 Audrey Maudoux
Audrey Maudoux Sandrine Vitry
Sandrine Vitry Aziz El-Amraoui
Aziz El-Amraoui
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Neurol. , 04 April 2022
Sec. Neuro-Otology
Volume 13 - 2022 | https://doi.org/10.3389/fneur.2022.816534
This article is part of the Research Topic New Approaches to Investigate Congenital Vestibular Disorders View all 7 articles
The inner ear is responsible for both hearing and balance. These functions are dependent on the correct functioning of mechanosensitive hair cells, which convert sound- and motion-induced stimuli into electrical signals conveyed to the brain. During evolution of the inner ear, the major changes occurred in the hearing organ, whereas the structure of the vestibular organs remained constant in all vertebrates over the same period. Vestibular deficits are highly prevalent in humans, due to multiple intersecting causes: genetics, environmental factors, ototoxic drugs, infections and aging. Studies of deafness genes associated with balance deficits and their corresponding animal models have shed light on the development and function of these two sensory systems. Bilateral vestibular deficits often impair individual postural control, gaze stabilization, locomotion and spatial orientation. The resulting dizziness, vertigo, and/or falls (frequent in elderly populations) greatly affect patient quality of life. In the absence of treatment, prosthetic devices, such as vestibular implants, providing information about the direction, amplitude and velocity of body movements, are being developed and have given promising results in animal models and humans. Novel methods and techniques have led to major progress in gene therapies targeting the inner ear (gene supplementation and gene editing), 3D inner ear organoids and reprograming protocols for generating hair cell-like cells. These rapid advances in multiscale approaches covering basic research, clinical diagnostics and therapies are fostering interdisciplinary research to develop personalized treatments for vestibular disorders.
Our sensory organs mediate and control our responses to the external and internal environment, continuously adapting to its various changes. The human inner ear, one of the major organs signaling to the central nervous system, consists of spatially interconnected fluid-filled ducts and chambers housing six different neurosensory suborgans essential for balance and hearing (Figure 1). The resulting labyrinth is one of the most morphologically elaborate tissues in vertebrates. Its intricate and fascinating organization and capacity to integrate sensory perceptions from multiple sources have, over the centuries, triggered considerable interest leading to research across many disciplines, including physics, developmental biology, neuroscience, behavioral cognition, genetics, organ evolution, molecular phylogeny, engineering, and clinical and translational medicine.
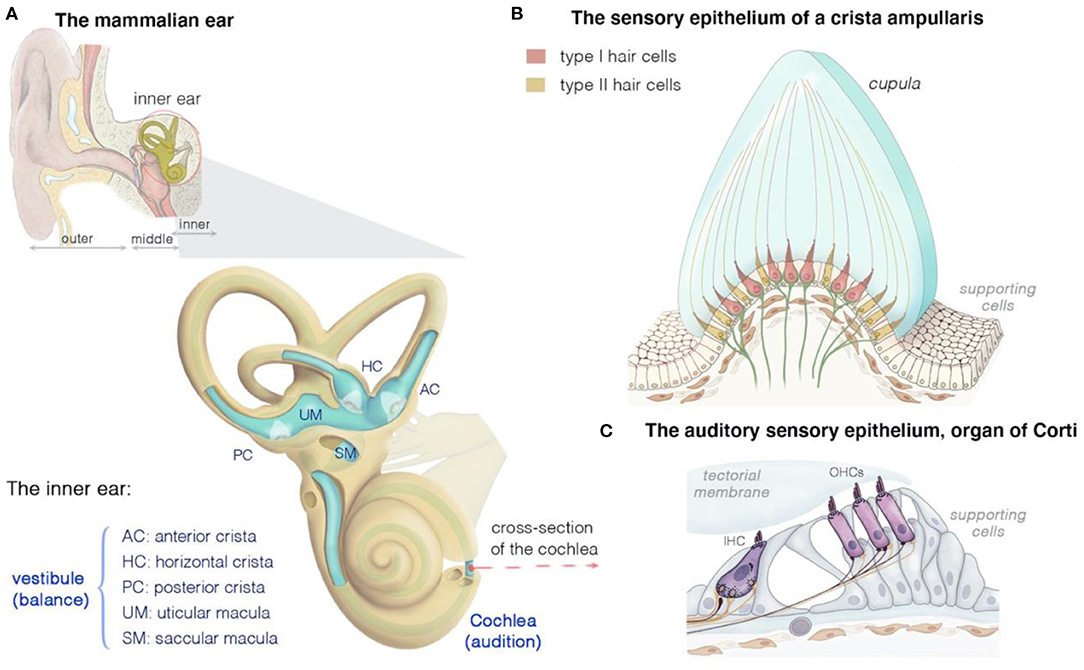
Figure 1. Structure and function of the mammalian inner ear, and of the organization of the vestibular and auditory sensory epithelia. (A) The inner ear houses both the hearing and the balance organs, the cochlea and the vestibule, respectively. The membranous labyrinth of the inner ear consists of three semicircular canals, each ending with an ampulla that form the anterior (AC), horizontal (HC), or posterior (PC) crista, two otolith organs (saccule and utricle), and the cochlea. The calyx- and bouton-like afferent innervation of hair cells is presented in green. (B) Cross section of a crista ampullaris, highlighting the organization of the type I and type II hair cells. All the crista hair cells have hair bundles displaying the same polarity orientation, extending into a gelatinous membrane called the cupula. Details of the macular sensory regions' organization are shown in Figure 2. (C) Cross section of the organ of Corti, showing the inner (IHC) and outer (OHCs) hair cells, associated afferent (green) and efferent (dark purple red) neurons and supporting cells.
Hearing and balance display tight anatomical and functional links. Both organs originated from a single otic vesicle that develops by the invagination of surface ectoderm adjacent to the developing hindbrain (1–5). However, changes in the needs of species in fluctuating environments have led to species-specific tissue and cellular adaptations. The auditory system has always been a subject of great interest and the focus of extensive research, due to its key role in the evolution of language and sound-based communication (1, 6). By comparison, at least until recently, the vestibular system has received much less attention. The inner ear, which first emerged about 500 million years ago, was initially composed exclusively of a gravitational organ that evolved to sense gravity, and then motion and balance (7). In humans, defects of hearing and/or balance, of peripheral and/or central origin, may manifest at any age, with varying degrees of severity. Vestibular dysfunction leads to one of the most common complaints in medicine, dizziness and/or vertigo, which affects 15–35% of the general population (8–11). In Europe, vestibular disorders of peripheral origin account for 3% of all medical consultations for patients over the age of 50 years and about 1% of the emergency cases seen at hospitals (12, 13). National questionnaires have estimated that the prevalence of dizziness/vertigo in children (3 years of age to adolescence) ranges from 5.3 to 8% (14, 15). However, these figures are probably underestimated, as young children tolerate dizziness better than adolescents and adults. In clinics specializing in the treatment of pediatric vestibular disorders, the prevalence of vestibular dysfunction in children referred for dizziness/vertigo ranges from 20 to 36.5% (16, 17). The prevalence of balance dysfunction increases with age; about one third of adults over the age of 65 years suffers from dizziness, resulting in frequent falls (18).
Over the last 20 years, an increasing awareness of the actual importance of the vestibular system has fueled substantial work that has shed light on the multifaceted contributions of this system. The inputs of the vestibular sensory organs extend far beyond the brainstem, reaching many cortical centers in the brain. Indeed, vestibular system activity supports numerous functions, from gaze stabilization and postural control to high-level cortical functions involving spatial cognition, including self-body perception, verticality perception, orientation, navigation, and spatial memory (19–22). The vestibular organs also influence postural blood pressure regulation, bone density and muscle composition via specific vestibulo-sympathetic pathways in the brainstem and mesencephalon and have been shown to act as a powerful synchronizer of circadian rhythms and play a substantial role in sleep-wake cycles (7). Untreated losses of vestibular function have a major effect on patient quality of life and a major socioeconomic impact on healthcare systems worldwide.
Despite the high level of medical need, the management of patients with vertigo and dizziness still suffers from a lack of diagnostic tools for accurately monitoring balance and its effects on the development and behavior of the individual. In children, the timely detection of vestibular dysfunction is crucial, to provide parents with appropriate counseling and for the initiation of appropriate vestibular rehabilitation. Biomarkers for the more accurate diagnosis of vestibular disorders and the selection of targeted therapies are needed but not yet available. The pathophysiological mechanisms underlying the various vestibular disorders also remain unknown and there are currently no curative solutions. Redundancy and compensatory mechanisms may operate in cases of vestibular organ dysfunction, but the magnitude and accuracy of peripheral and central multisensory and behavioral substitution processes, and of the plasticity mechanisms sustaining the restoration of activity remain unclear (23).
In this review, we address these issues with the aim of providing an integrative view of the development and function of the vestibular system, taking into account much of the knowledge accumulated to date in animal models and human individuals with and without vestibular disorders. A precise understanding of organ evolution, development, physiology, and behavior will pave the way for better diagnostic tools and the precise and accurate definition and classification of vestibular disorders. Together, these discoveries should help guide patient management and the search for effective rehabilitation and treatment strategies.
The inner ear provides a remarkably rich context for studying cell fate and tissue organogenesis. Over the last two decades, a wealth of information has been obtained concerning how a single otic vesicle gives rise to distinct subsensory regions containing different assemblies of highly specialized hair cells, neurons, and supporting cells.
Gene expression data, lineage tracing, fate-mapping studies, and the characterization of animal models that display abnormal development or dysfunction in the vestibular and cochlear organs have all proved essential for unraveling the key mechanisms underlying inner ear development (2, 4, 24–26). Studies in fish, amphibian, chicken, and mouse animal models, either normal or with defects, have provided considerable insight into the signals and pathways governing the identity, proliferation, and differentiation of inner ear cells, and tissue planar polarity. Specific details about these various aspects of development are provided by many excellent recent reviews (27–29). Remarkable progress has been made in our understanding of how different morphogens and transcription factors cooperate to establish anteroposterior and dorsoventral organ patterning, mediate cell-fate commitment into sensory, non-sensory, and neurogenic components, and orchestrate correct cellular remodeling and assembly during organ morphogenesis. Various genes, including different factors (e.g., retinoid acids, cyclins), morphogens (e.g. FGF, BMP, Notch, Wnt), transcription factors and key molecules (e.g., Atoh1, Gfi1, prox1, Lfng, Sox2, and Ngn1, NeuroD, Tbx1, Gata, Lmx, Emx, Shh, Eya1, Six1, Delta-like 1, Jagged-1 or Tlx3/Rnx) (27, 28, 30–34) have been implicated in these processes. Neurotrophins (BDNF, TrkB, TrkC, NT3), chemoattractants (e.g., Ntn1), repellants (slits, semaphorins and ephrins) and their receptors have also been shown to play a fundamental role in early guidance decisions and the wiring of the hair-cell circuitry and associated cochlear and vestibular neuronal connections (35–38). These factors, operating alone or in specific combinations, act in a precise, well-orchestrated sequence, at specific timepoints and positions within the organ, to facilitate the sequential cell commitment, differentiation, and tissue integration of each cell type in the appropriate position. Further studies are required to improve our understanding of the interdependent sequences of the developmental programs required to build the intricate subsensory regions, each with a specific nano-scale microarchitecture adapted to a specific sensory need.
The inner ear houses the sensory organ for hearing, the cochlea, and the organ responsible for balance, the vestibule (Figures 1A–C) (25, 39–43). The scala media of the three fluid-filled turns of the cochlea houses the auditory sensory epithelium, the organ of Corti (Figure 1C) (25, 39–43). Five subregions can be distinguished in the mammalian vestibular apparatus: three semicircular canals, and two otolithic organs (Figure 1A) (25, 39–43). The semicircular canals—three tubular structures filled with endolymph—are responsible for dynamic equilibrium, responding to rotational movements (angular accelerations) of the head. The horizontal/lateral, posterior, and anterior/superior canals are oriented orthogonally to each other, each contributing to the sense of balance in a different direction (4, 25, 43, 44). The two otolith organs, the utricle and saccule, account for static equilibrium through their response to different movements of the head (4, 25, 43, 44). They are both essential to the detection of the gravity vector and represent static tilt with respect to gravity. The saccule, which is vertically oriented, responds to linear accelerations in the vertical plane, whereas the utricle, which lies horizontally, detects lateral displacements (Figure 2A).
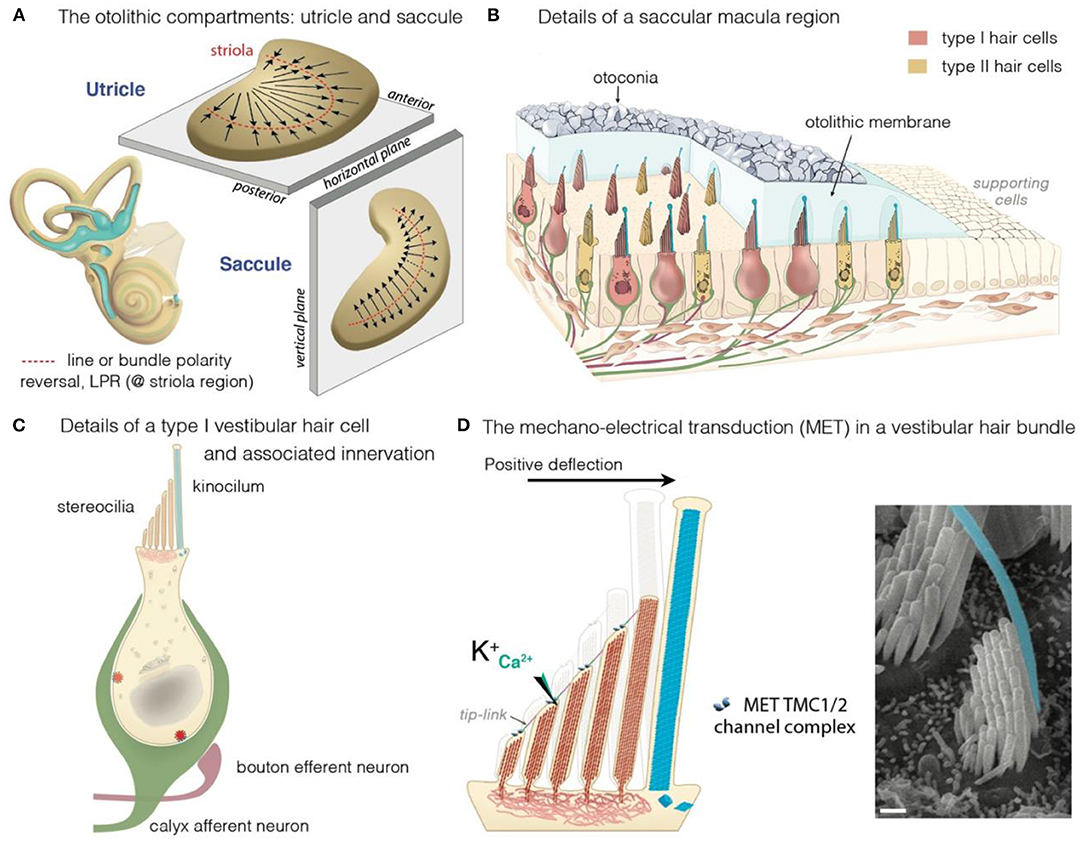
Figure 2. The vestibular maculae, the hair cells and motion-induced mechano-electrical transduction. (A) Illustration of the morphological and the polarization of hair cells in the utricular and saccular maculae: the black arrows indicate the orientation of the hair bundles. In the utricle, hair cells are oriented with their stereociliary bundles pointing toward the line of polarity reversal (LPR, in red). In the saccule, the stereociliary bundles point away from the LPR. (B–D) Overview of a macular sensory region (B), type I vestibular hair cells (VHCs) (C), and the hair bundle (D). (B) The VHCs extend their hair bundles into the overlying otolithic membrane; an extracellular matrix embedded with otoconia. The kinocilium is colored in cyan. Apart from difference in shape, type I and type II hair cells can also differ according to their innervation. Afferent (green) and efferent (dark purple red) are shown. (B,C) The VHCs are innervated by three types of afferent neurons: The calyx-only class of neurons makes contacts with type I hair cells, essentially in the striola region; dimorphic neurons contact both types of hair cells throughout the sensory epithelium, forming calyces with type I hair cells and boutons with type II hair cells; bouton-only afferents only contact type II hair cells located outside of the central striola region. (D) The basic transduction mechanism is the same in the auditory and vestibular hair cells: a mechanical stimulus bends the stereociliary bundle toward the kinocilium (artificially colored in cyan). The bundle deflection stretches the tip-link, causing an influx of K+ and Ca2+ ions into the transducing stereocilia. The ensuing change in cell membrane receptor potential leads to glutamate release, generating electrical signals in afferent neurons (green in B,C) forming the VIIIth cranial nerve.
The mechanosensitive hair cells are located in the crista ampullaris of the semicircular canals (Figures 1A,B), and the maculae of the utricle and saccule (Figures 2A,B) (4, 25, 43, 44). Both organs have type I and type II hair cells, but the otolithic and canal organs have a specific organization (45–48). The stereocilia of crista hair cells are embedded in a glycoprotein-rich gelatinous mass, the cupula, that spans the width of the ampulla, thereby acting as a barrier to endolymph circulation (Figure 1B). The stereocilia of all the hair bundles within the same crista are oriented in the same direction. When the head turns in the plane of one of the semicircular canals, the endolymph in the canal also moves, exerting a force across the cupula. This leads to a deformation of the cupula that is transmitted to the hair bundles of the crista, which are displaced, triggering hair-cell stimulation.
In the maculae of the utricle and saccule, an axis of mirror symmetry in the central region, the striola, divides the organ into two subsets of hair cells with hair bundles of opposite polarizations (4, 25, 43, 44). The stereocilia of the macular hair cells are included in a gelatinous layer, the otolithic membrane, covered by a layer of calcium crystals called the otoconia (Figure 2A). Linear accelerations produce a direct displacement of the otoconia and underlying structures. The otoconia are much heavier than the underlying structures, and this difference in mass causes the otolithic membrane to lag behind the maculae, thereby setting up responses to changes in velocity (acceleration) rather than velocity itself. This shift between the otoconia and the sensory epithelium results in the deflection of the hair bundles embedded in the gelatinous layer, triggering hair-cell stimulation (Figure 2D).
