can be amenable to curative surgery, and the identification of a genetic mutation may enable effective targeted treatment (3, 6).
The diagnosis and management of epilepsy in children pose unique challenges. Misdiagnosis is common, as paroxysmal events are often confused with epilepsy in children (7), and, in neonates, diagnosis is often impossible without continuous electroencephalography monitoring (8, 9). Seizures in infancy and childhood can have detrimental effects on behavior and cognition, and early, effective treatment is therefore essential (3, 6, 10, 11). However, antiseizure medication (ASM) can itself negatively affect neurodevelopment (6, 11, 12). Benefits and side effects should therefore be carefully weighed when choosing ASMs for children.
Because of the methodological and ethical challenges associated with randomized, placebo-controlled trials in children, ASMs are typically first tested and licensed in adults, and it may take years before a new drug is approved in children and infants (6, 13–16). Off-label ASM use in children is therefore common (17–20). Recognizing that this poses risks to treated children and may further complicate the conduct of placebo-controlled trials and delay pediatric drug approval, regulators now accept extrapolation of efficacy data for focal-onset seizures from adults to children ≥2 or ≥4 years old because the pathophysiology of focal-onset seizures and their responsiveness to ASMs are considered largely comparable in both populations (13, 15, 21, 22). However, as the etiology of epilepsy in adults differs from that in children, this extrapolation might be too simplistic, and some experts advocate etiology-driven treatment decisions. Tolerability and safety cannot be extrapolated and should be evaluated in prospective studies that may be single-arm and open-label (13, 15, 21, 22). Effects on behavior and cognition are particularly important to assess, but data are still limited (6, 12). Furthermore, dosing regimens cannot be extrapolated to children (particularly newborns and young infants) because pharmacokinetic properties of drugs (absorption, drug distribution, metabolism, and excretion) evolve with age due to physiological and anatomical changes (23).
In this review, we summarize the efficacy evidence that was used to approve the pediatric indication (patients <18 years) of focal-onset seizures for the ASMs available in Europe and highlight challenges related to the extrapolation of adult data to children. We also elaborate on the importance of etiology- and syndrome-driven research and treatment decisions, using examples of childhood epilepsy syndromes characterized by predominantly focal-onset seizures.
Pharmacological Treatment of Focal-Onset Seizures in Children and Adolescents
ASMs for Focal-Onset Seizures in Children and Adolescents
In Europe, 17 ASMs are indicated for the adjunctive treatment of focal-onset seizures in children and/or adolescents (Table 1, Figure 1). Of these, 11 are also approved as monotherapy. Indications differ in terms of the lower age limit at which treatment can be given, with only the first-generation ASMs (carbamazepine, phenobarbital, phenytoin, primidone, and valproate), vigabatrin and levetiracetam indicated to treat focal-onset seizures in infants (<1 year; the last two only as adjunctive treatment). For second-generation ASMs (gabapentin, lamotrigine, levetiracetam, oxcarbazepine, tiagabine, topiramate, vigabatrin, and zonisamide), licensure in children or adolescents was based on randomized, controlled trials in the pediatric population (Table 1) (66–75). The four most recent drug approvals for focal-onset seizures in children <12 years old (i.e., for the third-generation ASMs eslicarbazepine acetate, lacosamide, brivaracetam, and perampanel) were granted by the European Medicines Agency (EMA) based on extrapolation of adult or adolescent efficacy data (Table 1) (76–79).
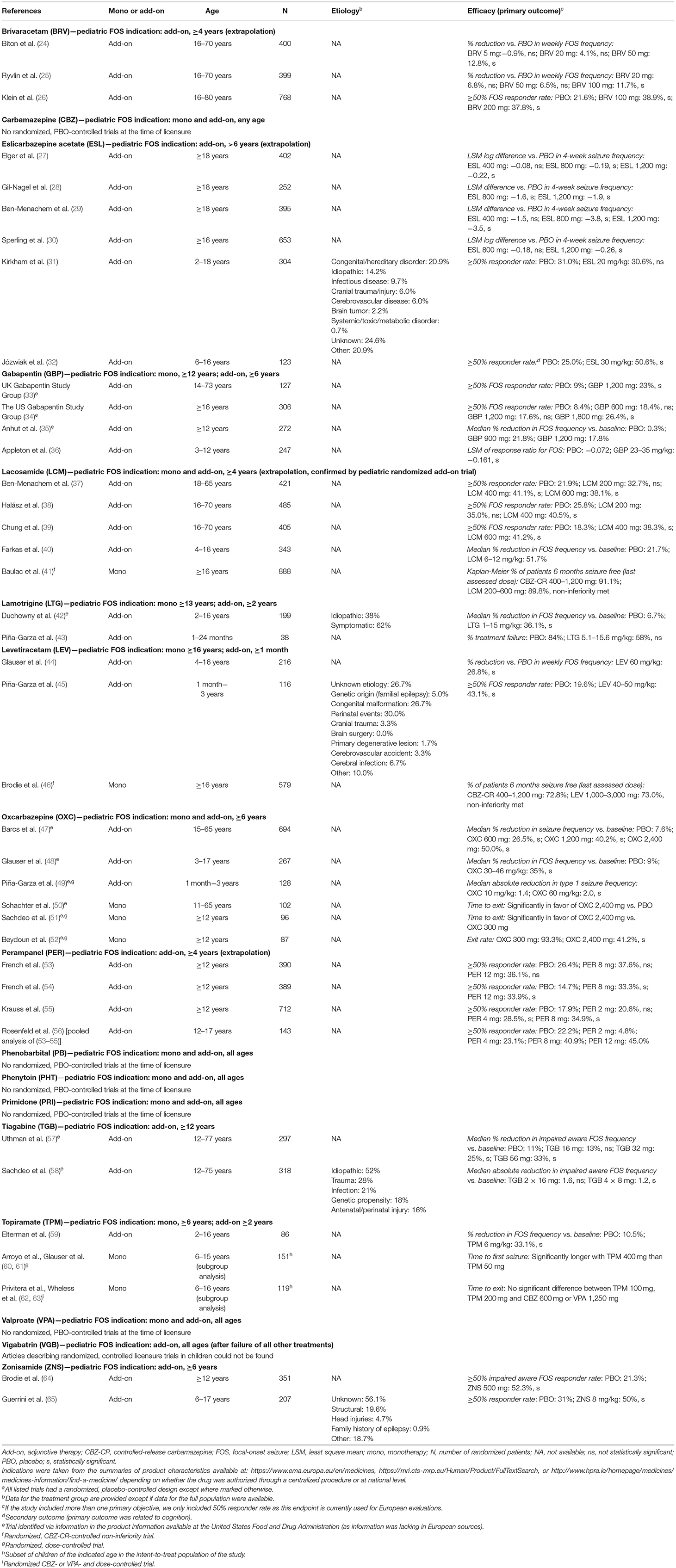
Table 1. Antiseizure medication with a pediatric indication for focal-onset seizures in Europe and summary of efficacy evidence from randomized, placebo-controlled trialsa used as basis for these indications.

Figure 1. Antiseizure medication approved in Europe for focal-onset seizures in children and adolescents. “Any age” includes neonatal use. Mono, approved for monotherapy; add-on, approved for adjunctive therapy; m, months of age; y, years of age. This figure only pertains to focal-onset seizures in children; some of these drugs are also indicated for the treatment of other seizure types and/or of focal-onset seizures in adults. Indications were taken from the summaries of product characteristics available at: https://www.ema.europa.eu/en/medicines, https://mri.cts-mrp.eu/Human/Product/FullTextSearch, or http://www.hpra.ie/homepage/medicines/medicines-information/find-a-medicine/ depending on whether the drug was authorized through a centralized procedure or at national level. In addition to the listed drugs, a small number of drugs have been approved in Europe for use in specific childhood epilepsy syndromes accompanied by focal-onset seizures (e.g., sulthiame for childhood epilepsy with centrotemporal spikes, everolimus for tuberous sclerosis complex, cannabidiol for tuberous sclerosis complex and Dravet syndrome, fenfluramine for Dravet syndrome).
