
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
SYSTEMATIC REVIEW article
Front. Neurol. , 11 March 2022
Sec. Epilepsy
Volume 13 - 2022 | https://doi.org/10.3389/fneur.2022.832380
 Jiangwei Ding1,2†
Jiangwei Ding1,2† Lei Wang1,3†
Lei Wang1,3† Zhe Jin1,4†Yuanyuan Qiang1
Zhe Jin1,4†Yuanyuan Qiang1 Wenchao Li1,3
Wenchao Li1,3 Yangyang Wang1,3
Yangyang Wang1,3 Changliang Zhu1,2
Changliang Zhu1,2 Shucai Jiang1,2
Shucai Jiang1,2 Lifei Xiao1,2Xiaoyan Hao5Xulei Hu1,2
Lifei Xiao1,2Xiaoyan Hao5Xulei Hu1,2 Xinxiao Li6*
Xinxiao Li6* Feng Wang1,7*
Feng Wang1,7* Tao Sun1,2*
Tao Sun1,2*Background: Dravet syndrome (DS) is a severe epileptic encephalopathy mainly caused by haploinsufficiency of the gene SCN1A, which encodes the voltage-gated sodium channel NaV1. 1 in the brain. While SCN1A mutations are known to be the primary cause of DS, other genes that may cause DS are poorly understood. Several genes with pathogenic mutations result in DS or DS-like phenotypes, which may require different drug treatment approaches. Therefore, it is urgent for clinicians, especially epilepsy specialists to fully understand these genes involved in DS in addition to SCN1A. Particularly for healthcare providers, a deep understanding of these pathogenic genes is useful in properly selecting and adjusting drugs in a more effective and timely manner.
Objective: The purpose of this study was to identify genes other than SCN1A that may also cause DS or DS-like phenotypes.
Methods: A comprehensive search of relevant Dravet syndrome and severe myoclonic epilepsy in infancy was performed in PubMed, until December 1, 2021. Two independent authors performed the screening for potentially eligible studies. Disagreements were decided by a third, more professional researcher or by all three. The results reported by each study were narratively summarized.
Results: A PubMed search yielded 5,064 items, and other sources search 12 records. A total of 29 studies published between 2009 and 2021 met the inclusion criteria. Regarding the included articles, seven studies on PCDH19, three on SCN2A, two on SCN8A, five on SCN1B, two on GABRA1, three on GABRB3, three on GABRG2, and three on STXBP1 were included. Only one study was recorded for CHD2, CPLX1, HCN1 and KCNA2, respectively. It is worth noting that a few articles reported on more than one epilepsy gene.
Conclusion: DS is not only identified in variants of SCN1A, but other genes such as PCDH19, SCN2A, SCN8A, SCN1B, GABRA1, GABRB3, GABRG2, KCNA2, CHD2, CPLX1, HCN1A, STXBP1 can also be involved in DS or DS-like phenotypes. As genetic testing becomes more widely available, more genes associated with DS and DS-like phenotypes may be identified and gene-based diagnosis of subtypes of phenotypes in this spectrum may improve the management of these diseases in the future.
Dravet syndrome (DS), also known as severe myoclonic epilepsy in infancy (SMEI), is a catastrophic developmental and epileptic encephalopathy with onset in infancy that was initially described and reported by Dravet in 1978 (1) to distinguish it from Lennox–Gastaut syndrome (LGS). As increasing reports found that patients did not present with myoclonic seizures and that seizures appeared not to be limited to infancy and childhood but persisted through adulthood (2), the International League Against Epilepsy named DS as a distinct syndrome in 1989 (3). The estimated incidence of DS ranges from 1 in 20,000 to 1 in 40,000 (4).
DS is often associated with developmental delays, severe cognitive deficits, sleep disorders, behavioral disorders, including autistic-like behavior, and increased risk of sudden unexpected death in epilepsy (SUDEP; nearly 20%) (5–7). DS is a severe developmental and epileptic encephalopathy typically caused by loss-of-function de novo mutations in the SCN1A gene that encodes the voltage-gated sodium channel isoform NaV1.1 (8–10).
The voltage-gated sodium channel comprises a primary α subunit (composed of six transmembrane domains, NaV1.1–NaV1.9, encoded by the genes SCN1A–SCN11A) and a secondary β subunit (a single transmembrane domain, β1–β4, encoded by the SCN1B–SCN4B genes (11). The α subunit forms the sodium channel pore and is the target of various antiepileptic drugs. The β subunits interact with α subunit and modulate the localization of alpha subunits and channel properties (10, 12).
NaV1.1 encoded by SCN1A is a membrane protein of 2,009 amino acids formed by four domains each containing six transmembrane segments. In the central nervous system, NaV1.1 is mainly expressed in cell bodies and dendrites, but also at the axon initial segments of some interneurons, which play a very important role in the generation and propagation of action potential (13–15). Mutations in the SCN1A gene lead to a significant decrease in sodium current in GABAergic interneurons, which influences GABA inhibitory function and leads to neuronal hyperexcitability and seizures (16, 17). In addition, reduced sodium current can also affect Purkinje cells, resulting in motor dysfunction, and can lead to behavioral problems and cognitive dysfunction (16, 18).
SCN1A mutation is the most common pathogenic factor of DS, and about 80% of DS patients have SCN1A mutations (19, 20). More than 1,800 pathogenic SCN1A variants have been identified to date (21). Most variants are de novo, but there are individuals who carry the SCNA1 mutation in one or both parents <10%; (19, 22). Patients with SCN1A mutations have varying manifestations, ranging from mild and drug-responsive epilepsy, such as genetic epilepsy with febrile seizures plus (GEFS+), to developmental and epileptic encephalopathy, including DS, West syndrome, and myoclonic-atonic epilepsy (23–25). Since about 80% of DS is caused by SCN1A mutations (20, 26), clinicians have sufficient knowledge of the association between SCN1A and DS, but DS caused by genes other than SCN1A are insufficiently understood and little is known about them (26). Herein, we review and discuss the latest evidence on other genes that are potentially involved in DS or DS-like phenotypes.
Using PubMed, we conducted a systematic literature search of studies published up to December 1, 2021, selecting research that examined genes other than SCN1A that are associated with DS. The search terms were selected from the thesaurus of the National Library of Medicine (Medical Subject Heading Terms, MeSH) and included the terms “Dravet syndrome” and “severe myoclonic epilepsy in infancy.” The final search equation was defined using the Boolean connectors “OR” following the formulation “Dravet syndrome” OR “severe myoclonic epilepsy in infancy.” The search included English or Chinese-language articles published from November 11, 1947, to November 27, 2021, and did not include any subheadings or tags (i.e., search fields “All fields”; Figure 1).
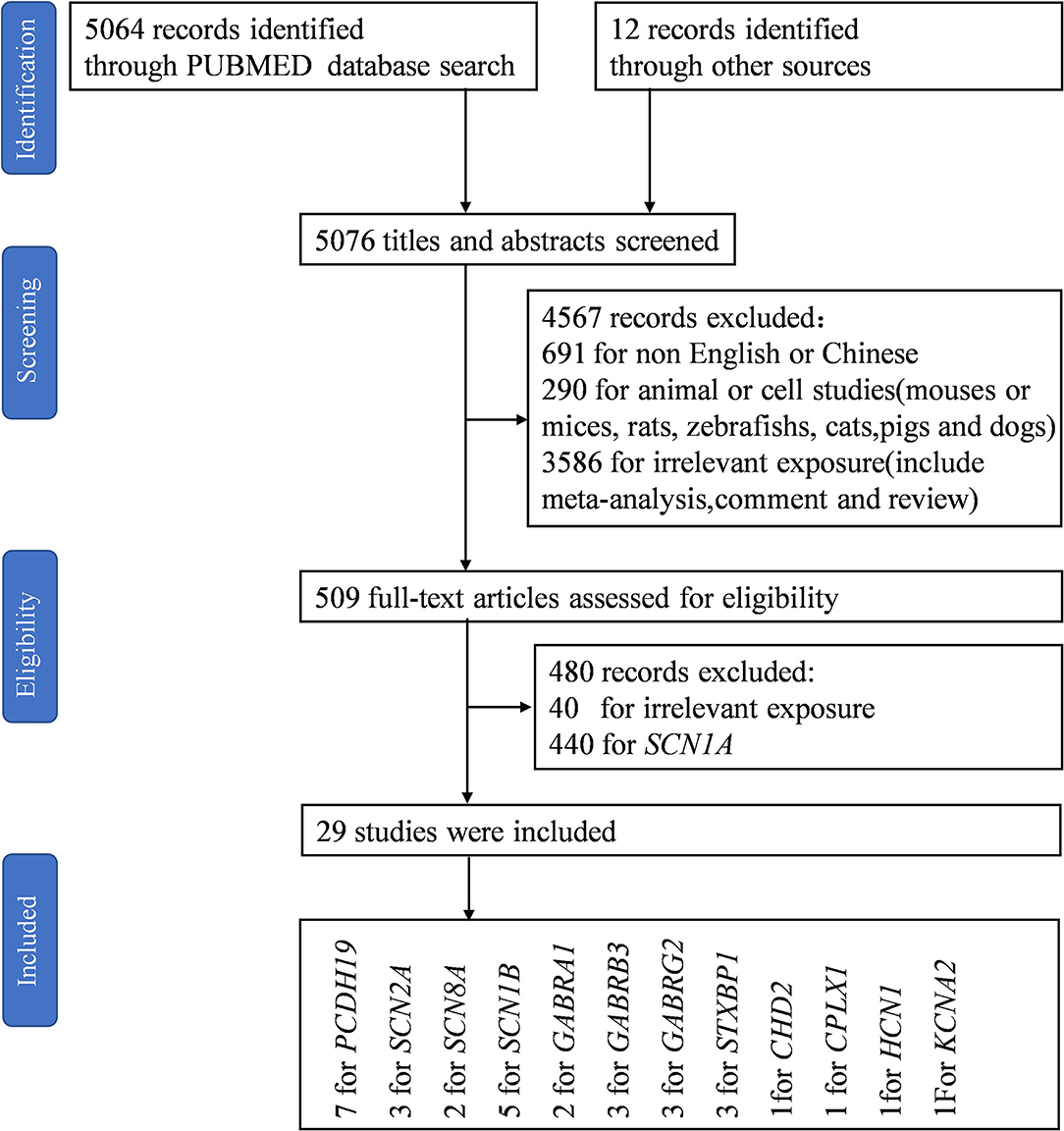
Figure 1. Flow diagram of included studies. “PCDH19, SCN8A, and STXBP1” and “GABRA1 and STXBP1” appeared in the same study, respectively (27, 28). Liu et al. reported the same epilepsy gene in two papers (29, 30).
