 Jun-Hong Geng
Jun-Hong Geng Yang Zheng
Yang Zheng Quan-Fu Li
Quan-Fu Li Qun Hou
Qun Hou Xiao-Hang Wang
Xiao-Hang Wang Yan Jiang
Yan Jiang- Department of Neurology, The First Affiliated Hospital of Zhejiang Chinese Medical University, Hangzhou, China
Background: Paroxysmal kinesigenic dyskinesia (PKD) is characterized by recurrent episodes of movement-induced motor attacks. PKD patients may have concomitant epilepsy. Differentiation between the two disorders and effective control of both diseases remain challenging.
Case Presentation: We present a Chinese girl with typical manifestations of PKD, who also suffered from generalized tonic-clonic seizure attacks at the same time. Genetic testing confirmed a PRRT2 mutation (c.649dupC). Oxcarbazepine was initially used, but withdrawn due to a hypersensitivity reaction. Levetiracetam was initiated afterwards, which was effective for seizures but failed to control her PKD symptoms. The addition of lacosamide (LCM) completely controlled her PKD symptoms.
Conclusion: This is the first case reporting the effectiveness of LCM for concomitant PKD and epilepsy. Our case proposes a novel alternative for such patients who are resistant or cannot tolerate conventional anti-sodium antiepileptics.
Introduction
Paroxysmal Kinesigenic Dyskinesia (PKD) is a rare disease characterized by recurrent, sudden attacks of dystonia, chorea, ballistic, and athetoid involuntary movements, which are triggered by sudden voluntary movements (1). The diagnosis of PKD is mainly based on features including movement-induced attacks with a short duration, reserved consciousness, a dramatic antiepileptic drug (AED) responsiveness and a positive family history or proline-rich transmembrane protein 2 (PRRT2) mutation (2). It has been reported that 8% of PKD had concomitant epilepsy (3). Despite its rarity, patients with concomitant PKD and epilepsy remain the biggest challenge, since a correct diagnosis remain difficult and a proportion were reported to be resistant to AEDs treatment (4, 5).
The first challenge in managing patients with both PKD and epilepsy lie in the diagnosis, given the similar presentations as paroxysmal recurrent motor attacks. Two-thirds of patients with PKD was misdiagnosed with epilepsy (3). The other challenge is treatment. Effective drugs remain unknown for patients with both PKD and epilepsy. Previous reports mostly use a trial-and-error strategy when choosing drugs, which might bring extra stress and economic burdens.
In this study, we described a 15-year-old Chinese girl, who carried a PRRT2 gene mutation, presenting with concomitant PKD and epilepsy. Her movement symptoms had an excellent response to LCM. We also summarized previous reports on concomitant PKD and epilepsy, hoping to address the diagnostic and therapeutic challenges in such patients.
Case Report
The 15-year-old girl was born at 38 weeks of gestation by vaginal delivery to a pair of non-consanguineous Chinese parents. She was the only child and her prenatal period was uneventful. Her growth and developmental milestones were normal.
At age of 5, she suffered from a seizure at sleep in the early morning, characterized by limb twitching and jerking, with loss of consciousness, which lasted for 3 min. She complained of weakness and headache after the attack. She was subsequently sent to the hospital where the electroencephalogram (EEG) and magnetic resonance imaging (MRI) were both non-revealing. At the age of 8, she presented with paroxysms of abnormal involuntary posturing of both hands, ranging from a few seconds to 1 min. The episodes were mostly triggered by sudden movement or emotional stress. Her consciousness was fully preserved during the attacks. Her family history was negative for seizures. However, her mother had dyskinesia after sudden movements when she was young, such as suddenly standing up or running. Her mother's symptoms completely disappeared after the age of 16. Additionally, her mother's younger sister and cousin had similar dyskinesia.
The girl went to a tertiary hospital and was diagnosed with PKD at the age of 10. She was initially treated with oxcarbazepine, which unfortunately, caused an allergic reaction in the form of skin rashes. Levetiracetam was initiated, with an initial dose of 500 mg twice daily. However, she still had paroxysmal dystonia despite the levetiracetam treatment, with a frequency of about once per week.
