 Yunchao Wang
Yunchao Wang Changhe Shi
Changhe Shi Yusheng LiWenkai YuSen WeiYu FanChengyuan MaoZhihua YangLulu YuZichen ZhaoShanshan LiYuan Gao*
Yusheng LiWenkai YuSen WeiYu FanChengyuan MaoZhihua YangLulu YuZichen ZhaoShanshan LiYuan Gao* Yuming Xu*
Yuming Xu*- Department of Neurology, The First Affiliated Hospital of Zhengzhou University, Zhengzhou University, Zhengzhou, China
Cerebral small vessel disease (CSVD) is a syndrome of clinical, neuroimaging, and neuropathological manifestations caused by disorders that affect small cerebral vessels. Although the pathogenesis of the disease remains unclear, some studies have demonstrated that genetic variants contribute to the development of CSVD. Our study aimed to explore the genetic characteristics of CSVD in the Chinese Han population. We enrolled 182 sporadic CSVD Chinese Han patients whose magnetic resonance imaging results showed grade 2-3 white matter lesions. Target region sequencing of seven monogenic CSVD-related genes, including NOTCH3, HTRA1, COL4A1, COL4A2, GLA, TREX1, and CTSA, was performed, and we identified pathogenic variants by screening the sequencing results and functional predictive analysis. All variants were predicted to be pathogenic by the SIFT Score, Polymorphism Phenotyping-2 score, Mutation Taster, Splice site score calculation, and MaxEntScan. All variants were validated in 300 healthy controls. In total, eight variants were identified in patients with CSVD, including five novel variants, c.1774C>T (NOTCH3), c.3784C>T (NOTCH3), c. 1207C>T (HTRA1), and c. 1274+1G> A (HTRA1), c.1937G>C (COL4A1) and three reported mutations. None of these variants were present in 300 healthy controls. No pathogenic variants in COL4A2, GLA, TREX1, and CTSA were detected. This study identified five novel variants in CSVD-related genes in Chinese Han patients with sporadic CSVD. Our results expand the genetic profile of CSVD.
Introduction
Cerebral small vessel disease (CSVD) is a syndrome of clinical, neuroimaging, and neuropathological manifestations caused by disorders that affect small cerebral vessels, including arteries, arterioles, capillaries and venules, in the brain (1). In the United States and Europe, CSVD accounts for 15–26% of cases of ischemic stroke (2–5), and the proportion ranges from 25 to 54% in Asia (6–10). Several studies have suggested that ethnic differences, vascular risk factors, genetic factors, and environmental sensitivities play an essential role in the mechanism and etiology of CSVD (11), with genetic factors being shown to have a significant effect on CSVD.
Most CSVD is sporadic (12), and Mendelian forms only account for 5% of CSVD patients (13). Monogenic CSVD has been proven to be caused by specific genetic mutations, including cerebral autosomal dominant arteriopathy with subcortical infarcts and leukoencephalopathy (CADASIL) (14, 15), cerebral autosomal recessive arteriopathy with subcortical infarcts and leukoencephalopathy (CARASIL) (15), COL4A1/2-related CSVD (16–18), Fabry's disease (19), retinal vasculopathy with cerebral leukodystrophy (RVCL) (14), and cathepsin A–related arteriopathy with strokes and leukoencephalopathy (CARASAL) (20). Overall, understanding genetic factors involved in CSVD will be helpful for improving the diagnosis and treatment of these monogenic diseases as well as sporadic CSVD.
In this study, we sequenced common monogenic CSVD-related genes, including NOTCH3, HTRA1, COL4A1/2, GLA, TREX1, and CTSA, in 182 Chinese Han patients with CSVD and 300 healthy subjects to investigate the characteristics of CSVD in the Chinese Han population.
Materials and Methods
Enrollment
We enrolled 182 unrelated patients with CSVD who were admitted to the Department of Neurology, First Affiliated Hospital of Zhengzhou University, from March 2017 to July 2018. The diagnosis of CSVD was performed by neurologists and neuroimaging (11). The inclusion criteria were as follows: (1) age over 18 years; (2) marked leukoencephalopathy defined as Fazekas grade 2 or grade 3 on neuroimaging (21). The exclusion criteria were as follows: (1) acute cerebral infarction lesions (the lesion diameter of diffusion-weighted imaging was more than 20 mm); (2) Acute cerebral hemorrhage, acute subarachnoid hemorrhage, history of cerebral vascular malformation or subarachnoid hemorrhage caused by cerebral aneurysm, or untreated cerebral aneurysm (diameter > 3 mm); (3) monogenic CSVD with known mutations; (4) dementia affected by confirmed neurodegenerative diseases, such as Alzheimer's disease (AD), Parkinson's Disease; (5) suffering from evident white matter lesions of non-vascular origin, such as multiple sclerosis, adult white matter dysplasia, and metabolic encephalopathy; (6) mental diseases diagnosed according to the Diagnostic and Statistical Manual of Mental Disorders V (DSM-V), intracranial infection, traumatic brain injury, or brain tumors. We recruited 300 healthy controls. This study was approved by the Ethics Committee of the First Affiliated Hospital of Zhengzhou University.
Magnetic Resonance Imaging
All patients with CSVD underwent imaging examination at the First Affiliated Hospital of Zhengzhou University. Demographic information, clinical symptoms, and imaging findings were recorded for all patients with CSVD. Neuroimaging was assessed independently by two trained neurologists. The severity of white matter hyperintensity was quantified on T2-weighted images or fluid-attenuated inversion recovery sequences of brain MRI; cerebral microbleeds were analyzed on T2*-weighted gradient-recalled echo or susceptibility-weighted sequences (11). The Fazekas scale was used to assess the severity of white matter hyperintensity.