Our objective was to investigate the infrequent or rare pathogenic genes of DS; thus, all genes associated with DS except SCN1A met the inclusion criteria. We excluded articles not written in English or Chinese, non-original work that was unrelated to human subjects, such as reviews, meta-analyses, animal or cell subjects, and experimental articles without information related to the question posed in this review. Records were screened by Jiangwei Ding and evaluated by Lei Wang with respect to the inclusion and exclusion criteria. Discrepancies were discussed and decided by Tao Sun or all three.
Of 5,076 references identified in the initial search in the electronic databases, the study selection process resulted in the inclusion of a total of 29 studies in the narrative synthesis (Figure 1).
The study characteristics included in this review are shown in Table 1, Figure 2. All studies involved DS or SMEI and were published from 2009 to 2021 in English or Chinese. Regarding the studies included, six studies on PCDH19 (27, 29–34), three on SCN2A (36–38), two on SCN8A (27, 39), five on SCN1B (40–44), two on GABRA1 (28, 45), three on GABRB3 (47–49), three on GABRG2 (50–52), and three on STXBP1 (27, 28, 46) were included. Only one study was recorded for CHD2 (54), CPLX1, HCN1, and KCNA2, respectively (35, 53, 55, 56). We mapped the available mutations onto the protein topologies (protein topologies from Protter - interactive protein feature visualization (ethz.ch).
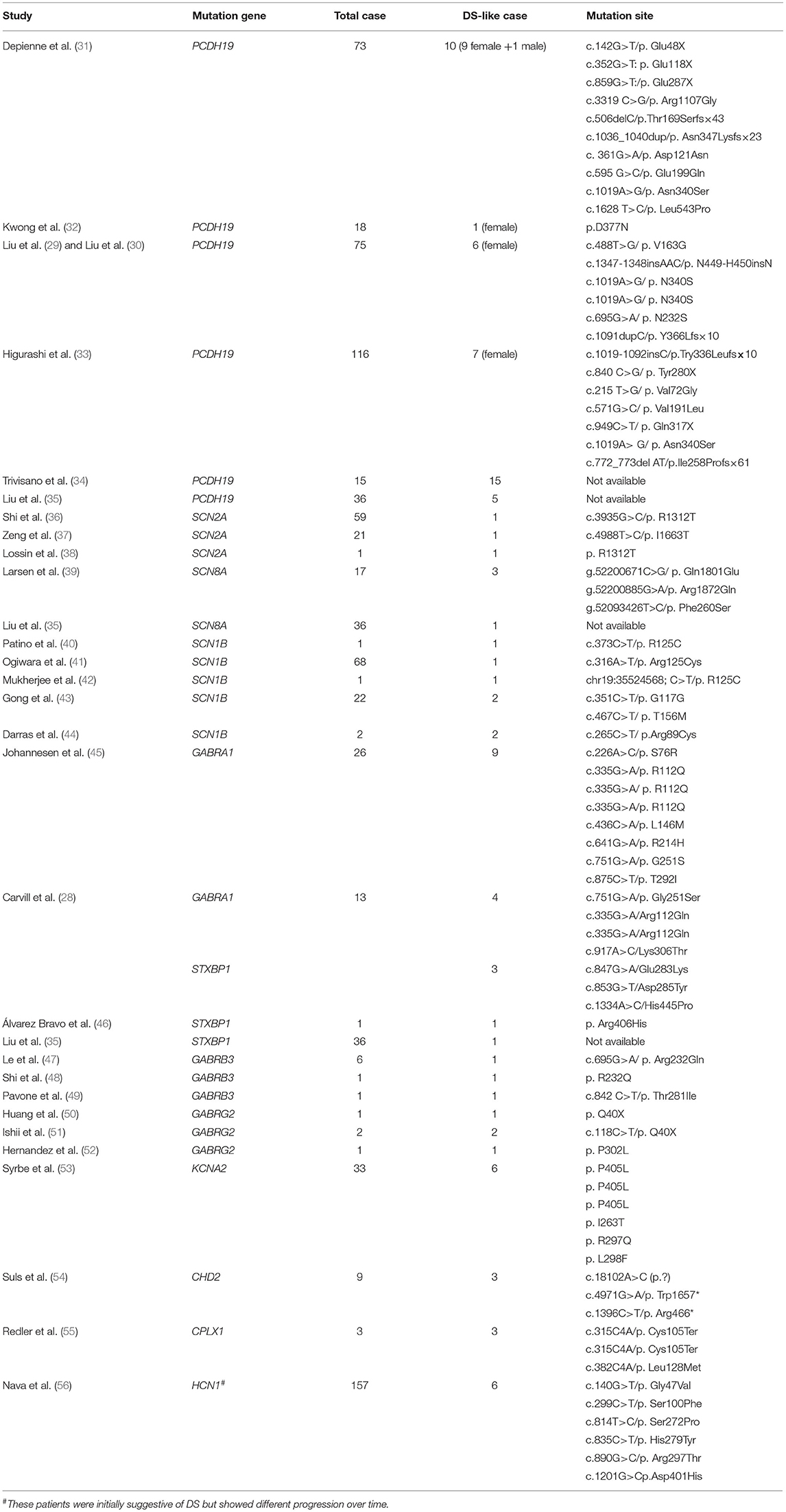
Table 1. The data of DS-like caused by multiple single-gene mutations that met the inclusion criteria were summarized.
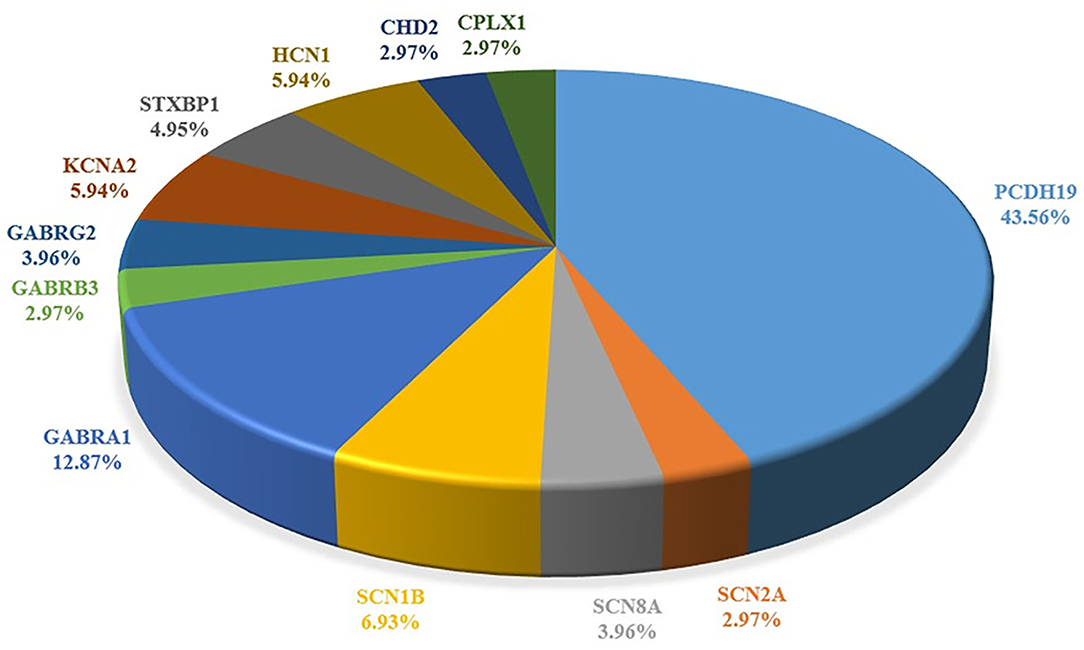
Figure 2. Summary of the pathogenic genes of Dravet syndrome. Comparison of proportion of each gene in literature reports.
DS is an early infantile epileptic and developmental encephalopathy and is mostly caused by loss-of-function mutations in the brain sodium channel NaV1.1 (6). SCN1A variants are known to cause a variety of other diseases, including epilepsy (Doose syndrome, epilepsy of infancy with migrating focal seizures, west syndrome, LGS, and Rett syndrome) and non-epileptic disorders (familial/sporadic hemiplegic migraines, autism spectrum disorder, sudden infant death syndrome, and arthrogryposis multiplex congenita) in addition to DS (24, 57–60). Conversely, could DS be caused by mutations in a variety of genes other than SCN1A? The answer seems to be clear from the data in the articles included in our study, which indicate that at least 13 genes, including SCN1A, have been identified as directly involved in DS to date.
According to our data, in addition to SCN1A, there are 12 genes that can cause DS or DS-Like phenotypes, among which PCDH19 is the most common, making it the second-most common pathogenic gene of DS. The other genes were three sodium channel–related genes (SCN2A, SCN8A, and SCN1B), one potassium ion channel–related gene (KCNA2), three gamma-aminobutyric acid receptors (GABAR) genes (GABRA2, GABRB3, and GABRG2), one cyclic nucleotide gated cation channel gene (HCN1), and other functional genes including CHD2, CPLX1, and STXBP1 (Table 1; Figure 2).
PCDH19, located on chromosome XQ22.1, encodes the protocadherin-19 and is the second-most pathogenic gene of DS, accounting for 23.4% of patients with SCN1A-negative DS (61). The product of PCDH19 is protocadherin-19, a single-channel transmembrane glycoprotein, which is the largest subgroup of the cadherin superfamily, and plays an important role in cell adhesion, dendritic self-avoidance, and axon guidance. PCDH19 consists of six exons and encodes 1,148 amino acids (Figure 3). This protein includes one signal peptide, six extracellular cadherin repeats, one transmembrane region, and one cytoplasmic region. Exon 1 encodes more than half of the proteins, including all extracellular and transmembrane domains. PCDH19 is highly conserved in humans, mice, zebrafish, and chickens, and is highly expressed in central nervous system tissues at different developmental stages, including the subventricular area, intermediate area, inferior plate, specific cerebral cortex, hippocampus, and cerebellum. It has been speculated that PCDH19 is involved in the establishment of neuronal connections and the signal transmission of synaptic membranes (62). PCDH19 gene mutation was first reported by Dibbens et al. (63) in seven families with epilepsy and intellectual disability confined to women. PCDH19-related DS main clinical feature is epilepsy with intellectual disability and onset in infancy or childhood. It is a special X-linked hereditary epilepsy, and the heterozygous female family members carrying the PCDH19 mutation are affected. Hemizygous men with the mutation are not affected. However, it has been subsequently reported that men with chimeric mutations of PCDH19 also develop the disease (31).