At the age of 15, she suffered from another seizure at sleep in the early morning, characterized by limb twitching and jerking with loss of consciousness, which lasted for around 3 min. Weakness and headache were reported after the attack. EEG at an outside institute showed the presence of scattered theta waves, sharp waves and sharp slow waves induced by hyperventilation. Long-term EEG showed intermittent bilateral frontal slowing. Neurologic examinations and brain MRI were normal at that time. Similarly, she experienced two grand mal seizures in the next month, both at sleep in the early morning. Dosage of levetiracetam was increased to 1,000 mg twice daily. However, the paroxysmal dystonia persisted. The diagnosis of concomitant PKD and epilepsy was considered. Genetic testing revealed a heterozygous frameshift mutation of c.649dupC (p.Arg217Profs*8) (Figure 1). LCM, at a dose of 50 mg twice daily, was added to the previous treatment. The patient responded excellently to the treatment. Neither dystonia nor seizures recurred after the LCM treatment (Figure 2). No adverse effects such as cardiac abnormalities and mental disorders were observed. The patient remained free of both PKD and seizures at the last follow-up 10 months after the initiation of LCM.
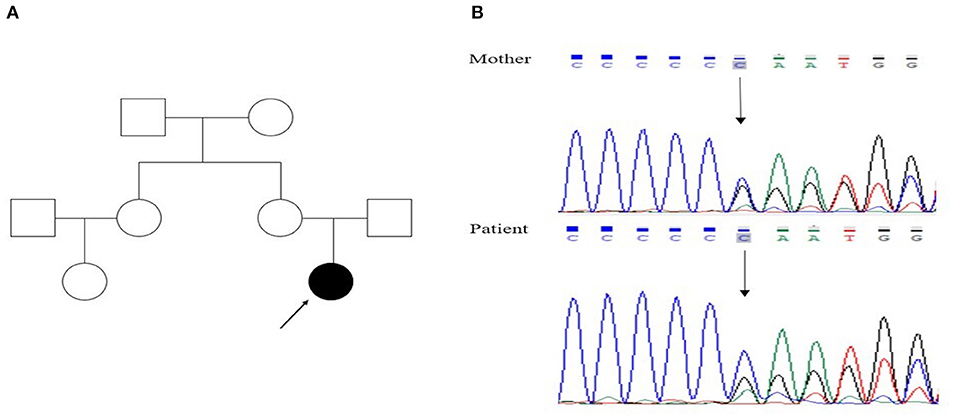
Figure 1. (A) Family pedigree of the proband (filled symbols represent the patient, open symbols normal persons, index case indicated by arrow); (B) genetic analysis showed that both the patient and her mother had a heterozygous mutation, c.649dupC, in the PRRT2 gene.
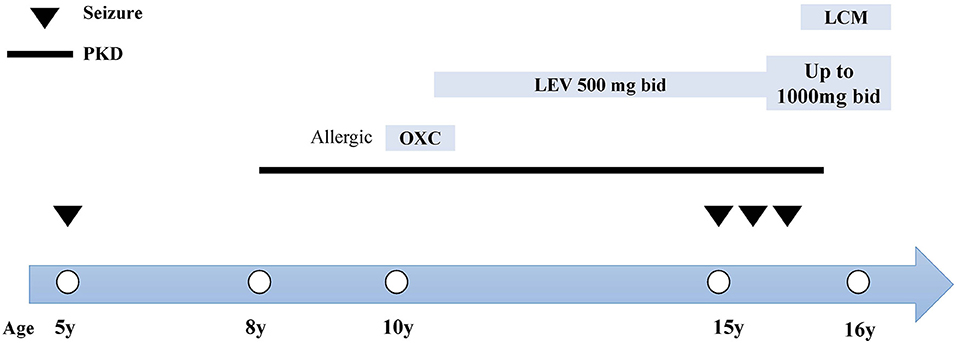
Figure 2. Clinical course and treatment of the case. The x-axis indicates the age of patient at each attack. Epileptic seizures are indicated by inverted triangles. The course of PKD is denoted by the black line. The name, dosage and timing of drug treatment are shown at the top of the diagram. bid, twice daily; LCM, lacosamide; LEV, levetiracetam; OXC, oxcarbazepine.
Discussion
Herein, we report a case of an adolescent female who had concomitant PKD and epilepsy with a PRRT2 mutation (c.649dupC). The diagnostic pearls for patients with episodic motor attacks are listed. We also discuss managements for patients with concomitant PKD and epilepsy. The excellent efficacy of even low-dose LCM highlights a new therapeutic option for such patients.
