 Greta Amore1
Greta Amore1 Ambra Butera1
Ambra Butera1 Giulia Spoto1
Giulia Spoto1 Giulia Valentini1
Giulia Valentini1 Maria Concetta Saia1
Maria Concetta Saia1 Vincenzo Salpietro2,3,4*
Vincenzo Salpietro2,3,4* Francesco Calì5
Francesco Calì5 Gabriella Di Rosa1
Gabriella Di Rosa1 Antonio Gennaro Nicotera1
Antonio Gennaro Nicotera1- 1Department of Human Pathology of the Adult and Developmental Age “Gaetano Barresi”, Unit of Child Neurology and Psychiatry, University of Messina, Messina, Italy
- 2Department of Neuromuscular Disorders, Institute of Neurology, University College London, London, United Kingdom
- 3Pediatric Neurology and Muscular Diseases Unit, Scientific Institute for Research, Hospitalization and Healthcare (IRCCS) Istituto Giannina Gaslini, Genoa, Italy
- 4Department of Neurosciences, Rehabilitation, Ophthalmology, Genetics, Maternal and Child Health, University of Genoa, Genoa, Italy
- 5Oasi Research Institute-Scientific Institute for Research, Hospitalization and Healthcare (IRCCS), Troina, Italy
Potassium Voltage-Gated Channel Subfamily Q Member 2 (KCNQ2) gene has been initially associated with “Benign familial neonatal epilepsy” (BFNE). Amounting evidence arising by next-generation sequencing techniques have led to the definition of new phenotypes, such as neonatal epileptic encephalopathy (NEE), expanding the spectrum of KCNQ2-related epilepsies. Pyridoxine (PN) dependent epilepsies (PDE) are a heterogeneous group of autosomal recessive disorders associated with neonatal-onset seizures responsive to treatment with vitamin B6 (VitB6). Few cases of neonatal seizures due to KCNQ2 pathogenic variants have been reported as successfully responding to VitB6. We reported two cases of KCNQ2-related neonatal epilepsies involving a 5-year-old male with a paternally inherited heterozygous mutation (c.1639C>T; p.Arg547Trp), and a 10-year-old female with a de novo heterozygous mutation (c.740C>T; p.Ser247Leu). Both children benefited from VitB6 treatment. Although the mechanisms explaining the efficacy of VitB6 in such patients remain unclear, this treatment option in neonatal-onset seizures is easily taken into account in Neonatal Intensive Care Units (NICUs). Further studies should be conducted to better define clinical guidelines and treatment protocols.
Introduction
Potassium Voltage-Gated Channel Subfamily Q Member 2 (KCNQ2) gene, first described by Singh et al. (1), is located on chromosome 20q13.3 and encodes for the voltage-gated potassium channel subunit Kv7.2. It has been traditionally related to “Benign familial neonatal epilepsy” (BFNE). This clinical entity often occurs within the first 2 weeks of life and is characterized by tonic/clonic early-onset seizures with apneic episodes and autonomic manifestations. Generally, it presents a self-limiting course for weeks/months with a regular neurodevelopmental outcome (2–4).
Amounting evidence arising from innovative genetic testing, and particularly next-generation sequencing (NGS) techniques, has revealed that different pathogenic variants of the same gene are responsible for several epileptic phenotypes; in particular, KCNQ2-related neonatal epileptic encephalopathy (NEE) contributed to expanding the spectrum of KCNQ2-related epilepsies (5). This is a severe phenotype, with an onset during the first days of life of intractable seizures (usually focal tonic), variably evolving into seizure freedom or a worsening/relapsing of the epileptic attacks over time, and an inevitable, though variable, adverse neurodevelopmental outcome (4–6). The electroencephalogram (EEG) may show a burst-suppression pattern or multifocal epileptiform abnormalities with background attenuation. This phenotype is mostly due to de novo missense variants of KCNQ2 causing a dominant-negative effect leading to a loss or a reduction of the M-current (a slow activating non-inactivating potassium current modulating the resting membrane potential) and less commonly to a gain of function effect, but not to a loss of function (haplo-insufficiency) (4, 7–9).
First described by Hunt et al. (10), pyridoxine (PN) dependent epilepsies (PDE) are a heterogeneous group of autosomal recessive disorders associated with neonatal-onset seizures not well-controlled with anti-epileptic drugs (AEDs) but responsive to large daily doses of vitamin B6 (VitB6) (11). PDE are caused by inborn errors of PN metabolism, usually due to pathogenic variants of ALDH7A1 gene (antiquitin). ALDH7A1 encodes for the alpha-aminoadipic semialdehyde (a-AASA) dehydrogenase, a key enzyme in the lysine metabolism, eventually implicated in the activation of VitB6 (12). Moreover, pyridoxamine5'-phosphate oxidase (PNPO) and pyridoxal 5′-phosphate binding protein (PLPBP) genes [both involved in the homeostasis of the active form of PN, namely pyridoxal 5′-phosphate (PLP)], have recently been related to variants of PDE more responsive to PLP than PN (13, 14).
The phenotypic spectrum associated with ALDHE mutation include: (1) Classic PDE-ALDH7A1 (with a dramatic early onset of prolonged or recurrent seizures of different types); (2) Atypical PDE-ALDH7A1 (including a late-onset, or seizures initially responding only to AEDs and eventually controllable only with PN/PLP several months later, or folic acid-responsive seizures, anyhow associated with variable degree of neurocognitive outcomes) (15). Clinical diagnosis is based on the demonstration of seizures ceasing after PN treatment, even after the elimination of all AEDs, and re-occurrence after its withdrawal (11, 16).
In addition to genetic testing, elevated levels of a-AASA in plasma and urine, and of pipecolic acid in plasma, urine and cerebrospinal fluid (CSF), as well as a high plasma-to-CSF PLP ratio, can be suggestive for VitB6 disorders (17, 18). However, a clear interpretation of these results is complicated; in fact, to date, no studies on CSF PLP levels in healthy newborns or infants are available, hence reference ranges derive from neurologically abnormal children undergoing CSF metabolite measurement for diagnostic workup (18).
Interestingly, PN and PLP have been proven to be effective in several early-onset seizures, not meeting the diagnosis of PDE, but instead falling within the category of “Pyridoxine-responsive epilepsy” (PRE) (19).
In this regard, they have been increasingly used in the clinical practice as add-on treatments in early-onset epilepsies, and it should be noted that few reports are today available specifically on KCNQ2-related neonatal epilepsies trialed with VitB6 (either in its inactive or active form) with variable outcomes (Table 1). Herein, we reported two new cases of KCNQ2-related neonatal epilepsy who benefited from VitB6 treatment.