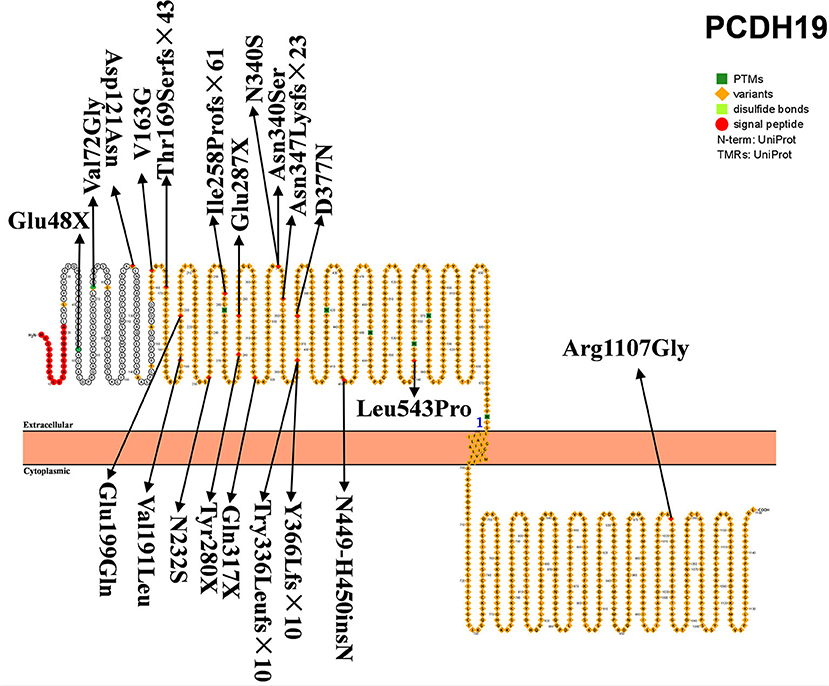
Figure 3. Schematic diagram of protein encoded by PCDH19, illustrating the location of the amino acids affected by the mutations identified in patients.
Depienne et al. found the mutation of PCDH19 in 15% of SCN1A-negative DS patients. This was the first time the relationship between PCDH19 and DS was described and analyzed, indicating that PCDH19 may be a DS or DS-like pathogenic gene (31). Subsequently, a large number of reports and descriptions of PCDH19 mutation causing DS were published (33, 34). A recent review showed that the incidence of DS with SCN1A mutation was significantly higher than that of DS patients with PCHDH19 mutation (76.6 vs. 23.4%), while the incidence of autism was higher in DS patients with PCDH19 variants [62.5 vs. 37.5%, respectively; (61)]. The patients with PCDH19 variance had obvious tendencies toward hyper-febrile seizures. Patients with PCDH19 and SCN1A mutations had very similar clinical characteristics, including the association of early febrile and non-febrile seizures, cluster seizures, developmental and language delays, behavioral disorders, and cognitive degradation (32). However, PCDH19 mutations rarely reveal status epilepticus, photosensitive seizures, clonic seizures, myoclonic seizures, and absence seizures. Furthermore, the seizures gradually decrease after school age, and the prognosis is generally better than that of DS. PCDH19 is currently considered to be the second-most common pathogenic gene for DS (but much less common than SCN1A).
The SCN2A gene, encoding voltage-gated sodium channel α subunit NaV1.2, is located on chromosome 2q24.3 and contains 26 exons and encodes 2,005 amino acids (Figure 4A). NaV1.2 is widely distributed in the brain and highly expressed in the cortex, hippocampus, striatum, and midbrain (64). SCN2A is the main pathogenic gene of benign familial neonatal–infantile epilepsy (BFNIE) (65). SCN2A mutations can lead to a wide clinical spectrum of epilepsy, such as benign familial neonatal epilepsy (BFNE), benign familial infantile epilepsy (BFIE), and developmental epileptic encephalopathy such as DS, Ohtahara syndrome, and epilepsy of infancy with migrating focal seizures (EIMFS), West syndrome, Doose Syndrome, LGS, and unclassified epileptic encephalopathy (38, 66, 67).
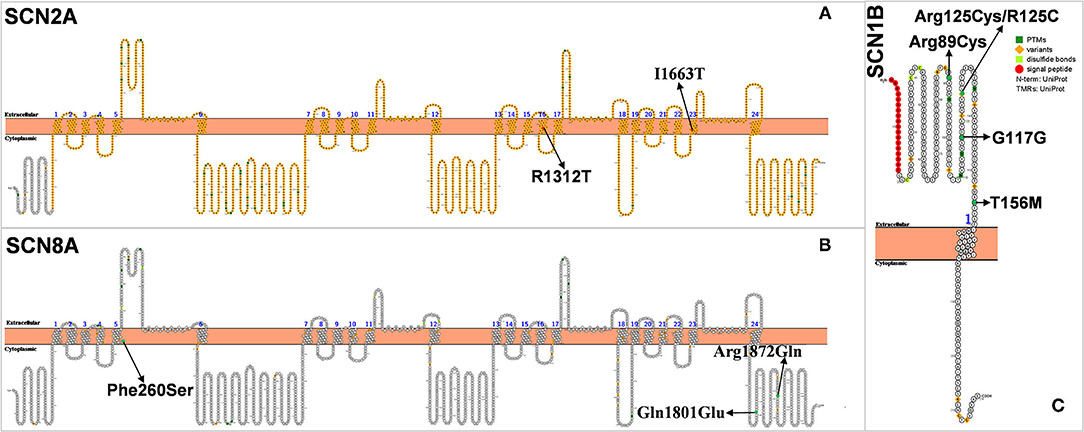
Figure 4. (A–C) Schematic diagram of protein encoded by SCN2A, SCN8A, and SCN1B, illustrating the location of the amino acids affected by the mutations identified in patients.
In 2009, Japanese scholars Shi et al. found for the first time three SCN2A missense mutations in 59 patients with DS, one of which was a de novo SCN2A mutation (36). In 2012, Lossin et al. also found that SCN2A mutation (c.3935G>C/p. R1312T) could cause DS. This mutation is thought to affect the position of arginine at the channel voltage sensor, which in turn causes conformational changes in the ion channel and opens the otherwise closed channel pore, which may be responsible for the pathogenesis of DS (38). Chinese scholars Zeng et al. retrospectively analyzed 21 patients with SCN2A mutation and found that one of them was diagnosed with DS (37). The gene mutation in this patient was inherited from his mother. The onset of the disease was at 6 months of life. The patient mainly had generalized tonic-clonic seizures (GTCS), myoclonic seizures (MS), and febrile seizures (FS). His symptoms did not improve at the 42-month follow-up, and his intellectual and motor development was delayed. These studies suggest that SCN2A is one of the epileptogenic genes in DS.
SCN8A, encoding voltage-gated sodium channel NaV1.6, is located on chromosome 12q13.13 (Figure 4B) (68). NaV1.6 is the main voltage-gated sodium channel in the mammalian central nervous system. It is highly concentrated at the axon hillock and plays an important role in depolarization during the generation of neuronal action potential (69). Sodium-channel sequences are highly conserved during evolution, and even small changes in biophysical properties can have significant effects in vivo (70). In 2012, Veeramah et al. first discovered and reported the mutation of SCN8A(c.5302A>G/p.Asn1768Asp), which is also the fifth sodium-channel gene involved in epilepsy (68). More than 140 cases of SCN8A mutation-associated epilepsy have been reported in children (71).
Within SCN8A-related encephalopathy, individuals have been diagnosed with syndromes including unclassified EE (epileptic encephalopathy), early infantile EE, LGS, West syndrome, and DS-like. Similar to DS, children with SCN8A epilepsy also present with speech problems and intellectual retardation/developmental delay (72). In 2015, Larsen et al. found three DS-like patients with SCN8A mutation (39). The core symptoms of the SCN8A-related DS-like phenotype are markedly different from those of the typical Dravet syndrome caused by the SCN1A mutation. The average age of onset of DS-like caused by SCN8A mutation in the three patients was 4.6 months (3, 7, and 4 months, respectively), earlier than that of the typical DS. Only one patient had fever-induced seizures and a few patients had convulsive seizures. In contrast, typical DS often starts with fever-induced seizures and rarely presents with convulsive seizures. The development of the patient is normal before the onset of seizures, but when seizures start occurring, mental, motor, and language skills show gradual retardation, and autistic symptoms similar to those of DS. Recently, Liu et al. found an SCN8A mutation in 36 DS patients but did not describe the symptoms (27).
DS is caused by a dominant loss-of-function mutation in SCN1A that leads to reduced NaV1.1 activity, resulting in insufficient excitability of GABAergic neurons, while the gain of function mutations in SCN8A can lead to severe epileptic encephalopathy subtypes through overactivation of the NaV1.6 channel and of glutamatergic neurons (73). These observations suggest that NaV1.1 and NaV1.6 represent two opposing aspects of the neural balance between inhibition and activation. This difference may be responsible for the effect of sodium channel blockers such as oxcarbazepine and lamotrigine on SCN8A-related epilepsy while exacerbating SCN1A-related epilepsy.
SCN8A can also be considered as a modifier gene of SCN1A-induced DS. To test this hypothesis, Martin and his colleagues generated Scn1a (+/-); Scn8a(med-jo/+) in double-heterozygous mice. They found that Scn1a (+/-); Scn8a(med-jo/+) mice had a similar susceptibility to epilepsy as wild-type mice, unlike Scn1a (+/-) mice. The Scn8a(med-jo) allele was also able to rescue the premature lethality of Scn1a(+/-) mice and extended the lifespan of Scn1a(-/-) mutants (74). Lenk et al. (75) recently used antisense oligonucleotide (ASO) to reduce the transcription of Scn8a in mice with epileptic encephalopathy and SCN1A-related DS and found a 25–50% reduction in delayed seizure onset and lethality, which was consistent with the results of Martin et al. (74, 75). It was further confirmed that SCN8A is not only an epilepsy gene, but also a modification gene of epilepsy.
SCN1B, encoding the voltage-gated sodium channel (VGSC) β1 non–pore-forming subunits, is the first gene confirmed to be associated with GEFS+ pathogenesis located on 19q13.11 (76, 77) (Figure 4C). The mutation of SCN1B plays an important role in the pathogenesis of febrile seizures plus spectrum diseases and is associated with epilepsy syndromes including febrile seizures (FS), GEFS+ (77, 78). The reported mutations of epileptogenic SCN1B are all missense mutations except for one splice-site mutation. Heterozygous missense mutations mostly cause FS or GEFS+ with mild symptoms, while homozygous missense mutations lead to DS severe symptoms (40–43).