Efficacy Evidence Used for Approval of the Pediatric Indication of ASMs for Focal-Onset Seizures
We aimed to summarize the efficacy data from randomized, controlled trials used to grant approval for the pediatric indication of focal-onset seizures for the ASMs available in Europe. To identify the pivotal studies used for licensure, we consulted the product information made available by the EMA or European national authorities, or—if insufficient information was available through these sources—by the United States Food and Drug Administration. We subsequently used the ClinicalTrials.gov register and PubMed to specifically search for articles that disclosed the results of these studies.
Table 1 summarizes study characteristics and primary efficacy outcomes. Supplementary Table 1 provides additional details on the study design, inclusion/exclusion criteria and other outcomes. Data from pivotal trials in adults are included if the pediatric indication was granted based on extrapolation from adult data.
Consistent with previous literature reviews on the pharmacotherapy of focal-onset seizures and other seizure types in children (80–82), our assessment shows that, among licensure trials, the number of high-quality, randomized, controlled trials performed in children <6 years old is limited, and efficacy data in infants and children <2 or <3 years are nearly absent (Table 1). Many studies included heterogeneous populations, comprising children (or adolescents) as well as adults, and did not analyze efficacy outcomes by age subgroup. Almost no randomized, controlled monotherapy trials have been performed in children with new-onset or untreated focal-onset seizures; instead, most trials assessed adjunctive therapy in patients who did not respond to one or multiple ASMs, and efficacy estimates in these trials were often low (as expected in refractory patients; Table 1, Supplementary Table 1). Moreover, only very few studies provided information on the etiology of epilepsy among enrolled patients, but none showed data segregated by etiology (and would have lacked power to conclude on such subgroup analyses). Likewise, these trials did not investigate specific childhood syndromes.
Safety Considerations of ASMs in Children and Adolescents
While efficacy data on the pharmacological treatment of focal-onset seizures can be extrapolated from adults to children ≥2 or ≥4 years old, safety and tolerability should be assessed in pediatric clinical trials, with an adequate representation of all age ranges (13, 15, 21, 22). The pediatric safety and tolerability profiles of ASMs have been reviewed extensively (12, 83–87). The most common adverse events associated with ASM use in children and adolescents are similar to those seen in adults and include somnolence, drowsiness, gastrointestinal disturbances, anorexia, irritability, and nervousness (83–85, 88). When choosing the optimal ASM for a patient, certain adverse events may weigh in more at different ages. For instance, weight gain, cosmetic side effects, and depression may be more relevant for older children and adolescents, as are side effects that influence the ability to drive a car (in older adolescents) or teratogenicity (in girls of childbearing potential). Of particular importance to younger children are changes in neurocognitive function (e.g., memory, intelligence, attention, language, and visuomotor coordination) and behavioral effects (e.g., hyperactivity, irritability, agitation, and aggression) (12, 86, 87). However, data from high-quality trials on the effects of ASMs on cognition and behavior are sparse, with conflicting results for some ASMs (12, 86, 87). Heterogeneity across studies in terms of trial design, dose titration, duration of follow-up and methods used to assess cognitive and behavioral outcomes makes it hard to assess and compare the impact of different ASMs (86). A report from the International League Against Epilepsy (ILAE) provided some general recommendations for ASM use in children based on behavioral and cognitive complications (87). Because of the risk of cognitive or behavioral adverse effects, the ILAE recommends careful monitoring of cognition in children receiving phenobarbital, phenytoin, topiramate and zonisamide, and of behavior in children treated with phenobarbital, valproate, gabapentin, topiramate, levetiracetam, and zonisamide. Conversely, the available evidence suggests limited positive effects of lamotrigine on cognition and behavior, and of levetiracetam on cognition (87).
From Seizure Type-Based to Etiology/Syndrome-Based Research and Treatment
As we concluded based on our overview of pivotal, randomized, controlled trials used for pediatric licensure, most studies evaluate the efficacy of ASMs against seizure types, irrespective of etiology or epilepsy syndrome (Table 1). However, owing to advances in molecular genetics and neuroimaging, there has been a gradual shift from “one-size-fits-all” treatment based on seizure type to more etiology- or syndrome-driven treatment (3, 89–91). An early etiological diagnosis is important, as prompt treatment with adequate drugs (or surgery) may improve the outcome of the child with epilepsy (89, 92–94). In pediatric epilepsy, there are notable examples illustrating the power of etiology- or syndrome-driven research. For example, cannabidiol and fenfluramine were rigorously tested and subsequently approved in Dravet syndrome (95–99), a well-defined epilepsy syndrome with a uniform genetic background. Such research was facilitated as both drugs had received orphan drug designation, which allows sponsors to benefit from incentives established to encourage the development and marketing of drugs to treat rare diseases (100).
In the following sections, we discuss examples of epilepsy syndromes with structural and genetic etiologies characterized by predominantly focal-onset seizures in children. These examples illustrate how etiology- or syndrome-driven research and treatment has been applied or could be considered.
Structural Etiologies
Structural etiologies may have a genetic basis, as is the case for various cortical malformations, or may be acquired, such as hypoxic-ischemic lesions, brain lesions after accidental or non-accidental trauma, infection, stroke or bleeding (5). In clinical practice, some children with structural epilepsies achieve good long-term seizure control with ASMs. In children with drug-resistant structural epilepsies, surgery is highly effective and should be considered as first-choice treatment in eligible patients (101). However, not all children with structural epilepsies are eligible for surgery, for instance, because of the extent of the epileptogenic zone or the vicinity of the lesion to the eloquent cortex. For structural epilepsies, to date, no dedicated drug trials have been performed. An exception is a study in patients with tuberous sclerosis complex (TSC) (102), a genetic-structural epilepsy with brain tubers causing focal-onset seizures. TSC is caused by mutations in the TSC1 or TSC2 gene, which result in overactivation of the mammalian target of rapamycin (mTOR) complex (103). A randomized, controlled trial showed that the mTOR inhibitor everolimus significantly decreased the number of focal-onset seizures in TSC (102, 104). This trial is a good example of etiology-driven research with precision medicine. Everolimus not only affects seizure frequency but also reduces the volume of brain and renal tumors seen in TSC, improves skin lesions and preserves renal function in patients with TSC (105–108).
Genetic Etiologies
The recognized non-structural focal childhood epilepsy syndromes are mainly genetic or presumed genetic. In most of these epilepsies, treatment is still empiric, purely symptomatic and not always based on results from randomized, controlled trials. However, many of these epilepsies are self-limiting and can be successfully treated, with complete seizure control and the possibility to stop medication after a few years of seizure freedom on medication (109).
In infancy, familial and non-familial epilepsies can be diagnosed (110). The most common genetic etiology for these epilepsies is a mutation in the PRRT2 gene (proline-rich transmembrane protein 2) (111). Mutations in other genes, e.g., SCN2A (sodium voltage-gated channel alpha subunit 2), KCNQ2 and KCNQ3 (potassium voltage-gated channel subfamily Q members 2 and 3) have also been identified (110). Seizures are typically focal, with (subtle) behavioral arrest, impaired awareness, automatism and eye/head turning (110), and are usually easily controlled with standard ASMs for focal-onset seizures (112). Although large, prospective trials are lacking, there is increasing evidence that classic sodium channel blockers (SCBs, e.g., carbamazepine) are highly efficacious in potassium channel epilepsies (e.g., due to KCNQ2 mutations) (112). In SCN2A epilepsies, effectiveness of SCBs depends on the mutation type: gain-of-function mutations, typically presenting early in life (<3 months), respond well to SCBs, while loss-of-function mutations, which often have a later onset, are better controlled with other ASMs and may worsen on SCBs (113–115). In SCN8A-related epilepsies, levetiracetam may also be associated with clinical worsening or regression (116). These are typical examples of precision medicine: the precise genetic background of the epilepsy predicts which drugs can be beneficial.