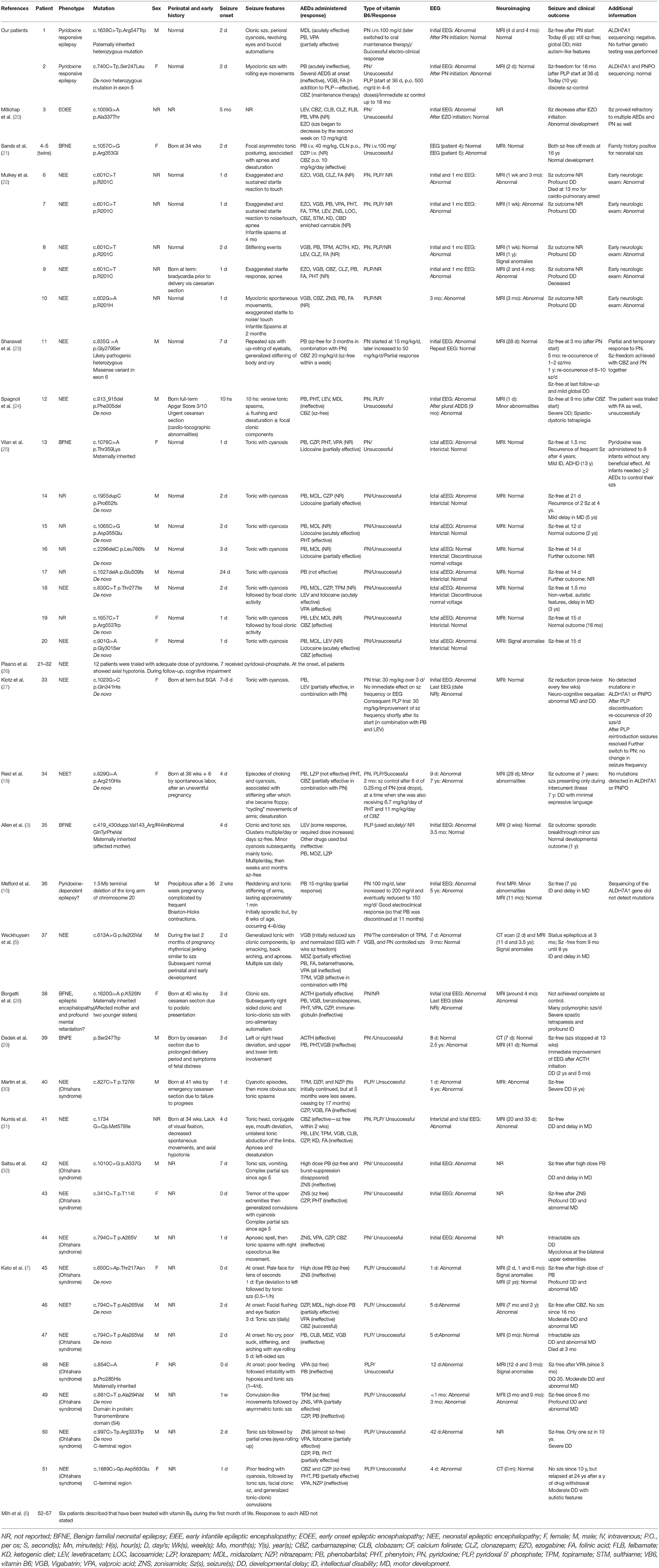
Table 1. Patients described in the literature who carry a KCNQ2 mutation and have been trialed with vitamin B6.
Case Reports
Case 1
A 6-year-old Caucasian boy was born at term by eutocic vaginal delivery from an uneventful pregnancy. Birth weight was 2,870 gr. Perinatal period was unremarkable and physical examination at birth normal. He was the second child of healthy related parents with common ancestors. The patient's family history was positive for epilepsy both in the maternal and paternal line; in particular, the father was diagnosed with a KCNQ2-related-epilepsy treated with Valproate (VPA) and Phenobarbital (Pb). It is to note that the father referred this data only at the end of our patient's genetic investigation.
On the second day of life, the patient presented clonic seizures at the limbs. The episodes resolved spontaneously. On the fourth day, he was hospitalized for the re-occurrence of seizures associated with perioral cyanosis. An electroencephalogram (EEG) showed polyspike wave complexes. Given the persistence of continuous clonic seizures, associated with perioral cyanosis, revolving eyes and buccal automatisms (sucking), midazolam by continuous intravenous infusion and intravenous boluses of Pb were administered. Brain magnetic resonance imaging (MRI) was normal. On the 10th day of life, oral Pb treatment was started, with seizure control for almost 3 months. Meanwhile, metabolic tests and the sequencing of KCNQ2 gene were performed.
At 3 months of life, recurrent convulsive seizures/status epilepticus occurred; thus, VPA was started at 10 mg/kg/bid, with subsequent titration up to 30 mg/kg/bid with transient remission of seizures. About a month later, focal clonic seizures at the limbs appeared, mainly with deviation of the eyes and cyanosis, lasting 60 s. An EEG showed “slight abnormalities of electrical brain activity in the left posterior areas.” Further brain MRI scans were normal. Given the age of the patient and the partial response to stabilized treatment with VPA (100 mg/bid) and intravenous Pb (7.5 mg/bid), we decided to start PN supplementation as an intramuscular formulation of the vitamin B complex, containing 100 mg/day of PN. Seizure cessation and disappearance of the EEG abnormalities were gained after few days of treatment. Maintenance treatment was carried on, in agreement with the patient's parents (who were hesitant about PN withdrawal, as initially planned), with an oral formulation of multivitamin B complex at a very low dosage of 5 ml/day, equivalent to 0.45 mg of PN, with a seizure-free interval of about a month. In the meanwhile, a Developmental Quotient (DQ) of 70 was measured by using the Brunet-Lezine developmental scale, detecting a neurodevelopmental delay. The next month, in concomitance to an infectious episode, he presented with a further epileptic seizure characterized by clonic movements of the limbs, revolving eyes, perioral cyanosis, lasting about 60 s and resolving spontaneously. Antiepileptic treatment was unchanged. No further seizures occurred thereafter, either during infectious episodes.
Throughout the following years, VPA first, and Pb later, were discontinued. Today the child is seizure-free and takes daily multivitamin B complex, also containing PN. Despite the global neurodevelopmental delay, he is able to walk alone, jump and climb; his expressive language is limited to simple and short phrases and characterized by dyslalia, whereas the comprehension is adequate. The child presents mild autism-like features (such as self-stimulatory, repetitive and stereotyped behaviors, and relational difficulties). He has also gained sphincteric control.