All these hair cells have a mechanosensitive organelle, the hair bundle, at their apical surface [see (49, 50)]. This organelle is the key to our ability to perceive sound and maintain balance (41, 42, 51, 52). Each hair bundle consists of 50–300 F-actin-filled stereocilia, arranged in a highly organized staircase pattern (Figure 2D). A special subset of interstereociliary links, the tip-links, connect the tip of each stereocilium to its adjacent tallest stereocilium and are present in all species (53). These links are essential for the mechano-electrical transduction (MET) process (54–56). The mechanical and geometric characteristics of the hair bundles differ between vestibular hair cells (VHCs), but their morphological and functioning polarity are similar in all vertebrate hair cells. Following hair-bundle deflection (positive direction, toward the tallest row), the increase in tip-link tension causes the MET channel, located at the tip of the stereocilia in all but the tallest rows of the hair bundle, to open (41, 42, 51, 52, 57). This opening of the MET channels results in an inward current, sustained principally by K+, but also by Ca2+ ions, causing membrane depolarization and regulating the probability of the MET channels opening (54, 55). In VHCs, it ultimately leads to the opening of the voltage-gated calcium channels at the basolateral membrane. The ensuing Ca2+ influx triggers the fusion of synaptic vesicles to the plasma membrane, the release of glutamate and, finally, the transmission of a signal to the brain, providing information about motion of the head and its orientation with respect to gravity (Figures 2C,D) (58).
Like the cochlea, the vestibule displays a high degree of architectural organization in all its compartments and subregions, with multiple parameters, including dimension, volume, liquid composition, radius of the canal, and vestibular vs. cochlear chambers, constructed to facilitate optimal responses to species-specific directional (gravity and motion displacements) and acoustic (frequency range) features (29, 44, 59). The construction of this diversity involves strict developmental programs that are finely tuned in time and space, and necessary for adaptation to the specific needs of aquatic, terrestrial and aerial species (1–5). Cross-species comparative morphological and molecular analyses of neurosensory epithelia from distantly related species may provide a wealth of information, highlighting essential organ and cell similarities and differences shedding light on the development and functioning of the inner ear suborgans.
The capacity of the inner ear to detect various stimuli in different environments has led to studies tracing the precise evolutionary origins of its compartments. Comparative anatomy studies based on fossil records, and cross-species studies, have provided a wealth of information about the role of convergent evolution in creating analogous ears adapted to specific needs in distantly-related taxa (26, 29, 60, 61). Recent reviews have provided detailed information about the development of the inner ear and changes during its evolution, and about the independent evolution of mechanosensitive cells to accommodate mechanical senses (26, 29, 60, 61). Focusing on vertebrates, we highlight here some major changes pertaining to the hearing and balance organs of the inner ear (Figure 3). Initially, at the start of its evolution, the inner ear consisted exclusively of vestibular epithelia (26, 29, 60). The fossils of the earliest jawed fishes, dating from more than 450 million years ago, show that these animals had inner ears consisting of three semicircular canals and the utricular and saccular organs—an organization that persists in their descendants, including sharks, bony fish, amphibians, reptiles, mammals, and birds (26, 29, 60, 61). The cellular organization of the neurosensory epithelium is similar in all vestibular organs, with highly specialized hair cells surrounded by supporting cells, and overlaid by an acellular gelatinous membrane (Figure 2B, mammalian maculae). Fish (which account for half of all living animals) have no dedicated auditory epithelium. They use their vestibular sensory organs (the saccular maculae in most teleost fishes) to perceive acoustic signals (26, 29, 60, 61). An organ exclusively dedicated to hearing, the basilar papilla, first emerged in amphibians about 380 million years ago, when vertebrates moved from an aquatic to a terrestrial environment (Figure 3) (26, 29, 60, 61). The hearing organ subsequently increased in size, leading to improvements in the resolution of sound frequencies for given ranges of species-specific perceived frequencies (26, 29, 60, 61). The spiral shape of the cochlea evolved later in mammals, about 120 million years ago, just before the therians split into the marsupial and placental lineages (Figure 3).
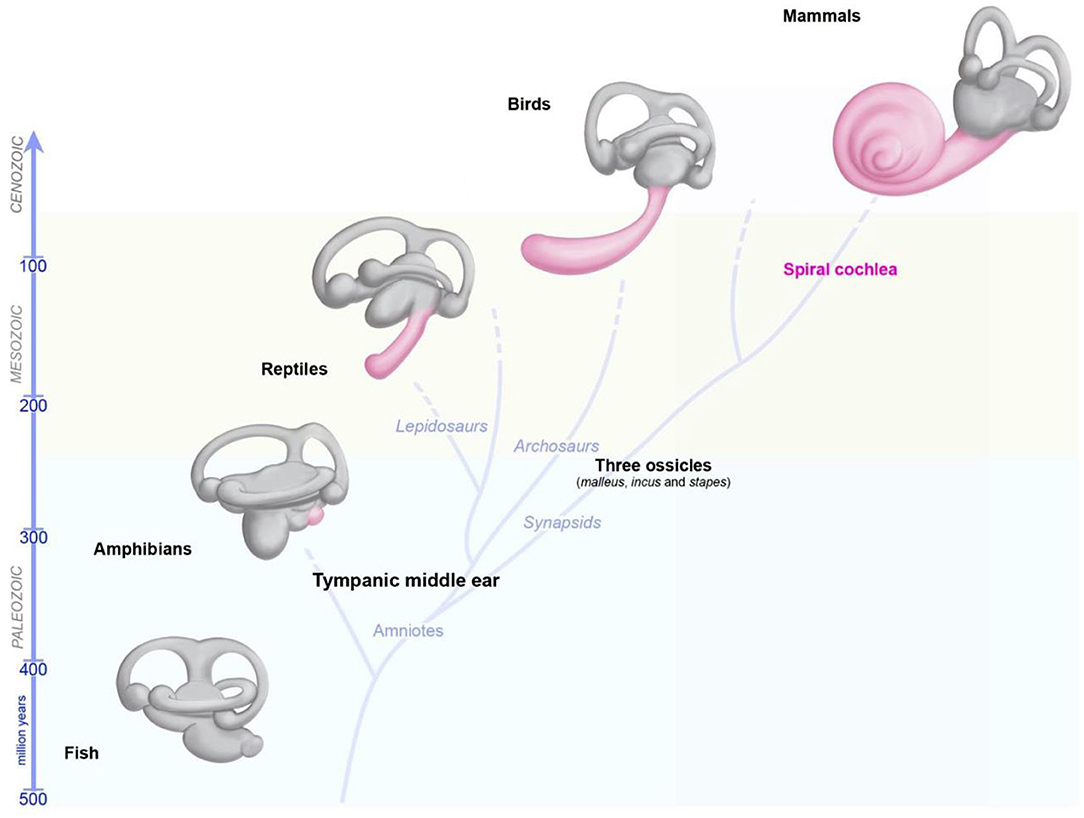
Figure 3. Evolution of the inner ear in vertebrates. The global organization of the five vestibular end organs (utricle, saccule, and the three semicircular canals) has changed very little from fish to current mammals. A sense organ dedicated exclusively to hearing, the papilla, appeared for the first time in amphibians, and then progressively elongates in reptile and birds, and form a spiral in modern mammals.
During these evolutionary transitions, pre-existing and new cells and structures appeared and evolved within the inner ear or became associated with it, thereby creating new settings, or further optimizing existing specializations. These major new additions included the appearance of the tympanal middle ear, the basilar and tectorial acellular membranes in the cochlea, the increasing specialization of the supporting cells, hair cells and the associated innervation, and continual changes in the geometry and composition of the hair bundles. In addition to the emergence of two types of hair cell in the vestibular apparatus, one crucial event was the appearance of the outer hair cells in the cochlea, a type of electromotile hair cells found only in mammals.
In the cochlea, hearing depends on 12,000–15,500 hair cells divided in two types: the outer hair cells (OHCs; 9,000–12,000 cells organized into three rows), which amplify sound stimuli, and the inner hair cells (IHCs; a single row of 3,000–3,500 cells), the genuine sensory cells responsible for transmitting sensory information to the central nervous system. The vestibular sensory epithelia in mammals also contain two types of hair cells—type I and type II—with different morphological, molecular, electrophysiological and synaptic features (Figures 1, 2) (45, 47, 62, 63). As in the cochlea, the hair cells and adjacent supporting cells are sealed together by tight adherens junctions, maintaining a clear separation between the endolymph bathing the apex of the cells, and the perilymph, in which the cell bodies are immersed. Type II hair cells are thought to have evolved first; they occur in all vertebrate vestibular epithelia, whereas type I hair cells are found only in amniotes (reptiles, birds, and mammals) [reviewed in (29)]. The regional distribution of both cell types varies between species; but type I cells are generally most abundant at the apex/center of the cristae ampullaris and in the central striola region of the maculae (47, 63, 64). Each of the sensory cell types is characterized by different anatomical features at the micro- and nano-scales, and these features are associated with functional outcomes in terms of specific mechanical properties, and receptor sensitivities (47, 62, 64–67). The seminal studies performed by Lewis Tilney over the course of a decade on hair bundles in the hearing organ of chickens described the precise sequential steps in hair bundle formation and maturation. According to their position in the chicken basilar papilla, the hair bundles have a specific number of stereocilia, with specific length, width, and arrangements (68–71). However, few studies to date have addressed the precise architecture of vestibular hair bundles in vestibular organs and their variations, or the variation of the position of these hair bundles in a given suborgan. Several groups have shown that the hair bundles of type I hair cells are wider than those of type II hair cells, with taller stereocilia, arranged to form a steeper bundle slope (44, 62, 72, 73). This architecture may translate into type I bundle responses displaying a phase advance relative to those of type II hair cells. Studies of the utricular macula have shown some differences between the hair bundles of type I hair cells in the striola and those in the lateral or medial extrastriolar regions (44, 62, 72). In particular, the striolar hair bundles in vertebrate otoconial organs are stiffer, which may improve the signaling of high head accelerations or high-frequency components of head movements (44, 72).
Type I and type II hair cells also display differences in innervation. The vestibular afferent neurons form three groups on the basis of terminal structure: bouton, pure calyceal (“cup-like” calretinin-positive terminals), and dimorphic (terminating in both calyceal and bouton endings) (Figures 2B,C). The calyceal afferent terminals surround the basolateral surface of one or more “pear-shaped” type I hair cells. Type II cells have a cylindrical body and connect to multiple afferent bouton synapses (Figures 2B,C). Both types of hair cells also receive cholinergic efferent innervation, directly to the soma of type II cells [shown in frog (74)] and via the afferent terminals for both type I and type II cells (Figures 2B,C) (46, 62, 66). Type II hair cells also have thinner and fewer stereocilia per hair bundle (45, 62). The type I hair cells have a relatively hyperpolarized resting membrane potential, −80 mV, dominated by a large, slowly activating K+ current (IK, L). Their primary current is a large non-activating K+ delayed inward rectifier, which allows these cells to produce rapid voltage responses and signal transmission (66). Type II hair cells have a more depolarized membrane potential and a smaller activation range (47, 66, 67). We still have much to learn about how calyceal synapses evolved, but one of the driving forces behind their evolution exclusively within the vestibular system may be the need for faster reflexes in a terrestrial environment (29, 47, 75). Further investigations of the relationships between synaptic features, and the geometric and mechanical properties of the hair bundles are required, to shed light on both phylogenic aspects of evolution and morphometric sets for functional outcomes.
As described above, changes in major cellular processes, protein profiles, and interacting regulatory pathways in the hair cells and adjacent cells have been necessary to accommodate the perception of increasingly high frequencies. One of the major consequences of these adaptive morphological and molecular changes was a loss of the ability of hair cells to regenerate (76). Interestingly, the ears of sharks, bony fish, amphibians, reptiles, and birds continue to produce hair cells throughout the life of the animal (77). New hair cells are produced continually as the hair-cell populations turn over, as in fish inner ears, or through the spontaneous replacement of damaged hair cells, as in chickens. In the avian cochlea, many mature supporting cells retain a latent capacity to de-differentiate. Upon hair cell damage or death, these supporting cells re-enter the cell cycle and differentiate to produce new hair cells or supporting cells (78, 79). However, for yet unknown reasons, the mature mammalian hearing organ displays an almost total loss of regenerative capacity. There is no compelling evidence for the existence of a regenerative process in the adult organ of Corti for the production of new, healthy, mature, functional hair cells. Nonetheless, several studies have reported that mammalian ears may harbor some reserve stem cell populations (59), although this “stemness potential” is limited in both time and space. A subset of supporting cells that can develop into new hair cells has been identified, mostly in young animals, before the onset of hearing (27, 59). In adult mammals, some vestibular type II hair cells also still have preserved regenerative abilities. After near complete elimination of all hair cells in diphtheria toxin receptor (DTR) mice exposed to dyptheria toxin, up to 40% of the type II hair cell population is regenerated, through regional trans-differentiation of supporting cells (80). Recent findings further support this regional segregation of hair-cell precursors with various hair-cell regeneration capacities. Regenerated hair cells derived from Plp1+ supporting cells (from the extrastriolar/peripheral region) display type II hair-cell properties. Lgr5+ supporting cells in the striolar region may have either type I or II hair-cell properties (32). However, whilst postnatally generated and regenerated hair cells acquire many somatic properties (e.g., morphology, hair-cell subtype markers, synaptic markers, nerve terminals, and basolateral conductance), the structural and functional MET properties of the hair bundle remain immature (32). Further studies are required to overcome these aberrant or incomplete maturation programs, why type I hair cells have less or no preserved regenerative abilities, and to confirm the stability over time and functional activity of regenerated cells and the absence of late degenerative processes after regeneration.
Major efforts are underway to unravel the intricacies of hair-cell regeneration in older mammals, through transcription factor supplementation to reprogram non-sensory inner ear cells to develop into hair cells in particular (31, 34). Regeneration is a tightly regulated, multistep process with high energy demands. Cell de-differentiation and activation of new differentiation cycle pathways trigger major changes in tissue organization, the remodeling of established cell-cell junctions, and a reshaping of proliferating or newly converted cell types. Cross-species comparisons of the mammalian ear and the regeneration-competent ears from non-mammals have highlighted inverse correlations of the ability of the inner ear organ to regenerate with organ complexity and the degree of structural and cellular specialization. The intricate organization of the mammalian hearing organ, with highly specialized hair cells, neurons and supporting cells, is unmatched in the animal kingdom. Studies of vestibular organs have also revealed a massive thickening of the circumferential F-actin belts supporting cell-cell junctions in mouse and human vestibular neurosensory epithelia (81–85). This cytoskeletal reinforcement, which occupies almost 90% of the cell at the intercellular junction, is further strengthened by high levels of adhesion proteins in adult mammals, as shown for the E-cadherin and P120 catenin proteins (81, 82). Conversely, similar studies in five classes of non-mammals revealed a lack of junction reinforcement, with the actin belts in the utricular supporting cells remaining thin, from hatching through adulthood. Cadherin levels at supporting cell junctions remain low (or absent) throughout life in fish, frogs, turtles, and chickens, consistent with the preserved capacity for regeneration in these species (83–85).
Further studies of the regeneration-competent ears of non-mammals, and comparisons between mammalian ears will probably help to identify potential permissive and limiting factors for hair-cell regeneration. These efforts should also address the issue of whether the re-activation of a lost signal in an already established and highly stabilized mature tissue, as in mammals, is feasible without potential late degenerative processes triggering deleterious consequences over time. With the advent of single-cell transcriptomics (scRNA-seq), evidence has been obtained for cell heterogeneity within the vestibular subcompartments (48, 86, 87). There are probably spatially different supporting cell types with different types of regenerative behavior. Careful attention should also be paid to the organization of the whole organ, and the crucial need to re-establish close correlations and connections between hair cells, neurons, and neighboring supporting cells.
Like hearing loss (49, 88), vestibular disorders may have multiple causes, including genetics, aging, drugs, immune and environmental factors (e.g., infections, noise exposure) (89). The prevalence of vestibular impairment is high in children and adults with sensorineural hearing loss (SNHL), between 20 and 70% (90, 91). It has been shown that 50% of children eligible for cochlear implantation have some sort of vestibular dysfunction, with up to 20% presenting complete bilateral vestibular impairment (92). Despite the frequent association of vestibular dysfunction with hearing loss, it remains extremely difficult to predict the type or severity of impairment from the characteristics of hearing loss (92). However, the etiology of the hearing loss seems to play a role, and abnormal vestibular responses are more frequently found in specific types of hearing loss (Figure 4; Supplementary Table S1).
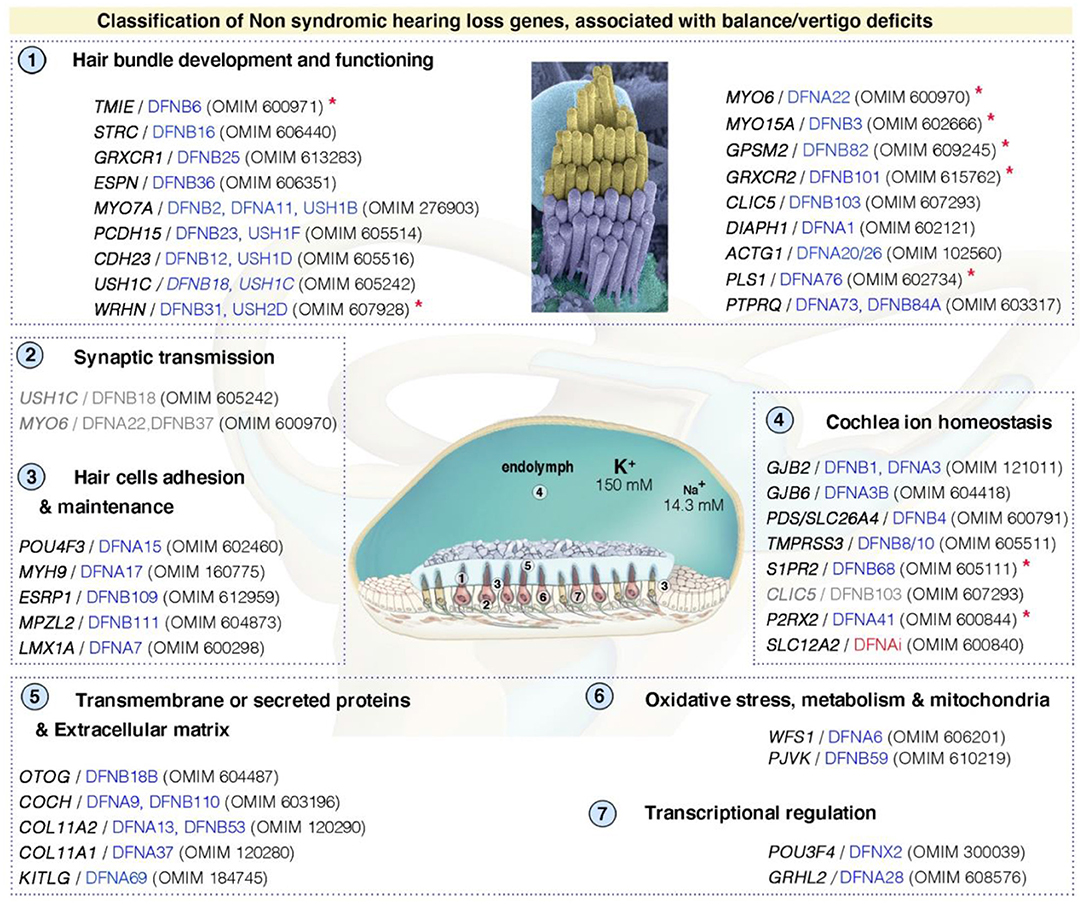
Figure 4. Functional stratification of genes/proteins causing isolated deafness hearing loss, combined with vestibular deficits. Only human genes responsible for non-syndromic deafness, and for which vestibular deficits have been reported based on clinical findings in affected patients (cf. OMIM numbers); and established role and characterization of corresponding animal models, are shown. These can be grouped into several functional categories: (1) hair bundle development and functioning, (2) synaptic transmission, (3) hair cell's adhesion and maintenance, (4) cochlea ion homeostasis, (5) transmembrane or secreted proteins and extracellular matrix, (6) oxidative stress, metabolism and mitochondrial defects, and (7) transcriptional regulation. DFNAi (red) denotes autosomal-dominant forms of deafness with undefined locus number. The genes/loci in gray denote that they share several functional categories. Similar stratification has been observed for deafness genes (49). Red asterisks indicate those included based on the presence of unambiguous vestibular deficits only in mouse models. More detailed information regarding the deafness causative genes is provided in Supplementary Table S1.