In contrast with these self-limiting syndromes, epilepsy of infancy with migrating focal seizures is usually a drug-resistant epilepsy, within the spectrum of epileptic encephalopathies. Focal seizures start independently in both hemispheres and can “migrate” from one cortical region to another in the same or the other hemisphere, with changing seizure semiology (110). While the cause is often unknown, different gene mutations have been linked with this syndrome. Gain-of-function mutations in KCNT1 (potassium sodium-activated channel subfamily T member 1) are among the more frequent etiologies (117), and quinidine, a partial KCNT1 antagonist, was tested as precision treatment in this syndrome. Case reports showed seizure reduction in some patients, but this effect was not consistent in all patients (118–122). No randomized, controlled trials have been performed to study the effect of quinidine in KCNT1-mutant epilepsy of infancy with migrating focal seizures, and a single randomized, controlled trial in six patients with a different childhood-onset epilepsy syndrome (severe autosomal dominant nocturnal frontal lobe epilepsy) due to KCNT1 mutations did not demonstrate efficacy (123).
The self-limiting childhood epilepsy syndromes, Panayiotopoulos syndrome and childhood occipital epilepsy (or Gastaut type) are presumed polygenic, but no associated genes have been identified thus far (110). Most patients respond favorably to common ASMs for focal-onset seizures, but no systematic treatment studies have been performed for these epilepsies. By contrast, treatment options for childhood epilepsy with centrotemporal spikes (CECTS), also known as Rolandic epilepsy, one of the best known and most common childhood focal epilepsy syndromes, have been explored in numerous clinical trials (124, 125). CECTS is self-limiting, with an onset between 3 and 14 years of age, characterized by brief, hemifacial seizures that may evolve to focal-to-bilateral tonic-clonic seizures if they occur at night. Seizures resolve during adolescence (110). A few randomized, controlled trials have been performed to assess ASMs in children with CECTS, none of which provided high-quality evidence (81, 124–126). A 2013 evidence review by the ILAE considered carbamazepine and valproate as possibly effective, and gabapentin, levetiracetam, oxcarbazepine and sulthiame as potentially effective monotherapy for CECTS (81). Some low-quality studies have reported that carbamazepine may induce negative myoclonus, atypical absences, drop attacks and electrical status epilepticus during sleep (127). A recent systematic review of seizure freedom rates in patients with CECTS suggested sulthiame, levetiracetam, or clobazam as first-line ASMs for CECTS (125), but highlighted—as in an earlier Cochrane review (124)—that there is insufficient evidence about the optimal ASM for children with CECTS (125). Two randomized, placebo-controlled trials showed that sulthiame (128) or gabapentin (126, 129) monotherapy were effective in preventing seizures in children with CECTS. The other available randomized trials assessing seizure control compared different ASMs [levetiracetam with oxcarbazepine (130, 131), clobazam with carbamazepine (132), topiramate with carbamazepine (133), and levetiracetam with sulthiame (134)]. Most did not show significant differences in seizure control between ASMs, except for one study showing a higher effectiveness of oxcarbazepine vs. levetiracetam (131). A few trials compared the effects of different ASMs on cognition: topiramate vs. carbamazepine (133), clobazam vs. carbamazepine (132), sulthiame vs. levetiracetam (135), and levetiracetam vs. oxcarbazepine (131). Only the levetiracetam-vs.-oxcarbazepine trial found a significant difference between groups, with oxcarbazepine having a superior effect on cognition (131). Low-quality evidence suggests that some children with CECTS do not need treatment with ASM (126). Some experts only consider treatment favorable in case of multiple seizures, early onset, the presence of language dysfunction, cognitive and/or neuropsychological disorder or focal-to-bilateral tonic-clonic seizures (127). Other experts consider that the decision to treat depends on the intensity of interictal epileptiform discharges and their possible negative impact on behavior and cognition (127). Considering the low seizure burden in these self-limiting childhood epilepsy syndromes, high-quality trials are needed to provide evidence on the frequency and type of seizures (i.e., focal aware vs. focal-to-bilateral tonic-clonic) for which ASM treatment should be considered, especially with respect to cognitive outcomes.
Discussion
With regulators now accepting extrapolation of efficacy data for focal-onset seizures from adults to children ≥2 or ≥4 years of age, the path to licensure of ASMs for this indication in the pediatric population has been simplified, and access to available ASMs can be accelerated. However, the sparsity of high-quality randomized, controlled trials in children, particularly in the very young, and the lack of etiology- or syndrome-specific data in such trials complicates the selection of the optimal ASM. In addition, high-quality data on the effects of ASMs on cognition and behavior are lacking, further complicating treatment decisions. While some childhood epilepsy syndromes respond well to most or several ASMs used for focal-onset seizures, a more etiology- or syndrome-driven approach is needed for others. Improvements in molecular genetics and imaging in recent years have made it possible to determine the etiology of epilepsy in an increasing number of cases, and thereby tailor research and treatment to the patient's needs. For epilepsy syndromes that qualify as rare diseases and drugs that received an orphan designation, various incentives have enabled high-quality randomized, controlled trials. Similar incentives to support research and drug development for more common epilepsy types and syndromes in children might be beneficial and ultimately enable more evidence-based treatment decisions. In addition, long-term follow-up of efficacy and safety in children enrolled in ongoing trials should be stimulated by international collaborations and registries. Study designs such as “N-of-1 trials” (double-blind, randomized crossover trials in single patients, in which the order of experimental and control treatments are randomly allocated to the patient) could provide high-level evidence of approved ASMs and reveal possible new treatment options in rare epilepsies (136). Finally, personalized medicine, or precision medicine, which combines multiple factors (e.g., genetics, age, gender, environment, and lifestyle of individuals) to identify the best way to prevent or treat disease via pharmacological and non-pharmacological treatments, may become more common in the future. Different forms of intervention may be embraced, from choosing the most suitable drug according to the type of epilepsy or expected adverse effects to gene therapy. An “ideal” epilepsy therapy would stop seizures and undo the changes caused by specific genetic mutations (137, 138).
Author Contributions
AA and LL: conceptualization, investigation, writing—original draft, and writing—review and editing. BC: conceptualization, investigation, and writing—review and editing. All authors contributed to the article and approved the submitted version.
Funding
This work received funding from Eisai Co., Ltd.
Conflict of Interest
AA declares having received fees from Eisai and Zogenix for participating in advisory boards and support from UCB and Zogenix for attending meetings. BC declares that Antwerp University Hospital may benefit financially from a royalty arrangement that is related to this research if Zogenix is successful in marketing its product, fenfluramine and also declares having received fees from Eisai, Brabant Pharma, Novartis, UCB, and Zogenix for participating in advisory boards. LL declares having received consultancy fees and payment or honoraria for lectures, presentations, speakers bureaus, manuscript writing, or educational events from Novartis, Zogenix, Eisai, LivaNova, UCB, and Epihunter, and in addition having a patent for the use of fenfluramine with potential royalties paid through the University of Leuven.
The authors declare that this work received funding from Eisai Co., Ltd. The funder had the following involvement with the work: payment for medical writing services and publishing costs.
Publisher's Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Acknowledgments
The authors thank Natalie Denef of Modis Life Sciences, Wavre, Belgium for providing medical writing support, which was funded by Eisai in accordance with Good Publication Practice (GPP3).
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fneur.2022.842276/full#supplementary-material
Abbreviations
ASM, antiseizure medication; CECTS, childhood epilepsy with centrotemporal spikes; EMA, European Medicines Agency; ILAE, International League Against Epilepsy; KCNQ2/KCNQ3, potassium voltage-gated channel subfamily Q members 2 and 3; KCNT1, potassium sodium-activated channel subfamily T member 1; mTOR, mammalian target of rapamycin; PRRT2, proline-rich transmembrane protein 2; SCB, sodium channel blocker; SCN2A, sodium voltage-gated channel alpha subunit 2; TSC, tuberous sclerosis complex.
