 Rui Ma
Rui Ma Jing Ye
Jing Ye Jiaqi Han1,2
Jiaqi Han1,2 Chaodong Wang
Chaodong Wang Yuping Wang
Yuping Wang- 1Department of Neurology, Xuanwu Hosptial, Captial Medical University, Beijing, China
- 2Beijing Key Laboratory of Neuromodulation, Beijing, China
- 3National Clinical Research Center for Geriatric Diseases, Beijing, China
Background: Hereditary orotic aciduria (HOA) is a rare genetic disorder of pyrimidine metabolism caused by variations in the uridine monophosphate synthetase (UMPS) gene and inheritance are autosomal recessive. Heterozygous UMPS mutations can also lead to orotic aciduria without clinical consequence.
Methods: We conducted molecular genetic analyses on proband using whole-exome sequencing (WES) and on 12 family members using Sanger sequencing for UMPS mutation. We analyzed the urine metabolites of family members carrying UMPS heterozygous variants with standard gas chromatography-mass spectrometry (GC-MS).
Results: We identified a novel UMPS mutation (c.517G>C) in a Chinese-origin of orotic aciduria pedigree. The proband presented with epilepsy and intellectual disability (ID). Other mutation carriers in our pedigree presented with mild orotic aciduria without relevant medical complaints except for the proband.
Conclusion: Our study further expanded the genotype of orotic aciduria and highlighted the probability of misdiagnosis in clinical practice.
Introduction
Hereditary orotic aciduria (HOA) (OMIM #258900) is a rare genetic disorder of pyrimidine metabolism characterized by early onset of megaloblastic anemia, global developmental delay, and failure to thrive, which is associated with massive urinary overexcretion of orotic acid (sometimes with orotic acid crystalluria) (1). Patients without megaloblastic anemia, but with additional manifestations such as epilepsy, have also been reported (2). The HOA is caused by variations in the uridine monophosphate synthetase (UMPS) gene with autosomal recessive inheritance. Heterozygous UMPS mutations can also lead to mild and isolated orotic aciduria without clinical consequence (1). Here, we discuss a mild orotic aciduria (OA) pedigree with a novel heterozygous UMPS mutation.
Materials and Methods
Editorial Policies and Ethical Considerations
This study was approved by the Xuanwu Hospital Capital Medical University, and all participants provided written informed consent.
Patients and Family Members
We investigated a large family with 36 members spanning four generations, originating from Beijing province, China (Figure 1A). Five family members, including the proband (IV-5), carried a heterozygous mutation in the UMPS gene. Family members without an UMPS mutation were included as controls.
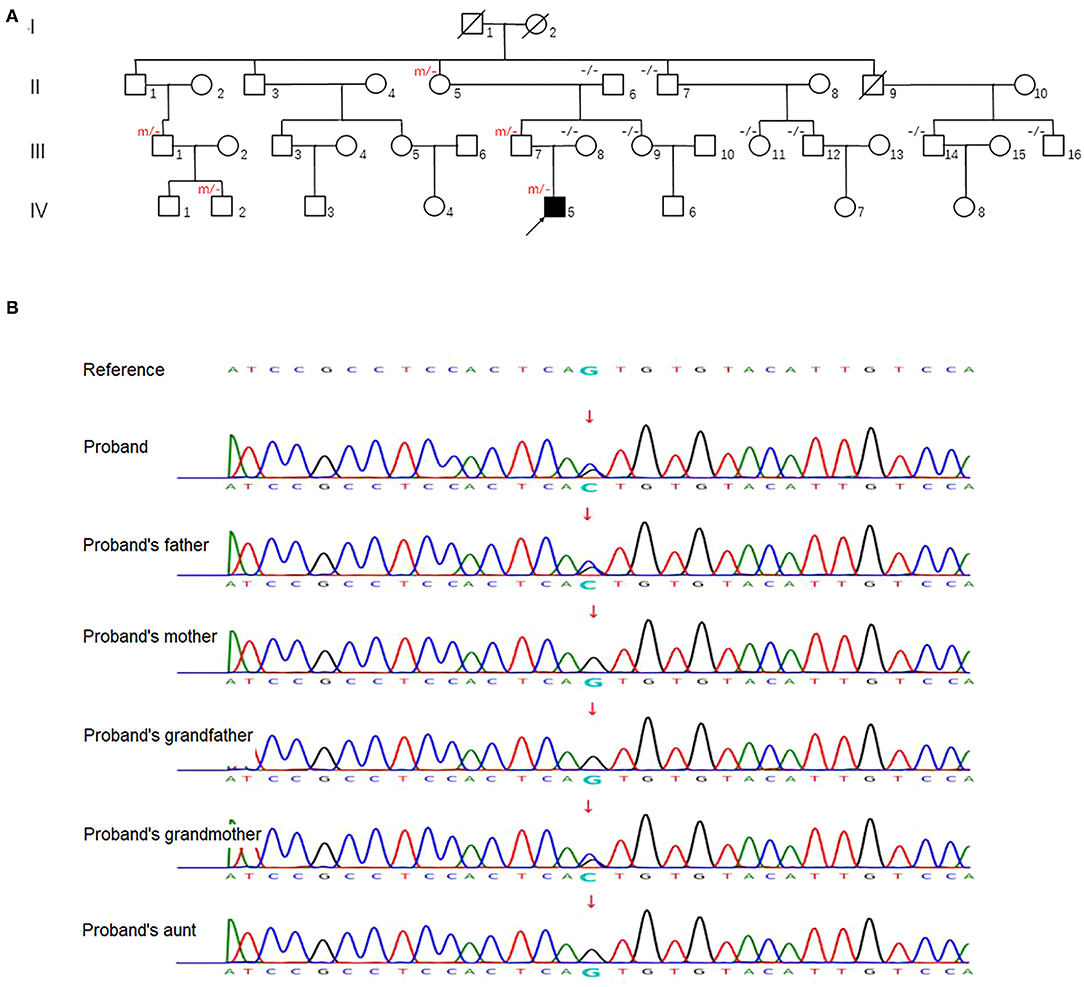
Figure 1. (A) Family pedigree. An arrow denotes the proband. “m/–” and “–/–” in the upper left indicates ascertained UMPS heterozygous mutation carrier and wildtype subject, respectively. (B) Sequence analysis of human UMPS gene in the proband and his family members. Representative sequence electropherograms are shown.
Genetic Tests