A multitude of clinical signs, such as spontaneous nystagmus, whirling vertigo, delayed postural and motor development, recurrent unexplained falling, poor balance on uneven ground or in situations of limited vision and oscillopsia (blurring of the vision with head movement) are indicative of vestibular involvement and should lead to a detailed evaluation of vestibular function. In case of oscillopsia or delayed postural and motor development bilateral vestibular loss should always be eliminated. However, some of these clinical signs (e.g., whirling vertigo, spontaneous nystagmus) may go unnoticed or be absent if both inner ears are damaged simultaneously (as in cases of systemic exposure to a vestibular toxin), if the damage is congenital or if it occurs in young children, as they tolerate dizziness better than adolescents and adults. For this reason, systematic vestibular testing should always be proposed if hearing loss is diagnosed. Complete vestibular assessment is easy to perform in clinical practice (even in young children under the age of 1 year) and includes an evaluation of canal function and otolithic function.
We briefly review here the different exploratory tests for vestibular function, and the vestibular dysfunctions observed in some forms of hearing loss.
Current clinical tests of vestibular function do not directly assess inner ear function. Instead, they evaluate the final motor output of the eyes or balance control systems. Five main sets of measurements are routinely recorded in clinics to evaluate the function of the vestibular organs and to determine the pattern specificities of vestibular disorders: c-VEMP, o-VEMP, caloric tests, rotatory chair test and head impulse tests (HITs) (92–94). cVEMP is recorded for the sternocleidomastoid muscle (SCM). This potential is a manifestation of the vestibulospinal reflex and saccular function. oVEMP is recorded for the extra-ocular muscles and is a manifestation of the VOR and utricular function. Canal function can be evaluated at 3 head velocities: at low frequency with bithermal caloric test (0.003 Hz); at middle frequency with rotatory tests (0.01–0.5 Hz) and at high frequency with the head impulse test (5–6 Hz). In the caloric test, warm and cooled water or air in the ear canal is used to stimulate the horizontal semicircular canal, causing nystagmus, which can be monitored by video tracking. The use of a rotatory chair with monitoring of the eye movement allows for the evaluation of the horizontal semicircular canal function at middle frequency. When tilting the axis of the chair rotation with respect to the gravitational axis, the otolith function can be tested (OVAR- off axis vertical testing). Head impulse testing can be used to test the function of all three semicircular canals at high frequency.
Compensatory mechanisms often occur in the central nervous system, with the re-adjustment of neural circuits and centers to overcome decreased input from the peripheral vestibular system. Vestibular assessments are not often included in routine phenotyping in clinics. Vestibular impairments present at birth, and moderate or progressive deficits may therefore be overlooked or undetectable due to the indirect nature of the information collected on questionnaires or through behavioral observations. It is important to note that even if compensatory mechanisms frequently occur, in particular by using vision and proprioception inputs, not all compensation strategies are adequate and in case of complete vestibular loss some functions can never be entirely compensated (e.g., gaze stabilization during movement). Moreover, all compensation mechanisms come with a cost in terms of attention and fatigue.
Animal models provide opportunities to obtain additional, more detailed information about vestibular functions, even in the absence of overt balance deficits (95–98). Behavioral tests in animals include the monitoring of behaviors suggestive of balance problems (e.g., head bobbing, wobbling, circling behavior, spatial misorientation, reaching and air-righting reflexes, gait, swimming tests, beam-crossing, platform or rotarod assays) (99). The recording of eye movements, such as the angular VOR (aVOR) and optokinetic response (OKR), have also been used to provide a more complete analysis of peripheral vestibular function (100, 101). If the OKR is preserved, the VOR deficits detected can be used to infer a peripheral abnormality, such as semicircular canal function (vestibular afferent response to earth-vertical axis head rotation, EVAR) or otolithic function (vestibular afferent response to off-vertical axis head rotation, OVAR). These eye movement recordings also reflect the input, integration and output components of nervous system function. Direct measurements of peripheral vestibular function through invasive tests can be performed on animals. For example, vestibular compound action potentials (also known as vestibular evoked potentials, VsEP) can be recorded from individual neural elements in animals. They reflect the direct activity of the vestibular nerve and central relays in response to linear head acceleration (102). Their measurement provides comparative values of response thresholds, peak latencies, and peak-to-peak amplitudes, and more precise indications of the sensitivity of the gravity receptor end organs, the utricle and the saccule (102, 103). Based on these animal research, the development of similar electrophysiological measurement of peripheral vestibular function has been initiated in humans (104). Such extensive, in-depth research on animal models is essential to improve our understanding of the correlation between vestibular symptoms and disease etiology and to facilitate the accurate evaluation of treatment strategies for protecting or restoring vestibular function (95–98, 105).
Congenital cytomegalovirus (CMV) infection is the main non-genetic cause of SNHL, implicated in 8% of cases of hearing loss of any degree of severity (106). It is estimated to be responsible for 20% of cases of hearing loss of unknown origin. Vestibular dysfunction is particularly frequent in patients with congenital CMV infection. A French retrospective study of auditory and vestibular assessments on 52 children with congenital CMV infection (107) showed that more than 90% of children with CMV-related hearing loss had a vestibular impairment and that a third of these children had a complete and bilateral loss of vestibular function (vestibular areflexia). However, no correlation was found between the severity of hearing loss and that of vestibular dysfunction. The pathogenetic hypothesis, derived from fetal and animal data, concerns the potassium regulation structures of the endolymphatic sector of the inner ear (108): the cochlear stria vascularis and vestibular dark cells, both of which are prime targets of CMV. Endolymphatic potential is essential for auditory sensory cell depolarization; lesions of the cochlear stria vascularis and vestibular dark cells may impair potassium recycling, thereby decreasing endolymphatic potential (109). Paradoxically, hair cells seem to be spared by viral infection. Hearing loss and vestibular disorders may therefore be due to a deregulation of potassium flow, with progressive decrease of endolymphatic potential explaining a delayed onset of hearing loss or a fluctuation of hearing loss with time.
Hearing loss is the most common complication of bacterial meningitis, affecting more than 50% of survivors of pneumococcal meningitis (110). Meningitis can also cause a loss of vestibular system function, potentially via the same pathophysiological mechanisms, which are assumed to involve bacterial invasion of the inner ear followed by inflammatory infiltration. Vestibular dysfunction is frequently associated with bacterial meningitis and can be severe, with up to 14% of patients developing vestibular areflexia in one or both ears (21, 111). In a population of patients with bilateral vestibulopathy, the etiology was reported to be meningitis in 5% of cases (112). A loss of vestibular function due to bacterial meningitis in young children may delay posturomotor development if meningitis occurs before the child begins to walk independently, even in the absence of neurological impairment (21).
Vestibular responses can be elicited by sound stimulation, and it is known that the otolith organs are the most sound-sensitive. There is increasing evidence that excessive noise can induce vestibular deficiency by damaging the VHC hair bundle and/or the vestibulo-spinal, VOR pathway (113, 114), the observed hearing threshold being related to VEMP responses (115). Two mechanisms are involved in the destruction of the end organs by noise: direct mechanical injury and metabolic damage to the organ of Corti leading to the degeneration of sensory elements. Nevertheless, clinical studies have reported a low incidence of clinical signs of vestibular malfunction in patients with noise-induced hearing loss (NIHL), probably because of the redundancy of the vestibular peripheral system and compensatory mechanisms (114). However, these compensatory mechanisms may weaken with aging and vestibular dysfunction may therefore become increasingly common with age in NIHL patients (114).
Presbycusis (age-related hearing loss) and presbystasis (age-related changes in the vestibular organ) are becoming increasingly frequent, particularly in developed countries, due to increasing life expectancy. These two systems may age at different rates in the same individual (116), but presbycusis and presbystasis have a number of mechanisms in common, including a dysfunction of inner ear components (e.g., hair cells, ganglion neurons, vascular cells) and age-related changes in the central nervous system (117, 118). It was suggested several years ago that presbycusis and presbystasis have a genetic basis, and genes potentially involved in the pathophysiology of both conditions have been identified (118).
SNHL is the most common audiovestibular symptom associated with systemic autoimmune diseases, followed by tinnitus and vertigo (119). The pathophysiology of inner ear involvement in autoimmune diseases remains unclear and may be related to circulating antibodies against several inner ear antigens (primary autoimmune inner ear diseases) or immune complex-mediated damage (inner ear involvement in systemic autoimmune diseases) (120). Immune complex deposition seems to be the major factor involved in cochlear and vestibular damage in systemic autoimmune diseases. The deposition of immune complexes reduces the caliber of the auditory arteries, leading to a decrease in blood flow, inducing an oxygen deficit and increase in the levels of reactive oxygen species responsible for damage to hair cells and spiral ganglion (121). The vestibular symptoms observed in systemic autoimmune diseases may be isolated, or part of a spectrum, as in Meniere's disease. However, many patients have asymptomatic vestibular involvement requiring an objective vestibular assessment. Treatment may be based on systemic steroids, antimetabolites, such as methotrexate, or intratympanic steroid treatment in some cases.
Drugs toxic to the vestibule are frequently used, especially in patients with serious infections or life-threatening tumors (122, 123), see also 1. Vestibulotoxicity can affect hair cells in the utricle, saccule, or semicircular canals, leading to diverse vestibular deficits2. The risk of developing a vestibular deficit depends principally on the dose and properties of the toxic drug, probably modulated by genetic predisposition. Some of the drugs that damage the vestibule also harm the cochlea and, therefore, also cause hearing loss and/or tinnitus, but others have toxic effects specific to the vestibular system, and vestibular dysfunction may be more difficult to diagnose. The main substances responsible for significant vestibular toxicity are antibiotics, particularly aminoglycosides, and chemotherapy agents, such as cisplatin. Aminoglycosides, especially streptomycin, neomycin and gentamicin, are the most toxic drugs to the inner ear and their vestibulotoxicity is even greater than their toxicity to the cochlea. For the vestibular system, there is no safe dose of gentamicin and no safe serum gentamicin concentration (124). The use of aminoglycosides can result in a dramatic loss of VHCs (123). At the subcellular level, these drugs induce changes including the hydropic and vacuolar degeneration of VHCs (125).
Cisplatin is a widely used anticancer agent with toxic effects on the sensory epithelia in the vestibular and cochlear systems. It has been reported not only to induce a marked decrease in hair cell density in the utricle, saccule, and ampullae, but also to result in an abnormal morphology and disorganization of the VHCs and their stereocilia bundles (126). Other substances, such as organic solvents, have toxic effects on the parts of the brain responsible for processing balance information, rather than on the vestibular system itself (127).
By performing a literature survey, including studies of the available mutant mice, along with the HHH Hereditary Hearing Loss database [https://hereditaryhearingloss.org, (128)] and the OMIM (Online Mendelian Inheritance in Man; https://www.omim.org/) database, we defined a list of genes for which defects lead to vestibular dysfunction (Figure 4; Supplementary Table S1). DFNA, DFNB, and DFNX denote isolated forms of human deafness with autosomal dominant (DFNA), recessive (DFNB) or X-linked (DFNX) transmission (128). Vestibular dysfunction can be suspected in 30% of the listed non-syndromic forms of SNHL (Supplementary Table S1). In fact, the prevalence of associated vestibular dysfunction seems to be higher for syndromic hearing loss (Supplementary Table S1). Indeed, the prevalence of vestibular dysfunction is probably underestimated because many studies fail to detect such dysfunctions due to their focus on other sensory modalities or the inherent difficulty of objective direct assessments of vestibular function. However, as mentioned above, tools for the clinical assessment of vestibular function are available and can provide a considerable amount of information about peripheral vestibular defects in patients with hearing loss. Additional studies are required, focusing on determining the real spectrum of vestibular dysfunction in hearing loss patients. Vestibular testing should be included as an integral part of all hearing loss evaluations. Some efforts have been made in this direction. For example, Belgium has implemented a vestibular defect screening test for children with hearing impairment (129) and the French Society for Otorhinolaryngology (SFORL) recommends a vestibular assessment for all children undergoing cochlear implantation in its latest guidelines on pediatric cochlear implantation (130).

Table 1. Gene therapies in animal models with vestibular deficits.
Our knowledge of the development and function of the vestibular sensory system owes much to experimental approaches with various vertebrate species. Standard clinical applications, such as the testing of VOR by head impulses or the quantification of otolith function by VEMP, are based on evidence gained from experiments in various animal models (97). Insight into the pathogenic mechanisms at the origin of hearing loss, sometimes associated with vestibular dysfunctions, has also been obtained through the study of animal models of genetic deficits underlying deafness in humans (95, 96, 131, 132). About 140 genes responsible for non-syndromic forms of deafness in humans and about 200 genes responsible for syndromic forms (defects of the inner ear and other organs, including in mice) have been reported (133). Mouse models exist for most of these human genes. A high degree of conservation of anatomical and functional organization has been reported between rodents and humans (95–98). As a result, the available mutant mice, most of which also display vestibular deficits (Table 1; Supplementary Table S1), have provided valuable information about the key pathways leading to vestibular dysfunction. In studies of organ patterning or MET, the use of more distantly related animal models, such as zebrafish, Drosophila, or C. elegans, has facilitated the identification of key conserved core biological features and highlighted species-specific features (88, 153–157).
Side-by-side comparisons in animal models have yielded major insight into the molecular, cellular and physiological underpinnings of human deafness and vestibulopathies. Figure 4 depicts some of the causal pathways involved in vestibular dysfunction, based on studies in animal models defective for genes associated with deafness in humans. As reported for hearing (49), the causal genes for vestibular dysfunction can be grouped into several categories affecting: (1) hair bundle development and functioning, (2) synaptic transmission, (3) hair cell adhesion and maintenance, (4) cochlea ion homeostasis, (5) transmembrane or secreted proteins and the extracellular matrix, (6) oxidative stress, metabolism, and mitochondrial defects, and (7) transcriptional regulation. Additional genes implicated in vestibular function are indicated in Table 1, based on clinical observations, and information from related mouse models [see also (103)]. The principal representative examples are described briefly below, highlighting the key roles of the hair bundle (Usher proteins and MET components), ion homeostasis (e.g., cochlin, pendrin), and acellular membranes (e.g., tectorins).
The vestibular and auditory hair cells have several anatomical and functional properties in common, due in particular, to the presence of a mechanosensitive hair bundle at their apical surface (Figures 1, 3). Unsurprisingly, therefore, defects of key structural and functional proteins of the hair bundle have been found to cause both hearing and balance deficits (Figure 4; Table 1). Here, we briefly describe some of these molecular and functional pathways affecting the links between stereocilia, the cytoskeleton, and the MET machinery.
Extensive studies on the pathophysiology of the Usher Syndrome, the major cause of combined vision and hearing loss, have provide valuable insight into the formation and functioning of hair bundles. Five main Usher type I genes have been identified, encoding the actin-based motor myosin VIIa (USH1B), the PDZ domain-containing submembrane protein harmonin (USH1C), the scaffold protein sans (USH1G), and two Ca2+-dependent adhesion proteins, cadherin-23 (USH1D) and protocadherin-15 (USH1F) (158, 159). All the available USH1 mutant mice faithfully reproduce the profound congenital deafness and bilateral vestibular dysfunction observed in human patients, as demonstrated by their typical circling behavior and the absence of VsEPs in USH1 mice. Studies of these mice, the properties of USH1 proteins and their interactions revealed key roles of this network in the hair bundle (158, 159). Together, cadherin-23 and protocadherin-15 make up the embryonic lateral USH1 links between stereocilia and the links between stereocilia and the kinocilium essential to the integrity of the early developing hair bundles (158, 159). These two cadherins also form the upper and lower parts, respectively, of the tip-link (160), the apical link that gates the MET channels. Harmonin and sans are scaffold proteins essential for the anchoring of the cadherin links to the stereocilia actin filaments. Myosin VIIa is also required for the transport of key hair bundle components (e.g., harmonin and protocadherin-15) into the stereocilia (158–162).
Usherin/USH2A- and Adgvr1/USH2C-deficient mice, which have defects of USH2 proteins, have no vestibular deficits, consistent with clinical observations in most human USH2A and USH2C patients (158, 159, 163). This suggests that the ankle links, fibrous links connecting the stereocilia at their base, are probably dispensable for hair bundle function in VHCs (163–165). Interestingly, whirlin, a PDZ scaffold protein similar to harmonin/USH1C, has been associated with DFNB31 isolated hearing loss (166) and Usher syndrome type 2D (167). Whirlin is present as two distinct isoforms, the long and short variants (WHRN-L and WHRN-S), and it has been shown that, in addition to contributing to the formation of ankle links, this protein is a major factor determining the height and staircase organization of the stereocilia (168–170). Whirlin colocalizes and interacts with two other causal proteins for deafness, MYO15A (DFNB3) and EPS8 (DFNB102), forming a tripartite complex that controls actin polymerization and elongation of the actin core at the tips of the stereocilia (168–170). An absence of both whirlin isoforms causes abnormally short stereocilia and profound deafness and vestibular dysfunction (166, 171). However, WHRN-S expression is sufficient to maintain stereociliary bundle morphology and function in a subset of hair cells, resulting in some auditory responses, but no overt vestibular dysfunction (171, 172). The occurrence of vestibular dysfunction in some USH2 patients warrants further investigation. Indeed, defective vestibular responses were reported in eight of eleven USH2 patients tested in one study, but the lack of a clear genetic diagnosis for these patients precluded the identification of a direct causal role of USH2 (171, 173).
In addition to late-onset and progressive vision and hearing loss, some USH3A patients also develop vestibular dysfunction with various patterns of onset, progression, and severity (159, 174). Defects of the USH3A protein, clarin-1, in mice have also been shown to cause variable vestibular deficits that manifest later in life (175). The precise role of clarin-1 in the MET complex and the origin of the variability of vestibular symptoms remain to be established (176, 177).
