
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Neurol. , 09 May 2022
Sec. Multiple Sclerosis and Neuroimmunology
Volume 13 - 2022 | https://doi.org/10.3389/fneur.2022.836337
 Qiling Ji1Huiqing Dong2*Hangil Lee3
Qiling Ji1Huiqing Dong2*Hangil Lee3 Zheng Liu2
Zheng Liu2 Yanna Tong1Kenneth Elkin3Yazeed Haddad3
Yanna Tong1Kenneth Elkin3Yazeed Haddad3 Xiaokun Geng1,3*
Xiaokun Geng1,3* Yuchuan Ding3
Yuchuan Ding3Objective: The present study sought to differentiate multiple sclerosis and neuromyelitis optica spectrum disorder patients at their first attack by describing and distinguishing their clinical features, radiographic characteristics, and immunologic characteristics of serum and cerebrospinal fluid.
Methods: We retrospectively studied 58 patients with multiple sclerosis (MS) and 52 patients with neuromyelitis optica spectrum disorder (NMOSD) by referencing brainstem lesions as the prodromal events. Their demographics and presentation at the time of the first attack was evaluated including their gender, age, clinical features of the first attack, the expanded disability status scale (EDSS), brainstem lesion(s) by head MRI, and immunological indices of serum and cerebrospinal fluid.
Results: The NMOSD group had more female patients (4.8 vs. 1.9, p < 0.05), and was older than the MS group (37.81 ± 16.60 vs. 27.57 ± 11.17, p < 0.001). NMOSD patients also had a significantly higher association with autoimmune diseases or positive autoimmune antibodies (p < 0.01). There was no significant difference in the EDSS scores between the two groups (p = 0.420). Central hiccups, vomiting, and pyramidal tract signs were more common in the NMOSD group than the MS group (p < 0.001, p < 0.001, p < 0.01), while eye movement abnormalities were more common with MS (p < 0.01). There were no significant differences in other clinical manifestations such as vertigo, diplopia, limb weakness, numbness, and eating difficulty. MS patients were more likely to have midbrain and pons imaging lesions (p < 0.001, p < 0.001), while NMOSD patients had more lesions in the medulla oblongata (p < 0.001). The lesions in the MS group were mostly located in the periphery, while those in the NMOSD group were centrally located (p < 0.001, p < 0.001). Patchy lesions were more common in MS patients (p < 0.001), while large lesions were more common in the NMOSD group (p < 0.001). Finally, serum AQP4 Ab was found only in the NMOSD group (p < 0.001).
Conclusion: Patients with MS and NMOSD have differentiating clinical manifestations at the time of their first brainstem lesions which include central hiccups, vomiting, pyramidal tract signs, and abnormal eye movements. Additionally, distinct imaging manifestations such as lesion location(s) and morphology may also aid in the development of pathognomonic criteria leading to timely initial diagnosis of MS and NMOSD.
Multiple sclerosis (MS) is a common demyelinating disease in the central nervous system (CNS) with a relapsing and remitting clinical course. Its incidence rate and morbidity are the highest among CNS demyelinating disorders. Previously, neuromyelitis optica (NMO) was considered as a subtype of MS that mainly affected the optic nerves and spinal cord. However, recent clinical, imaging, and immunologic data indicated that NMO is a distinct disease with a unique pathogenesis (1, 2). The discovery of anti-aquaporin-4 (AQP4) antibody (Ab) was essential for the evolution of NMO diagnostic criteria, leading to its reclassification as a member of a larger group of disorders called neuromyelitis optica spectrum disorders (NMOSD) (3).
Most lesions in MS patients involve the spinal cord, paraventricular region, cerebral cortex and subcortex, infratentorial region, and optic nerves. Although there are some similarities in NMOSD patients as they also have lesions in the spinal cord and the optic nerves, they are distinguished by involvement of area postrema, diencephalon, and posterior cortex. Both can involve the brainstem and exhibit corresponding clinical manifestations. Although they have some distinguishing clinical and imaging manifestations, they have significant overlap, making their differentiation challenging (1, 4).
As the pathogeneses of MS and NMOSD differ widely, their prognoses and treatment approaches are also consequently unique; hence, it is crucial to distinguish them at time of onset to optimize their care (3, 5–11). However, it is often difficult to distinguish between MS and NMOSD at their initial presentation with mainly brainstem involvement due to their overlapping clinical and radiographic manifestations. Although immunological indices are helpful for their differentiation, obtaining them at the time of disease onset is often unfeasible. Clinical symptoms and imaging are typically the most readily available information to clinicians, so it would be particularly helpful to be able to distinguish the two by those information alone.
Therefore, we compared and analyzed the clinical features and imaging manifestations of MS and NMOSD in patients when they first presented with brainstem lesion symptoms, aiming to delineate their unique characteristics to distinguish the two diseases at symptom onset. This work paves the way to expedite proper treatment administration and improve patient prognosis in the future.