DNA Extraction and Next-Generation Sequencing
Genomic DNA was isolated from 2 ml peripheral blood collected following the manufacturer's standard procedure. For each of the seven genes examined, both coding and non-coding (regulatory) regions were included in the sequencing target. The regulatory genomic regions comprised the promoter region (defined as 2 kb upstream of the transcription start site), 5′ untranslated region (5′ UTR), and intron-exon boundaries (50 bp). After sequencing primer hybridization, base incorporation was carried out using a HiSeq 2000 (Illumina, Inc., San Diego, CA) following the manufacturer's standard sequencing protocols for 101 cycles of sequencing per read to generate paired-end reads, including 100 bp at each end and six bp of the index tag.
Bioinformatics Analysis
Raw sequencing data were demultiplexed into individual Fastq read files with Illumina's bcl2fastq v2.16.0.10 based on unique index pairs. Low-quality (Q < 15) reads/bases were trimmed using the fastx tool. High-quality reads were aligned to the NCBI human reference genome (hg19) using Burrows Wheeler Aligner (BWA) software, which can build assemblies by mapping short reads to a reference genome using default parameters (22). SNV (single-nucleotide variant) calling was performed using both GATK (23) and Varscan (24), and the called SNV data were then combined. The SNV data in each sample were first compared to all variations annotated in dbSNP147 along with data from the 1000 Genomes Project. After this analysis, all newly identified variations were fully annotated. All filtering and annotation were performed using ANNOVAR (25). Variants that had a depth of coverage <10 were removed. The functional effect of non-synonymous SNVs was assessed by the SIFT score (http://sift.jcvi.org/www/SIFT), PolyPhen-2 (http://genetics.bwh.harvard.edu/pph2/) and Mutation Taster (http://www.mutationtaster.org) (26–28). Splice site mutations were analyzed using the bioinformatics tools SSS (http://rulai.cshl.edu/new_alt_exon_db2/HTML/score.html) and MaxEntScan (http://hollywood.mit.edu/burgelab/maxent/Xmaxentscan_scoreseq_acc.html). Finally, we analyzed the variants according to the American College of Medical Genetics (ACMG) practice guidelines.
Any SNV recorded in dbSNP147 with a minor allele frequency of ≥1% in the 1000 Genomes Project database, ≥1% in our dataset, and with missing calls in <10% of subjects was considered a single-nucleotide polymorphism and included in subsequent individual-variant association analysis; SNPs failing the Hardy-Weinberg equilibrium test at a significance level of 0.0001 were removed. SNVs in genes not reported in dbSNP and with a Global minor allele frequency (MAF) <0.01 in the 1000 Genomes Project database (http://www.1000genomes.org/home), the ExAC03 dataset (Exome Aggregation Consortium) (https://www.ncbi.nlm.nih.gov/bioproject/PRJEB8661), the esp6500 dataset (NHLBI GO Exome Sequencing Project Frequency of 6500 samples of full exon sequencing) (http://evs.gs.washington.edu/EVS/) and the GnomAD dataset (http://gnomad.broadinstitute.org) were considered to be rare variants and were included for subsequent gene-level burden analysis.
Validation by Sanger Sequencing
Sanger sequencing was performed to confirm the remaining eight candidate variants among the seven samples identified by NGS. Specific primers for the corresponding exons of NOTCH3, HTRA1, and COL4A1 were designed using Primer 6 Software. All these exons and exon-intron boundaries were amplified from genomic DNA by polymerase chain reaction (PCR). The purified PCR products were genotyped by Sanger sequencing. DNASTAR Lasergene MegAlign (v7.1.0) and Chromas (v2.33) were used to conduct sequence alignment. The primers used for all variants are shown in Supplementary Table 1.
Results
Variant Filtering
In total, 495 SNVs (single-nucleotide variants) were identified initially on the basis of NGS, with a mean target depth of 116 ×. For the first step, 379 variants located in introns (except for splice site variants) and synonymous variants were excluded. Considering the specific ethnicity being studied, the remaining 116 variants were then filtered based on the 1000 Genomes Project database, the ExAC03 dataset, the esp6500 dataset, and the GnomAD dataset to exclude those with minor allele frequencies >1% (41 variants). Among the 75 remaining variants, four were frameshift variants, 50 synonymous variants, and 21 splice-site variants. All four frameshift variants were excluded through validation by Sanger sequencing. Among the 50 synonymous variants, 18 were excluded due to their benign in silico prediction based on validation by Sanger sequencing, the SIFT score, the PolyPhen-2 score, and Mutation Taster. For the 21 splice site variants, 20 were excluded according to validation by Sanger sequencing, Mutation Taster, SSS, and MaxEntScan. Based on the amino acid composition of the coding region and metabolic pathway, eight variants in 3 genes were ultimately retained after the strict filtering process (Figure 1). The variants considered pathogenic are missense or splice site variants at highly conserved sites. The amino acids influenced by these seven missense variants are highly evolutionally conserved among the different species (Supplementary Figure 1).