The relationship between SCN1B and DS has been the focus since Patino et al. reported that the first patient with SCN1B mutant showed DS-like symptoms (40). They reported a case of homozygous SCN1B mutation in an infant with heat-dependent seizures triggered by vaccination and fever, consistent with some of the characteristics of DS. However, the age of onset at 3 months, abnormal electrical discharge on EEG, psychomotor deterioration at 5 months, and eventual death from respiratory pneumonia at about 14 months all preceded and were more severe than DS (40). Mukherjee et al. also reported on a patient with an SCN1B mutation (42). The patient had an onset of GTCS from 3 months of age after vaccination. Seizures then recurred at 4 months of age due to fever, by which time a global developmental delay had occurred. The age of onset of epilepsy and cognitive dysfunction in this patient were prematurely earlier than that of DS. According to the diagnosis of DS, the onset is about 6–12 months, and cognitive functioning may deteriorate until 1–2 years old. However, the authors suggest that, as the child was having post-vaccinal convulsions, a possible diagnosis of DS was considered, which is clearly inappropriate (42). Aeby et al. recently reported a patient with SCN1B mutation who was hypotonic at birth and presented with multiple seizures (multifocal myoclonus, focal seizures, and myoclonic status epilepticus) at 2.5–3 months of age, associated with fever. The patient's auditory brainstem response (ABR) showed bilateral hearing loss. After taking fenfluramine, the frequency of seizures was significantly reduced, and status epilepticus disappeared after 2 years of follow-up. The authors diagnosed it as early infantile developmental and epileptic encephalopathy (DEE) rather than DS (79).
Nevertheless, some cases with SCN1B mutations that could be considered as DS have been reported. Ogiwara et al. found one case with a homozygous SCN1B mutation in 67 patients with DS but without SCN1A and SCN2A mutations (41). Gong et al. also discovered two SCN1B mutations in 22 DS patients without SCN1A mutations (43). Recently, Darras et al. identified two DS patients with SCN1B mutations. However, these two patients had mild symptoms that differed markedly from the previous cases reported by Patino and Mukherjee et al. (40, 42). O'Malley et al. reported that adult Scn1b knockout mice showed DS-like phenotype such as epilepsy and SUDEP (80). In summary, SCN1B mutation is not a common cause of DS-like, if any (81).
The KCNA2 gene encoding the Kv1.2 channel is located on chromosome 1p13.3, with a total length of about 3,100 bp, containing three exons and two introns, and encoding 499 amino acids (Figure 5). The Kv1.2 channel consists of four subunits with six transmembrane (S1–S6). The transmembrane segments S1–S4 form a voltage sensor domain, and S5–S6 form a pore region containing selective filters and gate ion flow (82, 83). Kv1.2 is mainly expressed in the axons and presynaptic terminals of neurons (84). In 2015, Pena and Coimbra (85) performed exome analysis in a 7-year-old boy who presented with ataxia and myoclonic epilepsy as an infant and identified a novel KCNA2 mutation (c.890C>A/p. Arg297Gln). In the same year, Syrbe et al. identified four different de novo variants in six patients (one mutation recurred three times independently) with epileptic encephalopathy, including one with clinical symptoms similar to DS (53). In animal model studies, Kcna2 knockout mice were found to be more prone to epilepsy and premature death than wild-type mice, which further confirms the important role of KCNA2 in the pathogenesis of epilepsy including DS (86).
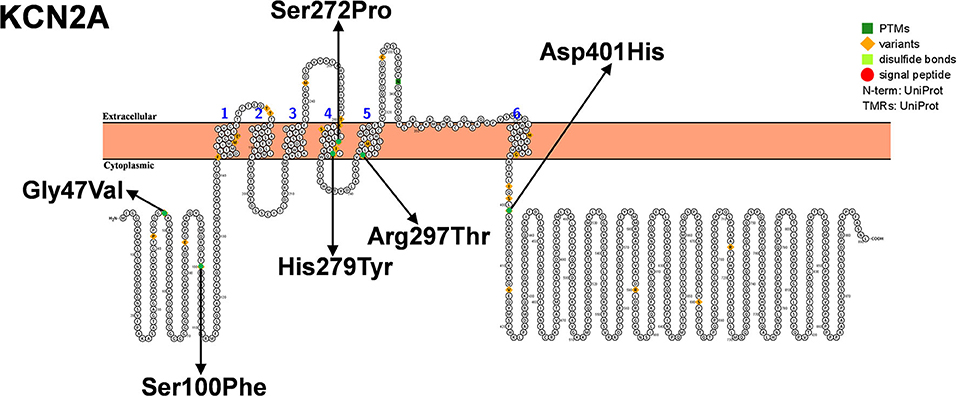
Figure 5. Schematic diagram of protein encoded by KCN2A, illustrating the location of the amino acids affected by the mutations identified in patients.
Gamma-aminobutyric acid receptors type A (GABAAR) are ligand-gated chloride ion channels that mediate synaptic inhibition in the brain. GABAAR is a hetero-pentameric protein complex composed of different subunits (α1-6, β1-3, γ1- 3, δ, ε, π, θ, and ρ1-3) (87, 88). The combination α1β2γ2 is thought to be the most abundant form of receptor in the brain. In 2002, Cossette et al. (89) first discovered a mutation of the GABRA1 encoding the α1 subunit (Ala322Asp) in an idiopathic generalized epilepsy (IGE) patient (Figure 6A). Furthermore, they constructed cells carrying Ala322Asp mutation and found that cells expressing the mutant GABAAR (α1Ala322Aspβ2γ2) had a lower amplitude of GABA-activated currents than those expressing the wild-type (α1β2γ2) receptor. Similarly, LaChance-Touchette et al. also found GABRA1 mutations in IGE (90). In a recent study, a GABRA1-deficient zebrafish model exhibited reduced activity and defective expression of other GABAAR subunits (91). In addition to IGE, GABRA1 mutations are also closely associated with Ohtahara syndrome and West syndrome, suggesting that GABRA1 is not only involved in mild genetic generalized epilepsies and febrile seizures, but is also associated with early-onset epileptic encephalopathies [EOEEs; (28, 92)].
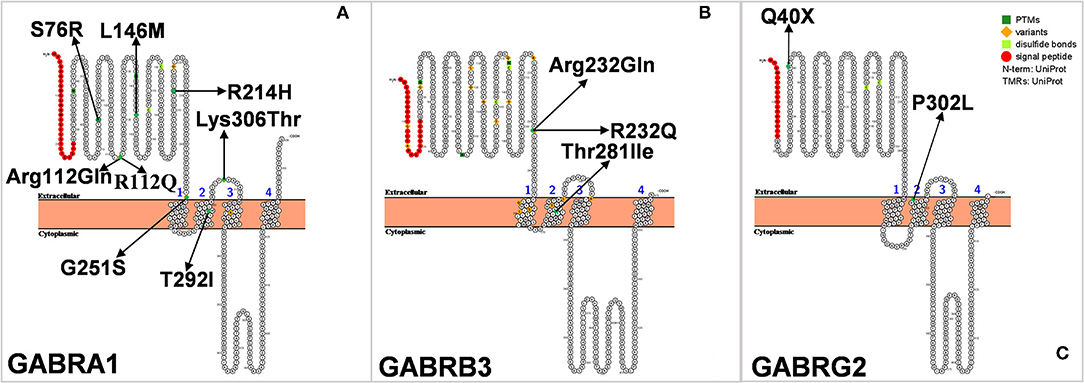
Figure 6. (A–C) Schematic diagram of protein encoded by GABRA1, GABRB3, and GABRG2, illustrating the location of the amino acids affected by the mutations identified in patients.
GABRA1 mutation is also rare in Dravet syndrome. In 2014, Carvill et al. sequenced the genes of seven patients with SCN1A-negative DS and found that four of them were associated with GABRA1 mutation. The four patients had an average age of onset of epilepsy at 9 months of age, and all their seizures were associated with fever. Two of the patients had developmental delays, which might have contributed to the diagnosis of DS, but their full clinical details have not been published (28). In 2016, Johannesen et al. evaluated 26 patients with epilepsy caused by GABRA1 mutation, of whom five were diagnosed with DS and four were diagnosed with DS-like. Febrile seizures are the first symptom in most patients diagnosed with DS, whereas tonic-clonic and hemiclonic seizures are the main symptoms of DS-like disorders. The age of onset of DS was later than that of DS-like disorder (9.5 months vs. 6.3 months) (45). All of the above studies directly verified the correlation between GABRA1 and DS or DS-like.
GABRB3 is located in the 15q11.2-q12 region and encodes theβ3 subunit of the GABAAR (Figure 6B). A large number of reports have indicated that the variation of GABRB3 may be related to the occurrence of childhood absence epilepsy (CAE) (93). In addition, the reduced expression of GABRB3 is also closely associated with Rett syndrome, Angelman syndrome, and autism spectrum disorders (94). The common characteristics of these diseases are epilepsy and neurodevelopmental delay. DS is a refractory epileptic encephalopathy, accompanied by febrile seizures, spontaneous epilepsy, intellectual disability, autism, and other complications. Could mutations in GABRB3 also cause DS? In 2017, Le et al. carried out exome sequencing on six patients with SCN1A-negative DS and found a new heterozygous missense variant of GABRB3, and thus GABRB3 was considered to be a candidate gene for DS (47). Recently, Piero et al. also found a novel GABRB3 missense mutation in an 18-year-old girl with DS (49). The patient experienced two febrile seizures when she was 7 months old. Electroencephalography revealed bilateral slow waves mainly in the occipital regions. The seizures were poorly controlled with oral antiepileptic drugs, and progressive rapid motor and cognitive impairments appeared after several months. While growing up, her social skills were still poor, and her intelligence quotient (IQ) was low (IQ = 42). These prompted the authors to make a diagnosis of DS. Unlike patients with typical DS, the patient was found to have dysmorphism of the spine with dorsal hyperkyphosis at the age of 15 months.