Genomic DNA was extracted from peripheral blood samples of the enrolled patient and their available family members using standard procedures. Variants were detected by next-generation sequencing using the NovaSeq600 platform by Berry Genomics (Beijing, China). Sanger sequencing with specific primers (5′-TTGAAGGTCACTGATGCCATAGT-3′ as the forward primer and 5′-GATTCGCTGCCACAAAGACA-3′ as the reverse primer) was performed to confirm the selected variant in the proband and other family members.
Metabolic Screening Tests
Urine metabolites were analyzed by standard gas chromatography-mass spectrometry (GC-MS) as previously described (3).
Results
Case Report
The patient is a 16-year-old Chinese man who presented with epilepsy and intellectual disability. Pregnancy and delivery were uneventful. His parents are not consanguineous, and they had no relevant medical complaints. The first seizure, at the age of eight, was a generalized tonic-clonic seizure (GTCS) after he fell asleep. The second attack came 5 years later, presenting with limb weakness, numbness, head involuntary movement, and impaired awareness after a fever to a maximum of 38.4°C. Paroxysmal unilateral limb numbness with face numbness, dizziness, double vision, and slurred speech occurred approximately twice a year after the age of 14. Wechsler Intelligence Scale for Children (WISC) was tested at the age of eight, indicating mild intelligent disability (intelligence quotient 68). Several electroencephalograms (EEG)s showed paroxysmal generalized spike and slow-wave, prominent in the posterior head. No abnormality was observed in the head MRI. The patient did not have blood count abnormalities, relevant hyperammonemia, or altered plasma amino acid profile except a reduced level of Vitamin B12 (161 pg/ml, reference range 180-914 pg/ml). The patient's parents refused to take antiepileptic drugs and uridine triacetate except for mecobalamin. None of these episodes occurred at a one-year follow-up.
Genetic and Metabolic Screening Analysis
After obtaining informed consent, blood samples were collected and processed for genomic DNA isolation. Whole-exome sequencing (WES) was performed using a trio-based approach (patient, mother, and father). The analysis demonstrated a previously unreported heterozygous mutation inherited from his father in the UMPS gene (c.517G>C, p.Val173Leu, reference sequence: NM_000373.3), which have not yet been reported in the literature. These sequence data have been submitted to the GenBank databases under accession number OK605588. Parents were clinically asymptomatic. Subsequent Sanger sequencing of all available paternal family members found that III-7, II-5, III-1, and IV-2 also carried UMPS variant (c.517G>C, p.V173L) with no neurological symptoms (Figures 1A,B). The mutation located in the orotate phosphoribosyltransferase domain was not found in The Genome Aggregation (GnomAD), The Exome Aggregation (ExAC), and 1,000 Genomes (1,000G) databases. Prediction tools [Sorting Intolerant from Tolerant (SIFT) and Polymorphism Phenotyping v2 (PolyPhen-2)] were suggested for c.517G>C (p.V173L), a minor effect on enzymatic activity. Mutation Taster considered the mutation disease-causing. Based on the American College of Medical Genetics and Genomics (ACMG) standards and guidelines, c.517G>C of the UMPS gene was predicted to be likely pathogenic (PS3+PM1+PM2). The location of the mutation in the schematic diagram shown in Figure 2.