References
1. GBD 2016 Epilepsy Collaborators. Global, regional, and national burden of epilepsy, 1990–2016: a systematic analysis for the Global Burden of Disease Study 2016. Lancet Neurol. (2019) 18:357–75. doi: 10.1016/S1474-4422(18)30454-X
2. Camfield P, Camfield C. Incidence, prevalence and aetiology of seizures and epilepsy in children. Epileptic Disord. (2015) 17:117–23. doi: 10.1684/epd.2015.0736
3. Wilmshurst JM, Berg AT, Lagae L, Newton CR, Cross JH. The challenges and innovations for therapy in children with epilepsy. Nat Rev Neurol. (2014) 10:249–60. doi: 10.1038/nrneurol.2014.58
4. Fisher RS. The new classification of seizures by the International League against Epilepsy 2017. Curr Neurol Neurosci Rep. (2017) 17:48. doi: 10.1007/s11910-017-0758-6
5. Scheffer IE, Berkovic S, Capovilla G, Connolly MB, French J, Guilhoto L, et al. ILAE classification of the epilepsies: position paper of the ILAE Commission for Classification and Terminology. Epilepsia. (2017) 58:512–21. doi: 10.1111/epi.13709
6. Pressler RM, Lagae L. Why we urgently need improved seizure and epilepsy therapies for children and neonates. Neuropharmacology. (2020) 170:107854. doi: 10.1016/j.neuropharm.2019.107854
7. Leibetseder A, Eisermann M, LaFrance WC Jr, Nobili L, von Oertzen TJ. How to distinguish seizures from non-epileptic manifestations. Epileptic Disord. (2020) 22:716–38. doi: 10.1684/epd.2020.1234
8. Shellhaas RA, Chang T, Tsuchida T, Scher MS, Riviello JJ, Abend NS, et al. The American Clinical Neurophysiology Society's guideline on continuous electroencephalography monitoring in neonates. J Clin Neurophysiol. (2011) 28:611–7. doi: 10.1097/WNP.0b013e31823e96d7
9. Shellhaas RA. Continuous long-term electroencephalography: the gold standard for neonatal seizure diagnosis. Semin Fetal Neonatal Med. (2015) 20:149–53. doi: 10.1016/j.siny.2015.01.005
10. Holmes GL. Effect of seizures on the developing brain and cognition. Semin Pediatr Neurol. (2016) 23:120–6. doi: 10.1016/j.spen.2016.05.001
11. Besag F, Aldenkamp A, Caplan R, Dunn DW, Gobbi G, Sillanpää M. Psychiatric and behavioural disorders in children with epilepsy: an ILAE task force report. Epileptic Disord. (2016) 18(Suppl.1):1–86. doi: 10.1684/epd.2016.0809
12. Moavero R, Santarone ME, Galasso C, Curatolo P. Cognitive and behavioral effects of new antiepileptic drugs in pediatric epilepsy. Brain Dev. (2017) 39:464–9. doi: 10.1016/j.braindev.2017.01.006
13. Arzimanoglou A, D'Cruz O, Nordli D, Shinnar S, Holmes GL. A review of the new antiepileptic drugs for focal-onset seizures in pediatrics: role of extrapolation. Paediatr Drugs. (2018) 20:249–64. doi: 10.1007/s40272-018-0286-0
14. Arzimanoglou A, Kalilani L, Anamoo MA, Cooney M, Golembesky A, Taeter C, et al. Role of observational studies in supporting extrapolation of efficacy data from adults to children with epilepsy - a systematic review of the literature using lacosamide as an example. Eur J Paediatr Neurol. (2019) 23:589–603. doi: 10.1016/j.ejpn.2019.05.002
15. Pellock JM, Arzimanoglou A, D'Cruz O, Holmes GL, Nordli D, Shinnar S. Extrapolating evidence of antiepileptic drug efficacy in adults to children ≥2 years of age with focal seizures: the case for disease similarity. Epilepsia. (2017) 58:1686–96. doi: 10.1111/epi.13859
16. Pellock JM, Carman WJ, Thyagarajan V, Daniels T, Morris DL, D'Cruz O. Efficacy of antiepileptic drugs in adults predicts efficacy in children: a systematic review. Neurology. (2012) 79:1482–9. doi: 10.1212/WNL.0b013e31826d5ec0
17. Franco V, Canevini MP, Capovilla G, De Sarro G, Galimberti CA, Gatti G, et al. Off-label prescribing of antiepileptic drugs in pharmacoresistant epilepsy: a cross-sectional drug utilization study of tertiary care centers in Italy. CNS Drugs. (2014) 28:939–49. doi: 10.1007/s40263-014-0189-8
18. Kuchenbuch M, Chemaly N, Henniene KM, Kaminska A, Chiron C, Nabbout R. Off-label use and manipulations of antiepileptic drugs in children: analysis of the outpatient prescriptions in a tertiary center. Epilepsy Behav. (2018) 82:133–9. doi: 10.1016/j.yebeh.2018.03.013
19. Silverstein FS, Ferriero DM. Off-label use of antiepileptic drugs for the treatment of neonatal seizures. Pediatr Neurol. (2008) 39:77–9. doi: 10.1016/j.pediatrneurol.2008.04.008
20. Schrier L, Hadjipanayis A, Stiris T, Ross-Russell RI, Valiulis A, Turner MA, et al. Off-label use of medicines in neonates, infants, children, and adolescents: a joint policy statement by the European Academy of Paediatrics and the European society for Developmental Perinatal and Pediatric Pharmacology. Eur J Pediatr. (2020) 179:839–47. doi: 10.1007/s00431-019-03556-9
21. U.S. Food and Drug Administration, Center for Drug Evaluation and Research. Drugs for Treatment of Partial Onset Seizures: Full Extrapolation of Efficacy From Adults to Pediatric Patients 2 Years of Age and Older. Guidance for Industry. (2019). Available online at: https://www.fda.gov/media/130449/download (accessed November 12, 2020).
22. European Medicines Agency. Guideline on Clinical Investigation of Medicinal Products in the Treatment of Epileptic Disorders. (2018). Available online at: https://www.ema.europa.eu/en/documents/scientific-guideline/draft-guideline-clinical-investigation-medicinal-products-treatment-epileptic-disorders-revision-3_en.pdf (accessed November 12, 2020).