Direct sequencing of the KCNQ2 (NM_172107.4; NP_742105.1) gene demonstrated a paternally inherited heterozygous mutation (c.1639C>T; p.Arg547Trp), leading to the replacement of Arginine with Tryptophan at position 547 of the protein and involving a conserved amino-acid of the protein predicted to have functional consequences on the protein (Figure 1). This variant had already been reported in the literature by Zara et al. (2) (as causative of a BFNE in a female patient with a maternal inheritance) and Lindy et al. (33) (no clinical data available). In silico analysis performed with bioinformatic tools predicted that mutation has a potential strong damaging effect on the structure/function of the KCNQ2 protein. ACMG standard criteria for assessment of pathogenicity of variants (Varsome) (34) should be considered as “Pathogenic” (Table 2).
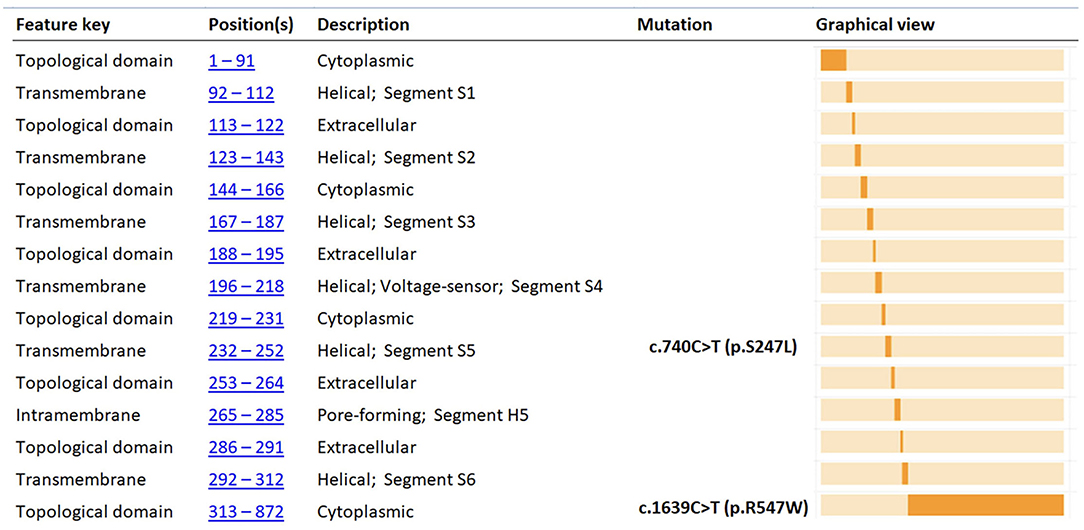
Figure 1. Functional domain position of p.R547W and p.S247L amino acid changes of KCNQ2 protein (adapted from https://www.uniprot.org). The figure shows the KCNQ2 functional domain position of the variants found in our two cases (p.R547W and p.S247L). In silico analysis (Table 2) predicted that the two mutations have potential strong damaging effect on the structure/function of the KCNQ2 protein.
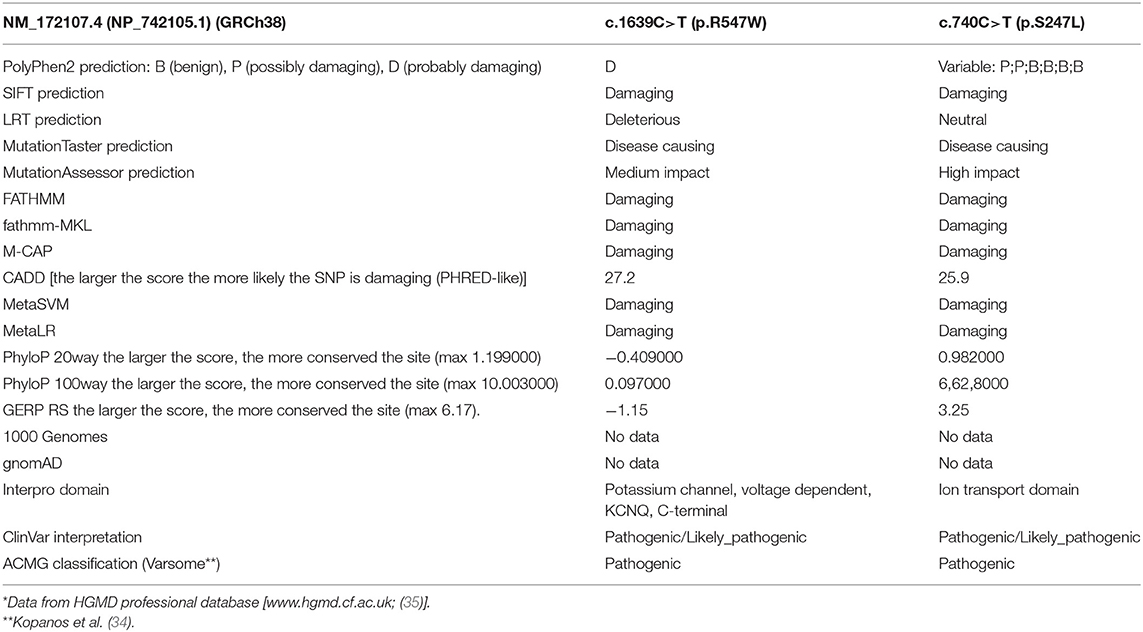
Table 2. In silico prediction of the KCNQ2 missense mutations*.
In the case of our patient, ALDH7A1 sequencing was later reported to be negative, and no further genetic testing was performed. This data prevented us the possibility to explain his positive response to PN and his clinical phenotype (that, despite the clinical continuum existing between BFNE and NEE, seems closer to the NEE phenotype), in which a possible role of other genes on his impaired neurodevelopment cannot be ruled out.
Case 2
A 10-year-old Caucasian female was born at term by eutocic vaginal delivery from an uneventful pregnancy. Birth weight was 3,400 gr. Prenatal and perinatal history was unremarkable. On the second day of life, the baby showed myoclonic seizures associated with sudden loss of muscle tone and rolling eye movements. The patient started treatment with Pb, without effects. The EEG showed a suppression-burst pattern. Brain MRI and routine metabolic investigations resulted to be normal. At day 36 of life, after unsuccessful therapeutic attempts with Pb and VPA, and the start of PN administration, she was admitted to the NICU without considerable response. Herein, she was started on oral PLP therapy (up to a dose of 500 mg/day divided in 4–6 administrations), achieving immediate seizure control. An extended metabolic workup was performed highlighting an elevation of pipecolic acid both in serum and urine, albeit the sequencing of ALDH7A1 and PNPO genes was normal. Given the good clinical response to PLP and the poor neurodevelopment performance (no achievement of motor milestones, marked muscular hypotonia, and visual disturbances), she received a diagnosis of “Focal epilepsy with pyridoxal phosphate-responsive seizures and global neurodevelopmental delay.”