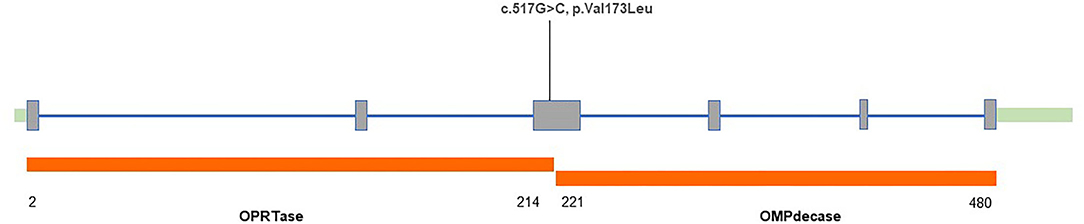
Figure 2. Structure of the human uridine monophosphate synthetase (UMPS) gene with variant found in our patient.
Further findings of a significant urinary excretion of orotate in our patient at the age of 16 (elevated 16.739 times of reference value) supported the diagnosis of orotic aciduria. Values of urinary excretion of orotate of family members carrying UMPS heterozygous variant are shown in Table 1. Except for III-1, our patient's paternal uncle, the ratios of analysis value to reference value of other family members with UMPS variant are more significant than one, indicating the diagnosis of orotic aciduria. According to the results of metabolic screening analysis, four family members (III-7, II-5, IV-2, and the proband) in this family can be diagnosed as orotic aciduria, although with no neurological symptoms.
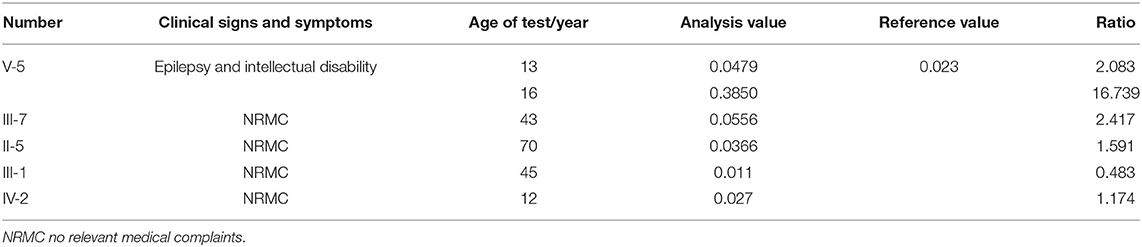
Table 1. Value of urinary excretion of orotate of family members carrying uridine monophosphate synthetase (UMPS) heterozygous variant.
Discussion
The HOA results from deficiencies in uridine monophosphate synthetase (UMPS), which consists of two domains of the enzyme: orotate phosphoribosyl pyrophosphate transferase (OPRTase, EC 2.4.2.10) and orotidine monophosphate decarboxylase (OMPdecase, EC 4.1.1.23). According to the values of urinary orotic acid and orotidine excretion, the type of UMPS deficiency can be presented in vivo (4): Type I is caused by the loss of both enzyme activities of the UMP enzyme; Type II is thought to be due to the specific inactivation of OMPdecase; Type III, orotic aciduria without megaloblastic anemia (OAWA), is expected to be also secondary to the inactivation of OMPdecase. Urinary orotidine levels were not available in our pedigree, so the type of OA cannot be further classified.
To the best of our knowledge, this UMPS nonsynonymous mutation (c.517G>C) had never been identified in the single-nucleotide polymorphism database (dbSNP), Leiden Open Variation Database (LOVD) (v3.1), and ClinVar (https://www.ncbi.nlm.nih.gov/clinvar/) databases. There are previous reports of patients with seizure-associated HOA (2, 5), thus, proposing a link between UMPS deficiency and epilepsy. Intellectual disability occurs in patients with OA with homozygous and heterozygous UPMS mutations (1). Moreover, our OA proband presented with epilepsy, intellectual disability, and vitamin B12 deficiency, which is partly consistent with previous reports. Wortmann et al. (1) reported 11 index cases and 18 healthy heterozygous mutation carriers, revealing high phenotypic heterogeneity of UMPS mutations. They thought that presentation of developmental delay or intellectual disability in their patient population might be due to ascertainment bias and in utero exposure to elevated levels of orotic acid and/or decreased pools of pyrimidines. However, our proband had a paternally derived mutation and did not support the utero exposure hypothesis. The other four UPMS mutation carriers in our family all presented with no neurologic complaints. Three of them had mild orotic aciduria, which also confirms the high phenotypic heterogeneity of UMPS. Significant urinary excretion of orotate was detected in those members who carried this mutation by urine metabolic screening in our research. Orotate is a substrate of orotate phosphoribosyl pyrophosphate transferase and orotidine monophosphate decarboxylase, which are encoded by the UMPS gene. Therefore, the excessive accumulation of orotate can reflect the impaired function of the UMPS gene and augment the value of this case report (6).
