 Alessandro Simonati
Alessandro Simonati Ruth E. Williams
Ruth E. Williams
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Neurol., 11 March 2022
Sec. Pediatric Neurology
Volume 13 - 2022 | https://doi.org/10.3389/fneur.2022.811686
This article is part of the Research TopicNeuronal Ceroid Lipofuscinosis: a Multidisciplinary UpdateView all 12 articles
The main aim of this review is to summarize the current state-of-art in the field of childhood Neuronal Ceroid Lipofuscinosis (NCL), a group of rare neurodegenerative disorders. These are genetic diseases associated with the formation of toxic endo-lysosomal storage. Following a brief historical review of the evolution of NCL definition, a clinically-oriented approach is used describing how the early symptoms and signs affecting motor, visual, cognitive domains, and including seizures, may lead clinicians to a rapid molecular diagnosis, avoiding the long diagnostic odyssey commonly observed. We go on to focus on recent advances in NCL research and summarize contributions to knowledge of the pathogenic mechanisms underlying NCL. We describe the large variety of experimental models which have aided this research, as well as the most recent technological developments which have shed light on the main mechanisms involved in the cellular pathology, such as apoptosis and autophagy. The search for innovative therapies is described. Translation of experimental data into therapeutic approaches is being established for several of the NCLs, and one drug is now commercially available. Lastly, we show the importance of palliative care and symptomatic treatments which are still the main therapeutic interventions.
The Neuronal Ceroid Lipofuscinoses (NCL) are neurodegenerative disorders, mostly of childhood onset. They form a heterogeneous group of lysosomal storage diseases (LSD) mainly affecting brain and retina (1). They are genetic disorders, and the first description of putative juvenile NCL was of four siblings in Norway with progressive visual loss, cognitive decline, seizures and premature death. This report remained unnoticed until 150 years later (2).
Cases of progressive visual loss with cognitive decline of infantile and/or childhood onset and a fatal outcome were grouped around the turn of the 19th century under the term “amaurotic familial idiocy”, coined by the American neurologist Sachs (3), and included the ocular manifestations described by the British ophthalmologist Tay (4). Several familial cases were described from different European countries. The contribution of neuropathology further characterized this group of diseases by describing the topography of brain abnormalities and the selective involvement of the cerebral and cerebellar cortices and of subcortical gray nuclei (5–9). Adult onset cases were reported by Kufs (10). The common appearance of swollen cerebral neurons (as well as retinal ganglion cells), whose topology was distorted because of the cytoplasmic engulfment with granular materials with similar staining properties was also described (11). Batten (12) and Spielmeyer (8) went on to outline some differences in the features and distribution of storage material between these entities, allowing division of the amaurotic familial idiocies into different clinical entities with different genetic backgrounds (13), specific biochemical properties (14, 15), selected neuropathological features (16), and eventually genetic markers toward the end of the last century. This was the beginnings of the classification and nosography of so-called storage diseases.
NCL are currently grouped under two major eponyms, Batten disease and Kufs disease. Batten disease refers to childhood NCLs, regardless the age of onset, whereas the term Kufs disease is assigned to the two major phenotypes of adult onset NCL [Kufs A and B (17)]. NCL definition relies on pathological criteria: the presence of autofluorescent lipofuscin and the characteristic cytosomes. Zeman and colleagues introduced the term “Neuronal Ceroid Lipofuscinosis” based on the histochemical and ultrastructural features (18). The identification of such markers allowed the NCLs to be further characterized and distinguished from other “amaurotic familial idiocies”, such as gangliosidoses (19).
Intracytoplasmic accumulation of autofluorescent material is a pathogenetic hallmark of the NCLs, and it is ascribed to the abnormal storage of ceroid, a pathologically derived material, with similar biochemical properties to lipofuscin, the “aging pigment” (20). The abnormal storage material is embedded within lysosomes, and its ultrastructural features probably reflect the chemical composition of the storage and the related aggregates (16). The biochemical composition of the storage is still only partially defined. A main component (subunit c of the mitochondrial ATP synthase) accumulates in the late infantile variants and in juvenile onset NCL; sphingolipid activating proteins (Saposins A and D) are enriched in two infantile onset forms (21, 22). Interestingly these components are detected in CNS tissue only, and they are not present in peripheral tissues, where the storage can be detected in several cell types ultrastructurally.