The GABRG2 gene, located on chromosome 5, encodes the γ2 subunit of GABAAR (Figure 6C), which plays an important role in receptor transport to the membrane and aggregation in the postsynaptic membrane. In 2002, Kananura et al. first found the splicing donor mutation of the GABRG2 gene (IVS6+2T → G) in a patient with childhood absence epilepsy and febrile seizures as the main clinical manifestations (95). We have previously found that mice selectively knocked out of Gabrg2 had temperature-dependent epilepsy (96). Up to now, about 20 mutations of GABRG2 have been reported, including Q40X, Q390X, R136X, W429X, Q40X, R136X, Q390X, and W429X. Mice carrying the GABRG2 mutation (P.R82Q) showed less cortical inhibition, spontaneous spiny discharges, and heat-induced seizures (97). GABRG2 mutations result in variable clinical phenotypes, ranging from mild symptoms for FS, CAE, and rolandic epilepsy (RE) to severe GEFS+ to epileptic encephalopathies (EEs) such as DS, West syndrome, and Doose syndrome, which may differ due to mutations at different sites. To date, only four cases of GABRG2 mutation causing DS have been reported, including a pair of twins (50–52). This is the only case occurring in twins that was reported in the literature with a simple symptom description. The patients came from a Japanese twin family and had experienced seizures since they were 2 months old. One of the twins died at 3 years old, and the other died at 5 months old. Interestingly, their father, who also had the same GABRG2 mutation, had no seizure episodes, while their mother, who did not carry the mutation, had several seizure episodes during childhood (51).
Chromodomain helicase DNA-binding (CHD) protein belongs to the SNF2-related ATPase superfamily, which uses the energy of ATP hydrolysis to change the position of nucleosomes and reorganize chromatin structures. CHD2 belongs to a subfamily of the CHD protein family, which plays a unique role in human brain development and function. The CHD2 gene is located on 15q26, with a total length of 128 951 bp, and its protein coding region contains 39 exons (Figure 7A) (98, 99). In 2009 Veredice et al. reported the first CHD2 mutation in a 30-month-old female infant with myoclonic encephalopathy (100). She began to have seizures at 6 months of age, presenting as distinct generalized clonic seizures of approximately 35 min duration, followed by seizures several times a day; seizures were also observed to be sensitive to intermittent light stimulation, which could be reduced by wearing sunglasses. In addition to epilepsy, she also showed mild intellectual disability [developmental quotient (DQ) = 67]. Carvill et al. conducted massive parallel resequencing of 19 known and 46 candidate genes in 500 patients with epileptic encephalopathy, among which six CHD2 mutations were found (101).
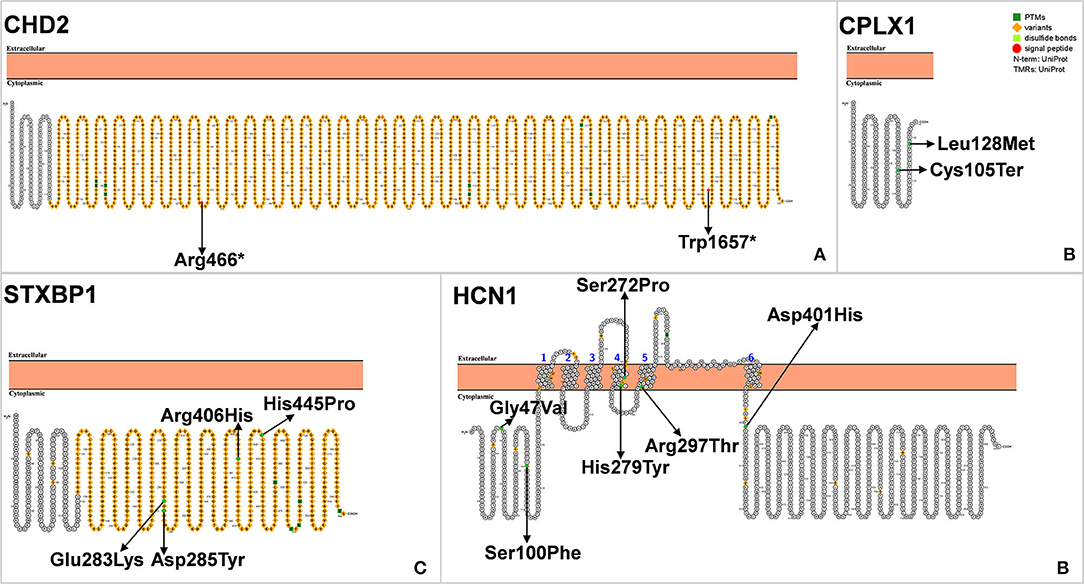
Figure 7. (A–D) Schematic diagram of protein encoded by CHD2, CPLX1, STXBP1, and HCN1A, illustrating the location of the amino acids affected by the mutations identified in patients.
Although CHD2 is associated with epilepsy, there have been few reports of CHD2 mutations in DS (54).
Unlike SCN1A mutation-associated DS, the onset of seizures in these patients was at more than 1 year of age (mean age: 26.6 months), and the seizures were heat-sensitive and accompanied by mild cognitive impairment. Some patients also show symptoms of ataxia and dysarthria (54). Suls et al. performed whole-exome sequencing on genomic DNA of the nine selected individuals with SCN1A-negative DS and both unaffected parents and found three CHD2 mutations. They then knocked down Chd2 in zebrafish using targeted morpholine antisense oligomers. Epileptiform discharges were recorded and confirmed on the Chd2-knockdown larvae, which were similar to seizures in affected patients (54). Therefore, CHD2 gene testing is recommended for DS with negative SCN1A gene mutation and accompanied by myoclonic seizures and intellectual disability. Current genetic testing methods (parallel sequencing) will significantly increase the probability of CHD2 mutant detection.
CPLX1 encodes complexin 1, a presynaptic small molecule protein that belongs to the highly conserved complexin protein family. CPLX1 forms a soluble N-ethylmaleimide–sensitive factor-attachment protein receptor (SNARE) complex in the central nervous system involved in the anchoring, pre-excitation, and fusion of axonal end vesicles (102, 103). The abnormal expression of CPLX1 is seen in several neurodegenerative and psychiatric disorders including schizophrenia, Alzheimer's disease, Huntington's disease, major depressive illness and bipolar disorder (103–105). Studies using knockout rodent models have shown that CPLX1 is associated in neurological and psychiatric disorders, such as social disorder ataxia and premature death (106, 107). Although CPLX1 is associated with a variety of neurological disorders, little is known about its association with epilepsy. In 2015, Karaca and colleagues found CPLX1 gene mutation in two patients with malignant epilepsy (108). In 2017, Redler et al. identified three CPLX1 mutations in patients with SMEI, which was the first reported association between SMEI and CPLX1 (Figure 7B) (55). Distinctly different from typical DS, SMEI caused by the CPLX1 mutation can result in non-febrile seizures, with an alarming number of seizures up to several hundred times per day. The most characteristic CPLX1 mutations are associated with mild craniofacial dysmorphisms.
Syntaxin-binding protein 1 (STXBP1), also known as Munc18-1 or SM protein, is a membrane transporter primarily expressed in the brain. This protein plays an important role in synaptic signal transduction by interacting SNARE to mediate the release of synaptic vesicles (109). STXBP1 is located on chromosome 9q34.11 and contains 20 exons. STXBP1 is composed of 594 amino acids and contains three domains, namely domains 1, 2 and 3, which form a bow structure (Figure 7C).
New heterozygous mutations of STXBP1 can cause early-onset epileptic encephalopathy and neurodevelopmental disorders. The STXBP1 mutation causes a wide range of febrile epileptic syndromes, ranging from simple febrile seizures to severe epileptic encephalopathy (110). In 2008, Saitsu et al. first identified four STXBP1 heterozygous missense mutations in four patients with Otahara syndrome (three males and one female) and identified STXBP1as the pathogenic gene (111). In seven SCN1A-negative DS patients, Carvill et al. found three patients with STXBP1 mutation, of which one patient presented with onset at >1 year old; the two other patients presented with both tonic and non-tonic seizures. All three patients presented with febrile seizures with severe cognitive impairment, which could not be distinguished from those in typical DS based on the published clinical data (28). Alvarez Bravo et al. performed genetic testing on a 19-year-old female patient with SCN1A-positive DS and also found that the patient had STXBP1 mutation. At 6 months old, the patient had a generalized seizure due to infection and fever. Cognitive retardation began at 2 years of age and became more severe at 6 years of age. She had a myoclonus triggered by sound and tactile stimuli at the age of 13 years. At the age of 12 years, she had a severe movement disorder. Unlike typical DS, this patient had the movement disorder Parkinson's disease (112). Patients with STXBP1 mutation have DS-like phenotypes regardless of SCN1A mutations, and STXBP1 has been included as a rare candidate gene for DS (28).
Hyperpolarization-activated cyclic nucleotide-gated channel 1 (HCN1) plays a very important role in regulating the resting membrane potential of neurons and transmitting synaptic information (113). Nava et al. found two HCN1 mutations in two proband. Three HCN1 mutants were found to be in a heterozygous state in 95 febrile French early infantile epileptic encephalopathy (EIEE) cohort patients. A novel c.835C>T (p. His279Tyr) mutation was found in a Dutch follow-up cohort of 62 patients. For all six patients with HCN1 mutation in EIEE, the initial suggested diagnosis was DS (Figure 7D) (56). It is worth noting that the patients showed different clinical manifestations that deviated from DS over time. These patients may present with features consistent with some fragments of DS in the early stages of the disease (114).
DS is not only identified in variations of SCN1A; mutations in other genes such as PCDH19, SCN2A, SCN8A, SCN1B, KCNA2, GABRA1, GABRB3, GABRG2, CHD2, CPLX1, HCN1A, and STXBP1 are rare but can also occur in DS-like phenotypes. Although SCN1A-related-Dravet syndrome currently accounts for most DS diagnoses and the relationship between SCN1A mutations and DS is highly specific (21), the diagnosis and treatment of other gene-related-Dravet-like syndromes should not be ignored. It is important to highlight that some phenotypes initially considered as DS have now been reclassified as DS-like ones in the spectrum of developmental and epileptic encephalopathies (21). As genetic testing becomes more widely available, more genes associated with DS or DS-like may be identified and gene-based diagnosis of different subtypes may improve the management of these diseases in the future.
The original contributions presented in the study are included in the article/supplementary material, further inquiries can be directed to the corresponding authors.
All authors listed have made a substantial, direct, and intellectual contribution to the work and approved it for publication.
This work was funded by National Natural Science Foundation of China, Grant/Award Numbers: 81971085 and 81860220; the Ningxia Hui Autonomous Region 13th Five-Year Plan Major Science and Technology Projects (2002170101); and the Advantages Discipline Group Project of Ningxia Medical University, Grant/Award Number: XY201511.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
DS, Dravet syndrome; EE, epileptic encephalopathy; LGS, lennox-gastaut syndrome; SMEI, severe myoclonic epilepsy in infancy.