23. Lu H, Rosenbaum S. Developmental pharmacokinetics in pediatric populations. J Pediatr Pharmacol Ther. (2014) 19:262–76. doi: 10.5863/1551-6776-19.4.262
24. Biton V, Berkovic SF, Abou-Khalil B, Sperling MR, Johnson ME, Lu S. Brivaracetam as adjunctive treatment for uncontrolled partial epilepsy in adults: a phase III randomized, double-blind, placebo-controlled trial. Epilepsia. (2014) 55:57–66. doi: 10.1111/epi.12433
25. Ryvlin P, Werhahn KJ, Blaszczyk B, Johnson ME, Lu S. Adjunctive brivaracetam in adults with uncontrolled focal epilepsy: results from a double-blind, randomized, placebo-controlled trial. Epilepsia. (2014) 55:47–56. doi: 10.1111/epi.12432
26. Klein P, Schiemann J, Sperling MR, Whitesides J, Liang W, Stalvey T, et al. A randomized, double-blind, placebo-controlled, multicenter, parallel-group study to evaluate the efficacy and safety of adjunctive brivaracetam in adult patients with uncontrolled partial-onset seizures. Epilepsia. (2015) 56:1890–8. doi: 10.1111/epi.13212
27. Elger C, Halász P, Maia J, Almeida L, Soares-da-Silva P. Efficacy and safety of eslicarbazepine acetate as adjunctive treatment in adults with refractory partial-onset seizures: a randomized, double-blind, placebo-controlled, parallel-group phase III study. Epilepsia. (2009) 50:454–63. doi: 10.1111/j.1528-1167.2008.01946.x
28. Gil-Nagel A, Lopes-Lima J, Almeida L, Maia J, Soares-da-Silva P. Efficacy and safety of 800 and 1,200 mg eslicarbazepine acetate as adjunctive treatment in adults with refractory partial-onset seizures. Acta Neurol Scand. (2009) 120:281–7. doi: 10.1111/j.1600-0404.2009.01218.x
29. Ben-Menachem E, Gabbai AA, Hufnagel A, Maia J, Almeida L, Soares-da-Silva P. Eslicarbazepine acetate as adjunctive therapy in adult patients with partial epilepsy. Epilepsy Res. (2010) 89:278–85. doi: 10.1016/j.eplepsyres.2010.01.014
30. Sperling MR, Abou-Khalil B, Harvey J, Rogin JB, Biraben A, Galimberti CA, et al. Eslicarbazepine acetate as adjunctive therapy in patients with uncontrolled partial-onset seizures: results of a phase III, double-blind, randomized, placebo-controlled trial. Epilepsia. (2015) 56:244–53. doi: 10.1111/epi.12894
31. Kirkham F, Auvin S, Moreira J, Gama H, Falcão AC, Rocha JF, et al. Efficacy and safety of eslicarbazepine acetate as adjunctive therapy for refractory focal-onset seizures in children: a double-blind, randomized, placebo-controlled, parallel-group, multicenter, phase-III clinical trial. Epilepsy Behav. (2020) 105:106962. doi: 10.1016/j.yebeh.2020.106962
32. Józwiak S, Veggiotti P, Moreira J, Gama H, Rocha F, Soares-da-Silva P. Effects of adjunctive eslicarbazepine acetate on neurocognitive functioning in children with refractory focal-onset seizures. Epilepsy Behav. (2018) 81:1–11. doi: 10.1016/j.yebeh.2018.01.029
33. UK Gabapentin Study Group. Gabapentin in partial epilepsy. Lancet. (1990) 335:1114–7. doi: 10.1016/0140-6736(90)91123-R
34. The US Gabapentin Study Group. No. 5. Gabapentin as add-on therapy in refractory partial epilepsy: a double-blind, placebo-controlled, parallel-group study. Neurology. (1993) 43:2292–8. doi: 10.1212/WNL.43.11.2292
35. Anhut H, Ashman P, Feuerstein TJ, Sauermann W, Saunders M, Schmidt B. Gabapentin (Neurontin) as add-on therapy in patients with partial seizures: a double-blind, placebo-controlled study. Int Gabapentin Study Group Epilepsia. (1994) 35:795–801. doi: 10.1111/j.1528-1157.1994.tb02513.x
36. Appleton R, Fichtner K, LaMoreaux L, Alexander J, Halsall G, Murray G, et al. Gabapentin as add-on therapy in children with refractory partial seizures: a 12-week, multicentre, double-blind, placebo-controlled study. Gabapentin Paediatric Study Group Epilepsia. (1999) 40:1147–54. doi: 10.1111/j.1528-1157.1999.tb00833.x
37. Ben-Menachem E, Biton V, Jatuzis D, Abou-Khalil B, Doty P, Rudd GD. Efficacy and safety of oral lacosamide as adjunctive therapy in adults with partial-onset seizures. Epilepsia. (2007) 48:1308–17. doi: 10.1111/j.1528-1167.2007.01188.x
38. Halász P, Kälviäinen R, Mazurkiewicz-Beldzińska M, Rosenow F, Doty P, Hebert D, et al. Adjunctive lacosamide for partial-onset seizures: efficacy and safety results from a randomized controlled trial. Epilepsia. (2009) 50:443–53. doi: 10.1111/j.1528-1167.2008.01951.x
39. Chung S, Sperling MR, Biton V, Krauss G, Hebert D, Rudd GD, et al. Lacosamide as adjunctive therapy for partial-onset seizures: a randomized controlled trial. Epilepsia. (2010) 51:958–67. doi: 10.1111/j.1528-1167.2009.02496.x
40. Farkas V, Steinborn B, Flamini JR, Zhang Y, Yuen N, Borghs S, et al. Efficacy and tolerability of adjunctive lacosamide in pediatric patients with focal seizures. Neurology. (2019) 93:e1212–26. doi: 10.1212/WNL.0000000000008126
41. Baulac M, Rosenow F, Toledo M, Terada K, Li T, De Backer M, et al. Efficacy, safety, and tolerability of lacosamide monotherapy versus controlled-release carbamazepine in patients with newly diagnosed epilepsy: a phase 3, randomised, double-blind, non-inferiority trial. Lancet Neurol. (2017) 16:43–54. doi: 10.1016/S1474-4422(16)30292-7
42. Duchowny M, Pellock JM, Graf WD, Billard C, Gilman J, Casale E, et al. A placebo-controlled trial of lamotrigine add-on therapy for partial seizures in children. Lamictal pediatric partial seizure study group. Neurology. (1999) 53:1724–31. doi: 10.1212/WNL.53.8.1724
43. Piña-Garza JE, Levisohn P, Gucuyener K, Mikati MA, Warnock CR, Conklin HS, et al. Adjunctive lamotrigine for partial seizures in patients aged 1 to 24 months. Neurology. (2008) 70:2099–108. doi: 10.1212/01.wnl.0000285493.08622.35
44. Glauser TA, Ayala R, Elterman RD, Mitchell WG, Van Orman CB, Gauer LJ, et al. Double-blind placebo-controlled trial of adjunctive levetiracetam in pediatric partial seizures. Neurology. (2006) 66:1654–60. doi: 10.1212/01.wnl.0000217916.00225.3a
45. Piña-Garza JE, Nordli DR Jr, Rating D, Yang H, Schiemann-Delgado J, Duncan B. Adjunctive levetiracetam in infants and young children with refractory partial-onset seizures. Epilepsia. (2009) 50:1141–9. doi: 10.1111/j.1528-1167.2008.01981.x
46. Brodie MJ, Perucca E, Ryvlin P, Ben-Menachem E, Meencke HJ. Comparison of levetiracetam and controlled-release carbamazepine in newly diagnosed epilepsy. Neurology. (2007) 68:402–8. doi: 10.1212/01.wnl.0000252941.50833.4a
47. Barcs G, Walker EB, Elger CE, Scaramelli A, Stefan H, Sturm Y, et al. Oxcarbazepine placebo-controlled, dose-ranging trial in refractory partial epilepsy. Epilepsia. (2000) 41:1597–607. doi: 10.1111/j.1499-1654.2000.001597.x
48. Glauser TA, Nigro M, Sachdeo R, Pasteris LA, Weinstein S, Abou-Khalil B, et al. Adjunctive therapy with oxcarbazepine in children with partial seizures. The Oxcarbazepine pediatric study group. Neurology. (2000) 54:2237–44. doi: 10.1212/WNL.54.12.2237
49. Piña-Garza JE, Espinoza R, Nordli D, Bennett DA, Spirito S, Stites TE, et al. Oxcarbazepine adjunctive therapy in infants and young children with partial seizures. Neurology. (2005) 65:1370–5. doi: 10.1212/01.wnl.0000186800.18456.72