After 18 months of seizure-free interval, epileptic tonic spasms occurred, both during sleep and wakefulness. The EEG showed multifocal epileptic discharges.
Low CSF folic acid levels were revealed; hence, folic acid was started (7.5 mg/bid, later increased at 7.5 mg/tid), in add-on to her previous therapy, with benefits in terms of number and intensity of epileptic seizures.
Despite the overall discrete seizure control, subsequent interictal EEG recordings showed marked diffuse abnormalities up to a quasi-periodic epileptiform pattern. The patient showed severe global neurodevelopmental delay at the follow-up.
Further genetic testing was performed. Array-CGH was not informative. Whereas, trios NGS (epilepsy panel) revealed a de novo heterozygous mutation in Exon 5 of the KCNQ2 gene (NM_172107.4; NP_742105.1): c.740C>T/(p.Ser247Leu), affecting a highly conserved aminoacidic region and predicted to be deleterious in silico programs (Figure 1). However, using the standard procedures for assessment of pathogenicity of variants (ACMG criteria—Varsome) (34), should be considered as “Pathogenic” (Table 2). This variant has already been reported in the literature and related to early-onset epileptic phenotypes with a variable degree of neurocognitive impairment (36–41). However, none of these reports shows evidence of VitB6 supplementation. Our patient was maintained on Vigabatrin, PLP, and Folic acid for several years. By the age of 10 years, given the occurrence of severe behavioral and sleep disorders, PN, Folic acid, and Vigabatrin were withdrawn, and maintenance therapy with carbamazepine, in add-on to Lorazepam and Promazine, was started.
To date, the child failed to achieve trunk control, independent standing or walking. She shows axial hypotonia and limbs hypertonia. Language is limited to vocalizations and she presents severe intellectual disability. In addition, behavioral disturbances are evident, with aggressive manifestations and sleep disorders. She displays a good clinical control, with occurrence of rare seizures (one episode/1–2 years).
Discussion
Herein, we reported two cases of KCNQ2-related epilepsy, a 5-year-old male with a paternally inherited heterozygous mutation (c.1639C>T; p.Arg547Trp), and a 10-year-old female with a de novo heterozygous mutation (c.740C>T; p.Ser247Leu), who benefited from PN treatment.
The first patient presented with neonatal epilepsy with an onset during the first week of life, which was later ascribed to a KCNQ2 pathogenic variant paternally inherited.
The same variant had already been described in the literature (2, 33) and reported as causative of a BFNE phenotype in a female patient with maternal inheritance (2).
Our patient, with his overall good seizure-responsivity despite impaired neurodevelopment, does not show all the typical characteristics of BFNE or NEE. Even if no evidence of an inborn error of VitB6 was available for our patient (ruling out the diagnosis of a PDE), his seizures, scarcely controlled by AEDs, revealed a good and immediate electro-clinical response to the initiation of intramuscular PN. Interestingly, this effect did not cease after switching to PN oral administration, neither after the discontinuation of AEDs, strengthening the diagnostic hypothesis of PN-responsive epilepsy (PRE).
The second patient, after a sudden and severe onset of neonatal seizures, requiring plural hospitalizations, and failed attempts with AEDs, such as Pb and VPA, and with PN too, demonstrated an immediate clinical benefit from the initiation of PLP, and, over time, to folic acid as well.
In this case, a de novo disease-causing KCNQ2 variant was detected (c.740C>T; p.Ser247Leu), and the good clinical response to PLP and folic acid were supported, respectively, by elevated levels of pipecolic acid both in serum and urine and by reduced CSF folates level. Moreover, genetic testing resulted negative for ALDH7A1 and PNPO pathological variants. These data supported the possibility to classify this clinical phenotype into the PRE spectrum.
Moreover, even though the clear effect of PLP reduced over time, other AEDs (i.e., Pb and VPA) proved to be ineffective, and the neurocognitive outcome of the patient is quite poor, she still presents an overall good clinical control, with seizures being years apart.
Summarizing, regardless of the several divergences between these 2 cases, both of them may fit into the category of PRE, particularly within the subgroup of KCNQ2-related ones.
Data from the literature have demonstrated the usefulness of VitB6, either as PN or, even better, as PLP, in certain types of neonatal-onset epilepsies (13, 19). In addition, recent reports have been unraveling a potential correlation between KCNQ2-related epilepsies and VitB6 responsiveness (18). To date, several cases of patients carrying a pathogenic variant of KCNQ2 trialed with VitB6 (either in the form of PN or PLP) have been reported in the literature. Unfortunately, the effects of VitB6 have not been accurately described for all of them. Additionally, several cases have been extracted from studies focusing on other than the KCNQ2-PN correlation, making it arduous to infer the exact impact of VitB6 on the clinical picture (6, 22, 26). Five reports have openly disclosed various degrees of a successful response to PN/PLP in KCNQ2-related epilepsies (5, 16, 18, 23, 27). Interestingly, despite their small number, all patients share a neuro-cognitive impairment in the lack of genetic data confirming/denying the PN-dependency/responsiveness, in line with ours. All cases are reported in Table 1 (and detailed in the complete version of the table—see Supplementary Material).
Currently, the etiopathogenic mechanism underlying the PN responsiveness of certain neonatal seizures, and particularly of those KCNQ2-related, remains unclear. However, some hypotheses have been proposed [(18); Figure 2].