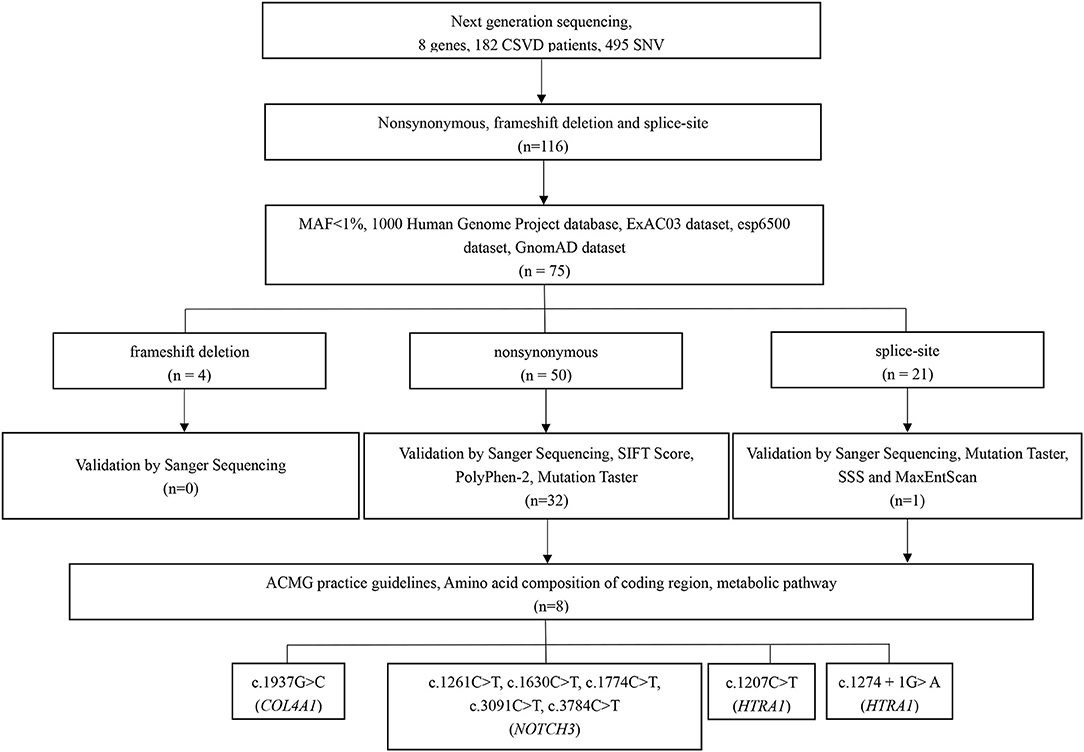
Figure 1. Filtration process and the remaining variants.
We identified eight variants in patients with CSVD, including c. 1261C>T (NOTCH3), c.1630C>T (NOTCH3), c.1774C>T (NOTCH3), c.3091C>T (NOTCH3), c.3784C>T (NOTCH3), c. 1207C>T (HTRA1), c. 1274 + 1G> A (HTRA1), and c.1937G>C (COL4A1). One patient carried c.3091C>T (NOTCH3) and c.1937G>C (COL4A1). The remaining six patients each carried one variant. Except for c. 1274 + 1G> A (HTRA1), the seven missense variants identified in this study involve highly evolutionarily conserved residues. No pathogenic variants in COL4A2, GLA, TREX1, and CTSA were detected. Moreover, the variants were absent in the 300 unrelated ethnically matched healthy controls. The results are presented in Table 1 and Figure 2. A comparison of clinical features between patients with pathogenic variants and those without pathogenic variants was presented in Supplementary Table 2.

Table 1. Eight pathogenic variants identified in CSVD-related genes in 7 SVD patients.
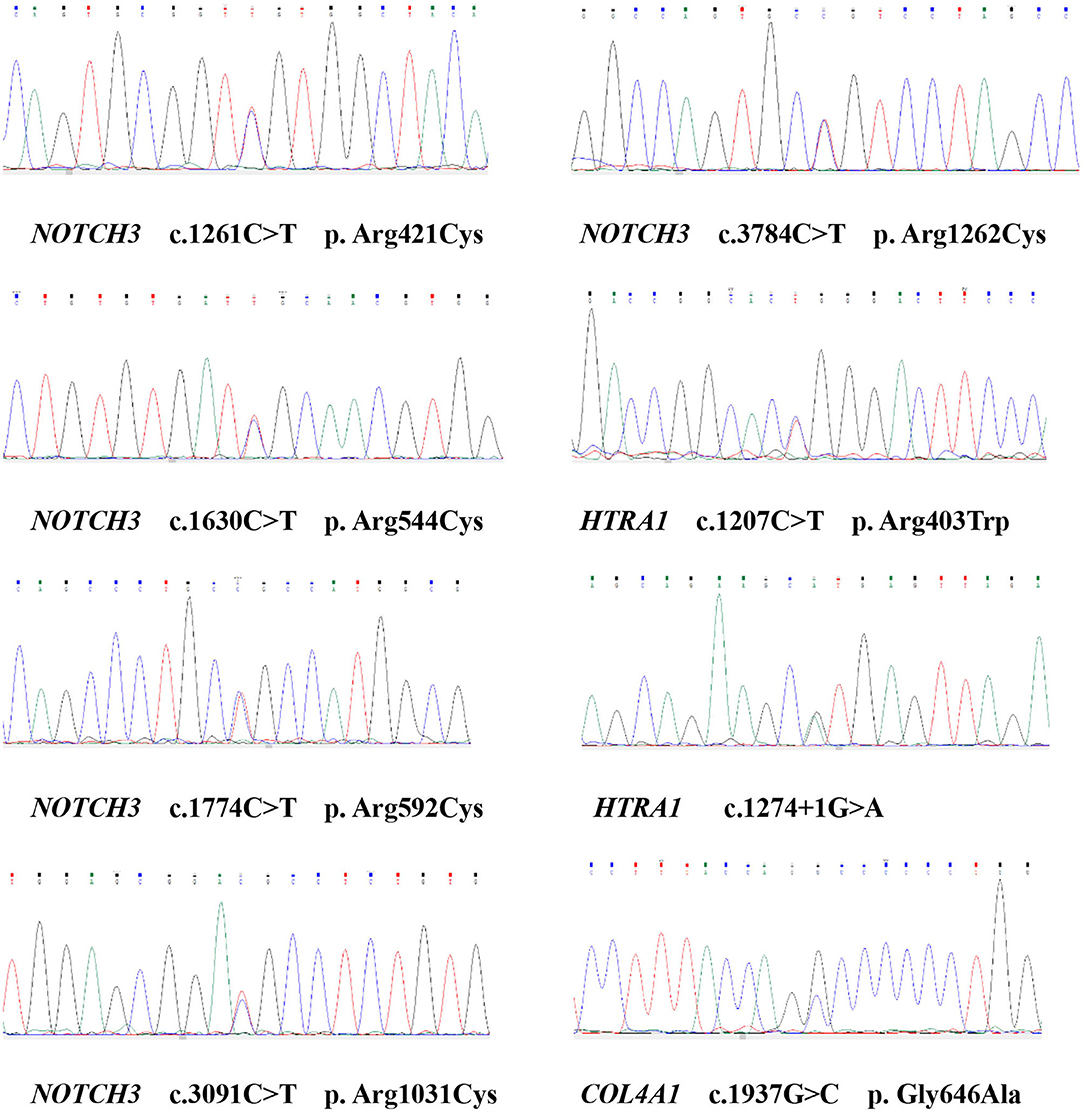
Figure 2. Sequencing chromatograms showed the variants of the NOTCH3, HTRA1, COL4A1.
NOTCH3
We found 22 non-synonymous and 16 synonymous variants. Among the 22 non-synonymous variants, the SIFT score, the PolyPhen-2 score, and the functional predictions of Mutation Taster indicated that five are pathogenic: c. 1261C>T (NOTCH3), c.1630C>T (NOTCH3), c.1774C>T (NOTCH3), c.3091C>T (NOTCH3), c.3784C>T (NOTCH3). These variants may cause the epidermal growth factor repeat (EGFR) to acquire a cysteine, causing a change in the abundance of cysteine residues. The frequencies of these five variants are low in the 1000 Genomes Project database, the ExAC03 dataset, the esp6500 dataset, and the GnomAD dataset (<1‰). Finally, we identified five NOTCH3 heterozygous variants, each of which was carried by one patient. In this study, the frequency of CSVD patients carrying the pathogenic NOTCH3 variants was 2.75% (Table 1).
These five patients exhibited varying degrees of cognitive impairment. One patient had hypertension and stroke, two patients had hypertension, stroke, and a family history of stroke. Two patients did not have any risk factors for cerebrovascular disease (Table 2).

Table 2. The clinical characteristics of the 7 SVD patients with pathogenic variants.
HTRA1