The present study included 58 patients diagnosed with multiple sclerosis(MS)and 52 patients with neuromyelitis optica spectrum disorders (NMOSD) who were admitted to Xuanwu Hospital Capital Medical University between January 2013 and June 2019. Patients with MS met the clinical diagnostic criteria of McDonald 2010 (12) and patients with NMOSD met the diagnostic criteria set by the International Panel for NMO Diagnosis (IPND) in 2015 (1). All patients presented initially with brainstem lesions and underwent MRI imaging, cerebrospinal fluid (CSF) testing, and serological testing within 1 month of symptom onset. MRI imaging included T1 weighted, T2 weighted, and T2 flair, with or without gadolinium contrast enhancement. Cerebrospinal fluid and serological testing included the cell-based transfer immunofluorescence assay (CBA) for AQP4 Ab detection and isoelectric focusing electrophoresis for cerebrospinal fluid specific oligoclonal band (SOB) detection. Patients with non-idiopathic demyelinating disease were excluded from the study.
This study is a retrospective analysis. Patients that met the respective diagnostic criteria with appropriate clinical data were included. A questionnaire was completed for each patient, which included the patient's gender, age, follow-up time, relapse times, presence of an autoimmune disease or autoimmune antibodies at the time of onset, EDSS score at onset, clinical signs and symptoms, and MRI images. Symptoms such as vertigo, pupillary miosis, nystagmus, diplopia, medial longitudinal fasciculus (MLF) syndrome, numbness of limbs, numbness of mouth and face, headache, dysphagia (coughing after eating), dysarthria (slurred speech), weakness of limbs, facial paralysis, central hiccups, central vomiting, pyramidal tract signs, sensory disturbance, ataxia, ophthalmoplegia (abnormal eye movements), and bulbar paralysis were regarded as manifestations of brainstem symptoms. On the MRI images, the lesion location, shape, size, and enhancement were noted. Immunological indices from serum and CSF were collected including the presence of cerebrospinal fluid specific oligoclonal bands (SOB), serum and cerebrospinal fluid AQP4 antibody, and myelin basic protein (MBP).
The SPSS 17.0 software was utilized for statistical analyses. Data with normal distribution were expressed as mean ± standard deviation and pairwise comparisons between groups were performed with the LSD t-test. The data of non-normal distribution was expressed as median with interquartile intervals. The Bonferroni correction was performed for the pairwise comparisons between groups. The Chi square test was used for categorical data. P <0.05 was used as the standard of statistical significance in all cases.
The NMOSD group had a statistically significant greater ratio of female patients as compared to the MS group, although there were no significant differences in follow-up times and episode frequencies. In the MS group, typical onset was from juvenile to middle-aged without any patients over 60 years old. NMOSD patients' onset were juvenile to elderly, but no patients over 65 years old were found; furthermore, NMOSD age of onset had a bimodal distribution with peaks at 16–30 years old and 61-65 years old (Table 1).
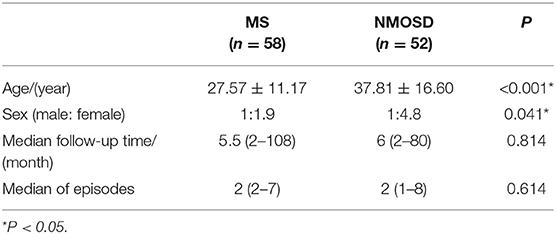
Table 1. General information of patients.
There were no statistically significant differences in the first EDSS score between the two groups. However, 15% of the NMOSD cohort was found to have an autoimmune disease or tested positive for an autoimmune antibody, while none from the MS group met these criteria (p = 0.006). There were several clinical symptoms with statistically significant differences between the two groups. The NMOSD group had more central hiccups (p < 0.001), central vomiting (p < 0.001), and pyramidal tract signs (p = 0.004), while the MS group had more MLF syndromes (p = 0.013) and ophthalmoplegias (p = 0.004) (Table 2).
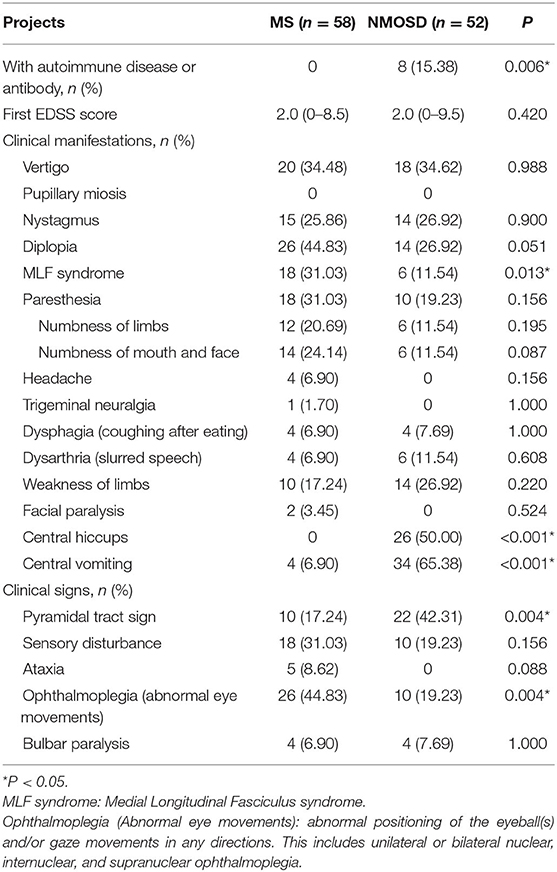
Table 2. General information and clinical features of patients.