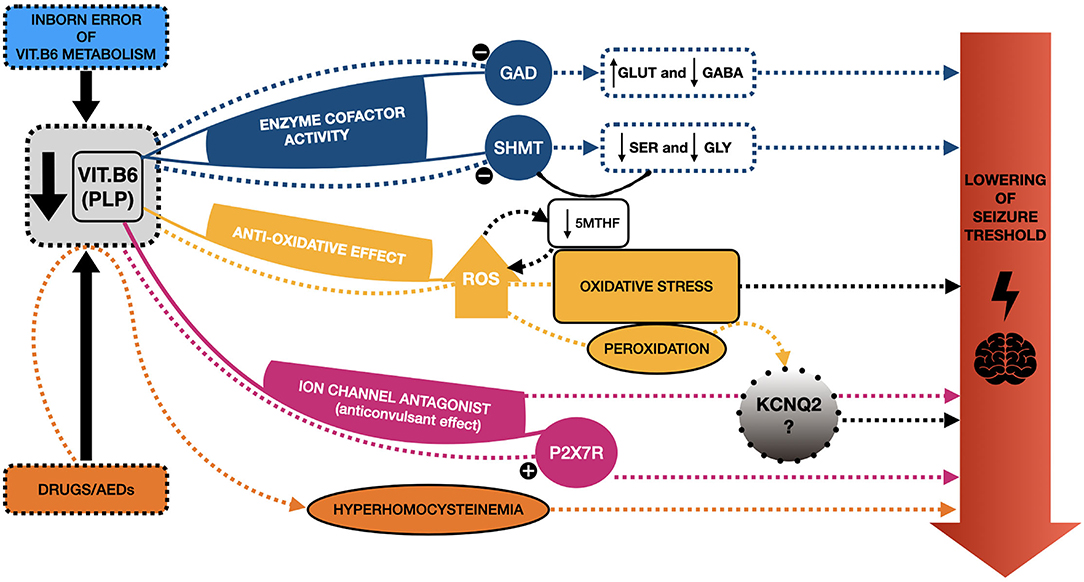
Figure 2. Schematic representation of the mechanisms proposed to explain seizure responsiveness to vitamin B6 and the potential link with KCNQ2. Several etiopathogenic mechanisms have been proposed to explain the responsiveness of certain neonatal seizures to vitamin B6 (VitB6), and particularly to its active form, pyridoxal 5′ phosphate (PLP). PLP acts as a cofactor of several enzymes, such as Glutamic acid decarboxylase (GAD), regulating the synthesis of gamma-amino butyric acid (GABA) from Glutamate (GLUT). A reduction of PLP may determine an imbalance between these two (in favor of GLUT), hence decreasing the seizure threshold. PLP is also involved in the synthesis of serine (SER) and glycine (GLY), modulating the serine hydroxymethyltransferase (SHMT) activity; thus, a reduction of PLP may determine lower levels of these two amino-acids, whose reduction has been related to epileptic phenotypes. SHMT is involved in the metabolic pathway, eventually leading to the formation of the main circulating form of folates, the 5-methyltetrahydrofolate (5-mTHF), a reduction of which has been reported in some patients suffering from pyridoxine-dependent epilepsies (PDE). Furthermore, the reduction of 5-mTHF may fall into another proposed mechanism underlying epilepsy, namely oxidative stress, with excessive production of reactive oxygen species (ROS). This may be a consequence of low levels of VitB6 (anti-oxidative agent) and, at the same time, a cause of folates depletion. Additionally, ROS contribute to the peroxidation of several molecules, also affecting ion channels, hence potentially KCNQ2. PLP also acts as an antagonist of specific ion channels, as P2X7R, with a possible anticonvulsant effect, which could not occur when PLP levels are low. A similar antagonist action on KCNQ2, with possible analogous effects, has been proposed. Finally, some drugs, especially antiepileptic ones (AEDs), may be responsible for the depletion of VitB6 and, indirectly, hyperhomocysteinemia, which is known to lower the seizure threshold. Overall, these mechanisms may explain the responsiveness of some neonatal seizures to VitB6 treatment and help shed light on the potential link with KCNQ2-related epilepsies.
First of all, VitB6, particularly as PLP, is notably implicated as a cofactor of hundreds of enzymatic reactions, having a role in several functions, such as in the metabolism of amino-acids and the synthesis of neurotransmitters (42). In this regard, PLP is notably a cofactor of glutamic acid decarboxylase (GAD), an enzyme involved in the synthesis of gamma-amino butyric acid (GABA) from glutamate (GLUT). These two are, respectively, the key inhibitory and excitatory neurotransmitters of the CNS, and an imbalance between them has long been considered among the molecular mechanisms underlying epilepsy (43). The reduction of GABA deriving from low PLP levels has been proposed as a potential mechanism in the genesis of PDE, but it is widespread among the authors the opinion that this cannot be the only one (43–45). Ramos et al. (45) have recently highlighted that conflicting data have been reported on GLUT and GABA levels in the CSF of patients suffering from VitB6 deficiency, supporting this hypothesis (45). In this regard, they carried out a study on a model system of VitB6-deficient Neuro-2a cells, revealing a significant reduction in the de novo synthesis of serine and, consequently, glycine, whose reduction has been related to epileptic phenotypes, suggesting a potential role of these two (45, 46).
Additionally, it is noteworthy to mention that glycine synthesis from serine depends on the enzyme serine hydroxymethyltransferase (SHMT), which requires PLP as a cofactor. SHMT is involved in the metabolic pathway which encompasses the enzyme methylenetetrahydrofolate reductase (MTHFR) as well, eventually leading to 5-methyltetrahydrofolate (5-mTHF) formation, the main circulating form of folate (45). This is particularly interesting since low levels of 5-mTHF in CSF have already been reported in our second patient and some PDE patients (18, 47).
In particular, the patient described by Reid et al. (18) and ours reported a positive response to VitB6 treatment, both in the presence of low folates and of different isolated lab findings suggestive for VitB6 disorders (respectively, a high plasma-to-CSF PLP ratio in the former, and an elevation of pipecolic acid both in serum and urine in the latter), though in the absence of a genetic confirmation of inborn errors of VitB6 metabolism. Although these findings cannot be fully explained, and no clear evidence on the topic is yet available, all this suggests not only a potential role of folates in PREs and PDEs, but also that there is much more to find out.
Besides, when it comes to folates, it is not possible to state whether their involvement in PDEs/PREs, and, in general, in epilepsy may be a causative factor, a consequence, or both. Their involvement in these conditions may be mutually related to another proposed mechanism underlying epilepsy, namely the excessive production of reactive oxygen species (ROS) (17, 18). Several studies have demonstrated an excessive ROS production in epilepsy, so they outgrow the capability of endogenous antioxidants to contrast their effect (43). Besides, the excess of ROS may determine a depletion of folates, which are known to exhibit antioxidant functions acting as ROS scavengers (18, 48).
Overall, high levels of ROS may have detrimental effects, including the peroxidation of structures and molecules (i.e., enzymes and components of cell membranes), potentially affecting ion channels indirectly as well (43). Moreover, given the demonstrated anti-oxidative effects of VitB6 (49) and the potential effect of ROS on ion channels, one could speculate that there may be a close, albeit yet unknown, relation between VitB6 and channelopathies, such as KCNQ2-related ones.
Anyhow, when ROS production is excessive, the result is a strong oxidative stress and an imbalance favoring the excitotoxicity (43).