Defects in another protein, the phosphatidylinositol phosphatase (PTPRQ), has been shown to affect vestibular function differently in humans and mice. PTPRQ, which is involved in DFNB84A and DFNA73 (128), is a component of the shaft links between stereocilia (178). PTPRQ-deficient mice have no overt behavioral signs of vestibular deficits, but detailed analyses have revealed subtle changes in swimming behavior associated with severe-to-profound macular receptor dysfunction, as confirmed by VsEP responses, and significant stereociliary bundle defects in the vestibular maculae (179). Like PTPRQ, other products of key deafness genes are located at the base of the stereocilia, where the shaft gradually narrows to form a conical taper, serving as the pivot point for deflection of the hair bundle upon stimulation. This includes myosin VI (MYO6/DFNA22), radixin (RDX), taperin (TPRN), chloride intracellular channel 5 (CLIC5/DFNB103) (103, 180, 181). VHCs of mutant mice lacking any one of these proteins display fused, thickened, and elongated stereocilia, accounting for the vestibular phenotype described (103, 180, 181).
The normal development and maintenance of stereociliary actin filaments is the key to hair cell function. Espin (ESPN/DFNB36), plastin-1 (PLS1/DFNA76) and fascin-2 (FSCN2) are three key actin filament crosslinkers that cooperate to produce the composite properties of the actin paracrystalline cores of the stereocilia (181). Espin is the major actin crosslinker, as mutant mice lacking this protein display typical abnormal circling behavior, probably due to a reduction of stereocilium diameter and length in both the auditory and vestibular hair cells (103, 181). By contrast, PLS1- and FSCN2-deficient mice displayed no overt behavioral traits, but the absence of either protein affected vestibular function and stereocilium structure, albeit with a markedly milder phenotype (181).
TMC1 and TMC2 have recently been identified as the core proteins of the MET channel complex, constituting the long-sought vertebrate MET channel subunits (182–185). The correct targeting of TMC1/2 pore-forming MET channel subunits to the tips of stereocilia (57) and their functional integrity involve key interactions with several other essential proteins (184, 185). In addition to the USH1 proteins described above, the lipoma HMGIC fusion partner-like 5 (LHFPL5, also known as TMHS, DFNB67), the transmembrane inner ear (TMIE, DFNB6), and the calcium and integrin-binding family member 2 (CIB2) proteins have been identified as components of the MET machinery at the tips of the transducing stereocilia in auditory and vestibular hair cells (186–189). Consistently, hair cells deficient for most of these proteins present a severe-to-complete loss of MET currents, leading to deafness and vestibular dysfunction (188, 190, 191). Of note, the lack of CIB2 causes profound hearing loss, but no signs of vestibular dysfunction have been observed (192–195), probably due to compensatory mechanisms involving CIB3 or other members of the same protein family (188, 196).
Mutations of SLC26A4 cause isolated DFNB4 hearing loss or a combination of congenital hearing loss, balance, and thyroid disorders known as Pendred syndrome (197, 198). SLC26A4 encodes pendrin, a CI/HCO3 anion exchanger crucial for fluid homeostasis in the inner ear (197, 199). Studies of Slc26a4-insufficient or -null mice have reported a clear disruption of sodium chloride and fluid absorption by the epithelial cells of the endolymphatic sac in these mice (198, 199). The impaired absorption of luminal Na+, Cl− and water leads to a swelling of the endolymphatic space, a feature also characteristic of a group of human inner ear malformations known as enlargement of vestibular aqueduct (EVA) disorders, and observed in clinical contexts such as Meniere's disease, experimental endolymphatic hydrops, and space motion sickness (198). While the etiology of abovementioned clinical entities is different, and the precise pathogenic link between the endolymphatic sac and inner ear dysfunction is unknown, alterations to pH, ionic composition, size, or the osmotic pressure of endolymph might constitute a common pathogenic pathway. Knock-in mouse models are being developed for studies of fluctuating EVA phenotypes, and to improve the management of patients with SLC26A4 defects. A panel of behavioral tests was recently used to document vestibular dysfunction in Slc26a4loop/loop mutant mice (200). These mice harbor a missense mutation (S408F) resulting in profound hearing loss, but they display various degrees of abnormality in vestibular behavior. The inner ears of all mutant mice with severe vestibular dysfunction display a pathological displacement of the otoconia into the crista ampullaris (200). This genetic predisposition for ectopic otoconia provides an excellent experimental model for studies of the pathophysiology of and possible treatment for benign paroxysmal positional vertigo (BPPV), a relatively common disorder in which the otoconia become detached and lodge in one of the semicircular canals (canalithiasis) or cupulae (cupulolithiasis).
No mutation of an otoconial component gene has ever been identified in humans. However, based on clinical symptoms in patients and studies in mouse, these structures seem clearly important for normal vestibular function. The prevalence of otoconium-related balance disorders is certainly largely underestimated. The morphology and composition of otoconia can be altered due to genetic and environmental factors, aging, ototoxic drugs, and head trauma (201). BPPV and aging are both high-risk factors for an abnormal dislocation of otoconia, causing an aberrant or ectopic distribution of otoconial debris, leading to dizziness or imbalance.
It is important to understand how otoconia are formed and how their attachment to macular acellular membranes is maintained if we wish to develop ways of influencing their regrowth/regeneration for the treatment of abnormalities. The structure and organization of the otoconia differ between mammals and fish, and even within vertebrates generally. Mammalian otoconia are unique in being the only calcium carbonate-containing biomineral found in normal healthy organs. The barrel-shaped calcitic crystals are partially embedded in the underlying gelatinous acellular membrane covering the neurosensory epithelium in the utricular and saccular maculae (see Figure 3). During head movement, the otoconia, which have a higher density than the surrounding endolymph, move relative to the sensory epithelium, leading to a sensing of either linear acceleration or the gravitational field (which is critical for spatial orientation and balance). In addition, by serving as a reservoir of ions, the otoconia may be involved in maintaining the homeostasis of the utricular and saccular endolymph, and the spatial and temporal gradients of calcium and other ions around the apical mechanosensitive hair bundles. Electrophysiological and behavioral studies have shown that the density of the crystals affects the magnitude of stimulus input to the sensory hair cells and that significant changes in crystal mass or location invariably leads to behavioral deficits.
Multiple, diverse pathways act in concert to ensure the normal biogenesis and maintenance of the otoconia. These pathways include various channel/pump proteins, enzymatic and trafficking processes [see Table 1 in (202)]. These components are widely distributed in the inner ear, including the hair cells, supporting cells, transitory epithelium, dark cells and epithelial cells lining vestibular cavities and the endolymphatic sac. Otoconial crystals have a distinct central core consisting of an organic matrix with a lower level of Ca2+, surrounded by largely inorganic peripheral zones with high Ca2+ concentrations. Otoconin-90, OC90, is the main component of otoconia (203); OC90 is extremely acidic, favoring the binding of calcium or calcium carbonate (CaCO3). This protein also interacts with and selectively recruits other otoconins (e.g., otolin-1, fetuin, osteopontin) to form the initial matrix facilitating the seeding process (204). The continuous addition of organic and inorganic components and the fusion of several minute crystallites ensure correct growth (201, 205). The core, periphery and external surface of the otoconia are interconnected with a fibrous material of various diameters and organizations. One of the major components of these fibers is otolin (also known as otolin-1), an inner-ear secreted glycoprotein that probably forms a collagen-like scaffold for optimal otoconium formation (201, 205).
The otoconial crystals are anchored to the sensory epithelia by the otoconial membrane, a honeycomb-like structure composed of collagenous proteins, such as otolin, and non-collagenous glycoproteins, including otogelin (206, 207), otogelin-like, α-tectorin, β-tectorin (208), and otoancorin (209). Otogelin is required for the anchoring of the otoconial membrane and cupula to the corresponding sensory epithelia. Otogelin-mutant mice have displaced acellular matrices and severe vestibular deficits (210). Mice lacking α-tectorin have few otoconial membranes, with fewer and larger otoconia, but no obvious vestibular behavioral deficits (208). By contrast, no structural or behavioral defects have yet been identified in mice lacking either β-tectorin or otoancorin (208, 211). Mutations of the genes encoding OTOG (otogelin, DFNB18B), OTOGL (otogelin-like, DFNB84B), and TECTA (α-tectorin, DFNA8/12, DFNB21) have been associated with hearing loss in humans, with occasional reports of vestibular hyporeflexia or vertigo in patients with OTOG or TECTA defects.
The development of otoconial biocrystals is regulated by various factors creating an appropriate environment for normal crystal seeding and growth, with correct concentrations of calcium, carbonate, and hydrogen ions in the endolymph above the utricular and saccular maculae. Severe otoconium losses, a complete absence of otoconia and otoconial abnormalities have been reported in mutant mice with an aberrant endolymph ionic composition (i.e., Ca2+, H+, , and Na+, K+, Cl−) (201, 202, 205). The list of key players required for the maintenance of crystal structure and the prevention of crystal degeneration include: (i) PMCA2, the plasma membrane Ca2+ ATPase 2 (ATP2B2) pump, the main source of Ca2+ ions for the formation and maintenance of otoconial crystals; (ii) the Cl−/ transporter pendrin, which secretes the bicarbonate required for CaCO3 formation and ensures a normal pH by regulating the levels of and/or Cl− ions; (iii) the alpha2 delta (α2δ1)-like ancillary subunit, which modulates Cav3 T-type CACHD1 channels by modulating voltage-gated calcium channels; this subunit contributes to the establishment of appropriate calcium concentrations in regions of otoconia formation (212); (iv) OTOP1, a multiple transmembrane (TM) protein that has been shown to regulate protein secretion and cellular calcium levels in vestibular supporting cells (213); (v) the membrane-bound enzymes NADPH oxidases (Noxs), NADPH oxidase 3 (Nox3), and associated proteins (p22, NOXO1), which produce reactive oxygen species, and contribute to calcium signaling; and (vi) zinc transporter 4 (SLC30A4), which is required for the normal expression of carbonic anhydrase (CA), which helps to maintain appropriate concentrations and pH (201, 205).
Additional studies are required to determine the precise mechanisms involving these proteins and to identify other key actors in disorders involving otoconia, otoconial membranes, and/or vestibular epithelia-related disorders. However, more specific, and appropriate clinical and experimental tools are required for thorough, in-depth evaluations of vestibular function, to elucidate the underlying mechanistic features. Naturally arising mutations in more than 340 genes have been reported to cause inner ear malformation or dysfunction in mice [Table S2 in (96)], but these genes only partially overlap with the more than 200 genes identified in humans. Ongoing programs, such as the International Mouse Phenotyping Consortium (IMPC), aiming to produce null alleles by deleting an early critical exon in each mouse gene (214, 215), will significantly increase the number of mutant mice with hearing and balance/vestibular deficits available (216–218). By early 2021, 9,719 knockout mice had been generated, 7,455 of which had been phenotyped through the IMPC comprehensive phenotyping pipeline including inner ear measurements (214), These mice are available to investigators worldwide (http://www.mousephenotype.org/) (215). More thorough studies of these models, with dedicated experimental tools for vestibular evaluations, should improve our understanding of the properties and singularities of vestibular organs.
Despite this high degree of medical need, as it was discussed in the introduction, the management of patients with vertigo and dizziness still suffers from a lack of diagnostic tools to guide therapeutic management and a lack of effective treatments. Current treatments for vestibular diseases are restricted to pharmacological treatments, rehabilitation, and surgery protocols. All aim to alleviate acute vestibular symptoms, enhance compensation, protect against symptoms aggravation, and promote vestibular rehabilitation. Innovative solutions with promising applications, such as vestibular implants and gene therapy approaches, are being tested in clinical trials.
Pharmacological approaches are frequently considered in the management of vestibular disorders. However, for most, the molecular basis of action remains unknown, efficacy is insufficient or even absent, and many anti-vertigo treatments have significant side effects. In the vestibular end organs, the drugs acting on the vestibular apparatus have diverse cellular targets, including the homeostasis of liquids and electrolytes in the inner ear, the regulation of blood flow, cell homeostasis and survival, and sensory processes related to vestibular information flow. In the vestibular nuclei, drugs act on homeostasis and cell survival, neurotransmitter receptors and ion channel modulation (219). For an in-depth review on the mechanism of action of the drugs used to treat vestibular disorders (219, 220).
Damage to the vestibular endorgans induces deafferentation of vestibular nucleus leading to a strong disorganization of the vestibular system. In the acute stage, such disturbances induce both static and dynamic vestibular deficits, causing postural, perceptual and oculomotor syndromes. Following vestibular damage, a spontaneous process, referred to as central compensation, allows substantial restoration of posture and balance in patients. It refers to all process by which the brain adapts to changes in inner ear vestibular organs function. The effectiveness of this process is highly case-dependent and its precise cellular and molecular mechanisms remain partially understood so far (221–224). What we know is that the central compensation mechanisms lead to the reorganization of the neuronal pathways and to the restoration of the activity in the ipsilateral and contralateral vestibular nuclei. Electrophysiological and behavioral investigation in animal models suggest that the compensation of the statis ocular motor and postural deficit results on the fast restoration of a balanced spontaneous resting activity in vestibular nuclei on both sides (through inhibition and facilitation mechanisms). The dynamic symptoms however require that the brain developpes new operating modes through different processes: synaptic remodeling, sensory substitution, or behavioral substitution. Dynamic symptoms improve more slowly and may never fully compensate. Vestibular rehabilitation is a physiotherapy program that accelerate compensation phenomena that are effective but slow and helps to correct inappropriate balance strategies. It is designed to promote vestibular compensation through adaptation, substitution, and habituation (225–227). Rehabilitation is often proposed in cases of acute unilateral vestibular deficits, inappropriate spontaneous compensation, or bilateral vestibular loss.
For patients with persistent defined vertigo symptoms after the exhaustion of pharmaceutical and other conservative treatment options suffering a severely impaired quality of life, surgical procedures are often the only remaining alternative. Function-preserving and destructive surgical procedures may be performed, with or without hearing preservation. However, there is heated debate among ENT specialists as to whether such surgery should be performed. All the available surgical procedures and their possible indications have been summarized in (228).
Apart from these traditional surgical procedures, approaches using electrical stimulation of the vestibule for restoring vestibular function have been developed. Currently, two technically different approaches for vestibular implants have been developed: vestibular co-stimulation with a cochlear implant (combined cochleo vestibular implant) or a stand-alone implant designed for intralabyrinthine stimulation in patients with bilateral vestibular loss but without profound sensorineural hearing loss.
The first vestibular implantation in humans was performed by the Maastricht-Geneva group in 2007, with a device derived from a cochlear implant and the placing of a single vestibular electrode on the posterior ampullary nerve (229). A new cochlear-vestibular implant prototype was developed a few years later 3, with a vestibular multichannel electrode associated with a special interface coupling a motion sensor for capturing the signals from a three-axis gyroscope (LYPR540AH; ST Microelectronics; Geneva, Switzerland) with the modulation of baseline electrical activity. Using this prototype, they showed that the vestibular implant could restore the VOR both at mid- (230) and high-frequency angular acceleration (231) (rotation test and head impulse test). They also showed that the implant could effectively suppress oscillopsia, one of the most frequent complaints in patients (232).
Another group from Johns Hopkins has worked on a promising standalone multichannel vestibular implant not combined with a cochlear implant. This device consists of a three-axis motion sensor, worn on the head, to record angular head velocity. It transmits the information, in the form of a modulation of the pulse rate and amplitude of stimulation, through the electrodes implanted in each semicircular canal (233). This group was the first to propose a method for vestibular stimulation 24-h-per-day, 7 days per week, preventing episodes of vertigo when the device is turned ON or OFF and allowing for use of the device outside the laboratory setting. Longitudinal follow-up at 6 months and 1 year after the unilateral implantation of a vestibular prosthesis for bilateral vestibular hypofunction revealed improvements over baseline for measurements relating to posture, gait, and quality of life, but hearing loss in the ear with the implant in all but one participant (234).
In parallel, Rubinstein et al. investigated a fundamentally different category of patients, using vestibular implants to treat patients with Meniere's disease. They designed their device to function as a “vestibular pacemaker” to treat Meniere's disease attacks (235). Unfortunately, although the vestibular stimulator successfully encoded vestibular information in Meniere's patients, it also led to a loss of hearing and vestibular function in the ear with the implant, which is a frequent side effect of vestibular implantation regardless of the technique and device used (236). In a more recent paper, they discussed results of their second-generation vestibular implant and they suggested that the etiology of vestibular loss could have a profound impact on sensitivity of vestibular afferents as opposed to cochlear afferents and that this could impact the feasibility of effective vestibular prosthetic devices (237).
In addition to these prototypes, designed to stimulate the semicircular canals, other efforts have been made to develop an implant for direct otolith stimulation (238).
The abovementioned rehabilitation treatments help to alleviate balance deficits and related symptoms, but curative treatments for vestibular disorders are lacking. The momentum gained in gene therapies over the last decade has spurred on the development of many applications in sensory organs. The inner ear, with its easy access, self-contained compartments, and immune-privileged status, is suitable for multiscale functional and behavioral evaluations of disease progression and beneficial outcomes of treatment. The first applications of gene therapy focused on gene replacement (or gene supplementation) therapy, in which a healthy copy of a defective gene is introduced into cells bearing the mutation. These studies essentially targeted key deafness genes small enough to be delivered with adeno-associated virus (AAV) vectors (maximum capacity of 4.7 kb for a single AAV) (49, 89, 239–241) (see Table 1). In recent years, new generations of AAVs with higher transduction rates for inner ear hair cells have been tested (139, 141, 146, 240, 242). Below, we detail some key attempts at cell-specific delivery for hearing and vestibular disorders.
Several recent studies have published promising results for the use of gene therapy to restore auditory and vestibular function in mouse models of Usher syndrome (159). Three of the mutant mice modeling Usher syndrome and displaying significant vestibular dysfunction have defects of genes small enough to fit into a single AAV: harmonin, sans, and whirlin (159). In Ush1c knock-in mutant mice, Pan et al. showed that a single injection of AAV2/Anc80L65-CMV-Ush1c via the round window membrane in neonatal animals (P0–P1) resulted in a mean rescue of auditory thresholds to within 20 dB of wild-type values for low-to mid-frequency hearing, whereas high-frequency hearing thresholds remained high (139). In a similar approach, a single injection of AAV8-Ush1g in Ush1g mutant mice totally restored vestibular function, as demonstrated by the restoration of VOR responses and a decrease in circling behavior (144). Focusing on whirlin, defects of which cause isolated hearing loss, and USH2D, Isgrig et al. showed that AAV8-CMV-Whrn, delivered via the posterior semicircular canal, successfully restored the morphology of stereociliary bundles and increased hair cell survival in both the treated cochlea and utricle of Whrnwi/wi mice (146). AAV-mediated whirlin expression was found to increase the length of the stereocilia in utricular hair cells and in the cochlear inner hair cells, and to promote hair cell survival in treated Whrnwi mice (146). Hearing recovery was only partial, with ABR thresholds at 60–70 dB SPL in some treated mutant mice, but vestibular function improved significantly, as demonstrated by a decrease in circling behavior and improvements in the performance of the animal in rotarod and swim tests. The posterior semicircular canal delivery approach resulted in better auditory and vestibular recovery than delivery via the cochlear round window, probably due to a higher hair-cell transduction efficiency following injection into the endolymphatic space (146). Other AAVs have been tested for improving viral transduction in the inner ear. They include exosome-associated AAVs (exo-AAV1, and exo-AAV9), which were used in a mutant mouse defective for LHFPL5, a key component of the MET machinery (141). Following the injection of exo-AAV1-CBA-Lhfpl5 into the inner ears of P1–P2 Lhfpl5 knockout mice, partial hearing recovery was observed (improvements of up to 30 dB SPL at frequencies of 4–22 kHz). Vestibular function also improved in the Lhfpl5 knockout mice, as shown by a decrease in head tossing and circling behaviors (141).