Patients in both groups underwent MRI imaging. In the MS group, there were 26 cases (44.83%) of midbrain lesions, 42 cases (72.41%) of pontine lesions, and 22 cases (37.93%) of medulla oblongata lesions; the NMOSD group had 8 cases (15.38%) of midbrain lesions, 18 cases (34.62%) of pontine lesions, and 46 cases (88.46%) of medulla oblongata lesions. The differences between the groups in all three brain regions were significant (p = 0.001, p <0.001, p <0.001). The number of MS patients with peduncle cerebri and brachium pontis (Middle cerebellar peduncle) lesions was significantly higher than the NMOSD group (p = 0.013, p = 0.003), while the reverse was true regarding lesions in cervical cord medulla junction and area postrema (AP) (p <0.001). MS lesions were peripherally located while the NMOSD lesions were centrally located, both of which were statistically significant (p <0.001, p <0.001). Moreover, the lesions in the MS group were mostly patchy, while those in the NMOSD group were mostly large (p <0.001, p <0.001). Importantly, four patients in the MS group showed enhancements while and none in the NMOSD group did, although this data did not achieve statistical significance (p = 0.055) (Table 3, Figures 1, 2).
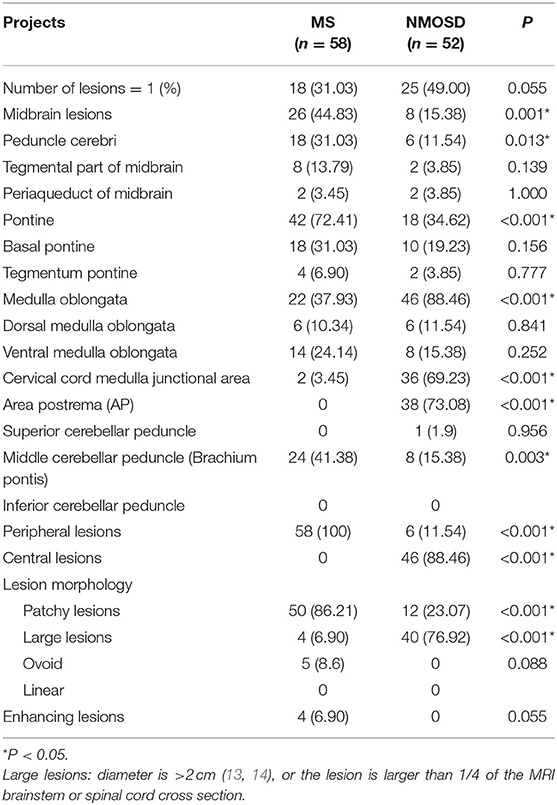
Table 3. MRI features of patients.
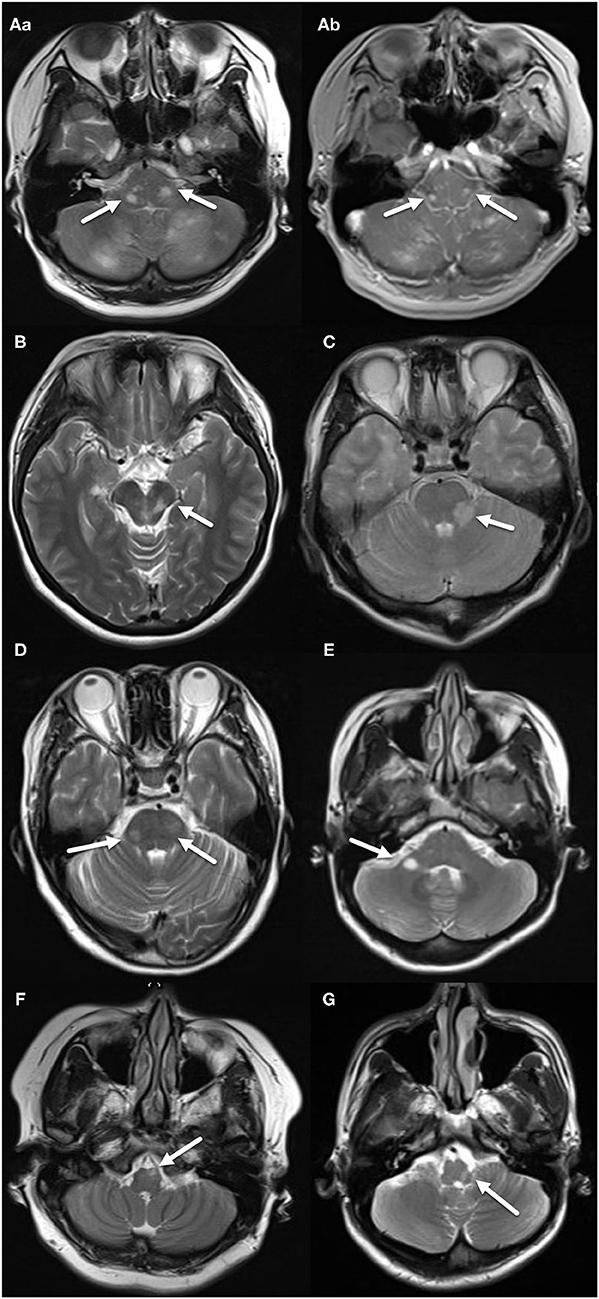
Figure 1. MRI features of the MS group. (A–a) Two patchy, abnormal signals located on both sides of pons, indicated by arrows. (A–b) Two nodular enhancing lesions located on both sides of pons, indicated by arrows. (B) Patchy, abnormal signal located in tegmentum of midbrain, indicated by an arrow. (C) Patchy, abnormal signal located in left brachium pontis, indicated by an arrow. (D) Two patchy, abnormal signals located on both sides of pons, indicated by arrows. (E) Ovoid, abnormal signal located in right brachium pontis, indicated by an arrow. (F) Patchy abnormal signal located in left ventral medulla oblongata, indicated by an arrow. (G) Patchy abnormal signal located in left dorsal medulla oblongata, indicated by an arrow.