We found three non-synonymous variants, six synonymous variants, one insertion frameshift variant, and four splice site variants. Among the three non-synonymous variants located in exons, the SIFT score, the PolyPhen-2 score, and the functional prediction of Mutation Taster predicted only p.R403W (HTRA1) to be pathogenic. The frequency of c. 1207C>T (HTRA1) in the 1000 Genomes Project database is 1.39‰, and the frequency of the gene in the ExAC03, esp6500, and GnomAD gene databases is very low (<1‰). Among the four splice site variants, SSS and MaxEntScan found that only c. 1274+1G> A (HTRA1) might affect the normal splicing of mRNA. As a result, the physiological function of serine protease one would be disrupted. c. 1274+1G> A (HTRA1) has a very low gene frequency (<1‰) in the ExAC03, esp6500, and GnomAD gene databases, indicating a rare variant. At the same time, Mutation Taster functional prediction showed that c. 1274+1G> A (HTRA1) is pathogenic. Finally, we identified two rare HTRA1 heterozygous variants; the relationship between these two variants and CSVD has never been reported, and each variant was carried by one patient. In this study, the frequency of CSVD patients carrying the pathogenic HTRA1 variants was 1.10% (Table 1).
Two patients displayed varying degrees of cognitive dysfunction. Both had hypertension and diabetes mellitus (DM). One patient had hyperhomocysteinemia, ischemic and hemorrhagic strokes, and the other had coronary artery disease (CAD). However, none of the patients had a family history of similar diseases (Table 2).
COL4A1
We found 11 non-synonymous variants and 18 synonymous variants. Only one synonymous variant would cause a change from glycine to alanine. SIFT score, PolyPhen-2 score, and Mutation Taster functional prediction showed that c.1937G>C (COL4A1) is pathogenic; the frequency of the variant is relatively low in the 1000 Genomes Project database, the ExAC03 dataset, the esp6500 dataset, and the GnomAD dataset (<1‰), indicating a rare variant. The frequency of the remaining non-synonymous variants is more than 1% in the 1000 Genomes Project database, and we believe that these variants are not likely to cause disease. Finally, we identified a rare heterozygous variant that has never been reported, which was carried by one patient. In this study, the frequency of CSVD patients carrying the pathogenic COL4A1 gene variants was 0.55% (Table 1).
This patient exhibited severe cognitive dysfunction, hypertension, recurrent ischemic stroke, and a severe family history of stroke. this patient also carried a pathogenic NOTCH3 variant (Table 2).