The first challenge in our case lies in the correct diagnosis of PKD and epilepsy. Both disorders can present with recurrent movements and both have a good initial response to anti-sodium-channel AEDs, though epilepsy patients would develop drug resistance later in disease course (6). To differentiate PKD and epilepsy, a careful history taking is vital. Certain ancillary testing including EEG, MRI and genetic testing would be helpful as well. PKD has an age of onset between 1 and 20 years of age with attacks consistently triggered by sudden movements, sometimes in the context of emotional, or physiologic stress (7). Kinesigenic paroxysmal movements in PKD are short and frequent. The patient has a reserved consciousness during attacks. Neurological examination and EEG are always normal (1). Epileptic seizures can mimic PKD as well, though seizure attacks are highly stereotyped and may occur during sleep. Mutations in SCN8A, CHRNA4, KCNT1, and DEPDC5 were associated with epilepsy (8, 9), while PRRT2 mutation was frequently related to PKD. In this patient, the movement-induced motor attacks are diagnosed with PKD, given the presence of triggers like sudden movements or emotional stress, the normal results on repeated MRI, a positive family history and the presence of PRRT2 mutation. The recurrent generalized tonic-clonic attacks all at night and presence of post-attack headache pointed toward the diagnosis of epilepsy. However, we did not further investigate the etiology of epilepsy considering the financial burdens of the patient. Herein, given a thorough history taking and evidence from EEG and genetic evidence, we diagnose the patient with concomitant PKD and epilepsy.
PKD is characterized by an exquisite response to antiepileptic drugs. First line treatments are carbamazepine, oxcarbazepine and phenytoin (1, 10). A dramatic response to antiepileptic drugs is seen in 98.4% of PRRT2-PKD patients (7). Treatment failure was mainly reported in homozygous or compound heterozygous PRRT2 mutation carriers (11, 12). Importantly, effective treatments to control both PKD and epilepsy remain undetermined, as shown in Table 1 (3–5, 13–19). Allergic reactions to certain AEDs like carbamazepine or oxcarbazepine further limited their use in such patients. Therapeutic options, both safe and efficacious, are urgently needed for those with concomitant PKD and epilepsy.
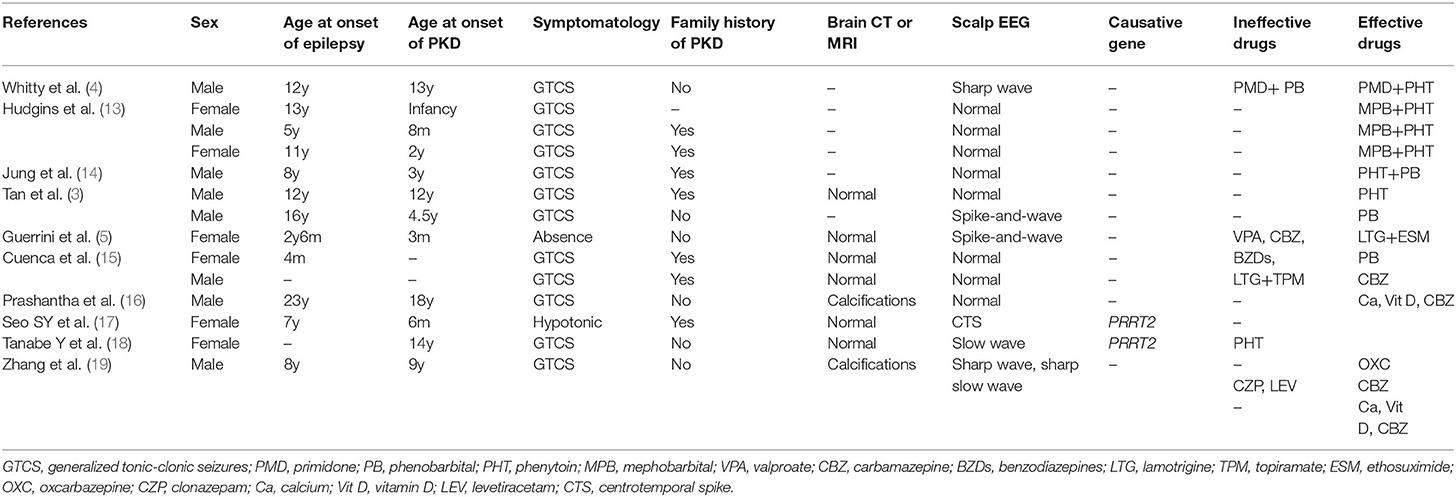
Table 1. Clinical characteristics of previously-reported patients with concomitant PKD and epilepsy.