50. Schachter SC, Vazquez B, Fisher RS, Laxer KD, Montouris GD, Combs-Cantrell DT, et al. Oxcarbazepine: double-blind, randomized, placebo-control, monotherapy trial for partial seizures. Neurology. (1999) 52:732–7. doi: 10.1212/WNL.52.4.732
51. Sachdeo R, Beydoun A, Schachter S, Vazquez B, Schaul N, Mesenbrink P, et al. Oxcarbazepine (Trileptal) as monotherapy in patients with partial seizures. Neurology. (2001) 57:864–71. doi: 10.1212/WNL.57.5.864
52. Beydoun A, Sachdeo RC, Rosenfeld WE, Krauss GL, Sessler N, Mesenbrink P, et al. Oxcarbazepine monotherapy for partial-onset seizures: a multicenter, double-blind, clinical trial. Neurology. (2000) 54:2245–51. doi: 10.1212/WNL.54.12.2245
53. French JA, Krauss GL, Biton V, Squillacote D, Yang H, Laurenza A, et al. Adjunctive perampanel for refractory partial-onset seizures: randomized phase III study 304. Neurology. (2012) 79:589–96. doi: 10.1212/WNL.0b013e3182635735
54. French JA, Krauss GL, Steinhoff BJ, Squillacote D, Yang H, Kumar D, et al. Evaluation of adjunctive perampanel in patients with refractory partial-onset seizures: results of randomized global phase III study 305. Epilepsia. (2013) 54:117–25. doi: 10.1111/j.1528-1167.2012.03638.x
55. Krauss GL, Serratosa JM, Villanueva V, Endziniene M, Hong Z, French J, et al. Randomized phase III study 306: adjunctive perampanel for refractory partial-onset seizures. Neurology. (2012) 78:1408–15. doi: 10.1212/WNL.0b013e318254473a
56. Rosenfeld W, Conry J, Lagae L, Rozentals G, Yang H, Fain R, et al. Efficacy and safety of perampanel in adolescent patients with drug-resistant partial seizures in three double-blind, placebo-controlled, phase III randomized clinical studies and a combined extension study. Eur J Paediatr Neurol. (2015) 19:435–45. doi: 10.1016/j.ejpn.2015.02.008
57. Uthman BM, Rowan AJ, Ahmann PA, Leppik IE, Schachter SC, Sommerville KW, et al. Tiagabine for complex partial seizures: a randomized, add-on, dose-response trial. Arch Neurol. (1998) 55:56–62. doi: 10.1001/archneur.55.1.56
58. Sachdeo RC, Leroy RF, Krauss GL, Drake ME Jr, Green PM, Leppik IE, et al. Tiagabine therapy for complex partial seizures. A dose-frequency study. The Tiagabine Study Group. Arch Neurol. (1997) 54:595–601. doi: 10.1001/archneur.1997.00550170069016
59. Elterman RD, Glauser TA, Wyllie E, Reife R, Wu SC, Pledger G. A double-blind, randomized trial of topiramate as adjunctive therapy for partial-onset seizures in children. Topiramate YP study group. Neurology. (1999) 52:1338–44. doi: 10.1212/WNL.52.7.1338
60. Arroyo S, Dodson WE, Privitera MD, Glauser TA, Naritoku DK, Dlugos DJ, et al. Randomized dose-controlled study of topiramate as first-line therapy in epilepsy. Acta Neurol Scand. (2005) 112:214–22. doi: 10.1111/j.1600-0404.2005.00485.x
61. Glauser TA, Dlugos DJ, Dodson WE, Grinspan A, Wang S, Wu SC. Topiramate monotherapy in newly diagnosed epilepsy in children and adolescents. J Child Neurol. (2007) 22:693–9. doi: 10.1177/0883073807303997
62. Privitera MD, Brodie MJ, Mattson RH, Chadwick DW, Neto W, Wang S. Topiramate, carbamazepine and valproate monotherapy: double-blind comparison in newly diagnosed epilepsy. Acta Neurol Scand. (2003) 107:165–75. doi: 10.1034/j.1600-0404.2003.00093.x
63. Wheless JW, Neto W, Wang S. Topiramate, carbamazepine, and valproate monotherapy: double-blind comparison in children with newly diagnosed epilepsy. J Child Neurol. (2004) 19:135–41. doi: 10.1177/08830738040190020901
64. Brodie MJ, Duncan R, Vespignani H, Solyom A, Bitenskyy V, Lucas C. Dose-dependent safety and efficacy of zonisamide: a randomized, double-blind, placebo-controlled study in patients with refractory partial seizures. Epilepsia. (2005) 46:31–41. doi: 10.1111/j.0013-9580.2005.14704.x
65. Guerrini R, Rosati A, Segieth J, Pellacani S, Bradshaw K, Giorgi L. A randomized phase III trial of adjunctive zonisamide in pediatric patients with partial epilepsy. Epilepsia. (2013) 54:1473–80. doi: 10.1111/epi.12233
66. European Medicines Agency. Keppra (levetiracetam): Overview, Authorisation Details, Product Information, Asessment History. (2021). Available online at: https://www.ema.europa.eu/en/medicines/human/EPAR/keppra (accessed May 14, 2021).
67. European Medicines Agency. Lamictal (lamotrigine) Referral. (2008). Available online at: https://www.ema.europa.eu/en/medicines/human/referrals/lamictal (accessed January 11, 2021).
68. European Medicines Agency. Topamax (topiramate) referral. (2009). Available online at: https://www.ema.europa.eu/en/medicines/human/referrals/topamax (accessed January 11, 2021).
69. European Medicines Agency. Zonegran (zonisamide): Overview, Authorisation Details, Product Information, Asessment History. (2021). Available online at: https://www.ema.europa.eu/en/medicines/human/EPAR/zonegran (accessed May 14, 2021).
70. European Medicines Agency. Neurontin (gabapentin) Referral. (2006). Available online at: https://www.ema.europa.eu/en/medicines/human/referrals/neurontin (accessed January 11, 2021).
71. European Commission Heads of Medicines Agencies. Trileptal (oxcarbazepine) in Mutual Recognition Information - Product Index (Finalized Product Information). (2021). Available online at: http://mri.cts-mrp.eu/Human/Product/Details/16892 (accessed March 30, 2021).
72. European Commission Heads of Medicines Agencies. Sabril (vigabatrin) in Mutual Recognition Information - Product Index. (2021). Available online at: http://mri.cts-mrp.eu/Human/Product/Details/23556 (accessed March 30, 2021).
73. European Commission Heads of Medicines Agencies. Gabitril (tiagabine) in Mutual Recognition Information - Product Index. (2021). Available online at: http://mri.cts-mrp.eu/Human/Product/Details/8742 (accessed March 30, 2021).
74. Health Products Regulatory Authority. Gabitril (tiagabine): Summary of Product Characteristics. (2021). Available online at: http://www.hpra.ie/img/uploaded/swedocuments/Licence_PA0749-199-003_12082020162355.pdf (accessed March 30, 2021).
75. Health Products Regulatory Authority. Sabril (vigabatrin): Summary of Product Characteristics. (2021). Available online at: http://www.hpra.ie/img/uploaded/swedocuments/Licence_PA0540-023-002_26012021140714.pdf (accessed March 30, 2021).
76. European Medicines Agency. Zebinix (eslicarbazepine acetate): Overview, Authorisation Details, Product Information, Asessment History. (2021). Available online at: https://www.ema.europa.eu/en/medicines/human/EPAR/zebinix (accessed May 14, 2021).
77. European Medicines Agency. Briviact (in Italy: Nubriveo) (brivaracetam): Overview, Authorisation Details, Product Information, Asessment History. (2021). Available online at: https://www.ema.europa.eu/en/medicines/human/EPAR/briviact-italy-nubriveo (accessed May 14, 2021).
s78. European Medicines Agency. Vimpat (lacosamide): Overview, Authorisation Details, Product Information, Asessment History. (2021). Available online at: https://www.ema.europa.eu/en/medicines/human/EPAR/vimpat (accessed May 14, 2021).
79. European Medicines Agency. Fycompa (perampanel): Overview, Authorisation Details, Product Information, Asessment History. (2021). Available online at: https://www.ema.europa.eu/en/medicines/human/EPAR/fycompa (accessed May 14, 2021).