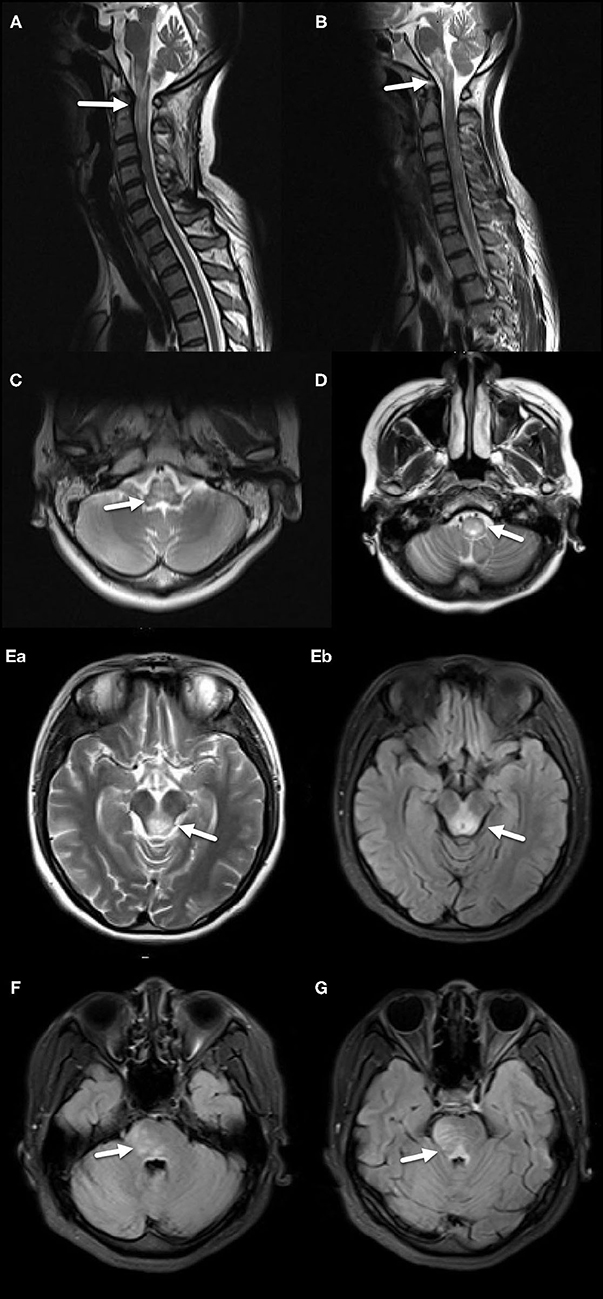
Figure 2. MRI features of the NMOSD group. (A) Diffuse, large abnormal signal extending from the junction of cervical spinal cord and medulla oblongata to the upper cervical spinal cord, including AP, indicated by an arrow. (B) Extensive diffuse, large abnormal signal in medulla oblongata and AP, indicated by an arrow. (C) Large abnormal signal located in central medulla oblongata, indicated by an arrow. (D) Large abnormal signal located in central lower medulla oblongata, indicated by an arrow. (E–a,b) Large abnormal signal around midbrain aqueduct, indicated by arrows. (F) Large abnormal signal located in pons, indicated by an arrow. (G) Large abnormal signal located in pons and around the fourth ventricle, indicated by an arrow.
CSF testing was performed in all patients. CSF from the NMOSD group had more WBCs than the MS group (P = 0.017). In the MS group, 58 patients were tested for oligoclonal bands (OB) + 24-h IgG synthesis rates, among which 50 (86.21%) were positive for the specific IgG oligoclonal band (SOB). A total of 48 patients with MS were tested for aquaporin-4 (AQP4) Abs in serum and cerebrospinal fluid, and all of them were negative. 16 MS patients were tested for Myelin basic protein (MBP), of which six were positive (37.5%). 52 patients in the NMOSD group were tested for OB + 24-h IgG synthesis rate, of which 8 (15.38%) were positive for the CSF-specific IgG oligoclonal band (SOB). The serums of 46 patients with NMOSD were tested for AQP4 Ab, of which 42 cases were positive (91.30%). Of the 46 patients with NMOSD who had their serums tested for AQP4 Ab, 36 cases were tested for AQP4 Ab in CSF and it was found that 20 cases were positive (55.56%), including one patient whose serum AQP4 Ab was negative. MBP was measured in 16 patients with NMOSD, which revealed eight (50%) positive cases (Table 4).