LCM is a third-generation antiepileptic drug, with unique mechanisms of enhancing the slow inactivation of voltage-gated sodium channels. Unlike other traditional sodium channel blockers such as phenytoin and carbamazepine which inhibit the fast inactivation of the channels, LCM mainly reduces the long-term availability of sodium channels for activation, resulting in the normalization of activation thresholds (20). Mathew et al. reported a 19-year-old boy with PKD, who was responsive to LCM treatment (at a dose of 50 mg twice daily) after a hypersensitivity reaction of oxcarbazepine (21). Another recent retrospective study included four children with PKD, one of them had a PRRT2 mutation (c.649dupC). The low dose of LCM showed an excellent efficacy in all children, regardless of the status of PRRT2 mutations (22). However, large sampled studies on the correlation between PRRT2 mutations and LCM response is lacking. Our patient with concomitant PRRT2-positive PKD and epilepsy had a good response to even low-dose LCM after an allergic reaction to oxcarbazepine and treatment failure with levetiracetam. Our case further demonstrated the efficacy of low-dose LCM in patients with concomitant PKD and epilepsy, offering an optimal and safe therapeutic option for such patients.
To our knowledge, this is the first case using LCM for concomitant PRRT2 PKD and epilepsy. We suggest that the use of low-dose LCM might be an efficacious, safe and tolerable option for such patients. Further studies with a large cohort are needed to confirm our findings, especially those who are allergic or unresponsive to carbamazepine and oxcarbazepine.
Data Availability Statement
The original contributions presented in the study are included in the article/supplementary files, further inquiries can be directed to the corresponding author.
Ethics Statement
The studies involving human participants were reviewed and approved by Ethics Committee of the First Affiliated Hospital of Zhejiang Chinese Medical University. Written informed consent to participate in this study was provided by the participants' legal guardian/next of kin.
Author Contributions
J-HG drafting of the article, clinical examination of the patient, and bibliographic search. YZ and YJ conceptualized and designed the study, coordinated and supervised data collection and analyzation, and critically reviewed and revised the manuscript. Q-FL, QH, and X-HW contributed to conception and design of the study. All authors contributed to the article and approved the submitted version.
Funding
This work was supported by the Natural Science Foundation of Zhejiang Province (CN) under Grant No. LY19H090001 and the Health Science and Technology Project of Zhejiang Province (CN) under Grant No. 2022KY228.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher's Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Acknowledgments
We warmly thank the patient and her family. J-HG wants to thank, in particular, the care and support from Yi-Kang Sun over passed years.
References
1. Bruno MK, Hallett M, Gwinn-Hardy K, Sorensen B, Considine E, Tucker S, et al. Clinical evaluation of idiopathic paroxysmal kinesigenic dyskinesia: new diagnostic criteria. Neurology. (2004) 63:2280–7. doi: 10.1212/01.wnl.0000147298.05983.50
2. Huang XJ, Wang SG, Guo XN, Tian WT, Zhan FX, Zhu ZY, et al. The phenotypic and genetic spectrum of paroxysmal kinesigenic dyskinesia in China. Mov Disord. (2020) 35:1428–37. doi: 10.1002/mds.28061
3. Tan LC, Tan AK, Tjia H. Paroxysmal kinesigenic choreoathetosis in Singapore and its relationship to epilepsy. Clin Neurol Neurosurg. (1998) 100:187–92. doi: 10.1016/s0303-8467(98)00038-9
4. Whitty CW, Lishman WA, Fitzgibbon JP. Seizures induced by movement:a form of reflex epilepsy. Lancet. (1964) 2:1403–6. doi: 10.1016/s0140-6736(64)91980-4
5. Guerrini R, Sanchez-Carpintero R, Deonna T, Santucci M, Bhatia KP, Moreno T, et al. Early-onset absence epilepsy and paroxysmal dyskinesia. Epilepsia. (2002) 43:1224–9. doi: 10.1046/j.1528-1157.2002.13802.x