2. Kanazawa Osamu. Medically intractable generalized tonic-clonic or clonic seizures in infancy. Epilepsy Behav. (1992) 5:143–8. doi: 10.1016/S0896-6974(05)80132-X
3. Dravet C. The core Dravet syndrome phenotype. Epilepsia. (2011) 2:3–9. doi: 10.1111/j.1528-1167.2011.02994.x
4. Anwar A, Saleem S, Patel UK, Arumaithurai K, Malik P. Dravet Syndrome: An Overview. Cureus. (2019) 11:e5006. doi: 10.7759/cureus.5006
5. Berkovic S. Epileptic encephalopathies of infancy: welcome advances. Lancet. (2019) 394:2203–4. doi: 10.1016/S0140-6736(19)31239-5
6. Bertuccelli M, Verheyen K, Hallemans A, Sander J, Ragona F, Bisiacchi P, et al. Deconstructing Dravet syndrome neurocognitive development: a scoping review. Epilepsia. (2021) 62:874–87. doi: 10.1111/epi.16844
7. Han S, Tai C, Westenbroek R, Yu F, Cheah C, Potter G, et al. Autistic-like behaviour in Scn1a+/- mice and rescue by enhanced GABA-mediated neurotransmission. Nature. (2012) 489:385–90. doi: 10.1038/nature11356
8. Zuberi SM, Brunklaus A, Birch R, Reavey E, Duncan J, Forbes GH. Genotype-phenotype associations in SCN1A-related epilepsies. Neurology. (2011) 76:594. doi: 10.1212/WNL.0b013e31820c309b
9. Wolff M, Cassé-Perrot C, Dravet C. Severe myoclonic epilepsy of infants (Dravet syndrome): natural history and neuropsychological findings. Epilepsia. (2006) 47(Suppl. 2):45–8. doi: 10.1111/j.1528-1167.2006.00688.x
10. Mantegazza M, Cestèle S, Catterall WA. Sodium channelopathies of skeletal muscle and brain. Physiol Rev. (2021) 101:1633–89. doi: 10.1152/physrev.00025.2020
11. Mantegazza M, Catterall WA. Voltage-gated Na(+) channels: structure, function, and pathophysiology. 4th ed. Noebels J, Avoli M, Rogawski MA, Olsen RW, Delgado-Escueta AV, editors. Jasper's Basic Mechanisms of the Epilepsies. Oxford University Press (2012).
12. Brackenbury W, Djamgoz M, Isom L. An emerging role for voltage-gated Na+ channels in cellular migration: regulation of central nervous system development and potentiation of invasive cancers. Neuroscientist. (2008) 14:571–83. doi: 10.1177/1073858408320293
13. Catterall W. Sodium channels, inherited epilepsy, and antiepileptic drugs. Annu Rev Pharmacol Toxicol. (2014) 54:317–38. doi: 10.1146/annurev-pharmtox-011112-140232
14. Escayg A, Goldin AL. Sodium channel SCN1A and epilepsy: Mutations and mechanisms. Epilepsia. (2010) 51:1650–8. doi: 10.1111/j.1528-1167.2010.02640.x
15. Mantegazza M. Broccoli V. SCN1A/Na(V) 11 channelopathies: Mechanisms in expression systems, animal models, and human iPSC models. Epilepsia. (2019) 60 (Suppl. 3):S25–38. doi: 10.1111/epi.14700
16. Catterall W, Kalume F. Oakley J. NaV11 channels and epilepsy. J Physiol. (2010) 588:1849–59. doi: 10.1113/jphysiol.2010.187484
17. Yu FH, Mantegazza M, Westenbroek RE, Robbins CA, Kalume F, Burton KA, et al. Reduced sodium current in GABAergic interneurons in a mouse model of severe myoclonic epilepsy in infancy. Nat Neurosci. (2006) 9:1142–9. doi: 10.1038/nn1754
18. Kalume F, Yu F, Westenbroek R, Scheuer T. Catterall W. Reduced sodium current in purkinje neurons from Nav11 mutant mice: implications for ataxia in severe myoclonic epilepsy in infancy. J Neurosci. (2007) 27:11065–74. doi: 10.1523/JNEUROSCI.2162-07.2007
19. Claes L, Del-Favero J, Ceulemans B, Lagae L, Van Broeckhoven C, De Jonghe P, et al. Novo mutations in the sodium-channel gene SCN1A cause severe myoclonic epilepsy of infancy. Am J Hum Genet. (2001) 68:1327–32. doi: 10.1086/320609
20. Marini C, Scheffer I, Nabbout R, Suls A, De Jonghe P, Zara F, et al. The genetics of Dravet syndrome. Epilepsia. (2011) 52(Suppl. 2):24–9. doi: 10.1111/j.1528-1167.2011.02997.x
21. Mei D, Cetica V, Marini C, Guerrini R. Dravet syndrome as part of the clinical and genetic spectrum of sodium channel epilepsies and encephalopathies. Epilepsia. (2019) 60(Suppl. 3):S2–7. doi: 10.1111/epi.16054
22. Myers K, Davey M, Ching M, Ellis C, Grinton B, Roten A, et al. Randomized controlled trial of melatonin for sleep disturbance in Dravet syndrome: the DREAMS study. J Clin Sleep Med. (2018) 14:1697–704. doi: 10.5664/jcsm.7376
23. Scheffer I, Berkovic S, Capovilla G, Connolly M, French J, Guilhoto L, et al. ILAE classification of the epilepsies: position paper of the ILAE commission for classification and terminology. Epilepsia. (2017) 58:512–21. doi: 10.1111/epi.13709
24. Scheffer I, Nabbout R. SCN1A-related phenotypes: epilepsy and beyond. Epilepsia. (2019) 60(Suppl. 3):S17–24. doi: 10.1111/epi.16386
25. Salgueiro-Pereira AR, Duprat F, Pousinha PA, Loucif A, Douchamps V, Regondi C, et al. A two-hit story: seizures and genetic mutation interaction sets phenotype severity in SCN1A epilepsies. Neurobiol Dis. (2019) 125:31–44. doi: 10.1016/j.nbd.2019.01.006
26. Steel D, Symonds J, Zuberi S, Brunklaus A. Dravet syndrome and its mimics: beyond SCN1A. Epilepsia. (2017) 58:1807–16. doi: 10.1111/epi.13889
27. Liu YH, Cheng YT, Tsai MH, Chou IJ, Hung PC, Hsieh MY, et al. Genetics and clinical correlation of Dravet syndrome and its mimics - experience of a tertiary center in Taiwan. Pediatr Neonatol. (2021) 62:550–8.
28. Carvill G, Weckhuysen S, McMahon J, Hartmann C, Møller R, Hjalgrim H, et al. GABRA1 and STXBP1: novel genetic causes of Dravet syndrome. Neurology. (2014) 82:1245–53. doi: 10.1212/WNL.0000000000000291
29. Liu A, Zhang Y, Xu X, Yang X, Yang Z, Wu Y, et al. [Genotype and phenotype of female Dravet syndrome with PCDH19 mutations]. Zhonghua er ke za zhi Chin J Pediatr. (2016) 54:327–31. doi: 10.3760/cma.j.issn.0578-1310.2016.05.004
30. Liu A, Xu X, Yang X, Jiang Y, Yang Z, Liu X, et al. The clinical spectrum of female epilepsy patients with PCDH19 mutations in a Chinese population. Clin Genet. (2017). 91:54–62. doi: 10.1111/cge.12846
31. Depienne C, Bouteiller D, Keren B, Cheuret E, Poirier K, Trouillard O, et al. Sporadic infantile epileptic encephalopathy caused by mutations in PCDH19 resembles Dravet syndrome but mainly affects females. PLoS Genet. (2009) 5:e1000381. doi: 10.1371/journal.pgen.1000381
32. Kwong A, Fung C, Chan S, Wong V. Identification of SCN1A and PCDH19 mutations in Chinese children with Dravet syndrome. PLoS ONE. (2012) 7:e41802. doi: 10.1371/journal.pone.0041802
33. Higurashi N, Shi X, Yasumoto S, Oguni H, Sakauchi M, Itomi K, et al. PCDH19 mutation in Japanese females with epilepsy. Epilepsy Res. (2012) 99:28–37. doi: 10.1016/j.eplepsyres.2011.10.014
34. Trivisano M, Pietrafusa N, Ciommo V, Cappelletti S, Palma L, Terracciano A, et al. PCDH19-related epilepsy and Dravet syndrome: face-off between two early-onset epilepsies with fever sensitivity. Epilepsy Res. (2016) 125:32–6. doi: 10.1016/j.eplepsyres.2016.05.015
35. Liu Y, Cheng Y, Tsai M, Chou I, Hung P, Hsieh M, et al. Genetics and clinical correlation of Dravet syndrome and its mimics - experience of a tertiary center in Taiwan. Pediatr Neonatol. (2021). doi: 10.1016/j.pedneo.2021.05.022
36. Shi X, Yasumoto S, Nakagawa E, Fukasawa T, Uchiya S, Hirose S. Missense mutation of the sodium channel gene SCN2A causes Dravet syndrome. Brain Dev. (2009) 31:758–62. doi: 10.1016/j.braindev.2009.08.009
37. Zeng Q, Zhang Y, Yang X, Zhang J, Liu A, Liu X, et al. [Phenotype study of SCN2A gene related epilepsy]. Zhonghua er ke za zhi Chin J pediatr. (2018) 56:518–23. doi: 10.3760/cma.j.issn.0578-1310.2018.07.009
38. Lossin C, Shi X, Rogawski M, Hirose S. Compromised function in the Na(v)1.2 Dravet syndrome mutation R1312T. Neurobiol Dis. (2012) 47:378–84. doi: 10.1016/j.nbd.2012.05.017