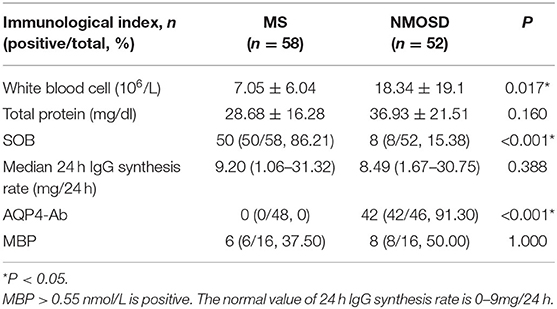
Table 4. Immunological index.
MS and NMOSD have been regarded as two distinct diseases, with distinct pathophysiology and clinical courses. The mechanism of MS is multifaceted, though it is generally regarded as an autoimmune disease induced by T cells. However, it has been revealed recently that B cells may play a greater role in MS, complicating the pathology (15, 16). The classic hallmark of MS is multifocal lesions in the central nervous system which vary in location and onset, exhibiting periods of remission. These lesions are characterized by demyelination, relative preservation of axons, inflammatory reactions, glial hyperplasia, and myelin regeneration. The clinical course of MS is diverse, and its lesions can accumulate in most parts of the central nervous system including white matter, optic nerve, brainstem, cerebellum, and spinal cord. On the other hand, NMOSD is a rare disease mediated by antibodies in the central nervous system and is related to serum aquaporin-4 immunoglobulin G antibody (AQP4-IgG). Contrary to MS, the pathophysiology of NMOSD is mediated by primary injury of astrocytes, while demyelination is a secondary manifestation of the disease (1, 17). Furthermore, NMOSD tends to show tropism in the optic nerve, AP, spinal cord, brainstem, diencephalon, and posterior part of the brain (1).
The present study found that both MS and NMOSD were more common in females compared to males, consistent with previous reports (17–21). However, we also found that NMOSD had a greater skew in the ratio of females to males. Additionally, NMOSD had a stronger association with autoimmune diseases and/or autoimmune antibodies (22–24) and involved the elderly population. Our findings concur with prior research which report that the average age of NMOSD patients in the world is between 32.6 and 47.5 years old, with patients over 50 years old accounting for about 25% of the incidence (19, 25, 26).
Previous studies have suggested that NMOSD patients have more serious dysfunction than patients with MS (14, 27), although our data did not find statistical difference in EDSS scores between the two groups at the first onset. This may be due to the fact that our EDSS scores were collected based on patient presentations at symptom onset as compared to the peak of their symptoms. Clinically, the present study found that MS had significantly greater association with MLF and ophthalmoplegia as compared to NMOSD, while NMOSD had greater association with central hiccups, central vomiting, and pyramidal tract signs. Although the MS group had headaches (4/58), paralysis (2/58), and trigeminal neuralgia (1/58) with none in the NMOSD, the difference did not reach statistical significance.
On MRI, MS had significantly more lesions in midbrain and pons compared to the NMOSD group, especially in peduncle cerebri. On the other hand, NMOSD had significantly more lesions in medulla oblongata, especially in the cervical cord medulla junction and AP. Although the MS group had exclusively peripheral lesions (58/58), the NMOSD group's lesions were mostly centrally located. These were consistent with the established knowledge that NMOSD lesions are classically located around ependyma. While the MS lesions were patchy and smaller, NMOSD lesions were large with blurry boundaries. 5 of 58 patients in the MS group had ovoid lesions with clearly defined borders. In the NMOSD group, there was no ovoid lesions with clear boundaries or any linear lesions, although previous reports showed that NMOSD patients had linear lesions (1, 13). There were also no linear lesions in the MS group.
Given that MS and NMOSD have distinct etiologies, pathogeneses, and prognoses, it is unsurprising that their current treatments differ. The main goal of MS therapy is to reduce the chance of recurrence and formation of new lesions, minimizing morbidity and disability (5, 6). On the other hand, NMOSD therapeutic guidelines aim to accelerate the recovery of acute exacerbations, prevent long-term relapse, and minimize chronic sequelae (4). Although the treatment plans of both aim to reduce disease recurrences, NMOSD therapy is generally comprised of immunosuppressive drugs, unlike the immunomodulators that are commonly used in MS (7). Some disease modifying drugs (DMDs) for MS, such as interferon-β, fingolimod, and natalizumab, have been found to be not only ineffective but actually aggravate the condition of NMOSD patients (4, 8, 9). Therefore, it is crucial to correctly diagnose these patients as soon as possible and provide the correct treatment.
Clinically, it is observed that some MS and NMOSD patients have partially overlapping symptoms when they initially present with brainstem lesions. Unsurprisingly, this makes diagnosis difficult, delaying early immunological intervention treatment and, therefore, patient prognosis. Thus, the present study sought to clarify diagnostic decision-making by characterizing the clinical manifestations and imaging features in patients who initially present with symptoms of brainstem involvement.