However, the limited packaging capacity of AAVs (<4.7 kb for a single AAV) limits the expansion of this delivery approach to many deafness-balance causal genes. Alternative approaches involving dual AAVs, or RNA-based therapies have also been explored to overcome this AAV limitation (49, 240, 243). Splice-switching antisense oligonucleotides (ASO) have been successfully used to target the USH1C messenger RNA (mRNA) transcribed in mice homozygous for the Ush1c c.216G > A mutation (135, 137). Recent studies include direct assessments of vestibular function based on vestibular sensory evoked potentials (VsEPs). VsEP responses are totally absent in untreated mice and were restored to near-normal levels after injection of the oligonucleotide into neonatal mice. More detailed explorations have shown that ASO-29 delivery to the inner ear improves cochlear hair-cell transduction currents, restores vestibular afferent irregularity, spontaneous firing rate, and sensitivity to head rotation, and successfully restores hearing thresholds and balance-related behaviors in USH1C mice (135, 137). Treatment at P5 and P15 was minimally effective at rescuing vestibular function, but vestibular recovery was nevertheless sufficient to support normal balance-related behaviors, suggesting positive therapeutic effects on balance (135, 137). Additional studies are underway to determine the best delivery approach to the inner ear, the optimal therapeutic window that enables the best beneficial outcomes, and stability of hearing and/or balance recovery overtime.
The recent advent of gene-editing techniques has paved the way for the development of gene therapy for rare diseases. Gene editing provides opportunities to obtain long-term therapeutic benefits regardless of the size of the gene or the nature of the mutation. This new strategy has already been tested and has yielded promising auditory outcomes for approaches targeting the TMC gene encoding the MET channel. AAV(Anc80L65)-mediated delivery of the SaCas9-KKH-gRNA complex efficiently prevented deafness in Bth mice for up to 1-year post injection (244). DPOAE thresholds in injected Tmc1Bth mice revealed the preservation of OHC function at lower frequencies (5–11 kHz) at 12 weeks of age, and up to 24 weeks of age in the surviving animals. In treated mice, the hair bundles of cochlear OHCs and IHCs (in the 8 and 16 kHz regions) and VHCs recovered a normal morphology after treatment, with minimal hair-cell loss. With steady progress and the improvement of gene-editing tools (new generations of CRISPR/Cas nucleases, base and prime editors) (49, 243, 245, 246), we anticipate increasing interest in the study of balance deficits in mouse models of vestibular dysfunction.
Attempts have been made to reactivate hair-cell regeneration in the inner ear, through Atoh1 overexpression (31), or the transplantation of human induced pluripotent stem cell (hiPSC) cells (247). The recent progress in hiPSC derived organoids make it possible to obtain 3D-models of human organ development and study the disease in human relevant cellular and genomic contexts (248–251). Despite some limitations to obtain mature cochlear tissues, current protocols can be used to obtain type I or type II hair cells in 3D-inner ear organoids (249, 251, 252). Further studies are needed to identify key pathways for optimal control of cell differentiation into supporting cells, neurons and/or hair cells according to downstream applications. Scalable hiPSC-based platforms can be designed to ensure a reliable source of cells for transplantation and cell therapy. Also, by modeling specific hearing and vestibular disorders, inner ear organoids can allow the establishment of clear molecular and cellular phenotypic features, which can be used as read-outs to test potential beneficial outcomes of regenerative or neuroprotective drugs to hearing and/or balance deficits. In this quest of curative treatments for hearing and/or balance disorders, combined information based on animal and human cellular models will likely help improve the potential beneficial outcomes of treatment in preclinical models, prior to clinical trials.
Clinical surveys worldwide clearly show that vestibular dysfunction is more prevalent than previously thought. This growing awareness of the importance of the vestibular system is timely and is driving efforts to dissect and understand the multiple contributions of the vestibular system. With current clinical tests for balance deficits, it is easy to perform a complete vestibular assessment in clinical practice, even in infants. Vestibular testing should always be proposed if hearing loss is discovered. However, current clinical tests can detect only gross vestibular behavioral defects, and it remains challenging to detect subtle changes in vestibular function and to identify the precise source of the vestibular deficits experienced by patients, in the otolithic organs, cristae, or beyond, in specific neuron subsets of the central vestibular system. More sensitive clinical tests and better software programs for data interpretation are required for accurate determinations of the extent and severity of the balance deficits detected. Various machine learning algorithms are now being implemented for image-based analysis and clinical diagnosis, to predict the presence of peripheral vestibular dysfunction from posturography parameters (253, 254). In this context, the development of new artificial intelligence (AI) tools will be required, to disentangle the intricate contributions of the vestibular, visual, and somatosensory systems, to facilitate the processing and integration of inputs and parameters from multiple sensory modalities (255). These tools may also have applications extending to the identification of clinical endophenotypes, facilitating the precise and accurate definition and classification of vestibular disorders, improving disease profiling and the prediction of disease progression, and guiding decisions about the type or utility of potential treatments.
Animal models presenting specific balance defects will continue to be instrumental in attempts to refine existing tests or to develop new exploratory paradigms adapting tests to the source of the deficit. In addition to the existing mouse models corresponding to most of the human deafness causal genes identified to date (49, 50), the development of many new models is anticipated, through ongoing international collaborative programs [see (132, 217); http://www.mousephenotype.org/]. These animal models are available to researchers worldwide. They allow a full evaluation of the function of all five vestibular endorgans, through the use of a battery of different methods, guaranteeing a high quality of vestibular testing, combined with morphomolecular investigations to ensure the correct interpretation of the scientific results. These models should improve our understanding of the properties and singular features of vestibular disorders, and provide further insight into the evolution, development, physiology and behavior of the vestibular system. These discoveries will also help guide patient management and the search for possible treatments, through studies of the time window for effective treatment, possible injection routes, and long-term treatment stability.
Gene therapy tools have progressed considerably [improved AAVs, new delivery strategies, emerging gene editing tools; see (49, 89, 239–241)] and there is an urgent need for their clinical application to hearing and balance disorders. Consistent with the considerable phylogenetic gap between vestibular and cochlear organs, the intermediate step toward successful gene therapies for the inner ear is now being achieved, at least in preclinical animal models presenting balance deficits. Thanks to recent progress in viral design, most currently available AAVs can mediate high and robust transduction rates of the VHCs in all vestibular compartments (139, 144, 146, 240, 256, 257), ensuring the efficient delivery of therapeutic agents to vestibular target cells. As described above, in almost all gene therapy studies carried out in deaf mouse mutants displaying vestibular deficits, normal balance function was successfully achieved, whereas outcomes for hearing were much more variable (49). Efforts are being made to improve the existing vectors and to identify new vectors providing better control and higher rates of auditory hair cell transduction, especially for the outer hair cells (highly specialized cells unique to mammals). Restoration of the unique properties of the OHCs, such as somatic electromotility or the well-organized shape and staircase pattern of stereocilia, and high-frequency hearing, remains challenging. In the quest for 3D organoids for inner ear organs, it remains difficult to obtain cochlear tissues, but major progress has been made toward reproducing the ontogeny of balance organs and production of vestibular-like hair cells. For these reasons, the vestibular system provides more opportunities for disentangling the signaling pathways for mammalian hair-cell regeneration, setting the stage for the implementation of treatment solutions for restoring normal balance. Gene therapies for balance deficits are now accessible and we expect an increasing number of successful proof of concept studies in animal models with balance deficits. Such studies can include research for possible combined tissue and engineering therapies, coupling the use of gene therapy with current prosthetic devices (hearing aids, cochlear and vestibular implants). Today's efforts to implement and improve these therapeutic interventions in animal models will better prepare their possible transfer into clinics and/or help implement rehabilitation and physical therapies for alleviating hearing and balance symptoms. The path is clear, but success can only be achieved through coordinated actions by scientists across many fields (AI, physics, virology, chemistry, immunology, molecular and cell biology, and physiology), along with joint efforts with AI researchers, engineers, audiologists, clinicians and all actors in the hearing and balance fields.
AM and AE-A wrote the first draft of the manuscript. AM, SV, and AE-A reviewed and critically revised the manuscript. All authors contributed to the article and approved the submitted version.
The work in the authors' laboratories was funded by the French National Research Agency (ANR), as part of the Second Investissements d'Avenir Program (light4deaf, ANR-15-RHUS-0001), and LabEx LIFESENSES (ANR-10-LABX-65), Fondation pour l'Audition (FPA-19-Stg), ANR-HearInNoise-(ANR-17-CE16-0017), LHW-Stiftung, Fondation de France, Fondation Maladies Rares, & Retina-France. We acknowledge the support of the Institut de l‘Audition by Fondation pour l'Audition.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
We thank Pr. Anna Lysakowski for critical review of the manuscript, and her helpful comments and suggestions.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fneur.2022.816534/full#supplementary-material
1. ^Available online at: https://balanceanddizziness.org/disorders/vestibular-disorders/vestibular-toxicity/.
2. ^Available online at: https://dizziness-and-balance.com/disorders/bilat/ototoxins.html.
3. ^Available online at: https://patentscope.wipo.int/search/en/detail.jsf?docId=WO2014118094.
1. Fritzsch B, Beisel KW, Jones K, Farinas I, Maklad A, Lee J, et al. Development and evolution of inner ear sensory epithelia and their innervation. J Neurobiol. (2002) 53:143–56. doi: 10.1002/neu.10098
2. Fekete DM, Wu DK. Revisiting cell fate specification in the inner ear. Curr Opin Neurobiol. (2002) 12:35–42. doi: 10.1016/S0959-4388(02)00287-8
3. Barald KF, Kelley MW. From placode to polarization: new tunes in inner ear development. Development. (2004) 131:4119–30. doi: 10.1242/dev.01339
4. Wu DK, Kelley MW. Molecular mechanisms of inner ear development. Cold Spring Harb Perspect Biol. (2012) 4:a008409. doi: 10.1101/cshperspect.a008409
5. Ankamreddy H, Bok J, Groves AK. Uncovering the secreted signals and transcription factors regulating the development of mammalian middle ear ossicles. Dev Dyn. (2020) 249:1410–24. doi: 10.1002/dvdy.260
6. Kelly MC, Chen P. Development of form and function in the mammalian cochlea. Curr Opin Neurobiol. (2009) 19:395–401. doi: 10.1016/j.conb.2009.07.010
7. Besnard S, Tighilet B, Chabbert C, Hitier M, Toulouse J, Le Gall A, et al. The balance of sleep: role of the vestibular sensory system. Sleep Med Rev. (2018) 42:220–8. doi: 10.1016/j.smrv.2018.09.001
8. Yardley L, Owen N, Nazareth I, Luxon L. Prevalence and presentation of dizziness in a general practice community sample of working age people. Br J Gen Pract. (1998) 48:1131–5.
9. Gopinath B, McMahon CM, Rochtchina E, Mitchell P. Dizziness and vertigo in an older population: the Blue Mountains prospective cross-sectional study. Clin Otolaryngol. (2009) 34:552–6. doi: 10.1111/j.1749-4486.2009.02025.x
10. Wiltink J, Tschan R, Michal M, Subic-Wrana C, Eckhardt-Henn A, Dieterich M, et al. Dizziness: anxiety, health care utilization and health behavior–results from a representative German community survey. J Psychosom Res. (2009) 66:417–24. doi: 10.1016/j.jpsychores.2008.09.012
11. Kovacs E, Wang X, Grill E. Economic burden of vertigo: a systematic review. Health Econ Rev. (2019) 9:37. doi: 10.1186/s13561-019-0258-2
12. Muller M, Goeldlin MB, Gaschen J, Sauter TC, Stock S, Wagner F, et al. Characteristics and resource needs in patients with vestibular symptoms: a comparison of patients with symptoms of unknown versus determined origin. BMC Emerg Med. (2020) 20:70. doi: 10.1186/s12873-020-00361-8
13. Goeldlin M, Gaschen J, Kammer C, Comolli L, Bernasconi CA, Spiegel R, et al. Frequency, aetiology, and impact of vestibular symptoms in the emergency department: a neglected red flag. J Neurol. (2019) 266:3076–86. doi: 10.1007/s00415-019-09525-4
14. Humphriss RL, Hall AJ. Dizziness in 10 year old children: an epidemiological study. Int J Pediatr Otorhinolaryngol. (2011) 75:395–400. doi: 10.1016/j.ijporl.2010.12.015
15. Li CM, Hoffman HJ, Ward BK, Cohen HS, Rine RM. Epidemiology of dizziness and balance problems in children in the United States: a population-based study. J Pediatr. (2016) 171:240–7 e1–3. doi: 10.1016/j.jpeds.2015.12.002
16. Sommerfleck PA, Gonzalez Macchi ME, Weinschelbaum R, De Bagge MD, Bernaldez P, Carmona S. Balance disorders in childhood: main etiologies according to age. Usefulness of the video head impulse test. Int J Pediatr Otorhinolaryngol. (2016) 87:148–53. doi: 10.1016/j.ijporl.2016.06.020
17. Wiener-Vacher SR, Quarez J, Priol AL. Epidemiology of vestibular impairments in a pediatric population. Semin Hear. (2018) 39:229–42. doi: 10.1055/s-0038-1666815
18. Iwasaki S, Yamasoba T. Dizziness and imbalance in the elderly: age-related decline in the vestibular system. Aging Dis. (2015) 6:38–47. doi: 10.14336/AD.2014.0128
19. Rine RM, Braswell J. A clinical test of dynamic visual acuity for children. Int J Pediatr Otorhinolaryngol. (2003) 67:1195–201. doi: 10.1016/j.ijporl.2003.07.004
20. Brandt T, Schautzer F, Hamilton DA, Bruning R, Markowitsch HJ, Kalla R, et al. Vestibular loss causes hippocampal atrophy and impaired spatial memory in humans. Brain. (2005) 128:2732–41. doi: 10.1093/brain/awh617
21. Wiener-Vacher SR, Obeid R, Abou-Elew M. Vestibular impairment after bacterial meningitis delays infant posturomotor development. J Pediatr. (2012) 161:246–51 e1. doi: 10.1016/j.jpeds.2012.02.009
22. Lopez C. The vestibular system: balancing more than just the body. Curr Opin Neurol. (2016) 29:74–83. doi: 10.1097/WCO.0000000000000286
23. Lacour M, Tighilet B. Plastic events in the vestibular nuclei during vestibular compensation: the brain orchestration of a “deafferentation” code. Restor Neurol Neurosci. (2010) 28:19–35. doi: 10.3233/RNN-2010-0509
24. Kwan T, White PM, Segil N. Development and regeneration of the inner ear. Ann N Y Acad Sci. (2009) 1170:28–33. doi: 10.1111/j.1749-6632.2009.04484.x
25. Groves AK, Fekete DM. Shaping sound in space: the regulation of inner ear patterning. Development. (2012) 139:245–57. doi: 10.1242/dev.067074
26. Fritzsch B, Elliott KL. Gene, cell, and organ multiplication drives inner ear evolution. Dev Biol. (2017) 431:3–15. doi: 10.1016/j.ydbio.2017.08.034
27. Iyer AA, Groves AK. Transcription factor reprogramming in the inner ear: turning on cell fate switches to regenerate sensory hair cells. Front Cell Neurosci. (2021) 15:660748. doi: 10.3389/fncel.2021.660748
28. Elliott KL, Pavlinkova G, Chizhikov VV, Yamoah EN, Fritzsch B. Development in the mammalian auditory system depends on transcription factors. Int J Mol Sci. (2021) 22:4189. doi: 10.3390/ijms22084189
29. Mackowetzky K, Yoon KH, Mackowetzky EJ, Waskiewicz AJ. Development and evolution of the vestibular apparatuses of the inner ear. J Anat. (2021) 239:801–28. doi: 10.1111/joa.13459
30. Bermingham NA, Hassan BA, Price SD, Vollrath MA, Ben-Arie N, Eatock RA, et al. Math1: an essential gene for the generation of inner ear hair cells. Science. (1999) 284:1837–41. doi: 10.1126/science.284.5421.1837
31. Sayyid ZN, Wang T, Chen L, Jones SM, Cheng AG. Atoh1 directs regeneration and functional recovery of the mature mouse vestibular system. Cell Rep. (2019) 28:312–24 e4. doi: 10.1016/j.celrep.2019.06.028
32. Wang T, Niwa M, Sayyid ZN, Hosseini DK, Pham N, Jones SM, et al. Uncoordinated maturation of developing and regenerating postnatal mammalian vestibular hair cells. PLoS Biol. (2019) 17:e3000326. doi: 10.1371/journal.pbio.3000326
33. Matern MS, Milon B, Lipford EL, McMurray M, Ogawa Y, Tkaczuk A, et al. GFI1 functions to repress neuronal gene expression in the developing inner ear hair cells. Development. (2020) 147:dev186015. doi: 10.1242/dev.186015
34. Lee S, Song JJ, Beyer LA, Swiderski DL, Prieskorn DM, Acar M, et al. Combinatorial Atoh1 and Gfi1 induction enhances hair cell regeneration in the adult cochlea. Sci Rep. (2020) 10:21397. doi: 10.1038/s41598-020-78167-8
35. Meyer AC, Moser T. Structure and function of cochlear afferent innervation. Curr Opin Otolaryngol Head Neck Surg. (2010) 18:441–6. doi: 10.1097/MOO.0b013e32833e0586
36. Appler JM, Goodrich LV. Connecting the ear to the brain: molecular mechanisms of auditory circuit assembly. Prog Neurobiol. (2011) 93:488–508. doi: 10.1016/j.pneurobio.2011.01.004
37. Fritzsch B, Pan N, Jahan I, Elliott KL. Inner ear development: building a spiral ganglion and an organ of Corti out of unspecified ectoderm. Cell Tissue Res. (2015) 361:7–24. doi: 10.1007/s00441-014-2031-5
38. Delacroix L, Malgrange B. Cochlear afferent innervation development. Hear Res. (2015) 330:157–69. doi: 10.1016/j.heares.2015.07.015
39. Lim DJ. Functional structure of the organ of Corti: a review. Hear Res. (1986) 22:117–46. doi: 10.1016/0378-5955(86)90089-4
40. Fettiplace R, Hackney CM. The sensory and motor roles of auditory hair cells. Nat Rev Neurosci. (2006) 7:19–29. doi: 10.1038/nrn1828
41. Hudspeth AJ, Corey DP. Sensitivity, polarity, and conductance change in the response of vertebrate hair cells to controlled mechanical stimuli. Proc Natl Acad Sci USA. (1977) 74:2407–11. doi: 10.1073/pnas.74.6.2407
42. Hudspeth AJ. How the ear's works work: mechanoelectrical transduction and amplification by hair cells. C R Biol. (2005) 328:155–62. doi: 10.1016/j.crvi.2004.12.003
43. Driver EC, Kelley MW. Development of the cochlea. Development. (2020) 147:dev162263. doi: 10.1242/dev.162263
44. Eatock RA, Lysakowski A. Mammalian vestibular hair cells. In: Eatock RA, Fay RR, Popper AN, editors. Vertebrate Hair Cells. New York, NY: Springer-Verlag (2006). p. 348–442.