39. Larsen J, Carvill G, Gardella E, Kluger G, Schmiedel G, Barisic N, et al. The phenotypic spectrum of SCN8A encephalopathy. Neurology. (2015) 84:480–9. doi: 10.1212/WNL.0000000000001211
40. Patino G, Claes L, Lopez-Santiago L, Slat E, Dondeti R, Chen C, et al. A functional null mutation of SCN1B in a patient with Dravet syndrome. J Neurosci. (2009) 29:10764–78. doi: 10.1523/JNEUROSCI.2475-09.2009
41. Ogiwara I, Nakayama T, Yamagata T, Ohtani H, Mazaki E, Tsuchiya S, et al. A homozygous mutation of voltage-gated sodium channel β(I) gene SCN1B in a patient with Dravet syndrome. Epilepsia. (2012) 53:e200–3. doi: 10.1111/epi.12040
42. Mukherjee D, Mukherjee S, Niyogi P, Mahapatra M. Dravet syndrome with SCN1B gene mutation: a rare entity. Neurol India. (2017) 65:801–3. doi: 10.4103/neuroindia.NI_1115_15
43. Gong J, Liao H, Long H, Li X, Long L, Zhou L, et al. SCN1B and SCN2B gene variants analysis in Dravet syndrome patients: analysis of 22 cases. Medicine. (2019) 98:e14974. doi: 10.1097/MD.0000000000014974
44. Darras N, Ha TK, Rego S, Martin PM, Barroso E, Slavotinek AM, et al. Developmental and epileptic encephalopathy in two siblings with a novel, homozygous missense variant in SCN1B. Am J Med Genet A. (2019) 179:2190–5. doi: 10.1002/ajmg.a.61344
45. Johannesen K, Marini C, Pfeffer S, Møller R, Dorn T, Niturad C, et al. Phenotypic spectrum of GABRA1: From generalized epilepsies to severe epileptic encephalopathies. Neurology. (2016) 87:1140–51. doi: 10.1212/WNL.0000000000003087
46. Aravindhan A, Shah K, Pak J. Veerapandiyan A. Early-onset epileptic encephalopathy with myoclonic seizures related to 9q333-q3411 deletion involving STXBP1 and SPTAN1 genes Epileptic Disord. (2018) 20:214–8. doi: 10.1684/epd.2018.0969
47. Le S, Le P, Le T, Kieu Huynh T, Hang Do T. A mutation in GABRB3 associated with Dravet syndrome. Am J Med Genet A. (2017) 173:2126–31. doi: 10.1002/ajmg.a.38282
48. Shi Y, Zhang Q, Cai K, Poliquin S, Shen W, Winters N, et al. Synaptic clustering differences due to different GABRB3 mutations cause variable epilepsy syndromes. Brain. (2019) 142:3028–44. doi: 10.1093/brain/awz250
49. Pavone P, Pappalardo X, Marino S, Sciuto L, Corsello G, Ruggieri M, et al. A novel GABRB3 variant in Dravet syndrome: case report and literature review. Mol Genet Genomic Med. (2020) 8:e1461. doi: 10.1002/mgg3.1461
50. Huang X, Tian M, Hernandez C, Hu N, Macdonald R. The GABRG2 nonsense mutation, Q40X, associated with Dravet syndrome activated NMD and generated a truncated subunit that was partially rescued by aminoglycoside-induced stop codon read-through. Neurobiol Dis. (2012) 48:115–23. doi: 10.1016/j.nbd.2012.06.013
51. Ishii A, Kanaumi T, Sohda M, Misumi Y, Zhang B, Kakinuma N, et al. Association of nonsense mutation in GABRG2 with abnormal trafficking of GABAA receptors in severe epilepsy. Epilepsy Res. (2014) 108:420–32. doi: 10.1016/j.eplepsyres.2013.12.005
52. Hernandez CC, Kong W, Hu N, Zhang Y, Shen W, Jackson L, et al. Altered channel conductance states and gating of GABA(A) receptors by a pore mutation linked to Dravet syndrome. eNeuro. (2017) 4:ENEURO.0251-16.2017. doi: 10.1523/ENEURO.0251-16.2017
53. Syrbe S, Hedrich U, Riesch E, Djémié T, Müller S, Møller R, et al. De novo loss- or gain-of-function mutations in KCNA2 cause epileptic encephalopathy. Nat Genet. (2015) 47:393–9. doi: 10.1038/ng.3239
54. Suls A, Jaehn J, Kecskés A, Weber Y, Weckhuysen S, Craiu D, et al. De novo loss-of-function mutations in CHD2 cause a fever-sensitive myoclonic epileptic encephalopathy sharing features with Dravet syndrome. Am J Hum Genet. (2013) 93:967–75. doi: 10.1016/j.ajhg.2013.09.017
55. Redler S, Strom T, Wieland T, Cremer K, Engels H, Distelmaier F, et al. Variants in CPLX1 in two families with autosomal-recessive severe infantile myoclonic epilepsy and ID. Eur J Hum Genet. (2017) 25:889–93. doi: 10.1038/ejhg.2017.52
56. Nava C, Dalle C, Rastetter A, Striano P, de Kovel C, Nabbout R, et al. De novo mutations in HCN1 cause early infantile epileptic encephalopathy. Nat Genet. (2014) 46:640–5. doi: 10.1038/ng.2952
57. Henriksen M, Ravn K, Paus B, von Tetzchner S, Skjeldal O. De novo mutations in SCN1A are associated with classic Rett syndrome: a case report. BMC Med Genet. (2018) 19:184. doi: 10.1186/s12881-018-0700-z
58. Jaber D, Gitiaux C, Blesson S, Marguet F, Buard D, Varela Salgado M, et al. De novo mutations of are responsible for arthrogryposis broadening the -related phenotypes. J Med Genet. (2020). doi: 10.1136/jmedgenet-2020-107166
59. Selmer K, Lund C, Brandal K, Undlien D, Brodtkorb E. SCN1A mutation screening in adult patients with Lennox-Gastaut syndrome features. Epilepsy Behav. (2009) 16:555–7. doi: 10.1016/j.yebeh.2009.08.021
60. Ding J, Li X, Tian H, Wang L, Guo B, Wang Y, et al. SCN1A mutation-beyond Dravet Syndrome: a systematic review and narrative synthesis. Front Neurol. (2021) 12:743726. doi: 10.3389/fneur.2021.743726
61. Rampazzo A, Dos Santos R, Maluf F, Simm R, Marson F, Ortega M, et al. Dravet syndrome and Dravet syndrome-like phenotype: a systematic review of the SCN1A and PCDH19 variants. Neurogenetics. (2021) 22:105–15. doi: 10.1007/s10048-021-00644-7
62. Homan C, Pederson S, To T, Tan C, Piltz S, Corbett M, et al. PCDH19 regulation of neural progenitor cell differentiation suggests asynchrony of neurogenesis as a mechanism contributing to PCDH19 girls clustering epilepsy. Neurobiol Dis. (2018) 116:106–19. doi: 10.1016/j.nbd.2018.05.004
63. Dibbens L, Tarpey P, Hynes K, Bayly M, Scheffer I, Smith R, et al. X-linked protocadherin 19 mutations cause female-limited epilepsy and cognitive impairment. Nat Genet. (2008) 40:776–81. doi: 10.1038/ng.149
64. Oliva M, Berkovic S, Petrou SJE. Sodium channels and the neurobiology of epilepsy. Epilepsia. (2012) 53:1849–59. doi: 10.1111/j.1528-1167.2012.03631.x
65. Heron S, Crossland K, Andermann E, Phillips H, Hall A, Bleasel A, et al. Sodium-channel defects in benign familial neonatal-infantile seizures. Lancet. (2002) 360:851–2. doi: 10.1016/S0140-6736(02)09968-3
66. Zerem A, Lev D, Blumkin L, Goldberg-Stern H, Michaeli-Yossef Y, Halevy A, et al. Paternal germline mosaicism of a SCN2A mutation results in Ohtahara syndrome in half siblings. Eur J Paediatr Neurol. (2014) 18:567–71. doi: 10.1016/j.ejpn.2014.04.008
67. Kim H, Yang D, Kim S, Kim B, Kim H, Lee J, et al. The phenotype and treatment of SCN2A-related developmental and epileptic encephalopathy. Epileptic Disord. (2020) 22:563–70. doi: 10.1684/epd.2020.1199
68. Veeramah K, O'Brien J, Meisler M, Cheng X, Dib-Hajj S, Waxman S, et al. De novo pathogenic SCN8A mutation identified by whole-genome sequencing of a family quartet affected by infantile epileptic encephalopathy and SUDEP. Am J Hum Genet. (2012) 90:502–10. doi: 10.1016/j.ajhg.2012.01.006
69. Solé L, Wagnon J, Akin E, Meisler M. Tamkun M. The MAP1B binding domain of Na16 Is required for stable expression at the axon initial segment. J Neurosci. (2019) 39:4238–51. doi: 10.1523/JNEUROSCI.2771-18.2019
70. O'Brien J., Meisler M. Sodium channel SCN8A (Nav16): properties and de novo mutations in epileptic encephalopathy and intellectual disability. Front Genet. (2013) 4:213. doi: 10.3389/fgene.2013.00213
71. Hammer MF, Wagnon JL, Mefford HC, Meisler MH. SCN8A-related epilepsy with encephalopathy[M]. Seattle, WA: University of Washington (2016). p. 1–18.