The differences in clinical and radiographical presentations described by this study may assist in differentiating between MS and NMOSD in clinical practice. Furthermore, much of our findings align with prior research. In terms of symptomatic presentations, previous research suggested that ophthalmoplegia is the most common abnormality in MS patients with brainstem lesions, specifically in the medial longitudinal fasciculus (28–30), while central hiccups and central vomiting are pathognomonic for NMOSD patients (31–33). These findings were consistent with the present study. Meanwhile, previous studies did not note that NMOSD patients had pyramidal symptoms. Our study found that both MS and NMOSD groups experienced pyramidal symptoms, with significantly greater association with NMOSD. On the other hand, previous studies had reported cases of pupillary miosis in patients with MS and NMOSD (34), but this phenomenon was not observed in the present study.
On MRI imaging, Jurynczyk et al. found that MS was distinct from myelin oligodendrocyte glycoprotein (MOG)-Antibody associated disease (MOGAD) and NMOSD, while it was difficult to distinguish MOGAD from NMOSD by imaging alone due to overlapping features. They further described that MS presented peripherally with ovoid lesions adjacent to the body of lateral ventricles (e.g., Dawson's fingers) or within the U fibers as T1 hypointense lesions. Meanwhile, they described MOGAD and NMOSD lesions as large, poorly demarcated (fluffy) brainstem lesions centrally located in pons and/or adjacent to the fourth ventricle, at the periaqueductal region or in area postrema. Overall, MOGAD/NMOSD had fewer lesions as compared to MS (13, 35). These aligned with our study's findings, which found that NMOSD has centrally located larger lesions in fewer quantities.
Similarly, Nakamura's group described that although both MS and NMO present with lesions in the corpus callosum, the qualities are distinctive. MS presented as small lesions at the corpus callosum, both acutely and chronically. Meanwhile, NMO only presented acutely as large, edematous legions with heterogeneous intensity, also described as a marble pattern. NMO lesions disappeared at the chronic stage (36). Meanwhile, in the spinal cord, MS presented peripherally in the lateral and posterior white matter regions, while NMO presented in the central gray matter; in the acute stage, NMO lesions covered more than half the cord (37). These descriptions aligned with our findings that MS tended to present as small, numerous lesions in the periphery, while NMO tended to present as large, fewer lesions in the central regions.
Matsumoto's group was able to tease apart MOGAD related imaging findings from NMOSD findings. They found that MOGAD had more subcortical white matter lesions of temporal lobe and cerebellar peduncle, and pyramidal and medial medulla lesions, while NMOSD had more lesions in dorsal medulla and area postrema. These aligned with our findings that NMOSD presented with lesions commonly in the medulla oblongata, although our data did not suggest that it was localized to a specific region as Matsumoto's group described (38).
Hayashida's group further elaborated on NMOSD's presentation in the spinal cord. They described that more than half of NMOSD lesions occupied the posterior and lateral columns, while only roughly a third was located in the anterior column, posterior horn, central portion, and anterior horn (39). Although these data agree with our findings that NMOSD lesions occupy gray matter, our study did not find the lesions to preferentially target white matter in the spinal cord columns.
In conclusion, it was found that most MS and NMOSD patients initially presenting with brainstem involvement are female, a fact which is magnified in NMOSD patients. NMOSD patients were more likely to be accompanied by autoimmune diseases or positive autoimmune antibody testing, and were more likely to exhibit central hiccups, central vomiting, and pyramidal tract signs. Lesions in NMOSD patients were mostly found in medulla oblongata, in the shape of large sheets, and were centrally located. Conversely, MS patients were more likely to experience ophthalmoplegia and MLF syndrome, and exhibit patchy, peripheral lesions mostly appearing on the midbrain and pons. Ultimately, these features may offer the means to create an algorithm that clinicians can follow in order to diagnose MS and NMOSD patients who present with brainstem lesions as the first manifestation, ultimately expediting initiation of treatment to improve overall prognosis.
The original contributions presented in the study are included in the article/supplementary material. Further inquiries can be directed to the corresponding author(s).
Ethical review and approval was not required for the study on human participants in accordance with the local legislation and institutional requirements. Written informed consent for participation was not required for this study in accordance with the national legislation and the institutional requirements. Written informed consent was not obtained from the individual(s) for the publication of any potentially identifiable images or data included in this article.