45. Lysakowski A, Goldberg JM. A regional ultrastructural analysis of the cellular and synaptic architecture in the chinchilla cristae ampullares. J Comp Neurol. (1997) 389:419–43. doi: 10.1002/(SICI)1096-9861(19971222)389:3<419::AID-CNE5>3.0.CO;2-33.0.CO;2-3
46. Eatock RA, Rusch A, Lysakowski A, Saeki M. Hair cells in mammalian utricles. Otolaryngol Head Neck Surg. (1998) 119:172–81. doi: 10.1016/S0194-5998(98)70052-X
47. Eatock RA. Specializations for fast signaling in the amniote vestibular inner ear. Integr Comp Biol. (2018) 58:341–50. doi: 10.1093/icb/icy069
48. Jan TA, Eltawil Y, Ling AH, Chen L, Ellwanger DC, Heller S, et al. Spatiotemporal dynamics of inner ear sensory and non-sensory cells revealed by single-cell transcriptomics. Cell Rep. (2021) 36:109358. doi: 10.1016/j.celrep.2021.109358
49. Delmaghani S, El-Amraoui A. Inner ear gene therapies take off: current promises and future challenges. J Clin Med. (2020) 9:2309. doi: 10.3390/jcm9072309
50. Safieddine S, El-Amraoui A, Petit C. The auditory hair cell ribbon synapse: from assembly to function. Annu Rev Neurosci. (2012) 35:509–28. doi: 10.1146/annurev-neuro-061010-113705
51. Roberts WM, Howard J, Hudspeth AJ. Hair cells: transduction, tuning, and transmission in the inner ear. Annu Rev Cell Biol. (1988) 4:63–92. doi: 10.1146/annurev.cb.04.110188.000431
52. Gillespie PG, Muller U. Mechanotransduction by hair cells: models, molecules, and mechanisms. Cell. (2009) 139:33–44. doi: 10.1016/j.cell.2009.09.010
53. Kachar B, Parakkal M, Kurc M, Zhao Y, Gillespie PG. High-resolution structure of hair-cell tip links. Proc Natl Acad Sci USA. (2000) 97:13336–41. doi: 10.1073/pnas.97.24.13336
54. Howard J, Hudspeth AJ. Compliance of the hair bundle associated with gating of mechanoelectrical transduction channels in the bullfrog's saccular hair cell. Neuron. (1988) 1:189–99. doi: 10.1016/0896-6273(88)90139-0
55. Pickles JO, Corey DP. Mechanoelectrical transduction by hair cells. Trends Neurosci. (1992) 15:254–9. doi: 10.1016/0166-2236(92)90066-H
56. Hackney CM, Furness DN. The composition and role of cross links in mechanoelectrical transduction in vertebrate sensory hair cells. J Cell Sci. (2013) 126:1721–31. doi: 10.1242/jcs.106120
57. Beurg M, Fettiplace R, Nam JH, Ricci AJ. Localization of inner hair cell mechanotransducer channels using high-speed calcium imaging. Nat. Neurosci. (2009) 12:553–8. doi: 10.1038/nn.2295
58. Moser T, Brandt A, Lysakowski A. Hair cell ribbon synapses. Cell Tissue Res. (2006) 326:347–59. doi: 10.1007/s00441-006-0276-3
59. Burns JC, Stone JS. Development and regeneration of vestibular hair cells in mammals. Semin Cell Dev Biol. (2017) 65:96–105. doi: 10.1016/j.semcdb.2016.11.001
60. Koppl C, Manley GA. A functional perspective on the evolution of the cochlea. Cold Spring Harb Perspect Med. (2019) 9:a033241. doi: 10.1101/cshperspect.a033241
61. Fettiplace R. Diverse mechanisms of sound frequency discrimination in the vertebrate cochlea. Trends Neurosci. (2020) 43:88–102. doi: 10.1016/j.tins.2019.12.003
62. Rusch A, Lysakowski A, Eatock RA. Postnatal development of type I and type II hair cells in the mouse utricle: acquisition of voltage-gated conductances and differentiated morphology. J Neurosci. (1998) 18:7487–501. doi: 10.1523/JNEUROSCI.18-18-07487.1998
63. McInturff S, Burns JC, Kelley MW. Characterization of spatial and temporal development of Type I and Type II hair cells in the mouse utricle using new cell-type-specific markers. Biol Open. (2018) 7:bio038083. doi: 10.1242/bio.038083
64. Desai SS, Zeh C, Lysakowski A. Comparative morphology of rodent vestibular periphery. I. Saccular utricular maculae. J Neurophysiol. (2005) 93:251–66. doi: 10.1152/jn.00746.2003
65. Sadeghi SG, Chacron MJ, Taylor MC, Cullen KE. Neural variability, detection thresholds, and information transmission in the vestibular system. J Neurosci. (2007) 27:771–81. doi: 10.1523/JNEUROSCI.4690-06.2007
66. Eatock RA, Songer JE. Vestibular hair cells and afferents: two channels for head motion signals. Annu Rev Neurosci. (2011) 34:501–34. doi: 10.1146/annurev-neuro-061010-113710
67. Curthoys IS, Grant JW, Pastras CJ, Frohlich L, Brown DJ. Similarities and differences between vestibular and cochlear systems - a review of clinical and physiological evidence. Front Neurosci. (2021) 15:695179. doi: 10.3389/fnins.2021.695179
68. Tilney LG, Saunders JC. Actin filaments, stereocilia, and hair cells of the bird cochlea. I. Length, number, width, and distribution of stereocilia of each hair cell are related to the position of the hair cell on the cochlea. J Cell Biol. (1983) 96:807–21. doi: 10.1083/jcb.96.3.807
69. Tilney LG, Tilney MS, Cotanche DA. New observations on the stereocilia of hair cells of the chick cochlea. Hear Res. (1988) 37:71–82. doi: 10.1016/0378-5955(88)90079-2
70. Tilney LG, Cotanche DA, Tilney MS. Actin filaments, stereocilia and hair cells of the bird cochlea. VI. How the number and arrangement of stereocilia are determined. Development. (1992) 116:213–26. doi: 10.1242/dev.116.1.213
71. Tilney LG, Tilney MS, DeRosier DJ. Actin filaments, stereocilia, and hair cells: how cells count and measure. Annu Rev Cell Biol. (1992) 8:257–74. doi: 10.1146/annurev.cb.08.110192.001353
72. Li A, Xue J, Peterson EH. Architecture of the mouse utricle: macular organization and hair bundle heights. J Neurophysiol. (2008) 99:718–33. doi: 10.1152/jn.00831.2007
73. Nam JH, Grant JW, Rowe MH, Peterson EH. Multiscale modeling of mechanotransduction in the utricle. J Neurophysiol. (2019) 122:132–50. doi: 10.1152/jn.00068.2019
74. Lysakowski A. Synaptic organization of the crista ampullaris in vertebrates. Ann NY Acad Sci. (1996) 781:164–82. doi: 10.1111/j.1749-6632.1996.tb15700.x
75. Govindaraju AC, Anna Lysakowski A, Eatock RA, Raphael RM. A biophysical model of nonquantal transmission at the vestibular hair cell-calyx synapse: KLV currents modulate fast electrical and slow K+ potentials in the synaptic cleft. bioRxiv [Preprint]. (2021) 469197. doi: 10.1101/2021.11.18.469197
76. Elliott KL, Fritzsch B, Duncan JS. Evolutionary and developmental biology provide insights into the regeneration of organ of corti hair cells. Front Cell Neurosci. (2018) 12:252. doi: 10.3389/fncel.2018.00252
77. Warchol ME. Sensory regeneration in the vertebrate inner ear: differences at the levels of cells and species. Hear Res. (2011) 273:72–9. doi: 10.1016/j.heares.2010.05.004
78. Corwin JT, Cotanche DA. Regeneration of sensory hair cells after acoustic trauma. Science. (1988) 240:1772–4. doi: 10.1126/science.3381100
79. Stone JS, Cotanche DA. Hair cell regeneration in the avian auditory epithelium. Int J Dev Biol. (2007) 51:633–47. doi: 10.1387/ijdb.072408js
80. Golub JS, Tong L, Ngyuen TB, Hume CR, Palmiter RD, Rubel EW, et al. Hair cell replacement in adult mouse utricles after targeted ablation of hair cells with diphtheria toxin. J Neurosci. (2012) 32:15093–105. doi: 10.1523/JNEUROSCI.1709-12.2012
81. Burns JC, Christophel JJ, Collado MS, Magnus C, Carfrae M, Corwin JT. Reinforcement of cell junctions correlates with the absence of hair cell regeneration in mammals and its occurrence in birds. J Comp Neurol. (2008) 511:396–414. doi: 10.1002/cne.21849
82. Luo WW, Wang XW, Ma R, Chi FL, Chen P, Cong N, et al. Junctional E-cadherin/p120-catenin is correlated with the absence of supporting cells to hair cells conversion in postnatal mice cochleae. Front Mol Neurosci. (2018) 11:20. doi: 10.3389/fnmol.2018.00020
83. Collado MS, Thiede BR, Baker W, Askew C, Igbani LM, Corwin JT. The postnatal accumulation of junctional E-cadherin is inversely correlated with the capacity for supporting cells to convert directly into sensory hair cells in mammalian balance organs. J Neurosci. (2011) 31:11855–66. doi: 10.1523/JNEUROSCI.2525-11.2011
84. Burns JC, Collado MS, Oliver ER, Corwin JT. Specializations of intercellular junctions are associated with the presence and absence of hair cell regeneration in ears from six vertebrate classes. J Comp Neurol. (2013) 521:1430–48. doi: 10.1002/cne.23250
85. Burns JC, Corwin JT. A historical to present-day account of efforts to answer the question: “what puts the brakes on mammalian hair cell regeneration?”. Hear Res. (2013) 297:52–67. doi: 10.1016/j.heares.2013.01.005
86. Wu M, Xia M, Li W, Li H. Single-cell sequencing applications in the inner ear. Front Cell Dev Biol. (2021) 9:637779. doi: 10.3389/fcell.2021.637779
87. Booth KT, Azaiez H, Jahan I, Smith RJH, Fritzsch B. Intracellular regulome variability along the organ of corti: evidence, approaches, challenges, and perspective. Front Genet. (2018) 9:156. doi: 10.3389/fgene.2018.00156
88. Nicolson T. Navigating hereditary hearing loss: pathology of the inner ear. Front Cell Neurosci. (2021) 15:660812. doi: 10.3389/fncel.2021.660812
89. Mei C, Dong H, Nisenbaum E, Thielhelm T, Nourbakhsh A, Yan D, et al. Genetics and the individualized therapy of vestibular disorders. Front Neurol. (2021) 12:633207. doi: 10.3389/fneur.2021.633207
90. Selz PA, Girardi M, Konrad HR, Hughes LF. Vestibular deficits in deaf children. Otolaryngol Head Neck Surg. (1996) 115:70–7. doi: 10.1016/S0194-5998(96)70139-0
91. Cushing SL, Papsin BC, Rutka JA, James AL, Gordon KA. Evidence of vestibular and balance dysfunction in children with profound sensorineural hearing loss using cochlear implants. Laryngoscope. (2008) 118:1814–23. doi: 10.1097/MLG.0b013e31817fadfa
92. Jacot E, Van Den Abbeele T, Debre HR, Wiener-Vacher SR. Vestibular impairments pre- and post-cochlear implant in children. Int J Pediatr Otorhinolaryngol. (2009) 73:209–17. doi: 10.1016/j.ijporl.2008.10.024
93. Coudert AVH, Ayari-Khalfallah S, Hermann R, Lina-Granade G, Truy E, Ionescu E. Vestibular assessment in cochlear implanted children: how to do? When to do? A review of literature. Curr Otorhinolaryngol Rep. (2017) 5:259–67. doi: 10.1007/s40136-017-0172-2
94. Janky KL, Rodriguez AI. Quantitative vestibular function testing in the pediatric population. Semin Hear. (2018) 39:257–74. doi: 10.1055/s-0038-1666817
95. Bowl MR, Dawson SJ. The mouse as a model for age-related hearing loss - a mini-review. Gerontology. (2015) 61:149–57. doi: 10.1159/000368399
96. Ohlemiller KK, Jones SM, Johnson KR. Application of mouse models to research in hearing and balance. J Assoc Res Otolaryngol. (2016) 17:493–523. doi: 10.1007/s10162-016-0589-1
97. Straka H, Zwergal A, Cullen KE. Vestibular animal models: contributions to understanding physiology and disease. J Neurol. (2016) 263(Suppl. 1):S10–23. doi: 10.1007/s00415-015-7909-y
98. Stone JS, Wisner SR, Bucks SA, Mellado Lagarde MM, Cox BC. Characterization of adult vestibular organs in 11 CreER mouse lines. J Assoc Res Otolaryngol. (2018) 19:381–99. doi: 10.1007/s10162-018-0676-6
99. Steel KP, Hardisty R. Assessing Hearing, Vision and Balance in Mice. What's Wrong With My Mouse? New Interplays Between Mouse Genes and Behavior. Washington, DC: Society for Neuroscience (1996). p. 26–38.
100. Iwashita M, Kanai R, Funabiki K, Matsuda K, Hirano T. Dynamic properties, interactions and adaptive modifications of vestibulo-ocular reflex and optokinetic response in mice. Neurosci Res. (2001) 39:299–311. doi: 10.1016/S0168-0102(00)00228-5
101. Migliaccio AA, Meierhofer R, Della Santina CC. Characterization of the 3D angular vestibulo-ocular reflex in C57BL6 mice. Exp Brain Res. (2011) 210:489–501. doi: 10.1007/s00221-010-2521-y
102. Jones SM, Subramanian G, Avniel W, Guo Y, Burkard RF, Jones TA. Stimulus and recording variables and their effects on mammalian vestibular evoked potentials. J Neurosci Methods. (2002) 118:23–31. doi: 10.1016/S0165-0270(02)00125-5
103. Jones SM, Jones TA. Genetics of peripheral vestibular dysfunction: lessons from mutant mouse strains. J Am Acad Audiol. (2014) 25:289–301. doi: 10.3766/jaaa.25.3.8
104. Brown DJ, Pastras CJ, Curthoys IS. Electrophysiological measurements of peripheral vestibular function-a review of electrovestibulography. Front Syst Neurosci. (2017) 11:34. doi: 10.3389/fnsys.2017.00034
105. Peusner KD, Bell NM, Hirsch JC, Beraneck M, Popratiloff A. Understanding the pathophysiology of congenital vestibular disorders: current challenges and future directions. Front Neurol. (2021) 12:708395. doi: 10.3389/fneur.2021.708395
106. Ogawa H, Suzutani T, Baba Y, Koyano S, Nozawa N, Ishibashi K, et al. Etiology of severe sensorineural hearing loss in children: independent impact of congenital cytomegalovirus infection and GJB2 mutations. J Infect Dis. (2007) 195:782–8. doi: 10.1086/511981
107. Bernard S, Wiener-Vacher S, Van Den Abbeele T, Teissier N. Vestibular disorders in children with congenital cytomegalovirus infection. Pediatrics. (2015) 136:e887–95. doi: 10.1542/peds.2015-0908
108. Teissier N, Delezoide AL, Mas AE, Khung-Savatovsky S, Bessieres B, Nardelli J, et al. Inner ear lesions in congenital cytomegalovirus infection of human fetuses. Acta Neuropathol. (2011) 122:763–74. doi: 10.1007/s00401-011-0895-y
109. Teissier N, Bernard S, Quesnel S, Van Den Abbeele T. Audiovestibular consequences of congenital cytomegalovirus infection. Eur Ann Otorhinolaryngol Head Neck Dis. (2016) 133:413–8. doi: 10.1016/j.anorl.2016.03.004
110. Worsoe L, Caye-Thomasen P, Brandt CT, Thomsen J, Ostergaard C. Factors associated with the occurrence of hearing loss after pneumococcal meningitis. Clin Infect Dis. (2010) 51:917–24. doi: 10.1086/656409
111. Rasmussen N, Johnsen NJ, Bohr VA. Otologic sequelae after pneumococcal meningitis: a survey of 164 consecutive cases with a follow-up of 94 survivors. Laryngoscope. (1991) 101:876–82. doi: 10.1288/00005537-199108000-00012
112. Strupp M, Feil K, Dieterich M, Brandt T. Bilateral vestibulopathy. Handb Clin Neurol. (2016) 137:235–40. doi: 10.1016/B978-0-444-63437-5.00017-0
113. Wang YP, Hsu WC, Young YH. Vestibular evoked myogenic potentials in acute acoustic trauma. Otol Neurotol. (2006) 27:956–61. doi: 10.1097/01.mao.0000231590.57348.4b
114. Stewart C, Yu Y, Huang J, Maklad A, Tang X, Allison J, et al. Effects of high intensity noise on the vestibular system in rats. Hear Res. (2016) 335:118–27. doi: 10.1016/j.heares.2016.03.002
115. Wang YP, Young YH. Vestibular-evoked myogenic potentials in chronic noise-induced hearing loss. Otolaryngol Head Neck Surg. (2007) 137:607–11. doi: 10.1016/j.otohns.2007.05.005
116. Enrietto JA, Jacobson KM, Baloh RW. Aging effects on auditory and vestibular responses: a longitudinal study. Am J Otolaryngol. (1999) 20:371–8. doi: 10.1016/S0196-0709(99)90076-5
117. Schuknecht HF, Gacek MR. Cochlear pathology in presbycusis. Ann Otol Rhinol Laryngol. (1993) 102:1–16. doi: 10.1177/00034894931020S101
118. Ciorba A, Hatzopoulos S, Bianchini C, Aimoni C, Skarzynski H, Skarzynski PH. Genetics of presbycusis and presbystasis. Int J Immunopathol Pharmacol. (2015) 28:29–35. doi: 10.1177/0394632015570819
119. Girasoli L, Cazzador D, Padoan R, Nardello E, Felicetti M, Zanoletti E, et al. Update on vertigo in autoimmune disorders, from diagnosis to treatment. J Immunol Res. (2018) 2018:5072582. doi: 10.1155/2018/5072582
120. Russo FY, Ralli M, De Seta D, Mancini P, Lambiase A, Artico, et al. Autoimmune vertigo: an update on vestibular disorders associated with autoimmune mechanisms. Immunol Res. (2018) 66:675–85. doi: 10.1007/s12026-018-9023-6
121. Ralli M, D'Aguanno V, Di Stadio A, De Virgilio A, Croce A, Longo L, et al. Audiovestibular symptoms in systemic autoimmune diseases. J Immunol Res. (2018) 2018:5798103. doi: 10.1155/2018/5798103
122. Cianfrone G, Pentangelo D, Cianfrone F, Mazzei F, Turchetta R, Orlando MP, et al. Pharmacological drugs inducing ototoxicity, vestibular symptoms and tinnitus: a reasoned and updated guide. Eur Rev Med Pharmacol Sci. (2011) 15:601–36.