Another possible mechanism may involve the recently demonstrated antagonist action of PLP toward P2X-receptors (and particularly the subtype P2X7R) (50). These receptors are a class of ligand-gated ion channels, activated by ATP, contributing to neuro- and glio-transmission and lately associated with epileptic conditions, such as status epilepticus (51). Since P2X7R antagonists have recently been reported as having anticonvulsant effects, this may apply as well to PLP, explaining at least partly its role in controlling seizures (52).
Given the above, Reid et al. (18) proposed that PLP may as well have a direct antagonist effect on ion channels, including the one encoded by KCNQ2. However, further evidence is needed to confirm this hypothesis.
Other potential mechanisms explaining PDE and PRE may relate to secondary defects of VitB6, for instance drug-induced ones. Several drugs, such as carbamazepine, phenytoin, and PB, may reduce VitB6 levels and indirectly lead to hyperhomocysteinemia (which is known to lower seizure threshold) (53, 54). It is likely that the same effect may apply to other AEDs and that it may partly explain the VitB6 responsiveness of certain patients, even when temporary. Unfortunately, it is not possible to confirm or deny this hypothesis for our patients.
Our study presents some limitations, such as its retrospective nature, with consequent possible information biases and lack of specific data (such as details about treatments and diagnostic assessment taking place in different centers from ours, or the correlation between AEDs modification and concomitant levels of VitB6 and homocysteine), as well as the small sample of patients, making it difficult to generalize our findings.
In the light of the above, further studies focusing on the correlation between types of AEDs used, treatment duration, concomitant plasma levels of VitB6, homocysteine and folates, and possibly considering the eventual disease-causing variants associated (and in particular KCNQ2 ones), may help unravel new evidence on the topic.
Prospective studies are needed to analyze and compare the effects of standardized treatment protocols with VItB6 in neonatal epilepsies in general and in those KCNQ2-related in particular.
All this is particularly important when considering the widespread resort to VitB6 trials in neonatal epilepsies in NICUs, where this treatment is quickly taken into account, though not devoid of risks. In fact, high doses of PN and PLP have been reported as causative of peripheral neuropathy and liver toxicity, respectively (14, 18). In our opinion, this sets some limits on the advisability of resorting to a blanket VitB6 treatment. Therefore, our suggestion would be to initiate it as soon as possible in all those cases in which clinical features and/or genetic testing and lab findings suggest, or, even better, clearly demonstrate a VitB6 disorder (preferably after excluding liver diseases and nerve conduction defects). Otherwise, we would instead reserve VitB6 treatment for peculiar situations, such as drug-resistant NEEs, including those with an already proven pathogenic KCNQ2 variant.
Conclusions
Despite the limits of our study, our data contribute to adding new evidence on the potential beneficial effect of VitB6 treatment in KCNQ2-neonatal epilepsies, in apparent lack of inborn errors of VitB6 metabolism. Further studies should be conducted to elucidate the mechanisms underlying the variability of VitB6 effects in these patients, to help discriminate whether to include or not KCNQ2 (besides ALDH7A1 and PNPO) in the genetic testing of neonatal-onset seizures responsive to PN/PLP, and, finally, to define appropriate clinical guidelines and treatment protocols.
Data Availability Statement
The datasets presented in this article are not readily available due to ethical and privacy restrictions. Requests to access the datasets should be directed to the corresponding author.
Ethics Statement
Ethical review and approval was not required for the study on human participants in accordance with the local legislation and institutional requirements. Written informed consent to participate in this study was provided by the participants' legal guardian/next of kin. Written informed consent was obtained from the minor(s)' legal guardian/next of kin for the publication of any potentially identifiable images or data included in this article. Written informed consent was obtained from the participant for the publication of this case report.
Author Contributions
AN and GD conceived planned and supervised the study. GA and AB wrote the first draft of the manuscript. GA, AB, and FC prepared the tables. GA and FC designed the figures. GS, GV, MS, and VS helped supervise the project. All authors contributed to manuscript revision, read, and approved the submitted version.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher's Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fneur.2022.826225/full#supplementary-material
References
1. Singh NA, Charlier C, Stauffer D, DuPont BR, Leach RJ, Melis R, et al. A novel potassium channel gene, KCNQ2, is mutated in an inherited epilepsy of newborns. Nat Genet. (1998) 18:25–9. doi: 10.1038/ng0198-25
2. Zara F, Specchio N, Striano P, Robbiano A, Gennaro E, Paravidino R, et al. Genetic testing in benign familial epilepsies of the first year of life: clinical and diagnostic significance. Epilepsia. (2013) 54:425–36. doi: 10.1111/epi.12089
3. Allen NM, Mannion M, Conroy J, Lynch SA, Shahwan A, Lynch B, et al. The variable phenotypes of KCNQ-related epilepsy. Epilepsia. (2014) 55:e99–105. doi: 10.1111/epi.12715
4. Spoto G, Saia MC, Amore G, Gitto E, Loddo G, Mainieri G, et al. Neonatal seizures: an overview of genetic causes and treatment options. Brain Sci. (2021) 11:1295. doi: 10.3390/brainsci11101295
5. Weckhuysen S, Mandelstam S, Suls A, Audenaert D, Deconinck T, Claes LRF, et al. KCNQ2 encephalopathy: Emerging phenotype of a neonatal epileptic encephalopathy. Ann Neurol. (2012) 71:15–25. doi: 10.1002/ana.22644
6. Milh M, Boutry-Kryza N, Sutera-Sardo J, Mignot C, Auvin S, Lacoste C, et al. Similar early characteristics but variable neurological outcome of patients with a de novo mutation of KCNQ2. Orphanet J Rare Dis. (2013) 8:80. doi: 10.1186/1750-1172-8-80
7. Kato M, Yamagata T, Kubota M, Arai H, Yamashita S, Nakagawa T, et al. Clinical spectrum of early onset epileptic encephalopathies caused by KCNQ2 mutation. Epilepsia. (2013) 54:1282–7. doi: 10.1111/epi.12200
8. Orhan G, Bock M, Schepers D, Ilina EI, Reichel SN, Löffler H, et al. Dominant-negative effects of KCNQ2 mutations are associated with epileptic encephalopathy. Ann Neurol. (2014) 75:382–94. doi: 10.1002/ana.24080
9. Miceli F, Soldovieri MV, Ambrosino P, De Maria M, Migliore M, Migliore R, Taglialatela M. Early-onset epileptic encephalopathy caused by gain-of-function mutations in the voltage sensor of Kv7.2 and Kv7.3 potassium channel subunits. J Neurosci. (2015) 35:3782–93. doi: 10.1523/JNEUROSCI.4423-14.2015
10. Hunt AD Jr, Stokes J Jr, Mccrory WW, Stroud HH. Pyridoxine dependency: report of a case of intractable convulsions in an infant controlled by pyridoxine. Pediatrics. (1954) 13:140–5.