The recognition of lysosomal involvement in the NCLs was followed by a huge research effort aiming to disentangle the pathogenetic mechanisms and to shed light on the cellular pathways and processes which are affected (see Section The Research Contribution to Knowledge).
The identification of lipopigment storage ultrastructurally became the main diagnostic tool for NCL, and cerebral biopsies represented the favored diagnostic approach. Evidence that storage material could be detected in readily accessible extra-neural tissues (such as skin, blood lymphocytes, skeletal muscle) led to an important shift from CNS to peripheral biopsies, a safer and more rapid pathway to diagnosis (23). Peripheral neurons, such as intramural ganglionic neurons of rectal mucosa, were also utilized for diagnostic purposes (24, 25). The impaired function of these cells was associated with gastrointestinal problems, including abdominal pain, constipation, and altered bowel motility in NCL patients. The involvement of the enteric nervous system was recently investigated in three NCL mouse models, which showed both impaired enteric functions and histological and ultrastructural findings consistent with neuronal loss and storage accumulation (26).
Based on clinical and pathological criteria, the NCLs were classified according to age of clinical onset and the ultrastructural features of the cytosomes. Their nomenclature was modified over time to include newly recognized variants [for example, infantile and early juvenile] and the adult form (16, 27, 28). Such classification has been valid for about four decades, and it is still useful in a clinical setting (Table 1). It has helped to target molecular genetic diagnostic investigations. The identification of cytosomes in the peripheral tissues of unusual cases, where the clinical phenotypes is not entirely consistent with known NCL genetic types, may lead to molecular analysis using NCL panels, sometimes obtaining diagnostic confirmation at molecular level (29). Major steps in NCL history are summarized in Figure 1.
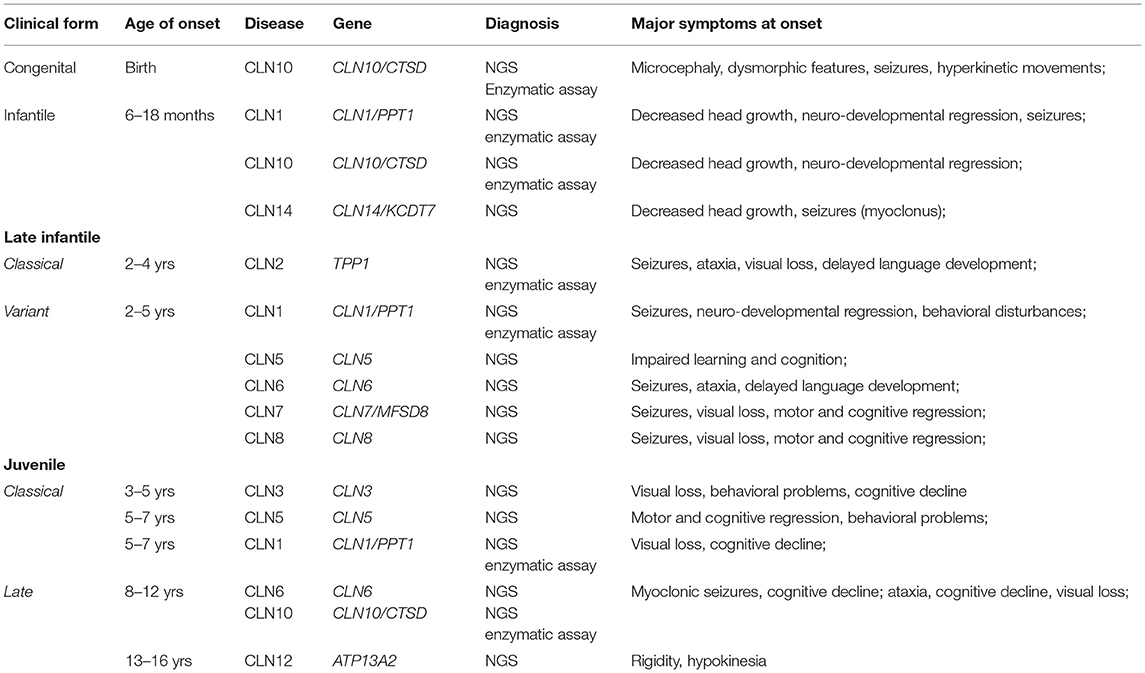
Table 1. NCL of childhood onset: clinical classification and major diagnostic procedure.

Figure 1. Major steps and achievements during nearly two centuries of Neuronal Ceroid Lipofuscinoses history are outlined, starting from the earliest clinical description to the present research which led to the first FDA/EMA approved treatment.