123. Wu P, Wu X, Zhang C, Chen X, Huang Y, Li H. Hair cell protection from ototoxic drugs. Neural Plast. (2021) 2021:4909237. doi: 10.1155/2021/4909237
124. Halmagyi GM, Fattore CM, Curthoys IS, Wade S. Gentamicin vestibulotoxicity. Otolaryngol Head Neck Surg. (1994) 111:571–4. doi: 10.1177/019459989411100506
125. Selimoglu E, Kalkandelen S, Erdogan F. Comparative vestibulotoxicity of different aminoglycosides in the Guinea pigs. Yonsei Med J. (2003) 44:517–22. doi: 10.3349/ymj.2003.44.3.517
126. Jiang L, Zheng Z, He Y. Progress in protecting vestibular hair cells. Arch Toxicol. (2021) 95:2613–23. doi: 10.1007/s00204-021-03067-3
127. Niklasson M, Tham R, Larsby B, Eriksson B. Effects of toluene, styrene, trichloroethylene, and trichloroethane on the vestibulo-and opto-oculo motor system in rats. Neurotoxicol Teratol. (1993) 15:327–34. doi: 10.1016/0892-0362(93)90034-L
128. Hereditary Hearing loss Homepage. Available online at: http://www.uia.ac.be/dnalab/hhh/ (accessed November 16, 2021).
129. Martens S, Dhooge I, Dhondt C, Vanaudenaerde S, Sucaet M, Rombaut L, et al. Vestibular Infant Screening (VIS)-Flanders: results after 1.5 years of vestibular screening in hearing-impaired children. Sci Rep. (2020) 10:21011. doi: 10.1038/s41598-020-78049-z
130. Simon F, Roman S, Truy E, Barone P, Belmin J, Blanchet C, et al. Guidelines (short version) of the French Society of Otorhinolaryngology (SFORL) on pediatric cochlear implant indications. Eur Ann Otorhinolaryngol Head Neck Dis. (2019) 136:385–91. doi: 10.1016/j.anorl.2019.05.018
131. Frejo L, Giegling I, Teggi R, Lopez-Escamez JA, Rujescu D. Genetics of vestibular disorders: pathophysiological insights. J Neurol. (2016) 263(Suppl. 1):S45–53. doi: 10.1007/s00415-015-7988-9
132. Potter PK, Bowl MR, Jeyarajan P, Wisby L, Blease A, Goldsworthy ME, et al. Novel gene function revealed by mouse mutagenesis screens for models of age-related disease. Nat Commun. (2016) 7:12444. doi: 10.1038/ncomms12444
133. a.h.m.j.o. Available online at: http://hereditaryhearingloss.org/ (accessed November 16, 2021).
134. Lentz JJ, Jodelka FM, Hinrich AJ, McCaffrey KE, Farris HE, Spalitta MJ, et al. Rescue of hearing and vestibular function by antisense oligonucleotides in a mouse model of human deafness. Nat Med. (2013) 19:345–50. doi: 10.1038/nm.3106
135. Vijayakumar S, Depreux FF, Jodelka FM, Lentz JJ, Rigo F, Jones TA, et al. Rescue of peripheral vestibular function in Usher syndrome mice using a splice-switching antisense oligonucleotide. Hum Mol Genet. (2017) 26:3482–94. doi: 10.1093/hmg/ddx234
136. Donaldson TN, Jennings KT, Cherep LA, McNeela AM, Depreux FF, Jodelka FM, et al. Antisense oligonucleotide therapy rescues disruptions in organization of exploratory movements associated with Usher syndrome type 1C in mice. Behav Brain Res. (2018) 338:76–87. doi: 10.1016/j.bbr.2017.10.012
137. Lentz JJ, Pan B, Ponnath A, Tran CM, Nist-Lund C, Galvin A, et al. Direct delivery of antisense oligonucleotides to the middle and inner ear improves hearing and balance in Usher Mice. Mol Ther. (2020) 28:2662–76. doi: 10.1016/j.ymthe.2020.08.002
138. Wang L, Kempton JB, Jiang H, Jodelka FM, Brigande AM, Dumont RA, et al. Fetal antisense oligonucleotide therapy for congenital deafness and vestibular dysfunction. Nucleic Acids Res. (2020) 48:5065–80. doi: 10.1093/nar/gkaa194
139. Pan B, Askew C, Galvin A, Heman-Ackah S, Asai Y, Indzhykulian AA, et al. Gene therapy restores auditory and vestibular function in a mouse model of Usher syndrome type 1c. Nat Biotechnol. (2017) 35:264–72. doi: 10.1038/nbt.3801
140. Wu X, Zhang L, Li Y, Zhang W, Wang J, Cai C, et al. Gene therapy via canalostomy approach preserves auditory and vestibular functions in a mouse model of Jervell and Lange-Nielsen syndrome type 2. Nat Commun. (2021) 12:697. doi: 10.1038/s41467-020-20808-7
141. Gyorgy B, Sage C, Indzhykulian AA, Scheffer DI, Brisson AR, Tan S, et al. Rescue of hearing by gene delivery to inner-ear hair cells using exosome-associated AAV. Mol Ther. (2017) 25:379–91. doi: 10.1016/j.ymthe.2016.12.010
142. Kim MA, Kim SH, Ryu N, Ma JH, Kim YR, Jung J, et al. Gene therapy for hereditary hearing loss by SLC26A4 mutations in mice reveals distinct functional roles of pendrin in normal hearing. Theranostics. (2019) 9:7184–99. doi: 10.7150/thno.38032
143. Takeda H, Miwa T, Kim MY, Choi BY, Orita Y, Minoda R. Prenatal electroporation-mediated gene transfer restores Slc26a4 knock-out mouse hearing and vestibular function. Sci Rep. (2019) 9:17979. doi: 10.1038/s41598-019-54262-3
144. Emptoz A, Michel V, Lelli A, Akil O, Boutet de Monvel J, Lahlou G, et al. Local gene therapy durably restores vestibular function in a mouse model of Usher syndrome type 1G. Proc Natl Acad Sci USA. (2017) 114:9695–700. doi: 10.1073/pnas.1708894114
145. Nist-Lund CA, Pan B, Patterson A, Asai Y, Chen T, Zhou W, et al. Improved TMC1 gene therapy restores hearing and balance in mice with genetic inner ear disorders. Nat Commun. (2019) 10:236. doi: 10.1038/s41467-018-08264-w
146. Isgrig K, Shteamer JW, Belyantseva IA, Drummond MC, Fitzgerald TS, Vijayakumar S, et al. Gene therapy restores balance and auditory functions in a mouse model of Usher Syndrome. Mol Ther. (2017) 25:780–91. doi: 10.1016/j.ymthe.2017.01.007
147. Staecker H, Praetorius M, Baker K, Brough DE. Vestibular hair cell regeneration and restoration of balance function induced by math1 gene transfer. Otol Neurotol. (2007) 28:223–31. doi: 10.1097/MAO.0b013e31802b3225
148. Schlecker C, Praetorius M, Brough DE, Presler RG Jr, Hsu C, et al. Selective atonal gene delivery improves balance function in a mouse model of vestibular disease. Gene Ther. (2011) 18:884–90. doi: 10.1038/gt.2011.33
149. Pfannenstiel SC, Praetorius M, Plinkert PK, Brough DE, Staecker H. Bcl-2 gene therapy prevents aminoglycoside-induced degeneration of auditory and vestibular hair cells. Audiol Neurootol. (2009) 14:254–66. doi: 10.1159/000192953
150. Jung JY, Avenarius MR, Adamsky S, Alpert E, Feinstein E, Raphael Y. siRNA targeting Hes5 augments hair cell regeneration in aminoglycoside-damaged mouse utricle. Mol Ther. (2013) 21:834–41. doi: 10.1038/mt.2013.18
151. Xu JC, Huang DL, Hou ZH, Guo WW, Sun JH, Zhao LD, et al. Type I hair cell regeneration induced by Math1 gene transfer following neomycin ototoxicity in rat vestibular sensory epithelium. Acta Otolaryngol. (2012) 132:819–28. doi: 10.3109/00016489.2012.673233
152. Suzuki M, Yagi M, Brown JN, Miller AL, Miller JM, Raphael Y. Effect of transgenic GDNF expression on gentamicin-induced cochlear and vestibular toxicity. Gene Ther. (2000) 7:1046–54. doi: 10.1038/sj.gt.3301180
153. Sollner C, Rauch GJ, Siemens J, Geisler R, Schuster SC, Muller U, et al. Mutations in cadherin 23 affect tip links in zebrafish sensory hair cells. Nature. (2004) 428:955–9. doi: 10.1038/nature02484
154. Gopal SR, Chen DH, Chou SW, Zang J, Neuhauss SC, Stepanyan R, et al. Zebrafish models for the mechanosensory hair cell dysfunction in Usher Syndrome 3 reveal that clarin-1 is an essential hair bundle protein. J Neurosci. (2015) 35:10188–201. doi: 10.1523/JNEUROSCI.1096-15.2015
155. Vona B, Mazaheri N, Lin SJ, Dunbar LA, Maroofian R, Azaiez H, et al. A biallelic variant in CLRN2 causes non-syndromic hearing loss in humans. Hum Genet. (2021) 140:915–31. doi: 10.1007/s00439-020-02254-z
156. Sheets L, Holmgren M, Kindt KS. How zebrafish can drive the future of genetic-based hearing and balance research. J Assoc Res Otolaryngol. (2021) 22:215–35. doi: 10.1007/s10162-021-00798-z
157. Toms M, Dubis AM, de Vrieze E, Tracey-White D, Mitsios A, Hayes M, et al. Clinical and preclinical therapeutic outcome metrics for USH2A-related disease. Hum Mol Genet. (2020) 29:1882–99. doi: 10.1093/hmg/ddaa004
158. Bonnet C, El-Amraoui A. Usher syndrome (sensorineural deafness and retinitis pigmentosa): pathogenesis, molecular diagnosis and therapeutic approaches. Curr Opin Neurol. (2012) 25:42–9. doi: 10.1097/WCO.0b013e32834ef8b2
159. Geleoc GGS, El-Amraoui A. Disease mechanisms and gene therapy for Usher syndrome. Hear Res. (2020) 394:107932. doi: 10.1016/j.heares.2020.107932
160. Kazmierczak P, Muller U. Sensing sound: molecules that orchestrate mechanotransduction by hair cells. Trends Neurosci. (2012) 35:220–9. doi: 10.1016/j.tins.2011.10.007
161. Grillet N, Xiong W, Reynolds A, Kazmierczak P, Sato T, Lillo C, et al. Harmonin mutations cause mechanotransduction defects in cochlear hair cells. Neuron. (2009) 62:375–87. doi: 10.1016/j.neuron.2009.04.006
162. Michalski N, Michel V, Caberlotto E, Lefevre GM, van Aken AF, Tinevez JY, et al. Harmonin-b, an actin-binding scaffold protein, is involved in the adaptation of mechanoelectrical transduction by sensory hair cells. Pflügers Arch. (2009) 459:115–30. doi: 10.1007/s00424-009-0711-x
163. Mathur P, Yang J. Usher syndrome: hearing loss, retinal degeneration and associated abnormalities. Biochim Biophys Acta. (2015) 1852:406–20. doi: 10.1016/j.bbadis.2014.11.020
164. Adato A, Lefevre G, Delprat B, Michel V, Michalski N, Chardenoux S, et al. Usherin, the defective protein in Usher syndrome type IIA, is likely to be a component of interstereocilia ankle links in the inner ear sensory cells. Hum Mol Genet. (2005) 14:3921–32. doi: 10.1093/hmg/ddi416
165. Michalski N, Michel V, Bahloul A, Lefèvre G, Barral J, Yagi H, et al. Molecular characterization of the ankle link complex in cochlear hair cells and its role in the hair bundle functioning. J Neurosci. (2007) 27:6478–88. doi: 10.1523/JNEUROSCI.0342-07.2007
166. Mburu P, Mustapha M, Varela A, Weil D, El-Amraoui A, Holme RH, et al. Defects in whirlin, a PDZ domain molecule involved in stereocilia elongation, cause deafness in the whirler mouse and families with DFNB31. Nat Genet. (2003) 34:421–8. doi: 10.1038/ng1208
167. Ebermann I, Scholl HP, Charbel Issa P, Becirovic E, Lamprecht J, Jurklies B, et al. A novel gene for Usher syndrome type 2: mutations in the long isoform of whirlin are associated with retinitis pigmentosa and sensorineural hearing loss. Hum Genet. (2007) 121:203–11. doi: 10.1007/s00439-006-0304-0
168. Delprat B, Michel V, Goodyear R, Yamasaki Y, Michalski N, El-Amraoui A, et al. Myosin XVa and whirlin, two deafness gene products required for hair bundle growth, are located at the stereocilia tips and interact directly. Hum Mol Genet. (2005) 14:401–10. doi: 10.1093/hmg/ddi036
169. Belyantseva IA, Boger ET, Naz S, Frolenkov GI, Sellers JR, Ahmed ZM, et al. Myosin-XVa is required for tip localization of whirlin and differential elongation of hair-cell stereocilia. Nat Cell Biol. (2005) 7:148–56. doi: 10.1038/ncb1219
170. Fettiplace R, Kim KX. The physiology of mechanoelectrical transduction channels in hearing. Physiol Rev. (2014) 94:951–86. doi: 10.1152/physrev.00038.2013
171. Mathur PD, Yang J. Usher syndrome and non-syndromic deafness: functions of different whirlin isoforms in the cochlea, vestibular organs, and retina. Hear Res. (2019) 375:14–24. doi: 10.1016/j.heares.2019.02.007
172. Ebrahim S, Ingham NJ, Lewis MA, Rogers MJC, Cui R, Kachar B, et al. Alternative splice forms influence functions of whirlin in mechanosensory hair cell stereocilia. Cell Rep. (2016) 15:935–43. doi: 10.1016/j.celrep.2016.03.081
173. Stemerdink M, Garcia-Bohorquez B, Schellens R, Garcia-Garcia G, Van Wijk E, Millan JM. Genetics, pathogenesis and therapeutic developments for Usher syndrome type 2. Hum Genet. (2021). doi: 10.1007/s00439-021-02324-w. [Epub ahead of print].
174. Adato A, Vreugde S, Joensuu T, Avidan N, Hamalainen R, Belenkiy O, et al. USH3A transcripts encode clarin-1, a four-transmembrane-domain protein with a possible role in sensory synapses. Eur J Hum Genet. (2002) 10:339–50. doi: 10.1038/sj.ejhg.5200831
175. Geng R, Geller SF, Hayashi T, Ray CA, Reh TA, Bermingham-McDonogh O, et al. Usher syndrome IIIA gene clarin-1 is essential for hair cell function and associated neural activation. Hum Mol Genet. (2009) 18:2748–60. doi: 10.1093/hmg/ddp210
176. Geng R, Melki S, Chen DH, Tian G, Furness DN, Oshima-Takago T, et al. The mechanosensory structure of the hair cell requires clarin-1, a protein encoded by Usher syndrome III causative gene. J Neurosci. (2012) 32:9485–98. doi: 10.1523/JNEUROSCI.0311-12.2012
177. Dulon D, Papal S, Patni P, Cortese M, Vincent PF, Tertrais M, et al. Clarin-1 gene transfer rescues auditory synaptopathy in model of Usher syndrome. J Clin Invest. (2018) 128:3382–401. doi: 10.1172/JCI94351
178. Goodyear RJ, Legan PK, Wright MB, Marcotti W, Oganesian A, Coats SA, et al. A receptor-like inositol lipid phosphatase is required for the maturation of developing cochlear hair bundles. J Neurosci. (2003) 23:9208–19. doi: 10.1523/JNEUROSCI.23-27-09208.2003
179. Goodyear RJ, Jones SM, Sharifi L, Forge A, Richardson GP. Hair bundle defects and loss of function in the vestibular end organs of mice lacking the receptor-like inositol lipid phosphatase PTPRQ. J Neurosci. (2012) 32:2762–72. doi: 10.1523/JNEUROSCI.3635-11.2012
180. Salles FT, Andrade LR, Tanda S, Grati M, Plona KL, Gagnon LH, et al. CLIC5 stabilizes membrane-actin filament linkages at the base of hair cell stereocilia in a molecular complex with radixin, taperin, myosin VI. Cytoskeleton. (2014) 71:61–78. doi: 10.1002/cm.21159
181. Jones SM, Johnson KR, Yu H, Erway LC, Alagramam KN, Pollak N, et al. A quantitative survey of gravity receptor function in mutant mouse strains. J Assoc Res Otolaryngol. (2005) 6:297–310. doi: 10.1007/s10162-005-0009-4
182. Pan B, Geleoc GS, Asai Y, Horwitz GC, Kurima K, Ishikawa K, et al. TMC1 and TMC2 are components of the mechanotransduction channel in hair cells of the mammalian inner ear. Neuron. (2013) 79:504–15. doi: 10.1016/j.neuron.2013.06.019
183. Kawashima Y, Kurima K, Pan B, Griffith AJ, Holt JR. Transmembrane channel-like (TMC) genes are required for auditory and vestibular mechanosensation. Pflugers Arch. (2015) 467:85–94. doi: 10.1007/s00424-014-1582-3
184. Corey DP, Akyuz N, Holt JR. Function and dysfunction of TMC channels in inner ear hair cells. Cold Spring Harb Perspect Med. (2019) 9:a033506. doi: 10.1101/cshperspect.a033506
185. Cunningham CL, Muller U. Molecular structure of the hair cell mechanoelectrical transduction complex. Cold Spring Harb Perspect Med. (2019) 9:a033167. doi: 10.1101/cshperspect.a033167
186. Holt JR, Pan B, Koussa MA, Asai Y. TMC function in hair cell transduction. Hear Res. (2014) 311:17–24. doi: 10.1016/j.heares.2014.01.001
187. Pan B, Akyuz N, Liu XP, Asai Y, Nist-Lund C, Kurima K, et al. TMC1 forms the pore of mechanosensory transduction channels in vertebrate inner ear hair cells. Neuron. (2018) 99:736–53 e6. doi: 10.1016/j.neuron.2018.07.033
188. Liang X, Qiu X, Dionne G, Cunningham CL, Pucak ML, Peng G, et al. CIB2 and CIB3 are auxiliary subunits of the mechanotransduction channel of hair cells. Neuron. (2021) 109:2131–49 e15. doi: 10.1016/j.neuron.2021.05.007
189. Zheng W, Holt JR. The mechanosensory transduction machinery in inner ear hair cells. Annu Rev Biophys. (2021) 50:31–51. doi: 10.1146/annurev-biophys-062420-081842
190. Xiong W, Grillet N, Elledge HM, Wagner TF, Zhao B, Johnson KR, et al. TMHS is an integral component of the mechanotransduction machinery of cochlear hair cells. Cell. (2012) 151:1283–95. doi: 10.1016/j.cell.2012.10.041
191. Zhao B, Wu Z, Grillet N, Yan L, Xiong W, Harkins-Perry S, et al. TMIE is an essential component of the mechanotransduction machinery of cochlear hair cells. Neuron. (2014) 84:954–67. doi: 10.1016/j.neuron.2014.10.041
192. Riazuddin S, Belyantseva IA, Giese AP, Lee K, Indzhykulian AA, Nandamuri SP, et al. Alterations of the CIB2 calcium- and integrin-binding protein cause Usher syndrome type 1J and nonsyndromic deafness DFNB48. Nat Genet. (2012) 44:1265–71. doi: 10.1038/ng.2426
193. Michel V, Booth KT, Patni P, Cortese M, Azaiez H, Bahloul A, et al. CIB2, defective in isolated deafness, is key for auditory hair cell mechanotransduction and survival. EMBO Mol Med. (2017) 9:1711–31. doi: 10.15252/emmm.201708087
194. Wang Y, Li J, Yao X, Li W, Du H, Tang M, et al. Loss of CIB2 causes profound hearing loss and abolishes mechanoelectrical transduction in mice. Front Mol Neurosci. (2017) 10:401. doi: 10.3389/fnmol.2017.00401
195. Booth KT, Kahrizi K, Babanejad M, Daghagh H, Bademci G, Arzhangi S, et al. Variants in CIB2 cause DFNB48 and not USH1J. Clin Genet. (2018) 93:812–21. doi: 10.1111/cge.13170
196. Giese APJ, Tang YQ, Sinha GP, Bowl MR, Goldring AC, Parker A, et al. CIB2 interacts with TMC1 and TMC2 and is essential for mechanotransduction in auditory hair cells. Nat Commun. (2017) 8:43. doi: 10.1038/s41467-017-00061-1
197. Griffith AJ, Wangemann P. Hearing loss associated with enlargement of the vestibular aqueduct: mechanistic insights from clinical phenotypes, genotypes, mouse models. Hear Res. (2011) 281:11–7. doi: 10.1016/j.heares.2011.05.009
198. Honda K, Griffith AJ. Genetic architecture and phenotypic landscape of SLC26A4-related hearing loss. Hum Genet. (2021). doi: 10.1007/s00439-021-02311-1. [Epub ahead of print].