In 2015, the U.S. Food and Drug Administration approved a treatment for HOA called uridine triacetate (Xuriden), which is a pyrimidine analog indicated for uridine replacement therapy (7). Patients who took the drug responded well (5). Although our proband only took mecobalamin, he was seizure-free for one year. We will continue to pay attention to the patient's condition and adjust the treatment plan according to the condition at any time.
Conclusion
Although extremely rare, hereditary orotic aciduria should be suspected in any child with neurological disorders, such as epilepsy and developmental delay, and with or without anemia. Our study characterizes a highly heterogeneous UMPS pedigree, expanding the mutational spectrum as we report one novel variant that is identified in the UMPS gene. Early diagnosis and treatment are essential for disease course.
Data Availability Statement
The datasets presented in this article are not readily available due to ethical and privacy restrictions. Requests to access the datasets should be directed to the corresponding author.
Ethics Statement
The studies involving human participants were reviewed and approved by the Ethical Committee of Xuanwu Hospital Capital Medical University. Written informed consent to participate in this study was provided by the participants' legal guardian/next of kin. Written informed consent was obtained from the individual(s), and minor(s)' legal guardian/next of kin, for the publication of any potentially identifiable images or data included in this article.
Author Contributions
RM: writing—original draft, visualization. JY: resources. JH: investigation. LG: writing—reviewing and editing, validation. CW: data curation and conceptualization. YW: supervision, project administration, and funding acquisition. All authors contributed to the article and approved the submitted version.
Funding
This study was supported by the National Key R&D Program (No. 2018YFC1314500, 2018YFC1314504) and the Clinical Cohort Study of Epilepsy Patient (2017YFC0907702).
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher's Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Acknowledgments
We are grateful for the family's cooperation and permission to publish this information.
References
1. Wortmann SB, Chen MA, Colombo R, Pontoglio A, Alhaddad B, Botto LD, et al. Mild orotic aciduria in UMPS heterozygotes: a metabolic finding without clinical consequences. J Inherit Metab Dis. (2017) 40:423–31. doi: 10.1007/s10545-017-0015-9
2. Grohmann K, Lauffer H, Lauenstein P, Hoffmann GF, Seidlitz G. Hereditary orotic aciduria with epilepsy and without megaloblastic anemia. Neuropediatrics. (2015) 46:123–5. doi: 10.1055/s-0035-1547341
3. Zhang C, Xu K, Dave UP, Wang Y, Matsumoto I. Inborn errors of metabolism discovered in Asian department of pediatrics and mental retardation research center. J Chromatogr B Biomed Sci Appl. (2000) 746:41–9. doi: 10.1016/S0378-4347(00)00087-6
4. Bailey CJ. Orotic aciduria and uridine monophosphate synthase: a reappraisal. J Inherit Metab Dis. (2009) 32:S227–33. doi: 10.1007/s10545-009-1176-y
5. Al Absi HS, Sacharow S, Al Zein N, Al Shamsi A, Al Teneiji A. Hereditary orotic aciduria (HOA): A novel uridine-5-monophosphate synthase (UMPS) mutation. Mol Genet Metab Rep. (2021) 26:100703. doi: 10.1016/j.ymgmr.2020.100703
Keywords: case report, orotic aciduria, UMPS, epilepsy, intellectual disability
Citation: Ma R, Ye J, Han J, Gao L, Wang C and Wang Y (2022) Case Report: A Novel Missense Mutation c.517G>C in the UMPS Gene Associated With Mild Orotic Aciduria. Front. Neurol. 13:819116. doi: 10.3389/fneur.2022.819116
Received: 20 November 2021; Accepted: 07 February 2022;
Published: 09 March 2022.
Edited by:
Fernando Cendes, State University of Campinas, BrazilReviewed by:
Ali Sazci, Okan University, TurkeyLi-Ping Zou, First Affiliated Hospital of Chinese PLA General Hospital, China
Copyright © 2022 Ma, Ye, Han, Gao, Wang and Wang. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Lehong Gao, Z2FvbGVob25nQHNpbmEuY29t; Chaodong Wang, Y2Rvbmd3YW5nMDFAMTI2LmNvbQ==; Yuping Wang, bWR3YW5neXBAc2luYS5jbg==
†These authors have contributed equally to this work and share senior authorship