6. Löscher W, Potschka H, Sisodiya SM, Vezzani A. Drug resistance in epilepsy: clinical impact, potential mechanisms, and new innovative treatment options. Pharmacol Rev. (2020) 72:606–38. doi: 10.1124/pr.120.019539
7. Huang XJ, Wang T, Wang JL, Liu XL, Che XQ, Li J, et al. Paroxysmal kinesigenic dyskinesia: clinical and genetic analyses of 110 patients. Neurology. (2015) 85:1546–53. doi: 10.1212/wnl.0000000000002079
8. de Gusmão CM, Garcia L, Mikati MA, Su S, Silveira-Moriyama L. Paroxysmal genetic movement disorders and epilepsy. Front Neurol. (2021) 12:648031. doi: 10.3389/fneur.2021.648031
9. Freitas ME, Ruiz-Lopez M, Dalmau J, Erro R, Privitera M, Andrade D, et al. Seizures and movement disorders: phenomenology, diagnostic challenges and therapeutic approaches. J Neurol Neurosurg Psychiatry. (2019) 90:920–8. doi: 10.1136/jnnp-2018-320039
10. Yang Y, Su Y, Guo Y, Ding Y, Xu S, Jiang Y, et al. Oxcarbazepine versus carbamazepine in the treatment of paroxysmal kinesigenic dyskinesia. Int J Neurosci. (2012) 122:719–22. doi: 10.3109/00207454.2012.715109
11. Labate A, Tarantino P, Viri M, Mumoli L, Gagliardi M, Romeo A, et al. Homozygous c.649dupC mutation in PRRT2 worsens the BFIS/PKD phenotype with mental retardation, episodic ataxia, and absences. Epilepsia. (2012) 53:e196–9. doi: 10.1111/epi.12009
12. Delcourt M, Riant F, Mancini J, Milh M, Navarro V, Roze E, et al. Severe phenotypic spectrum of biallelic mutations in PRRT2 gene. J Neurol Neurosurg Psychiatry. (2015) 86:782–5. doi: 10.1136/jnnp-2014-309025
13. Hudgins RL, Corbin KB. An uncommon seizure disorder: familial paroxysmal choreoathetosis. Brain. (1966) 89:199–204. doi: 10.1093/brain/89.2.199
14. Jung SS, Chen KM, Brody KA. Paroxysmal choreoathetosis. Report of Chinese cases. Neurology. (1973) 23:749–55. doi: 10.1212/wnl.23.7.749
15. Cuenca-Leon E, Cormand B, Thomson T, Macaya A. Paroxysmal kinesigenic dyskinesia and generalized seizures: clinical and genetic analysis in a Spanish pedigree. Neuropediatrics. (2002) 33:288–93. doi: 10.1055/s-2002-37079
16. Prashantha DK, Pal PK. Pseudohypoparathyroidism manifesting with paroxysmal dyskinesias and seizures. Mov Disord. (2009) 24:623–4. doi: 10.1002/mds.22382
17. Seo SY, You SJ. Paroxysmal kinesigenic dyskinesia in a patient with a PRRT2 mutation and centrotemporal spike discharges on electroencephalogram: case report of a 10-year-old girl. Korean J Pediatr. (2016) 59:S157–60. doi: 10.3345/kjp.2016.59.11.S157
18. Tanabe Y, Taira T, Shimotake A, Inoue T, Awaya T, Kato T, et al. An adult female with proline-rich transmembrane protein 2 related paroxysmal disorders manifesting paroxysmal kinesigenic choreoathetosis and epileptic seizures. Rinsho Shinkeigaku. (2019) 59:144–8. doi: 10.5692/clinicalneurol.cn-001228
19. Zhang C, Zhou X, Feng M, Yue W. Paroxysmal dyskinesia and epilepsy in pseudohypoparathyroidism. Mol Genet Genomic Med. (2020) 8:e1423. doi: 10.1002/mgg3.1423
20. Rogawski MA, Tofighy A, White HS, Matagne A, Wolff C. Current understanding of the mechanism of action of the antiepileptic drug lacosamide. Epilepsy Res. (2015) 110:189–205. doi: 10.1016/j.eplepsyres.2014.11.021
21. Mathew T, Aroor S, Nadig R, Sarma GR. Lacosamide in paroxysmal kinesigenic dyskinesia. Mov Disord. (2012) 27:801–2. doi: 10.1002/mds.24928
Keywords: lacosamide, paroxysmal kinesigenic dyskinesia, epilepsy, movement disorder, PRRT2
Citation: Geng J-H, Zheng Y, Li Q-F, Hou Q, Wang X-H and Jiang Y (2022) Case Report: A Case of Concomitant Paroxysmal Kinesigenic Dyskinesia and Epilepsy: Can We Treat Two Birds With One Stone? Front. Neurol. 13:826897. doi: 10.3389/fneur.2022.826897
Received: 01 December 2021; Accepted: 11 January 2022;
Published: 02 February 2022.
Edited by:
Fernando Cendes, State University of Campinas, BrazilReviewed by:
Claudio De Gusmao, Boston Children's Hospital and Harvard Medical School, United StatesCarlos R. M. Rieder, Federal University of Health Sciences of Porto Alegre, Brazil
Copyright © 2022 Geng, Zheng, Li, Hou, Wang and Jiang. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Yan Jiang, amlhbmd5YW5AemNtdS5lZHUuY24=