An axial classification system was proposed, including seven axes, to obtain information necessary to better categorize each NCL form, according to the specific items which characterize them (30) and represents a useful diagnostic tool for research purposes.
NCL are genetic diseases (13). All childhood and most adult NCLs are inherited as autosomal recessive diseases. There is only one dominantly transmitted adult-onset form, Parry disease, associated with mutations in CLN4.
A new age in the NCL history started during the last decade of the 20th century. The first human NCL genes, CLN1 and CLN3, were identified in 1995 by positional cloning (31, 32). The identification of the remaining eleven NCL genes occurred within the following decade, making use also of naturally occurring animal disease models. The pathogenicity of identified mutations was proven by different approaches in vivo (eg. using knock-out invertebrate and mammalian models) and/or in vitro.
The consequences of the genetic advances are multifaceted. From a clinical perspective it allowed a new gene-based classification and NCL nomenclature (33), provided a powerful tool for diagnosis, and helped clinicians recognize both phenotypic variability and heterogeneity within most NCL genetic disease types (34). The classical phenotypes are generally associated with the most common mutations in each gene, whereas variant forms may arise from “private” mutations. Phenotypic variability can be observed even within some families, which, for example, may lead to differences in survival among siblings. Advances in NCL genetics also gave the opportunity to re-evaluate NCL epidemiology and to recognize the worldwide distribution of this group of diseases (see below Section Epidemiology and Registries). The identification of the NCL genes and their related products helped to begin to identify altered cellular processes and patho-mechanisms leading to cell death and progressive neurodegeneration which characterizes all NCLs. The cross-talk between different cell compartments (eg lysosomes and endoplasmic reticulum, lysosomes and mitochondria) has been described in some NCL forms (35–38) and has helped to shed light on some clinical features which cannot be ascribed to the mutated NCL protein only. Animal and cellular models have been generated to expand our knowledge of the pathology, and so the first disease modifying and therapeutic agents are now becoming available for clinical trials (39). The first such agent was approved for clinical use in 2017 following a pivotal phase I/II clinical trial.
As shown in Table 2, allelic adult-onset variants are observed in several childhood forms (40). In most however, the adult phenotype shows clinical features which are consistent with the classical childhood form and are differentiated only by the age of onset. The well-defined adult onset NCLs are still named by classical eponyms [Kufs A, Kufs B and Parry diseases]. They are associated with pathogenic mutations of CLN6 [Kufs A disease, (41)] and with three other genes (CLN4, CLN11, CLN13) whose mutations give rise exclusively to adult-onset phenotypes (42–45).
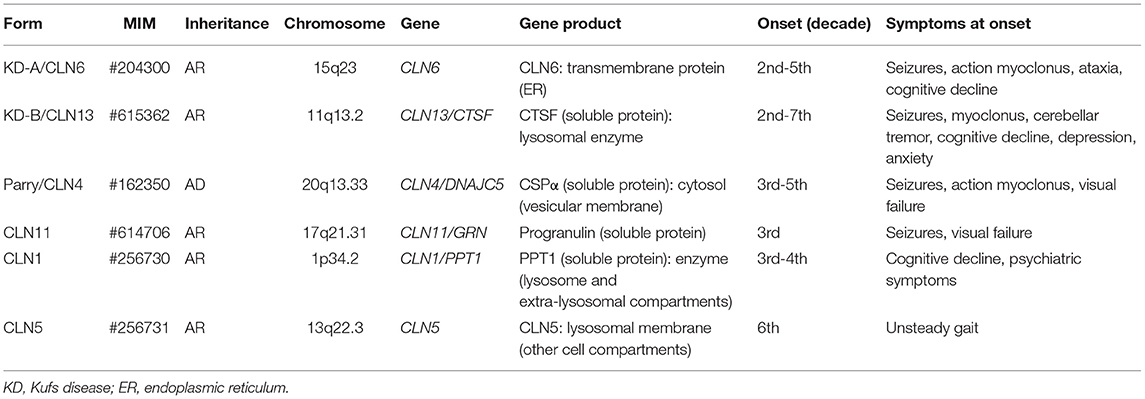
Table 2. Characterization of adult onset NCL.