72. Gardella E, Marini C, Trivisano M, Fitzgerald M, Alber M, Howell K, et al. The phenotype of SCN8A developmental and epileptic encephalopathy. Neurology. (2018) 91:e1112–e24. doi: 10.1212/WNL.0000000000006199
73. Meisler M. SCN8A encephalopathy: mechanisms and models. Epilepsia. (2019) 60(Suppl. 3):S86–S91. doi: 10.1111/epi.14703
74. Martin M, Tang B, Papale L, Yu F, Catterall W, Escayg A. The voltage-gated sodium channel Scn8a is a genetic modifier of severe myoclonic epilepsy of infancy. Hum Mol Genet. (2007) 16:2892–9. doi: 10.1093/hmg/ddm248
75. Lenk G, Jafar-Nejad P, Hill S, Huffman L, Smolen C, Wagnon J, et al. Scn8a antisense oligonucleotide is protective in mouse models of SCN8A encephalopathy and Dravet syndrome. Ann Neurol. (2020) 87:339–46. doi: 10.1002/ana.25676
76. O'Malley HA, Isom LL. Sodium channel β subunits: emerging targets in channelopathies. Annu Rev Physiol. (2015) 77:481–504. doi: 10.1146/annurev-physiol-021014-071846
77. Scheffer IE, Harkin LA, Grinton BE, Dibbens LM, Turner SJ, Zielinski MA, et al. Temporal lobe epilepsy and GEFS+ phenotypes associated with SCN1B mutations. Brain. (2007) 130:100–9. doi: 10.1093/brain/awl272
78. Audenaert D, Claes L, Ceulemans B, Löfgren A, Van Broeckhoven C, De Jonghe P, et al. Deletion in SCN1B is associated with febrile seizures and early-onset absence epilepsy. Neurology. (2003) 61:854–6. doi: 10.1212/01.wnl.0000080362.55784.1c
79. Aeby A, Sculier C, Bouza AA, Askar B, Lederer D, Schoonjans AS, et al. SCN1B-linked early infantile developmental and epileptic encephalopathy. Ann Clin Transl Neurol. (2019) 6:2354–67. doi: 10.1002/acn3.50921
80. O'Malley HA, Hull JM, Clawson BC, Chen C, Owens-Fiestan G, Jameson MB, et al. Scn1b deletion in adult mice results in seizures and SUDEP. Ann Clin Transl Neurol. (2019) 6:1121–6. doi: 10.1002/acn3.785
81. Kim YO, Dibbens L, Marini C, Suls A, Chemaly N, Mei D, et al. Do mutations in SCN1B cause Dravet syndrome? Epilepsy Res. (2013) 103:97–100. doi: 10.1016/j.eplepsyres.2012.10.009
82. Jan L, Jan Y. Voltage-gated potassium channels and the diversity of electrical signalling. J Physiol. (2012) 590:2591–9. doi: 10.1113/jphysiol.2011.224212
83. Tombola F, Pathak M, Isacoff E. Voltage-sensing arginines in a potassium channel permeate and occlude cation-selective pores. Neuron. (2005) 45:379–88. doi: 10.1016/j.neuron.2004.12.047
84. Gu C, Jan Y, Jan L. A conserved domain in axonal targeting of Kv1 (Shaker) voltage-gated potassium channels. Science. (2003) 301:646–9. doi: 10.1126/science.1086998
85. Pena S, Coimbra R. Ataxia and myoclonic epilepsy due to a heterozygous new mutation in KCNA2: proposal for a new channelopathy. Clin Genet. (2015) 87:e1–3. doi: 10.1111/cge.12542
86. Brew H, Gittelman J, Silverstein R, Hanks T, Demas V, Robinson L., et al. Seizures and reduced life span in mice lacking the potassium channel subunit Kv12, but hypoexcitability and enlarged Kv1 currents in auditory neurons. J Neurophysiol. (2007) 98:1501–25. doi: 10.1152/jn.00640.2006
87. Evenseth LSM, Gabrielsen M, Sylte I. The GABA(B) receptor-structure, ligand binding and drug development. Molecules. (2020) 25:3093. doi: 10.3390/molecules25133093
88. Maramai S, Benchekroun M, Ward SE, Atack JR. Subtype selective γ-aminobutyric acid type A receptor (GABA(A)R) modulators acting at the benzodiazepine binding site: an update. J Med Chem. (2020) 63:3425–46. doi: 10.1021/acs.jmedchem.9b01312
89. Cossette P, Liu L, Brisebois K, Dong H, Lortie A, Vanasse M, et al. Mutation of GABRA1 in an autosomal dominant form of juvenile myoclonic epilepsy. Nat Genet. (2002) 31:184–9. doi: 10.1038/ng885
90. Lachance-Touchette P, Brown P, Meloche C, Kinirons P, Lapointe L, Lacasse H, et al. Novel α1 and γ2 GABAA receptor subunit mutations in families with idiopathic generalized epilepsy. Eur J Neurosci. (2011) 34:237–49. doi: 10.1111/j.1460-9568.2011.07767.x
91. Reyes-Nava N, Yu H, Coughlin C, Shaikh T, Quintana A. Abnormal expression of GABAA receptor sub-units and hypomotility upon loss of gabra1 in zebrafish. Biol Open. (2020). 9:bio051367. doi: 10.1242/bio.051367
92. Kodera H, Ohba C, Kato M, Maeda T, Araki K, Tajima D, et al. De novo GABRA1 mutations in Ohtahara and west syndromes. Epilepsia. (2016) 57:566–73. doi: 10.1111/epi.13344
93. Urak L, Feucht M, Fathi N, Hornik K. Fuchs K. A GABRB3 promoter haplotype associated with childhood absence epilepsy impairs transcriptional activity. Hum Mol Genet. (2006) 15:2533–41. doi: 10.1093/hmg/ddl174
94. Samaco R, Hogart A, LaSalle J. Epigenetic overlap in autism-spectrum neurodevelopmental disorders: MECP2 deficiency causes reduced expression of UBE3A and GABRB3. Hum Mol Genet. (2005) 14:483–92. doi: 10.1093/hmg/ddi045
95. Kananura C, Haug K, Sander T, Runge U, Gu W, Hallmann K, et al. A splice-site mutation in GABRG2 associated with childhood absence epilepsy and febrile convulsions. Arch Neurol. (2002) 59:1137–41. doi: 10.1001/archneur.59.7.1137
96. Li X, Guo S, Xu S, Chen Z, Wang L, Ding J, et al. Neocortex- and hippocampus-specific deletion of Gabrg2 causes temperature-dependent seizures in mice. Cell Death Dis. (2021) 12:553. doi: 10.1038/s41419-021-03846-x
97. Reid C, Kim T, Phillips A, Low J, Berkovic S, Luscher B, et al. Multiple molecular mechanisms for a single GABAA mutation in epilepsy. Neurology. (2013) 80:1003–8. doi: 10.1212/WNL.0b013e3182872867
98. Wilson M, Henshall D, Byrne S, Brennan G. CHD2-related CNS pathologies. Int J Mol Sci. (2021) 22:588. doi: 10.3390/ijms22020588
99. Woodage T, Basrai M, Baxevanis A, Hieter P, Collins F. Characterization of the CHD family of proteins. Proc Natl Acad Sci U S A. (1997) 94:11472–7. doi: 10.1073/pnas.94.21.11472
100. Veredice C, Bianco F, Contaldo I, Orteschi D, Stefanini M, Battaglia D, et al. Early onset myoclonic epilepsy and 15q26 microdeletion: observation of the first case. Epilepsia. (2009) 50:1810–5. doi: 10.1111/j.1528-1167.2009.02078.x
101. Carvill G, Heavin S, Yendle S, McMahon J, O'Roak B, Cook J, et al. Targeted resequencing in epileptic encephalopathies identifies de novo mutations in CHD2 and SYNGAP1. Nat Genet. (2013) 45:825–30. doi: 10.1038/ng.2646
102. Archer DA, Graham ME, Burgoyne RD. Complexin regulates the closure of the fusion pore during regulated vesicle exocytosis. J Biol Chem. (2002) 277:18249–52. doi: 10.1074/jbc.C200166200
103. Lu W, Zhang Y, Fang X, Fan W, Tang W, Cai J, et al. Genetic association analysis of microRNA137 and its target complex 1 with schizophrenia in Han Chinese. Rep. (2017) 7:15084. doi: 10.1038/s41598-017-15315-7
104. Nie L, Wei G, Peng S, Qu Z, Yang Y, Yang Q, et al. Melatonin ameliorates anxiety and depression-like behaviors and modulates proteomic changes in triple transgenic mice of Alzheimer's disease. BioFactors. (2017) 43:593–611. doi: 10.1002/biof.1369
105. Morton A, Faull R, Edwardson J. Abnormalities in the synaptic vesicle fusion machinery in Huntington's disease. Brain Res Bull. (2001) 56:111–7. doi: 10.1016/S0361-9230(01)00611-6
106. Drew C, Kyd R, Morton A. Complexin 1 knockout mice exhibit marked deficits in social behaviours but appear to be cognitively normal. Hum Mol Genet. (2007) 16:2288–305. doi: 10.1093/hmg/ddm181
107. Xu Y, Zhao X, Liu J, Wang Y, Xiong L, He X, et al. Complexin i knockout rats exhibit a complex neurobehavioral phenotype including profound ataxia and marked deficits in lifespan. Pflugers Arch. (2020) 472:117–33. doi: 10.1007/s00424-019-02337-5
108. Karaca E, Harel T, Pehlivan D, Jhangiani S, Gambin T, Coban Akdemir Z, et al. Genes that affect brain structure and function identified by rare variant analyses of mendelian neurologic disease. Neuron. (2015) 88:499–513. doi: 10.1016/j.neuron.2015.09.048
109. Swanson D, Steel J, Valle D. Identification and characterization of the human ortholog of rat STXBP1, a protein implicated in vesicle trafficking and neurotransmitter release. Genomics. (1998) 48:373–6. doi: 10.1006/geno.1997.5202
110. Barcia G, Chemaly N, Gobin S, Milh M, Van Bogaert P, Barnerias C, et al. Early epileptic encephalopathies associated with STXBP1 mutations: could we better delineate the phenotype? Eur J Med Genet. (2014) 57:15–20. doi: 10.1016/j.ejmg.2013.10.006
111. Saitsu H, Kato M, Mizuguchi T, Hamada K, Osaka H, Tohyama J, et al. De novo mutations in the gene encoding STXBP1 (MUNC18-1) cause early infantile epileptic encephalopathy. Nat Genet. (2008) 40:782–8. doi: 10.1038/ng.150
112. Álvarez Bravo G, Yusta Izquierdo A. The adult motor phenotype of Dravet syndrome is associated with mutation of the STXBP1 gene and responds well to cannabidiol treatment. Seizure. (2018) 60:68–70. doi: 10.1016/j.seizure.2018.06.010
113. Lee C, MacKinnon R. Structures of the human HCN1 hyperpolarization-activated channel. Cell. (2017) 168:111–20.e11. doi: 10.1016/j.cell.2016.12.023
Keywords: Dravet syndrome, severe myoclonic epilepsy in infancy, SCN1A, PCDH19, epilepsy gene
Citation: Ding J, Wang L, Jin Z, Qiang Y, Li W, Wang Y, Zhu C, Jiang S, Xiao L, Hao X, Hu X, Li X, Wang F and Sun T (2022) Do All Roads Lead to Rome? Genes Causing Dravet Syndrome and Dravet Syndrome-Like Phenotypes. Front. Neurol. 13:832380. doi: 10.3389/fneur.2022.832380
Received: 09 December 2021; Accepted: 26 January 2022;
Published: 11 March 2022.
Edited by:
Kette D. Valente, Universidade de São Paulo, BrazilReviewed by:
Massimo Mantegazza, UMR7275 Institut de Pharmacologie Moléculaire et Cellulaire (IPMC), FranceCopyright © 2022 Ding, Wang, Jin, Qiang, Li, Wang, Zhu, Jiang, Xiao, Hao, Hu, Li, Wang and Sun. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Tao Sun, c3VudGFvX254bXVAMTYzLmNvbQ==; Feng Wang, bnh3d2FuZ0AxNjMuY29t; Xinxiao Li, bHh4OTg1QDE2My5jb20=
†These authors have contributed equally to this work
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.