QJ, ZL, and YT prepared the manuscript. HD, YD, and XG designed the study. YD, KE, YH, and HL revised the manuscript. All authors reviewed the manuscript and provided the final approval for the manuscript.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
1. Wingerchuk DM, Banwell B, Bennett JL, Cabre P, Carroll W, Chitnis T, et al. International consensus diagnostic criteria for neuromyelitis optica spectrum disorders. Neurology. (2015) 85:177–89. doi: 10.1212/WNL.0000000000001729
2. Kawachi I, Lassmann H. Neurodegeneration in multiple sclerosis and neuromyelitis optica. J Neurol Neurosurg Psychiatry. (2017) 88:137–45. doi: 10.1136/jnnp-2016-313300
3. Fujihara K. Neuromyelitis optica spectrum disorders: still evolving and broadening. Curr Opin Neurol. (2019) 32:385–94. doi: 10.1097/WCO.0000000000000694
4. Thompson AJ, Banwell BL, Barkhof F, Carroll WM, Coetzee T, Comi G, et al. Diagnosis of multiple sclerosis: 2017 revisions of the mcdonald criteria. Lancet Neurol. (2018) 17:162–73. doi: 10.1016/S1474-4422(17)30470-2
5. Vargas DL, Tyor WR. Update on disease-modifying therapies for multiple sclerosis. J Investig Med. (2017) 65:883–91. doi: 10.1136/jim-2016-000339
6. Doshi A, Chataway J. Multiple sclerosis, a treatable disease. Clin Med (Lond). (2016) 16:s53–9. doi: 10.7861/clinmedicine.16-6-s53
7. Kimbrough DJ, Fujihara K, Jacob A, Lana-Peixoto MA, Leite MI, Levy M, et al. Treatment of neuromyelitis optica: review and recommendations. Mult Scler Relat Disord. (2012) 1:180–87. doi: 10.1016/j.msard.2012.06.002
8. Kuchling J, Paul F. Visualizing the central nervous system: imaging tools for multiple sclerosis and neuromyelitis optica spectrum disorders. Front Neurol. (2020) 11:450. doi: 10.3389/fneur.2020.00450
9. Kleiter I, Gold R. Present and future therapies in neuromyelitis optica spectrum disorders. Neurotherapeutics. (2016) 13:70–83. doi: 10.1007/s13311-015-0400-8
10. Borisow N, Mori M, Kuwabara S, Scheel M, Paul F. Diagnosis and treatment of nmo spectrum disorder and mog-encephalomyelitis. Front Neurol. (2018) 9:888. doi: 10.3389/fneur.2018.00888
11. Habek M. Evaluation of brainstem involvement in multiple sclerosis. Expert Rev Neurother. (2013) 13:299–311. doi: 10.1586/ern.13.18
12. Polman CH, Reingold SC, Banwell B, Clanet M, Cohen JA, Filippi M, et al. Diagnostic criteria for multiple sclerosis: 2010 revisions to the mcdonald criteria. Ann Neurol. (2011) 69:292–302. doi: 10.1002/ana.22366
13. Jurynczyk M, Geraldes R, Probert F, Woodhall MR, Waters P, Tackley G, et al. Distinct brain imaging characteristics of autoantibody-mediated cns conditions and multiple sclerosis. Brain. (2017) 140:617–27. doi: 10.1093/brain/aww350
14. Tatekawa H, Sakamoto S, Hori M, Kaichi Y, Kunimatsu A, Akazawa K, et al. Imaging differences between neuromyelitis optica spectrum disorders and multiple sclerosis: a multi-institutional study in Japan. AJNR Am J Neuroradiol. (2018) 39:1239–47. doi: 10.3174/ajnr.A5663
15. Klineova S, Lublin FD. Clinical course of multiple sclerosis. Cold Spring Harb Perspect Med. (2018) 8. doi: 10.1101/cshperspect.a028928
16. Reich DS, Lucchinetti CF, Calabresi PA. Multiple sclerosis. N Engl J Med. (2018) 378:169–80. doi: 10.1056/NEJMra1401483
17. Huda S, Whittam D, Bhojak M, Chamberlain J, Noonan C, Jacob A. Neuromyelitis optica spectrum disorders. Clin Med (Lond). (2019) 19:169–76. doi: 10.7861/clinmedicine.19-2-169
18. Cook LJ, Rose JW, Alvey JS, Jolley AM, Kuhn R, Marron B, et al. Collaborative international research in clinical and longitudinal experience study in nmosd. Neurol Neuroimmunol Neuroinflamm. (2019) 6:e583. doi: 10.1212/NXI.0000000000000583
19. Wallin MT, Culpepper WJ, Campbell JD, Nelson LM, Langer-Gould A, Marrie RA, et al. The prevalence of ms in the united states: a population-based estimate using health claims data. Neurology. (2019) 92:e1029–40. doi: 10.1212/WNL.0000000000007035
20. Asseyer S, Masuda H, Mori M, Bellmann-Strobl J, Ruprecht K, Siebert N, et al. Aqp4-igg autoimmunity in japan and germany: differences in clinical profiles and prognosis in seropositive neuromyelitis optica spectrum disorders. Mult Scler J Exp Transl Clin. (2021) 7:20552173211006862. doi: 10.1177/20552173211006862
21. Hor JY, Asgari N, Nakashima I, Broadley SA, Leite MI, Kissani N, et al. Epidemiology of neuromyelitis optica spectrum disorder and its prevalence and incidence worldwide. Front Neurol. (2020) 11:501. doi: 10.3389/fneur.2020.00501
22. Trebst C, Jarius S, Berthele A, Paul F, Schippling S, Wildemann B, et al. Update on the diagnosis and treatment of neuromyelitis optica: recommendations of the neuromyelitis optica study group (nemos). J Neurol. (2014) 261:1–16. doi: 10.1007/s00415-013-7169-7