199. Everett LA, Belyantseva IA, Noben-Trauth K, Cantos R, Chen A, Thakkar SI, et al. Targeted disruption of mouse Pds provides insight about the inner-ear defects encountered in Pendred syndrome. Hum Mol Genet. (2001) 10:153–61. doi: 10.1093/hmg/10.2.153
200. Dror AA, Taiber S, Sela E, Handzel O, Avraham KB. A mouse model for benign paroxysmal positional vertigo with genetic predisposition for displaced otoconia. Genes Brain Behav. (2020) 19:e12635. doi: 10.1111/gbb.12635
201. Lundberg YW, Xu Y, Thiessen KD, Kramer KL. Mechanisms of otoconia and otolith development. Dev Dyn. (2015) 244:239–53. doi: 10.1002/dvdy.24195
202. Lundberg YW, Zhao X, Yamoah EN. Assembly of the otoconia complex to the macular sensory epithelium of the vestibule. Brain Res. (2006) 1091:47–57. doi: 10.1016/j.brainres.2006.02.083
203. Verpy E, Leibovici M, Petit C. Characterization of otoconin-95, the major protein of murine otoconia, provides insights into the formation of these inner ear biominerals. Proc Natl Acad Sci USA. (1999) 96:529–34. doi: 10.1073/pnas.96.2.529
204. Zhao X, Yang H, Yamoah EN, Lundberg YW. Gene targeting reveals the role of Oc90 as the essential organizer of the otoconial organic matrix. Dev Biol. (2007) 304:508–24. doi: 10.1016/j.ydbio.2007.01.013
205. Stooke-Vaughan GA, Obholzer ND, Baxendale S, Megason SG, Whitfield TT. Otolith tethering in the zebrafish otic vesicle requires Otogelin and alpha-Tectorin. Development. (2015) 142:1137–45. doi: 10.1242/dev.116632
206. El-Amraoui A, Cohen-Salmon M, Petit C, Simmler MC. Spatiotemporal expression of otogelin in the developing and adult mouse inner ear. Hear Res. (2001) 158:151–9. doi: 10.1016/S0378-5955(01)00312-4
207. Cohen-Salmon M, El-Amraoui A, Leibovici M, Petit C. Otogelin: a glycoprotein specific to the acellular membranes of the inner ear. Proc Natl Acad Sci USA. (1997) 94:14450–5. doi: 10.1073/pnas.94.26.14450
208. Goodyear RJ, Richardson GP. Extracellular matrices associated with the apical surfaces of sensory epithelia in the inner ear: molecular and structural diversity. J Neurobiol. (2002) 53:212–27. doi: 10.1002/neu.10097
209. Zwaenepoel I, Mustapha M, Leibovici M, Verpy E, Goodyear R, Liu XZ, et al. Otoancorin, an inner ear protein restricted to the interface between the apical surface of sensory epithelia and their overlying acellular gels, is defective in autosomal recessive deafness DFNB22. Proc Natl Acad Sci USA. (2002) 99:6240–5. doi: 10.1073/pnas.082515999
210. Simmler MC, Cohen-Salmon M, El-Amraoui A, Guillaud L, Benichou JC, Petit C, et al. Targeted disruption of otog results in deafness and severe imbalance. Nat Genet. (2000) 24:139–43. doi: 10.1038/72793
211. Petit C, Richardson G. Linking deafness genes to hair-bundle development and function. Nat Neurosci. (2009) 12:703–10. doi: 10.1038/nn.2330
212. Tian C, Johnson KR, Lett JM, Voss R, Salt AN, Hartsock JJ, et al. CACHD1-deficient mice exhibit hearing and balance deficits associated with a disruption of calcium homeostasis in the inner ear. Hear Res. (2021) 409:108327. doi: 10.1016/j.heares.2021.108327
213. Kim E, Hyrc KL, Speck J, Lundberg YW, Salles FT, Kachar B, et al. Regulation of cellular calcium in vestibular supporting cells by otopetrin 1. J Neurophysiol. (2010) 104:3439–50. doi: 10.1152/jn.00525.2010
214. Brown SDM. Advances in mouse genetics for the study of human disease. Hum Mol Genet. (2021) 30:274–84. doi: 10.1093/hmg/ddab153
215. Birling MC, Yoshiki A, Adams DJ, Ayabe S, Beaudet AL, Bottomley J, et al. A resource of targeted mutant mouse lines for 5,061 genes. Nat Genet. (2021) 53:416–9. doi: 10.1038/s41588-021-00825-y
216. Bowl MR, Brown SDM. Genetic landscape of auditory dysfunction. Hum Mol Genet. (2018) 27:R130–5. doi: 10.1093/hmg/ddy158
217. Bowl MR, Simon MM, Ingham NJ, Greenaway S, Santos L, Cater H, et al. A large scale hearing loss screen reveals an extensive unexplored genetic landscape for auditory dysfunction. Nat Commun. (2017) 8:886. doi: 10.1038/s41467-017-00595-4
218. Ingham NJ, Pearson SA, Vancollie VE, Rook V, Lewis MA, Chen J, et al. Mouse screen reveals multiple new genes underlying mouse and human hearing loss. PLoS Biol. (2019) 17:e3000194. doi: 10.1371/journal.pbio.3000194
219. Soto E, Vega R, Sesena E. Neuropharmacological basis of vestibular system disorder treatment. J Vestib Res. (2013) 23:119–37. doi: 10.3233/VES-130494
220. Soto E, Vega R. Neuropharmacology of vestibular system disorders. Curr Neuropharmacol. (2010) 8:26–40. doi: 10.2174/157015910790909511
221. Pfaltz CR, Kamath R. Central compensation of vestibular dysfunction. I. Peripheral lesions. Pract Otorhinolaryngol. (1970) 32:335–49. doi: 10.1159/000274957
222. Smith PF, Curthoys IS. Mechanisms of recovery following unilateral labyrinthectomy: a review. Brain Res Brain Res Rev. (1989) 14:155–80. doi: 10.1016/0165-0173(89)90013-1
223. Curthoys IS. Vestibular compensation and substitution. Curr Opin Neurol. (2000) 13:27–30. doi: 10.1097/00019052-200002000-00006
224. Lacour M, Helmchen C, Vidal PP. Vestibular compensation: the neuro-otologist's best friend. J Neurol. (2016) 263(Suppl. 1):S54–64. doi: 10.1007/s00415-015-7903-4
225. Han BI, Song HS, Kim JS. Vestibular rehabilitation therapy: review of indications, mechanisms, key exercises. J Clin Neurol. (2011) 7:184–96. doi: 10.3988/jcn.2011.7.4.184
226. Deveze A, Bernard-Demanze L, Xavier F, Lavieille JP, Elziere M. Vestibular compensation and vestibular rehabilitation. Current concepts and new trends. Neurophysiol Clin. (2014) 44:49–57. doi: 10.1016/j.neucli.2013.10.138
227. Hall CD, Herdman SJ, Whitney SL, Cass SP, Clendaniel RA, Fife TD, et al. Vestibular rehabilitation for peripheral vestibular hypofunction: an evidence-based clinical practice guideline: from The American Physical Therapy Association Neurology Section. J Neurol Phys Ther. (2016) 40:124–55. doi: 10.1097/NPT.0000000000000120
228. Volkenstein S, Dazert S. Recent surgical options for vestibular vertigo. GMS Curr Top Otorhinolaryngol Head Neck Surg. (2017) 16:Doc01. doi: 10.3205/cto000140
229. Guyot JP, Sigrist A, Pelizzone M, Feigl GC, Kos MI. Eye movements in response to electrical stimulation of the lateral and superior ampullary nerves. Ann Otol Rhinol Laryngol. (2011) 120:81–7. doi: 10.1177/000348941112000202
230. Perez Fornos A, Guinand N, van de Berg R, Stokroos R, Micera S, Kingma H, et al. Artificial balance: restoration of the vestibulo-ocular reflex in humans with a prototype vestibular neuroprosthesis. Front Neurol. (2014) 5:66. doi: 10.3389/fneur.2014.00066
231. Guinand N, Van de Berg R, Cavuscens S, Ranieri M, Schneider E, Lucieer F, et al. The video head impulse test to assess the efficacy of vestibular implants in humans. Front Neurol. (2017) 8:600. doi: 10.3389/fneur.2017.00600
232. Guinand N, Van de Berg R, Cavuscens S, Stokroos R, Ranieri M, Pelizzone M, et al. Restoring visual acuity in dynamic conditions with a vestibular implant. Front Neurosci. (2016) 10:577. doi: 10.3389/fnins.2016.00577
233. Boutros PJ, Schoo DP, Rahman M, Valentin NS, Chow MR, Ayiotis AI, et al. Continuous vestibular implant stimulation partially restores eye-stabilizing reflexes. JCI Insight. (2019) 4:e128397. doi: 10.1172/jci.insight.128397
234. Chow MR, Ayiotis AI, Schoo DP, Gimmon Y, Lane KE, Morris BJ, et al. Posture, gait, quality of life, and hearing with a vestibular implant. N Engl J Med. (2021) 384:521–32. doi: 10.1056/NEJMoa2020457
235. Rubinstein JT, Bierer S, Kaneko C, Ling L, Nie K, Oxford T, et al. Implantation of the semicircular canals with preservation of hearing and rotational sensitivity: a vestibular neurostimulator suitable for clinical research. Otol Neurotol. (2012) 33:789–96. doi: 10.1097/MAO.0b013e318254ec24
236. Phillips JO, Ling L, Nie K, Jameyson E, Phillips CM, Nowack AL, et al. Vestibular implantation and longitudinal electrical stimulation of the semicircular canal afferents in human subjects. J Neurophysiol. (2015) 113:3866–92. doi: 10.1152/jn.00171.2013
237. Rubinstein JT, Ling L, Nowack A, Nie K, Phillips JO. Results from a second-generation vestibular implant in human subjects: diagnosis may impact electrical sensitivity of vestibular afferents. Otol Neurotol. (2020) 41:68–77. doi: 10.1097/MAO.0000000000002463
238. Ramos de Miguel A, Falcon Gonzalez JC, Ramos Macias A. Vestibular response to electrical stimulation of the otolith organs. Implications in the development of a vestibular implant for the improvement of the sensation of gravitoinertial accelerations. J Int Adv Otol. (2017) 13:154–61. doi: 10.5152/iao.2017.4216
239. Sayyid ZN, Kim GS, Cheng AG. Molecular therapy for genetic and degenerative vestibular disorders. Curr Opin Otolaryngol Head Neck Surg. (2018) 26:307–11. doi: 10.1097/MOO.0000000000000477
240. Askew C, Chien WW. Adeno-associated virus gene replacement for recessive inner ear dysfunction: progress and challenges. Hear Res. (2020) 394:107947. doi: 10.1016/j.heares.2020.107947
241. Geleoc GS, Holt JR. Sound strategies for hearing restoration. Science. (2014) 344:1241062. doi: 10.1126/science.1241062
242. Bankoti K, Generotti C, Hwa T, Wang L, O'Malley BW Jr, et al. Advances and challenges in adeno-associated viral inner-ear gene therapy for sensorineural hearing loss. Mol Ther Methods Clin Dev. (2021) 21:209–36. doi: 10.1016/j.omtm.2021.03.005
243. Botto C, Dalkara D, El-Amraoui A. Progress in gene editing tools and their potential for correcting mutations underlying hearing and vision loss. Front Gene Edit. (2021) 3:737632. doi: 10.3389/fgeed.2021.737632
244. Gyorgy B, Nist-Lund C, Pan B, Asai Y, Karavitaki KD, Kleinstiver BP, et al. Allele-specific gene editing prevents deafness in a model of dominant progressive hearing loss. Nat Med. (2019) 25:1123–30. doi: 10.1038/s41591-019-0500-9
245. Omichi R, Shibata SB, Morton CC, Smith RJH. Gene therapy for hearing loss. Hum Mol Genet. (2019) 28:R65–79. doi: 10.1093/hmg/ddz129
246. Niggemann P, Gyorgy B, Chen ZY. Genome and base editing for genetic hearing loss. Hear Res. (2020) 394:107958. doi: 10.1016/j.heares.2020.107958
247. Taura A, Nakashima N, Ohnishi H, Nakagawa T, Funabiki K, Ito J, et al. Regenerative therapy for vestibular disorders using human induced pluripotent stem cells (iPSCs): neural differentiation of human iPSC-derived neural stem cells after in vitro transplantation into mouse vestibular epithelia. Acta Otolaryngol. (2016) 136:999–1005. doi: 10.1080/00016489.2016.1183169
248. Koehler KR, Nie J, Longworth-Mills E, Liu XP, Lee J, Holt JR, et al. Generation of inner ear organoids containing functional hair cells from human pluripotent stem cells. Nat Biotechnol. (2017) 35:583–9. doi: 10.1038/nbt.3840
249. Roccio M, Edge ASB. Inner ear organoids: new tools to understand neurosensory cell development, degeneration and regeneration. Development. (2019) 146:dev177188. doi: 10.1242/dev.177188
250. Kubota M, Scheibinger M, Jan TA, Heller S. Greater epithelial ridge cells are the principal organoid-forming progenitors of the mouse cochlea. Cell Rep. (2021) 34:108646. doi: 10.1016/j.celrep.2020.108646
251. Stojkovic M, Han D, Jeong M, Stojkovic P, Stankovic KM. Human induced pluripotent stem cells and CRISPR/Cas-mediated targeted genome editing: platforms to tackle sensorineural hearing loss. Stem Cells. (2021) 39:673–96. doi: 10.1002/stem.3353
252. van der Valk WH, Steinhart MR, Zhang J, Koehler KR. Building inner ears: recent advances and future challenges for in vitro organoid systems. Cell Death Differ. (2021) 28:24–34. doi: 10.1038/s41418-020-00678-8
253. Kamogashira T, Fujimoto C, Kinoshita M, Kikkawa Y, Yamasoba T, Iwasaki S. Prediction of vestibular dysfunction by applying machine learning algorithms to postural instability. Front Neurol. (2020) 11:7. doi: 10.3389/fneur.2020.00007
254. Tama BA, Kim DH, Kim G, Kim SW, Lee S. Recent advances in the application of artificial intelligence in otorhinolaryngology-head and neck surgery. Clin Exp Otorhinolaryngol. (2020) 13:326–39. doi: 10.21053/ceo.2020.00654
255. Starkov D, Strupp M, Pleshkov M, Kingma H, van de Berg R. Diagnosing vestibular hypofunction: an update. J Neurol. (2021) 268:377–85. doi: 10.1007/s00415-020-10139-4
256. Landegger LD, Pan B, Askew C, Wassmer SJ, Gluck SD, Galvin A, et al. A synthetic AAV vector enables safe and efficient gene transfer to the mammalian inner ear. Nat Biotechnol. (2017) 35:280–4. doi: 10.1038/nbt.3781
Keywords: balance disorders, vestibulopathy, rare diseases, animal models, pathophysiology and gene therapy, deafness
Citation: Maudoux A, Vitry S and El-Amraoui A (2022) Vestibular Deficits in Deafness: Clinical Presentation, Animal Modeling, and Treatment Solutions. Front. Neurol. 13:816534. doi: 10.3389/fneur.2022.816534
Received: 16 November 2021; Accepted: 23 February 2022;
Published: 04 April 2022.
Edited by:
Kenna Peusner, George Washington University, United StatesReviewed by:
James Phillips, University of Washington, United StatesCopyright © 2022 Maudoux, Vitry and El-Amraoui. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Sandrine Vitry, c2FuZHJpbmUudml0cnlAcGFzdGV1ci5mcg==; Aziz El-Amraoui, YXppei5lbC1hbXJhb3VpQHBhc3RldXIuZnI=
†These authors share senior authorship
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.