Discussion
Eight heterozygous variants were identified in this study, including c. 1261C>T (NOTCH3), c. 1630C>T (NOTCH3), c. 1774C>T (NOTCH3), c. 3091C>T (NOTCH3), c. 3784C>T (NOTCH3), c. 1207C>T (HTRA1), c. 1274 + 1G> A (HTRA1), and c. 1937G>C (COL4A1). Among them, c. 3091C>T (NOTCH3) and c. 1937G>C (COL4A1) were carried by the same patient; for the remaining six variants, each was carried by one patient. We did not detect pathogenic variants in COL4A2, GLA, TREX1, or CTSA, and no pathogenic variants were found in 300 healthy controls.
CADASIL is one of the most common single-gene disorders of cerebral small blood vessels caused by mutations in the NOTCH3 gene on chromosome 19q12 (29, 30). At present, there are more than 500 CADASIL families reported in the world, and more than 230 disease-causing mutations have been detected in the NOTCH3 gene (31, 32). The mutations are mainly concentrated in the exon 2-24 region (33–35). In this study, five heterozygous variants were found in exon 8, exon 11, exon 19, and exon 23. Opherk et al. reported the mutation c. 1261C>T (NOTCH3) in a retrospective study of 411 CADASIL patients in 2004 (32, 36), and Oberstein et al. (37) described the mutation c.1630C>T (NOTCH3) based on direct sequence analysis of an abnormal NOTCH3 gene in patients from 11 families in 1999. Joutel et al. (33) reported the c. 3091C>T (NOTCH3) mutation in 50 unrelated white patients with a strong suspicion of CADASIL in 1997. c. 1774C>T (NOTCH3) and c. 3784C>T (NOTCH3) have not been reported before. The five heterozygous variants are distributed in the EGFR domains of the NOTCH3 gene and are associated with CADASIL. These variants cause the EGFR domain to acquire another cysteine, resulting in a change in the number of this amino acid (29, 30, 38). According to electron microscopy (EM), the pathognomonic feature of CADASIL is the accumulation of granular osmiophilic material (GOM) in indentations of vascular smooth muscle cells (VSMCs) or in the extracellular space in close vicinity to VSMCs, leading to changes in cerebral vascular structure and function, such as leukoaraiosis, ischemic cerebrovascular disease, and cognitive impairment (1, 39). We also found some variants that did not affect cysteines, but the results of functional prediction suggest that the variants may cause disease. Previously, it has been reported that some variants detected in CADASIL patients do not cause changes in cysteines (40, 41). Moreover, it is interesting that some common NOTCH3 gene SNPs can cause CADASIL and increase the risk of age-related WMH in patients with hypertension (42). Therefore, in some cases, CADASIL appears to be derived from atypical NOTCH3 mutation that does not affect cysteine. The pathogenicity of these variants is currently controversial (15, 43, 44) and needs further research.
In addition, CARASIL is a very rare autosomal recessive CSVD caused by biallelic mutations of the HTRA1 gene (high-temperature requirement protease A1) (45). In this study, we found two variants, p. R403W and c. 1274+1G>A, that completely or partially abolish HTRA1 protease activity. The missense variant is located in the C-terminal PDZ domain, which participates in central regulation. As the serine protease 1 (HTRA1) chaperone (PDZ domain tightly closed) and proteolysis (PDZ domain open) functions are regulated by the PDZ domain, the variant affects these activities of the protein encoded by HTRA1. c. 1274+1G > A is located in the intron near exon 8. The variant affects the initial nucleotide of the splice donor site, impacting the normal splicing of mRNA and impairing the physiological function of HTRA1. To date, 44 mutations have been reported in this gene, most of which are missense and heterozygous variants (46). Although the exact mechanism responsible for a single heterozygous HTRA1 mutation causing CSVD is still not fully clear, impaired HTRA1 protease function seems to be a core feature of HTRA1-related autosomal dominant CSVD, and relevant in vitro functional assays may help to evaluate the pathogenicity of HTRA1 variants.
The COL4A1 and COL4A2 genes are located on chromosome 13q34 and encode collagen type IV alpha 1 (COL4A1) and collagen type IV alpha 2 (COL4A2), respectively (47). Type IV collagen is the main component of all basement membranes in humans. Mutations in the COL4A1 gene contribute to a broad spectrum of disorders involving a specific phenotype comprising hereditary angiopathy, nephropathy, aneurysms, and cramps (HANAC) (47). Dominant missense mutations in the COL4A1 gene result in a rare familial stroke characterized by deep ICH, lacunar ischemic stroke, and WMH (48, 49). More than 30 types of mutations have been reported in the COL4A1 gene thus far, and the number is rapidly increasing (18). COL4A1 and COL4A2 assemble to form a heterotrimeric helix with a constant 2:1 ratio [proα1(IV)] 2 [proα2 (IV)]. The triple-helical domain consists of a triple amino acid repeat sequence, Gly-Xaa-Yaa, where Xaa and Yaa can be any residue. This sequence is interrupted by several motifs containing cysteine residues, which are essential in providing flexibility to the collagen IV network and possible binding sites for intermolecular cross-linking (18, 50, 51). In this study, we found a new variant, namely, c.1937G>C. This variant causes glycine to become an alanine, leading to a change in the structure of the heterotrimeric helix in the basement membrane. It destroys the structure and stability of the vascular basement membrane, causing destruction of the vessel wall structure and a series of vascular diseases. Although COL4A1 gene mutations are known to lead to instability and defects in the basement membrane, the exact mechanism linking mutations and phenotypes is presently unknown.