23. Jarius S, Ruprecht K, Wildemann B, Kuempfel T, Ringelstein M, Geis C, et al. Contrasting disease patterns in seropositive and seronegative neuromyelitis optica: a multicentre study of 175 patients. J Neuroinflammation. (2012) 9:14. doi: 10.1186/1742-2094-9-14
24. Adawi M, Bisharat B, Bowirrat A. Systemic lupus erythematosus (sle) complicated by neuromyelitis optica (nmo - devic's disease): clinic-pathological report and review of the literature. Clin Med Insights Case Rep. (2014) 7:41–7. doi: 10.4137/CCRep.S15177
25. Pandit L, Asgari N, Apiwattanakul M, Palace J, Paul F, Leite MI, et al. Demographic and clinical features of neuromyelitis optica: a review. Mult Scler. (2015) 21:845–53. doi: 10.1177/1352458515572406
26. Quek AM, McKeon A, Lennon VA, Mandrekar JN, Iorio R, Jiao Y, et al. Effects of age and sex on aquaporin-4 autoimmunity. Arch Neurol. (2012) 69:1039–43. doi: 10.1001/archneurol.2012.249
27. Matthews L, Kolind S, Brazier A, Leite MI, Brooks J, Traboulsee A, et al. Imaging surrogates of disease activity in neuromyelitis optica allow distinction from multiple sclerosis. PLoS ONE. (2015) 10:e0137715. doi: 10.1371/journal.pone.0137715
28. Zabad RK, Stewart R, Healey KM. Pattern recognition of the multiple sclerosis syndrome. Brain Sci. (2017) 7. doi: 10.3390/brainsci7100138
29. Nerrant E, Tilikete C. Ocular motor manifestations of multiple sclerosis. J Neuroophthalmol. (2017) 37:332–40. doi: 10.1097/WNO.0000000000000507
30. Miller DH, Weinshenker BG, Filippi M, Banwell BL, Cohen JA, Freedman MS, et al. Differential diagnosis of suspected multiple sclerosis: a consensus approach. Mult Scler. (2008) 14:1157–74. doi: 10.1177/1352458508096878
31. Kremer L, Mealy M, Jacob A, Nakashima I, Cabre P, Bigi S, et al. Brainstem manifestations in neuromyelitis optica: a multicenter study of 258 patients. Mult Scler. (2014) 20:843–47. doi: 10.1177/1352458513507822
32. Cheng C, Jiang Y, Lu X, Gu F, Kang Z, Dai Y, et al. The role of anti-aquaporin 4 antibody in the conversion of acute brainstem syndrome to neuromyelitis optica. BMC Neurol. (2016) 16:203. doi: 10.1186/s12883-016-0721-1
33. Popescu BF, Lennon VA, Parisi JE, Howe CL, Weigand SD, Cabrera-Gómez JA, et al. Neuromyelitis optica unique area postrema lesions: nausea, vomiting, and pathogenic implications. Neurology. (2011) 76:1229–37. doi: 10.1212/WNL.0b013e318214332c
34. Uludag I F, Sariteke A, Öcek L, Zorlu Y, Sener U, Tokuçoglu F, et al. Neuromyelitis optica presenting with horner syndrome: a case report and review of literature. Mult Scler Relat Disord. (2017) 14:32–4. doi: 10.1016/j.msard.2017.03.011
35. Juryńczyk M, Tackley G, Kong Y, Geraldes R, Matthews L, Woodhall M, et al. Brain lesion distribution criteria distinguish ms from aqp4-antibody nmosd and mog-antibody disease. J Neurol Neurosurg Psychiatry. (2017) 88:132–36. doi: 10.1136/jnnp-2016-314005
36. Nakamura M, Misu T, Fujihara K, Miyazawa I, Nakashima I, Takahashi T, et al. Occurrence of acute large and edematous callosal lesions in neuromyelitis optica. Mult Scler. (2009) 15:695–700. doi: 10.1177/1352458509103301
37. Nakamura M, Miyazawa I, Fujihara K, Nakashima I, Misu T, Watanabe S, et al. Preferential spinal central gray matter involvement in neuromyelitis optica. An MRI study. J Neurol. (2008) 255:163–70. doi: 10.1007/s00415-008-0545-z
38. Matsumoto Y, Misu T, Mugikura S, Takai Y, Nishiyama S, Kuroda H, et al. Distinctive lesions of brain mri between mog-antibody-associated and aqp4-antibody-associated diseases. J Neurol Neurosurg Psychiatry. (2020). doi: 10.1136/jnnp-2020-324818. [Epub ahead of print].
Keywords: brainstem, multiple sclerosis, neuromyelitis optica spectrum disease, demyelination, image characterization
Citation: Ji Q, Dong H, Lee H, Liu Z, Tong Y, Elkin K, Haddad Y, Geng X and Ding Y (2022) Clinical Characteristics and Outcomes of Multiple Sclerosis and Neuromyelitis Optica Spectrum Disorder With Brainstem Lesions as Heralding Prodrome. Front. Neurol. 13:836337. doi: 10.3389/fneur.2022.836337
Received: 13 January 2022; Accepted: 28 February 2022;
Published: 09 May 2022.
Edited by:
Todd Hardy, Concord Repatriation General Hospital, AustraliaReviewed by:
Katsuhisa Masaki, University of Chicago Medical Center, United StatesCopyright © 2022 Ji, Dong, Lee, Liu, Tong, Elkin, Haddad, Geng and Ding. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Huiqing Dong, eHVlNzMxM0AxMjYuY29t; Xiaokun Geng, eGdlbmdAY2NtdS5lZHUuY24=
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.