80. Arya R, Glauser TA. Pharmacotherapy of focal epilepsy in children: a systematic review of approved agents. CNS Drugs. (2013) 27:273–86. doi: 10.1007/s40263-013-0048-z
81. Glauser T, Ben-Menachem E, Bourgeois B, Cnaan A, Guerreiro C, Kalviainen R, et al. Updated ILAE evidence review of antiepileptic drug efficacy and effectiveness as initial monotherapy for epileptic seizures and syndromes. Epilepsia. (2013) 54:551–63. doi: 10.1111/epi.12074
82. Rosati A, De Masi S, Guerrini R. Antiepileptic drug treatment in children with epilepsy. CNS Drugs. (2015) 29:847–63. doi: 10.1007/s40263-015-0281-8
83. Stevens CE, Stafstrom CE. Pharmacotherapy for focal seizures in children and adolescents. Drugs. (2018) 78:1321–37. doi: 10.1007/s40265-018-0959-6
84. Moavero R, Pisani LR, Pisani F, Curatolo P. Safety and tolerability profile of new antiepileptic drug treatment in children with epilepsy. Expert Opin Drug Saf. (2018) 17:1015–28. doi: 10.1080/14740338.2018.1518427
85. Mohd-Tahir NA, Li SC. Meta-analyses of newer antiepileptic drugs as adjunct for treatment of focal epilepsy in children. Epilepsy Res. (2018) 139:113–22. doi: 10.1016/j.eplepsyres.2017.11.007
86. Lagae L. The importance of assessing behaviour and cognition in antiepileptic drug trials in children and adolescents. Acta Neurol Belg. (2017) 117:425–32. doi: 10.1007/s13760-016-0734-y
87. Aldenkamp A, Besag F, Gobbi G, Caplan R, Dunn DW, Sillanpää M. Psychiatric and behavioural disorders in children with epilepsy (ILAE task force report): adverse cognitive and behavioural effects of antiepileptic drugs in children. Epileptic Disord. (2016) 18 (Suppl. 1):S55–67. doi: 10.1684/epd.2016.0817
88. Brodie MJ. Tolerability and safety of commonly used antiepileptic drugs in adolescents and adults: a clinician's overview. CNS Drugs. (2017) 31:135–47. doi: 10.1007/s40263-016-0406-8
89. Chen DY, Chowdhury S, Farnaes L, Friedman JR, Honold J, Dimmock DP, et al. Rapid diagnosis of KCNQ2-associated early infantile epileptic encephalopathy improved outcome. Pediatr Neurol. (2018) 86:69–70. doi: 10.1016/j.pediatrneurol.2018.06.002
90. Perucca P, Perucca E. Identifying mutations in epilepsy genes: impact on treatment selection. Epilepsy Res. (2019) 152:18–30. doi: 10.1016/j.eplepsyres.2019.03.001
91. Kearney H, Byrne S, Cavalleri GL, Delanty N. Tackling epilepsy with high-definition precision medicine: a review. J Am Med Assoc Neurol. (2019) 76:1109–16. doi: 10.1001/jamaneurol.2019.2384
92. Cornet MC, Cilio MR. Genetics of neonatal-onset epilepsies. Handb Clin Neurol. (2019) 162:415–33. doi: 10.1016/B978-0-444-64029-1.00020-5
93. Holthausen H, Pieper T, Kudernatsch M. Towards early diagnosis and treatment to save children from catastrophic epilepsy – focus on epilepsy surgery. Brain Dev. (2013) 35:730–41. doi: 10.1016/j.braindev.2013.05.003
94. Nariai H, Duberstein S, Shinnar S. Treatment of epileptic encephalopathies: current state of the art. J Child Neurol. (2018) 33:41–54. doi: 10.1177/0883073817690290
95. Devinsky O, Cross JH, Laux L, Marsh E, Miller I, Nabbout R, et al. Trial of cannabidiol for drug-resistant seizures in the dravet syndrome. N Engl J Med. (2017) 376:2011–20. doi: 10.1056/NEJMoa1611618
96. Devinsky O, Patel AD, Thiele EA, Wong MH, Appleton R, Harden CL, et al. Randomized, dose-ranging safety trial of cannabidiol in Dravet syndrome. Neurology. (2018) 90:e1204–11. doi: 10.1212/WNL.0000000000005254
97. Miller I, Scheffer IE, Gunning B, Sanchez-Carpintero R, Gil-Nagel A, Perry MS, et al. Dose-ranging effect of adjunctive oral cannabidiol vs. placebo on convulsive seizure frequency in dravet syndrome: a randomized clinical trial. J Am Med Assoc Neurol. (2020) 77:613–21. doi: 10.1001/jamaneurol.2020.0073
98. Lagae L, Sullivan J, Knupp K, Laux L, Polster T, Nikanorova M, et al. Fenfluramine hydrochloride for the treatment of seizures in Dravet syndrome: a randomised, double-blind, placebo-controlled trial. Lancet. (2019) 394:2243–54. doi: 10.1016/S0140-6736(19)32500-0
99. Nabbout R, Mistry A, Zuberi S, Villeneuve N, Gil-Nagel A, Sanchez-Carpintero R, et al. Fenfluramine for treatment-resistant seizures in patients with Dravet syndrome receiving stiripentol-inclusive regimens: a randomized clinical trial. J Am Med Assoc Neurol. (2020) 77:300–8. doi: 10.1001/jamaneurol.2019.4113
100. Hall AK, Carlson MR. The current status of orphan drug development in Europe and the US. Intractable Rare Dis Res. (2014) 3:1–7. doi: 10.5582/irdr.3.1
101. Widjaja E, Jain P, Demoe L, Guttmann A, Tomlinson G, Sander B. Seizure outcome of pediatric epilepsy surgery: systematic review and meta-analyses. Neurology. (2020) 94:311–21. doi: 10.1212/WNL.0000000000008966
102. French JA, Lawson JA, Yapici Z, Ikeda H, Polster T, Nabbout R, et al. Adjunctive everolimus therapy for treatment-resistant focal-onset seizures associated with tuberous sclerosis (EXIST-3): a phase 3, randomised, double-blind, placebo-controlled study. Lancet. (2016) 388:2153–63. doi: 10.1016/S0140-6736(16)31419-2
103. Moavero R, Mühlebner A, Luinenburg MJ, Craiu D, Aronica E, Curatolo P. Genetic pathogenesis of the epileptogenic lesions in Tuberous Sclerosis Complex: therapeutic targeting of the mTOR pathway. Epilepsy Behav. (2021) 107713. doi: 10.1016/j.yebeh.2020.107713
104. Curatolo P, Franz DN, Lawson JA, Yapici Z, Ikeda H, Polster T, et al. Adjunctive everolimus for children and adolescents with treatment-refractory seizures associated with tuberous sclerosis complex: post-hoc analysis of the phase 3 EXIST-3 trial. Lancet Child Adolesc Health. (2018) 2:495–504. doi: 10.1016/S2352-4642(18)30099-3
105. Krueger DA, Care MM, Holland K, Agricola K, Tudor C, Mangeshkar P, et al. Everolimus for subependymal giant-cell astrocytomas in tuberous sclerosis. N Engl J Med. (2010) 363:1801–11. doi: 10.1056/NEJMoa1001671
106. Bissler JJ, Kingswood JC, Radzikowska E, Zonnenberg BA, Frost M, Belousova E, et al. Everolimus for angiomyolipoma associated with tuberous sclerosis complex or sporadic lymphangioleiomyomatosis (EXIST-2): a multicentre, randomised, double-blind, placebo-controlled trial. Lancet. (2013) 381:817–24. doi: 10.1016/S0140-6736(12)61767-X
107. Bissler JJ, Budde K, Sauter M, Franz DN, Zonnenberg BA, Frost MD, et al. Effect of everolimus on renal function in patients with tuberous sclerosis complex: evidence from EXIST-1 and EXIST-2. Nephrol Dial Transplant. (2019) 34:1000–8. doi: 10.1093/ndt/gfy132
108. Franz DN, Budde K, Kingswood JC, Belousova E, Sparagana S, de Vries PJ, et al. Effect of everolimus on skin lesions in patients treated for subependymal giant cell astrocytoma and renal angiomyolipoma: final 4-year results from the randomized EXIST-1 and EXIST-2 studies. J Eur Acad Dermatol Venereol. (2018) 32:1796–803. doi: 10.1111/jdv.14964
109. Guerrini R, Pellacani S. Benign childhood focal epilepsies. Epilepsia. (2012) 53(Suppl.4):9–18. doi: 10.1111/j.1528-1167.2012.03609.x
110. International League Against Epilepsy. EpilepsyDiagnosis.org - Diagnostic manual. Epilepsy Syndromes. (2021). Available online at: https://www.epilepsydiagnosis.org/syndrome/epilepsy-syndrome-groupoverview.html (accessed March 30, 2021).