Conclusion
Our study aimed to explore the genetic characteristics of the sporadic CSVD in the Chinese Han population. The results expand the genetic profile of CSVD. We explored the status of multiple known monogenic CSVD genes in the sporadic population. The results of our study suggest that variants in these genes may be associated with CSVD in the sporadic population, providing substantial evidence for individuals susceptible to sporadic CSVD. This study also reminds clinicians to pay attention to the screening of genetic factors in sporadic patients with CSVD. This will help improve the diagnosis and treatment of sporadic CSVD. However, due to the small number of patients included in this study and they all came from the same hospital, there was a certain degree of bias. The pathogenic mechanisms involved need to be further studied in cell and animal models to provide a basis for further research and treatment.
Data Availability Statement
The original contributions presented in the study are included in the article/Supplementary Material, further inquiries can be directed to the corresponding authors.
Ethics Statement
The studies involving human participants were reviewed and approved by Ethics Committee of First Affiliated Hospital of Zhengzhou University. The patients/participants provided their written informed consent to participate in this study. Written informed consent was obtained from the individual(s) for the publication of any potentially identifiable images or data included in this article.
Author Contributions
YW, YX, and YG contributed to the conception and design of the study. WY, LY, ZZ, and SL contributed to the acquisition. YW drafted the text and prepared the figures. YW, YX, YG, CS, YL, WY, SW, YF, CM, and ZY contributed to the analysis of data. All authors critically reviewed the manuscript and approved it as submitted.
Funding
This work was supported by Henan Science and Technology Innovation Talent Project in 2015 to YX (Grant Number 154200510017), National Key Research and Development Program of China to YG (Grant Number 2018YFC1311303), the National Natural Science Foundation of China to YX (Grant Numbers U1904207, 81530037, and 91849115), and the Non-profit Central Research Institute Fund of Chinese Academy of Medical Sciences (Grant Number 2020-PT310-01).
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher's Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Acknowledgments
We would like to thank the patients and normal controls for their participation in this study.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fneur.2022.829438/full#supplementary-material
References
1. Pantoni L. Cerebral small vessel disease: from pathogenesis and clinical characteristics to therapeutic challenges. Lancet Neurol. (2010) 9:689–701. doi: 10.1016/S1474-4422(10)70104-6
2. White H, Boden-Albala B, Wang C, Elkind MS, Rundek T, Wright CB, et al. Ischemic stroke subtype incidence among whites, blacks, and Hispanics: the Northern Manhattan Study. Circulation. (2005) 111:1327–31. doi: 10.1161/01.CIR.0000157736.19739.D0
3. Sacco S, Marini C, Totaro R, Russo T, Cerone D, Carolei A, et al. population-based study of the incidence and prognosis of lacunar stroke. Neurology. (2006) 66:1335–8. doi: 10.1212/01.wnl.0000210457.89798.0e
4. Chamorro A, Sacco RL, Mohr JP, Foulkes MA, Kase CS, Tatemichi TK, et al. Clinical-computed tomographic correlations of lacunar infarction in the stroke data bank. Stroke. (1991) 22:175–81. doi: 10.1161/01.STR.22.2.175
5. Petty GW, Brown RD Jr, Whisnant JP, Sicks JD, O'Fallon WM, Wiebers DO Ischemic stroke subtypes: a population-based study of incidence and risk factors. Stroke. (1999) 30:2513–6. doi: 10.1161/01.STR.30.12.2513
6. Jung KH, Lee SH, Kim BJ Yu KH, Hong KS, Lee BC, et al. Secular trends in ischemic stroke characteristics in a rapidly developed country: results from the Korean stroke registry study (secular trends in Korean stroke). Circ Cardiovasc Qual Outcomes. (2012) 5:327–34. doi: 10.1161/CIRCOUTCOMES.111.963736
7. Syed NA, Khealani BA, Ali S, Hasan A, Akhtar N, Brohi H, et al. Ischemic stroke subtypes in Pakistan: the Aga Khan University stroke data bank. J Pakistan Med Assoc. (2003) 53:584−8
8. Tsai CF, Thomas B, Sudlow CL. Epidemiology of stroke and its subtypes in Chinese vs. white populations: a systematic review. Neurology. (2013) 81:264–72. doi: 10.1212/WNL.0b013e31829bfde3
9. Turin TC, Kita Y, Rumana N, Nakamura Y, Takashima N, Ichikawa M, et al. Ischemic stroke subtypes in a Japanese population: takashima stroke registry, 1988-2004. Stroke. (2010) 41:1871–6. doi: 10.1161/STROKEAHA.110.581033