11. Baxter P. Pyridoxine-dependent and pyridoxine-responsive seizures. Dev Med Child Neurol. (2001) 43:416–20. doi: 10.1017/s0012162201000779
12. Mills PB, Struys E, Jakobs C, Plecko B, Baxter P, Baumgartner M, et al. Mutations in antiquitin in individuals with pyridoxine-dependent seizures. Nat Med. (2006) 12:307–9. doi: 10.1038/nm1366
13. Wang HS, Kuo MF, Chou ML, Hung PC, Lin KL, Hsieh MY, et al. Pyridoxal phosphate is better than pyridoxine for controlling idiopathic intractable epilepsy. Arch Dis Child. (2005) 90:512–5. doi: 10.1136/adc.2003.045963
14. Mills PB, Camuzeaux SS, Footitt EJ, Mills KA, Gissen P, Fisher L, et al. Epilepsy due to PNPO mutations: genotype, environment and treatment affect presentation and outcome. Brain. (2014) 137(Pt 5):1350–60. doi: 10.1093/brain/awu051
15. Gospe SM Jr. Pyridoxine-dependent epilepsy – ALDH7A1. In: Adam MP, Ardinger HH, Pagon RA, Wallace SE, Bean LJH, Gripp KW, editors. GeneReviews®. Seattle, WA: University of Washington (1993–2022). Available online at: https://www.ncbi.nlm.nih.gov/books/NBK1486/
16. Mefford HC, Cook J, Gospe SM Jr. Epilepsy due to 20q13.33 subtelomere deletion masquerading as pyridoxine-dependent epilepsy. Am J Med Genet Part A. (2012) 158A:3190–5. doi: 10.1002/ajmg.a.35633
17. Footitt EJ, Heales SJ, Mills PB, Allen GF, Oppenheim M, Clayton PT. Pyridoxal 5'-phosphate in cerebrospinal fluid; factors affecting concentration. J Inherit Metab Dis. (2011) 34:529–38. doi: 10.1007/s10545-011-9279-7
18. Reid ES, Williams H, Stabej P, James C, Ocaka L, Bacchelli C, et al. Seizures due to a KCNQ2 mutation: treatment with vitamin B6. JIMD Rep. (2016) 27:79–84. doi: 10.1007/8904_2015_460
19. Ohtahara S, Yamatogi Y, Ohtsuka Y. Vitamin B(6) treatment of intractable seizures. Brain Dev. (2011) 33:783–9. doi: 10.1016/j.braindev.2011.01.010
20. Millichap JJ, Park KL, Tsuchida T, Ben-Zeev B, Carmant L, Flamini R, et al. KCNQ2 encephalopathy: features, mutational hot spots, and ezogabine treatment of 11 patients. Neurol Genet. (2016) 2:e96. doi: 10.1212/NXG.0000000000000096
21. Sands TT, Balestri M, Bellini G, Mulkey SB, Danhaive O, Bakken EH, et al. Rapid and safe response to low-dose carbamazepine in neonatal epilepsy. Epilepsia. (2016) 57:2019–30. doi: 10.1111/epi.13596
22. Mulkey SB, Ben-Zeev B, Nicolai J, Carroll JL, Grønborg S, Jiang YH, et al. Neonatal nonepileptic myoclonus is a prominent clinical feature of KCNQ2 gain-of-function variants R201C and R201H. Epilepsia. (2017) 58:436–45. doi: 10.1111/epi.13676
23. Sharawat IK, Kasinathan A, Sahu JK, Sankhyan N. Response to carbamazepine in KCNQ2 related early infantile epileptic encephalopathy. Indian J Pediatr. (2019) 86:301–2. doi: 10.1007/s12098-018-2796-8
24. Spagnoli C, Salerno GG, Iodice A, Frattini D, Pisani F, Fusco C. KCNQ2 encephalopathy: a case due to a de novo deletion. Brain Dev. (2018) 40:65–8. doi: 10.1016/j.braindev.2017.06.008
25. Vilan A, Mendes Ribeiro J, Striano P, Weckhuysen S, Weeke LC, Brilstra E, et al. A distinctive ictal amplitude-integrated electroencephalography pattern in newborns with neonatal epilepsy associated with KCNQ2 mutations. Neonatology. (2017) 112:387–93. doi: 10.1159/000478651
26. Pisano T, Numis AL, Heavin SB, Weckhuysen S, Angriman M, Suls A, et al. Early and effective treatment of KCNQ2 encephalopathy. Epilepsia. (2015) 56:685–91. doi: 10.1111/epi.12984
27. Klotz KA, Lemke JR, Korinthenberg R, Jacobs J. Vitamin B6-responsive epilepsy due to a novel KCNQ2 mutation. Neuropediatrics. (2017) 48:199–204. doi: 10.1055/s-0037-1601857
28. Borgatti R, Zucca C, Cavallini A, Ferrario M, Panzeri C, Castaldo P, et al. A novel mutation in KCNQ2 associated with BFNC, drug resistant epilepsy, and mental retardation. Neurology. (2004) 63:57–65. doi: 10.1212/01.wnl.0000132979.08394.6d
29. Dedek K, Fusco L, Teloy N, Steinlein OK. Neonatal convulsions and epileptic encephalopathy in an Italian family with a missense mutation in the fifth transmembrane region of KCNQ2. Epilepsy Res. (2003) 54:21–7. doi: 10.1016/S0920-1211(03)00037-8
30. Martin HC, Kim GE, Pagnamenta AT, Murakami Y, Carvill GL, Meyer E, et al. Clinical whole-genome sequencing in severe early-onset epilepsy reveals new genes and improves molecular diagnosis. Hum Mol Genet. (2014) 23:3200–11. doi: 10.1093/hmg/ddu030
31. Numis AL, Angriman M, Sullivan JE, Lewis AJ, Striano P, Nabbout R, et al. KCNQ2 encephalopathy: delineation of the electroclinical phenotype and treatment response. Neurology. (2014) 82:368–70. doi: 10.1212/WNL.0000000000000060
32. Saitsu H, Kato M, Koide A, Goto T, Fujita T, Nishiyama K, et al. Whole exome sequencing identifies KCNQ2 mutations in Ohtahara syndrome. Ann Neurol. (2012) 72:298–300. doi: 10.1002/ana.23620
33. Lindy AS, Stosser MB, Butler E, Downtain-Pickersgill C, Shanmugham A, Retterer K, et al. Diagnostic outcomes for genetic testing of 70 genes in 8565 patients with epilepsy and neurodevelopmental disorders. Epilepsia. (2018) 59:1062–71. doi: 10.1111/epi.14074
34. Kopanos C, Tsiolkas V, Kouris A, Chapple CE, Albarca Aguilera M, Meyer R, et al. VarSome: the human genomic variant search engine. Bioinformatics. (2019) 35:1978–80. doi: 10.1093/bioinformatics/bty897