111. Heron SE, Grinton BE, Kivity S, Afawi Z, Zuberi SM, Hughes JN, et al. PRRT2 mutations cause benign familial infantile epilepsy and infantile convulsions with choreoathetosis syndrome. Am J Hum Genet. (2012) 90:152–60. doi: 10.1016/j.ajhg.2011.12.003
112. Kuersten M, Tacke M, Gerstl L, Hoelz H, Stülpnagel CV, Borggraefe I. Antiepileptic therapy approaches in KCNQ2 related epilepsy: a systematic review. Eur J Med Genet. (2020) 63:103628. doi: 10.1016/j.ejmg.2019.02.001
113. Sanders SJ, Campbell AJ, Cottrell JR, Moller RS, Wagner FF, Auldridge AL, et al. Progress in understanding and treating SCN2A-mediated disorders. Trends Neurosci. (2018) 41:442–56. doi: 10.1016/j.tins.2018.03.011
114. Brunklaus A, Du J, Steckler F, Ghanty II, Johannesen KM, Fenger CD, et al. Biological concepts in human sodium channel epilepsies and their relevance in clinical practice. Epilepsia. (2020) 61:387–99. doi: 10.1111/epi.16438
115. Wolff M, Johannesen KM, Hedrich UBS, Masnada S, Rubboli G, Gardella E, et al. Genetic and phenotypic heterogeneity suggest therapeutic implications in SCN2A-related disorders. Brain. (2017) 140:1316–36. doi: 10.1093/brain/awx054
116. Schreiber JM, Tochen L, Brown M, Evans S, Ball LJ, Bumbut A, et al. A multi-disciplinary clinic for SCN8A-related epilepsy. Epilepsy Res. (2020) 159:106261. doi: 10.1016/j.eplepsyres.2019.106261
117. Barcia G, Fleming MR, Deligniere A, Gazula VR, Brown MR, Langouet M, et al. De novo gain-of-function KCNT1 channel mutations cause malignant migrating partial seizures of infancy. Nat Genet. (2012) 44:1255–9. doi: 10.1038/ng.2441
118. Mikati MA, Jiang YH, Carboni M, Shashi V, Petrovski S, Spillmann R, et al. Quinidine in the treatment of KCNT1-positive epilepsies. Ann Neurol. (2015) 78:995–9. doi: 10.1002/ana.24520
119. Bearden D, Strong A, Ehnot J, DiGiovine M, Dlugos D, Goldberg EM. Targeted treatment of migrating partial seizures of infancy with quinidine. Ann Neurol. (2014) 76:457–61. doi: 10.1002/ana.24229
120. Abdelnour E, Gallentine W, McDonald M, Sachdev M, Jiang YH, Mikati MA. Does age affect response to quinidine in patients with KCNT1 mutations? Report of three new cases and review of the literature. Seizure. (2018) 55:1–3. doi: 10.1016/j.seizure.2017.11.017
121. Numis AL, Nair U, Datta AN, Sands TT, Oldham MS, Patel A, et al. Lack of response to quinidine in KCNT1-related neonatal epilepsy. Epilepsia. (2018) 59:1889–98. doi: 10.1111/epi.14551
122. Yoshitomi S, Takahashi Y, Yamaguchi T, Oboshi T, Horino A, Ikeda H, et al. Quinidine therapy and therapeutic drug monitoring in four patients with KCNT1 mutations. Epileptic Disord. (2019) 21:48–54. doi: 10.1684/epd.2019.1026
123. Mullen SA, Carney PW, Roten A, Ching M, Lightfoot PA, Churilov L, et al. Precision therapy for epilepsy due to KCNT1 mutations: a randomized trial of oral quinidine. Neurology. (2018) 90:e67–72. doi: 10.1212/WNL.0000000000004769
124. Tan HJ, Singh J, Gupta R, de Goede C. Comparison of antiepileptic drugs, no treatment, or placebo for children with benign epilepsy with centro temporal spikes. Cochrane Database Syst Rev. (2014) 2014:Cd006779. doi: 10.1002/14651858.CD006779.pub2
125. Gerstl L, Willimsky E, Rémi C, Noachtar S, Borggräfe I, Tacke M. A systematic review of seizure-freedom rates in patients with benign epilepsy of childhood with centrotemporal spikes receiving antiepileptic drugs. Clin Neuropharmacol. (2021) 44:39–46. doi: 10.1097/WNF.0000000000000435
126. Glauser T, Ben-Menachem E, Bourgeois B, Cnaan A, Chadwick D, Guerreiro C, et al. ILAE treatment guidelines: evidence-based analysis of antiepileptic drug efficacy and effectiveness as initial monotherapy for epileptic seizures and syndromes. Epilepsia. (2006) 47:1094–120. doi: 10.1111/j.1528-1167.2006.00585.x
127. Hughes JR. Benign epilepsy of childhood with centrotemporal spikes (BECTS): to treat or not to treat, that is the question. Epilepsy Behav. (2010) 19:197–203. doi: 10.1016/j.yebeh.2010.07.018
128. Rating D, Wolf C, Bast T. Sulthiame as monotherapy in children with benign childhood epilepsy with centrotemporal spikes: a 6-month randomized, double-blind, placebo-controlled study. Sulthiame Study Group Epilepsia. (2000) 41:1284–8. doi: 10.1111/j.1528-1157.2000.tb04606.x
129. Bourgeois B, Brown LW, Pellock JM, Buroker M, Greiner M, Garofalo EA, et al. Gabapentin (Neurontin) monotherapy in children with benign childhood epilepsy with centrotemporal spikes (BECTS): a 36-week, doubleblind, placebo-controlled study. Epilepsia. (1998) 39:163.
130. Coppola G, Franzoni E, Verrotti A, Garone C, Sarajlija J, Operto FF, et al. Levetiracetam or oxcarbazepine as monotherapy in newly diagnosed benign epilepsy of childhood with centrotemporal spikes (BECTS): an open-label, parallel group trial. Brain Dev. (2007) 29:281–4. doi: 10.1016/j.braindev.2006.09.008
131. Suo GH, Zheng YQ, Wu YJ, Tang JH. Effects of levetiracetam and oxcarbazepine monotherapy on intellectual and cognitive development in children with benign epilepsy with centrotemporal spikes. Acta Neurol Belg. (2021) 121:1265–73. doi: 10.1007/s13760-021-01613-5
132. Andrade R, García-Espinosa A, Machado-Rojas A, García-González ME, Trápaga-Quincoses O, Morales-Chacón LM. A prospective, open, controlled and randomised study of clobazam vs. carbamazepine in patients with frequent episodes of Rolandic epilepsy. Rev Neurol. (2009) 49:581–6. doi: 10.33588/rn.4911.2009525
133. Kang HC, Eun BL, Wu Lee C, Ku Moon H, Kim JS, Wook Kim D, et al. The effects on cognitive function and behavioral problems of topiramate compared to carbamazepine as monotherapy for children with benign rolandic epilepsy. Epilepsia. (2007) 48:1716–23. doi: 10.1111/j.1528-1167.2007.01160.x
134. Borggraefe I, Bonfert M, Bast T, Neubauer BA, Schotten KJ, Maßmann K, et al. Levetiracetam vs. sulthiame in benign epilepsy with centrotemporal spikes in childhood: a double-blinded, randomized, controlled trial (German HEAD Study). Eur J Paediatr Neurol. (2013) 17:507–14. doi: 10.1016/j.ejpn.2013.03.014
135. Tacke M, Gerstl L, Heinen F, Heukaeufer I, Bonfert M, Bast T, et al. Effect of anticonvulsive treatment on neuropsychological performance in children with BECTS. Eur J Paediatr Neurol. (2016) 20:874–9. doi: 10.1016/j.ejpn.2016.07.015
136. Margolis A, Giuliano C. Making the switch: from case studies to N-of-1 trials. Epilepsy Behav Rep. (2019) 12:100336. doi: 10.1016/j.ebr.2019.100336
137. Balestrini S, Chiarello D, Gogou M, Silvennoinen K, Puvirajasinghe C, Jones WD, et al. Real-life survey of pitfalls and successes of precision medicine in genetic epilepsies. J Neurol Neurosurg Psychiatry. (2021) 92:1044–52. doi: 10.1136/jnnp-2020-325932
Keywords: focal-onset seizure, epilepsy, children, antiseizure medication, syndrome, etiology
Citation: Aeby A, Ceulemans B and Lagae L (2022) Treatment of Focal-Onset Seizures in Children: Should This Be More Etiology-Driven? Front. Neurol. 13:842276. doi: 10.3389/fneur.2022.842276
Received: 23 December 2021; Accepted: 24 January 2022;
Published: 07 March 2022.
Edited by:
Pasquale Parisi, Sapienza University of Rome, ItalyReviewed by:
Pasquale Striano, University of Genoa, ItalyDario Pruna, G. Brotzu Hospital, Italy
Gaetano Terrone, University of Naples Federico II, Italy
Copyright © 2022 Aeby, Ceulemans and Lagae. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Alec Aeby, YWxlYy5hZWJ5QGh1ZGVyZi5iZQ==