10. Kubo M, Hata J, Doi Y, Tanizaki Y, Iida M, Kiyohara Y. Secular trends in the incidence of and risk factors for ischemic stroke and its subtypes in Japanese population. Circulation. (2008) 118:2672–8. doi: 10.1161/CIRCULATIONAHA.107.743211
11. Wardlaw JM, Smith EE, Biessels GJ, Cordonnier C, Fazekas F, Frayne R, et al. Neuroimaging standards for research into small vessel disease and its contribution to ageing and neurodegeneration. Lancet Neurol. (2013) 12:822–38. doi: 10.1016/S1474-4422(13)70124-8
12. Longstreth WT. Brain vascular disease overt and covert. Stroke. (2005) 36:2062–3. doi: 10.1161/01.STR.0000179040.36574.99
13. Søndergaard CB, Nielsen JE, Hansen CK, Christensen H. Hereditary cerebral small vessel disease and stroke. Clin Neurol Neurosurg. (2017) 155:45–57. doi: 10.1016/j.clineuro.2017.02.015
14. Falcone GJ, Malik R, Dichgans M, Rosand J. Current concepts and clinical applications of stroke genetics. Lancet Neurol. (2014) 13:405–18. doi: 10.1016/S1474-4422(14)70029-8
15. Tikka S, Baumann M, Siitonen M, Pasanen P, Pöyhönen M, Myllykangas L, et al. CADASIL and CARASIL. Brain Pathol. (2014) 24:525–44. doi: 10.1111/bpa.12181
16. Jeanne M, Gould DB. Genotype-phenotype correlations in pathology caused by collagen type IV alpha 1 and 2 mutations. Matrix Biol. (2017) 57–58:29–44. doi: 10.1016/j.matbio.2016.10.003
17. Mao M, Alavi MV, Labelle-Dumais C, Gould DB. Type IV collagens and basement membrane diseases: cell biology and pathogenic mechanisms. Curr Top Membr. (2015) 76:61–116. doi: 10.1016/bs.ctm.2015.09.002
18. Kuo DS, Labelle-Dumais C, Gould DB. COL4A1 and COL4A2 mutations and disease: insights into pathogenic mechanisms and potential therapeutic targets. Hum Mol Genet. (2012) 21:R97–110. doi: 10.1093/hmg/dds346
19. Brady RO, Gal AE, Bradley RM, Martensson E, Warshaw AL, Laster L. Enzymatic defect in Fabry's disease. Ceramidetrihexosidase deficiency. N Engl J Med. (1967) 276:1163–7. doi: 10.1056/NEJM196705252762101
20. Bugiani M, Kevelam SH, Bakels HS, Waisfisz Q. Ceuterick-de Groote C, Niessen HW, et al. Cathepsin A-related arteriopathy with strokes and leukoencephalopathy (CARASAL). Neurology. (2016) 87:1777–86. doi: 10.1212/WNL.0000000000003251
21. Fazekas F, Barkhof F, Wahlund LO, Pantoni L, Erkinjuntti T, Scheltens P, et al. CT and MRI rating of white matter lesions. Cerebrovasc Dis. (2002) 13(Suppl. 2):31–6. doi: 10.1159/000049147
22. Li H, Durbin R. Fast and accurate long-read alignment with Burrows-Wheeler transform. Bioinformatics. (2010) 26:589–95. doi: 10.1093/bioinformatics/btp698
23. McKenna A, Hanna M, Banks E, Sivachenko A, Cibulskis K, Kernytsky A, et al. The genome analysis toolkit: a MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res. (2010) 20:1297–303. doi: 10.1101/gr.107524.110
24. Koboldt DC, Zhang Q, Larson DE, Shen D, McLellan MD, Lin L, et al. VarScan 2: somatic mutation and copy number alteration discovery in cancer by exome sequencing. Genome Res. (2012) 22:568–76. doi: 10.1101/gr.129684.111
25. Wang K, Li M, Hakonarson H. ANNOVAR functional annotation of genetic variants from high-throughput sequencing data. Nucleic Acids Res. (2010) 38:e164. doi: 10.1093/nar/gkq603
26. Ng PC, Henikoff S. SIFT Predicting amino acid changes that affect protein function. Nucleic Acids Res. (2003) 31:3812–4. doi: 10.1093/nar/gkg509
27. Adzhubei IA, Schmidt S, Peshkin L, Ramensky VE, Gerasimova A, Bork P, et al. A method and server for predicting damaging missense mutations. Nat Methods. (2010) 7:248–9. doi: 10.1038/nmeth0410-248
28. Schwarz JM, Rödelsperger C, Schuelke M, Seelow D. MutationTaster evaluates disease-causing potential of sequence alterations. Nat Methods. (2010) 7:575–6. doi: 10.1038/nmeth0810-575
29. Joutel A, Corpechot C, Ducros A, Vahedi K, Chabriat H, Mouton P, et al. Notch3 mutations in CADASIL, a hereditary adult-onset condition causing stroke and dementia. Nature. (1996) 383:707–10. doi: 10.1038/383707a0
30. Tournier-Lasserve E, Joutel A, Melki J, Weissenbach J, Lathrop GM, Chabriat H, et al. Cerebral autosomal dominant arteriopathy with subcortical infarcts and leukoencephalopathy maps to chromosome 19q12. Nat Genet. (1993) 3:256–9. doi: 10.1038/ng0393-256
31. Adib-Samii P, Brice G, Martin RJ, Markus HS. Clinical spectrum of CADASIL and the effect of cardiovascular risk factors on phenotype: study in 200 consecutively recruited individuals. Stroke. (2010) 41:630–4. doi: 10.1161/STROKEAHA.109.568402
32. Opherk C, Peters N, Herzog J, Luedtke R, Dichgans M. Long-term prognosis and causes of death in CADASIL: a retrospective study in 411 patients. Brain J Neurol. (2004) 127:2533–9. doi: 10.1093/brain/awh282
33. Joutel A, Vahedi K, Corpechot C, Troesch A, Chabriat H, Vayssière C, et al. Strong clustering and stereotyped nature of Notch3 mutations in CADASIL patients. Lancet. (1997) 350:1511–5. doi: 10.1016/S0140-6736(97)08083-5