35. Stenson PD, Mort M, Ball EV, Chapman M, Evans K, Azevedo L, et al. The Human Gene Mutation Database (HGMD®): optimizing its use in a clinical diagnostic or research setting. Hum Genet. (2020) 139:1197–207. doi: 10.1007/s00439-020-02199-3
36. Baldridge D, Heeley J, Vineyard M, Manwaring L, Toler TL, Fassi E, et al. The Exome clinic and the role of medical genetics expertise in the interpretation of exome sequencing results. Genet Med. (2017) 19:1040–8. doi: 10.1038/gim.2016.224
37. Freibauer A, Jones K. KCNQ2 mutation in an infant with encephalopathy of infancy with migrating focal seizures. Epilept Disord. (2018) 20:541–4. doi: 10.1684/epd.2018.1011
38. Palmer EE, Schofield D, Shrestha R, Kandula T, Macintosh R, Lawson JA, et al. Integrating exome sequencing into a diagnostic pathway for epileptic encephalopathy: evidence of clinical utility and cost effectiveness. Mol Genet Genomic Med. (2018) 6:186–99. doi: 10.1002/mgg3.355
39. Lee IC, Chang TM, Liang JS, Li SY. KCNQ2 mutations in childhood nonlesional epilepsy: variable phenotypes and a novel mutation in a case series. Mol Genet Genomic Med. (2019) 7:e00816. doi: 10.1002/mgg3.816
40. Papuc SM, Abela L, Steindl K, Begemann A, Simmons TL, Schmitt B, et al. The role of recessive inheritance in early-onset epileptic encephalopathies: a combined whole-exome sequencing and copy number study. Eur J Hum Genet. (2019) 27:408–21. doi: 10.1038/s41431-018-0299-8
41. Malerba F, Alberini G, Balagura G, Marchese F, Amadori E, Riva A, et al. Genotype-phenotype correlations in patients with de novo KCNQ2 pathogenic variants. Neurol Genet. (2020) 6:e528. doi: 10.1212/NXG.0000000000000528
42. Parra M, Stahl S, Hellmann H. Vitamin B6 and its role in cell metabolism and physiology. Cells. (2018) 7:84. doi: 10.3390/cells7070084
43. Kim JE, Cho KO. Functional nutrients for epilepsy. Nutrients. (2019) 11:1309. doi: 10.3390/nu11061309
44. Gospe SM Jr, Olin KL, Keen CL. Reduced GABA synthesis in pyridoxine-dependent seizures. Lancet. (1994) 343:1133–4. doi: 10.1016/s0140-6736(94)90236-4
45. Ramos RJ, Pras-Raves ML, Gerrits J, van der Ham M, Willemsen M, Prinsen H, et al. Vitamin B6 is essential for serine de novo biosynthesis. J Inherit Metab Dis. (2017) 40:883–91. doi: 10.1007/s10545-017-0061-3
46. Almannai M, El-Hattab AW. Inborn errors of metabolism with seizures: defects of glycine and serine metabolism and cofactor-related disorders. Pediatr Clin North Am. (2018) 65:279–99. doi: 10.1016/j.pcl.2017.11.007
47. van Karnebeek CD Tiebout SA Niermeijer J Poll-The Poll-The BT Ghani A Coughlin CR II . Pyridoxine-dependent epilepsy: an expanding clinical spectrum. Pediatric Neurol. (2016) 59:6–12. doi: 10.1016/j.pediatrneurol.2015.12.013
48. Joshi R, Adhikari S, Patro BS, Chattopadhyay S, Mukherjee T. Free radical scavenging behavior of folic acid: evidence for possible antioxidant activity. Free Radic Biol Med. (2001) 30:1390–9. doi: 10.1016/s0891-5849(01)00543-3
49. Chumnantana R, Yokochi N, Yagi T. Vitamin B6 compounds prevent the death of yeast cells due to menadione, a reactive oxygen generator. Biochim Biophys Acta. (2005) 1722:84–91. doi: 10.1016/j.bbagen.2004.11.013
50. Thériault O, Poulin H, Thomas GR, Friesen AD, Al-Shaqha WA, Chahine M. Pyridoxal-5'-phosphate (MC-1), a vitamin B6 derivative, inhibits expressed P2X receptors. Can J Physiol Pharmacol. (2014) 92:189–96. doi: 10.1139/cjpp-2013-0404
51. Henshall DC, Diaz-Hernandez M, Miras-Portugal MT, Engel T. P2X receptors as targets for the treatment of status epilepticus. Front Cell Neurosci. (2013) 7:237. doi: 10.3389/fncel.2013.00237
52. Jimenez-Pacheco A, Mesuret G, Sanz-Rodriguez A, Tanaka K, Mooney C, Conroy R, et al. Increased neocortical expression of the P2X7 receptor after status epilepticus and anticonvulsant effect of P2X7 receptor antagonist A-438079. Epilepsia. (2013) 54:1551–61. doi: 10.1111/epi.12257
53. Schwaninger M, Ringleb P, Winter R, Kohl B, Fiehn W, Rieser PA, et al. Elevated plasma concentrations of homocysteine in antiepileptic drug treatment. Epilepsia. (1999) 40:345–50. doi: 10.1111/j.1528-1157.1999.tb00716.x
Keywords: KCNQ2, neonatal epilepsy, vitamin B6, pyridoxine, pyridoxal 5 phosphate, pyridoxine-dependent epilepsy (PDE), pyridoxine-responsive epilepsy
Citation: Amore G, Butera A, Spoto G, Valentini G, Saia MC, Salpietro V, Calì F, Di Rosa G and Nicotera AG (2022) KCNQ2-Related Neonatal Epilepsy Treated With Vitamin B6: A Report of Two Cases and Literature Review. Front. Neurol. 13:826225. doi: 10.3389/fneur.2022.826225
Received: 30 November 2021; Accepted: 28 February 2022;
Published: 25 March 2022.
Edited by:
Marco Carotenuto, University of Campania Luigi Vanvitelli, ItalyReviewed by:
Gaetan Lesca, Université Claude Bernard Lyon 1, FranceHirokazu Oguni, TMG Asaka Medical Center, Japan
Copyright © 2022 Amore, Butera, Spoto, Valentini, Saia, Salpietro, Calì, Di Rosa and Nicotera. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Vincenzo Salpietro, di5zYWxwaWV0cm9AdWNsLmFjLnVr