34. Dichgans M. Cerebral autosomal dominant arteriopathy with subcortical infarcts and leukoencephalopathy: phenotypic and mutational spectrum. J Neurol Sci. (2002) 203–204:77–80. doi: 10.1016/S0022-510X(02)00270-8
35. Testi S, Malerba G, Ferrarini M, Ragno M, Pradotto L, Mauro A, et al. Mutational and haplotype map of NOTCH3 in a cohort of Italian patients with cerebral autosomal dominant arteriopathy with subcortical infarcts and leukoencephalopathy (CADASIL). J Neurol Sci. (2012) 319:37–41. doi: 10.1016/j.jns.2012.05.025
36. Peters N, Opherk C, Bergmann T, Castro M, Herzog J, Dichgans M. Spectrum of mutations in biopsy-proven CADASIL: implications for diagnostic strategies. Arch Neurol. (2005) 62:1091–4. doi: 10.1001/archneur.62.7.1091
37. Oberstein SA, Ferrari MD, Bakker E, van Gestel J, Kneppers AL, Frants RR, et al. Diagnostic Notch3 sequence analysis in CADASIL: three new mutations in Dutch patients. Dutch CADASIL research group. Neurology. (1999) 52:1913–5. doi: 10.1212/WNL.52.9.1913
38. Chabriat H, Joutel A, Dichgans M, Tournier-Lasserve E, Bousser MG. Cadasil. Lancet Neurol. (2009) 8:643–53. doi: 10.1016/S1474-4422(09)70127-9
39. Joutel A, Monet-Leprêtre M, Gosele C, Baron-Menguy C, Hammes A, Schmidt S, et al. Cerebrovascular dysfunction and microcirculation rarefaction precede white matter lesions in a mouse genetic model of cerebral ischemic small vessel disease. J Clin Investig. (2010) 120:433–45. doi: 10.1172/JCI39733
40. Mizuno T, Muranishi M, Torugun T, Tango H, Nagakane Y, Kudeken T, et al. Two Japanese CADASIL families exhibiting Notch3 mutation R75P not involving cysteine residue. Internal Med. (2008) 47:2067–72. doi: 10.2169/internalmedicine.47.1391
41. Muiño E, Gallego-Fabrega C, Cullell N, Carrera C, Torres N, Krupinski J, et al. Systematic review of cysteine-sparing NOTCH3 missense mutations in patients with clinical suspicion of CADASIL. Int J Mol Sci. (2017) 18:1964. doi: 10.3390/ijms18091964
42. Schmidt H, Zeginigg M, Wiltgen M, Freudenberger P, Petrovic K, Cavalieri M, et al. Genetic variants of the NOTCH3 gene in the elderly and magnetic resonance imaging correlates of age-related cerebral small vessel disease. Brain J Neurol. (2011) 134:3384–97. doi: 10.1093/brain/awr252
43. Quattrone A, Mazzei R. Cysteine-sparing notch3 mutations: cadasil or cadasil variants? Neurology. (2009) 72:2135–6; author reply 6. doi: 10.1212/01.wnl.0000349699.12456.06
44. Scheid R, Heinritz W, Leyhe T, Thal DR, Schober R, Strenge S, et al. Cysteine-sparing notch3 mutations: cadasil or cadasil variants? Neurology. (2008) 71:774–6. doi: 10.1212/01.wnl.0000324928.44694.f7
45. Hara K, Shiga A, Fukutake T, Nozaki H, Miyashita A, Yokoseki A, et al. Association of HTRA1 mutations and familial ischemic cerebral small-vessel disease. N Engl J Med. (2009) 360:1729–39. doi: 10.1056/NEJMoa0801560
46. Lee YC, Chung CP, Chao NC, Fuh JL, Chang FC, Soong BW, et al. Characterization of Heterozygous HTRA1 mutations in Taiwanese patients with cerebral small vessel disease. Stroke. (2018) 49:1593–601. doi: 10.1161/STROKEAHA.118.021283
47. Plaisier E, Gribouval O, Alamowitch S, Mougenot B, Prost C, Verpont MC, et al. COL4A1 mutations and hereditary angiopathy, nephropathy, aneurysms, and muscle cramps. N Engl J Med. (2007) 357:2687–95. doi: 10.1056/NEJMoa071906
48. Lanfranconi S, Markus HS. COL4A1 mutations as a monogenic cause of cerebral small vessel disease: a systematic review. Stroke. (2010) 41:e513–8. doi: 10.1161/STROKEAHA.110.581918
49. Yamamoto Y, Craggs L, Baumann M, Kalimo H, Kalaria RN. Review: molecular genetics and pathology of hereditary small vessel diseases of the brain. Neuropathol Appl Neurobiol. (2011) 37:94–113. doi: 10.1111/j.1365-2990.2010.01147.x
50. Khoshnoodi J, Pedchenko V, Hudson BG. Mammalian collagen IV. Microsc Res Tech. (2008) 71:357–70. doi: 10.1002/jemt.20564
Keywords: mutations, monogenic, cerebral small vessel disease, Chinese Han population, genetic study
Citation: Wang Y, Shi C, Li Y, Yu W, Wei S, Fan Y, Mao C, Yang Z, Yu L, Zhao Z, Li S, Gao Y and Xu Y (2022) Genetic Study of Cerebral Small Vessel Disease in Chinese Han Population. Front. Neurol. 13:829438. doi: 10.3389/fneur.2022.829438
Received: 05 December 2021; Accepted: 02 March 2022;
Published: 25 March 2022.
Edited by:
Andreas Charidimou, Massachusetts General Hospital and Harvard Medical School, United StatesReviewed by:
Dongsheng Fan, Peking University Third Hospital, ChinaDao Wen Wang, Huazhong University of Science and Technology, China
Copyright © 2022 Wang, Shi, Li, Yu, Wei, Fan, Mao, Yang, Yu, Zhao, Li, Gao and Xu. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Yuming Xu, eHV5dW1pbmdAenp1LmVkdS5jbg==; Yuan Gao, YmFtYm9vNTAwOEBzaW5hLmNvbQ==