The identification of childhood NCL genes is also contributing to the identification of potential genetic signatures for neurodegeneration in adulthood. A number of NCL genes share mutations or are allelic with mutations contributing to common adulthood neurodegenerative disease, such as Alzheimer's Disease, Fronto-Temporal Dementia, Parkinson's Disease (46–48). Moreover, lysosomal dysfunction and dysregulated autophagy which are observed in CLN3 and CLN6 disease (49, 50) are also seen in most forms of late-adulthood neurodegeneration (51, 52).
Advances in NCL genetics were accompanied by new opportunities to establish diagnosis at a biochemical level; enzymatic assays became available for four lysosomal enzymes: CTSD, CTSF, PPT1, and TPP1 (53–55). The gold standard for diagnosis is to require genetic confirmation by detecting both disease causing alleles. Therefore, when detection of the common disease causing mutations is not confirmed in the presence of a biochemical impairment, the diagnostic search for changes in cryptic gene regions (introns, untranslated regions, etc.) is extended. The requirement to establish a molecular diagnosis is of a primary importance for families helping to inform prognostic counseling, for antenatal diagnosis and, more recently, to judge suitability for “innovative” treatments where they are available. The recent availability of NGS technology has changed the clinical approach to NCL diagnosis, which relies on a straightforward molecular approach, leaving the neuropathological and ultrastructural investigations to cases which remain unsettled, even after molecular analysis (Figure 2).
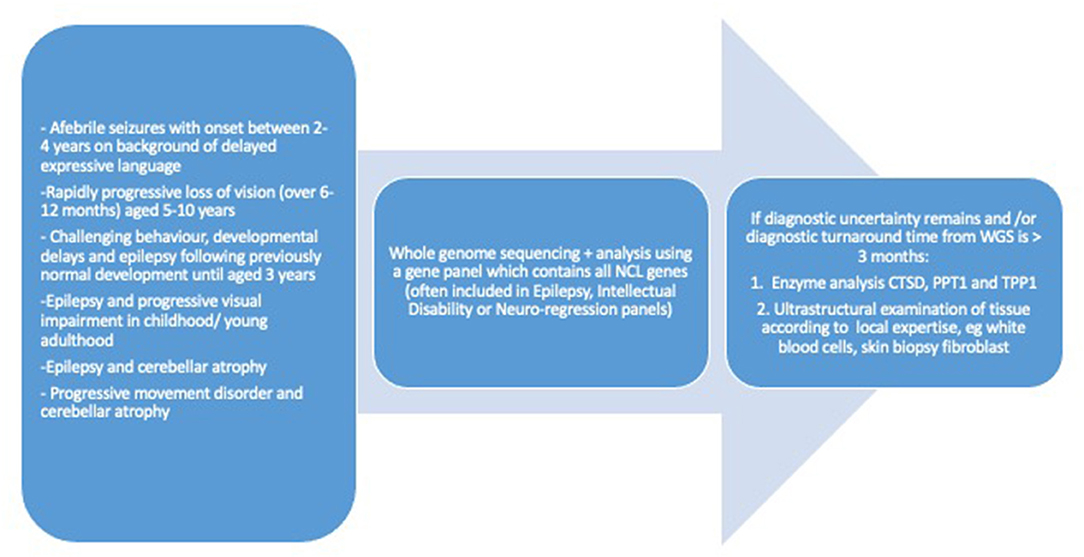
Figure 2. Diagnostic work up for neuronal ceroid lipofuscinosis of childhood onset.
NCL are rare diseases which are distributed world-wide. The higher prevalence of selected NCL forms in restricted geographical areas has some historical relevance and may also reflect the early progress in molecular diagnosis in some countries (31, 56, 57). Epidemiological data indicate an incidence of 1–3/100.000 and a prevalence of about 2–4/1.000.000 (58–62). These figures refer to Western countries where access to molecular diagnosis has become standard of care over the last 10–15 years. More detailed epidemiological study is necessary to improve awareness of these diseases, the efficacy of genetic counseling, to plan appropriate services, and to facilitate access to “new” treatments. In Western countries CLN3 disease (juvenile NCL) is the most common form, followed by the classical late infantile form, CLN2 disease which is more frequent in Southern Europe and the Mediterranean region. Ultrarare childhood NCL are CLN10, CLN12, CLN14. A major contribution to the dissemination of knowledge comes from disease registries and databases. The independent International NCL Database (led by Dr A Schulz in Hamburg, Germany; Clinical trials identifier NCT04613089, an extension of former NCL DEM-CHILD patient database) is collecting static and dynamic clinical data of NCL patients from 19 countries from Europe, North and South America and Asia. The UCL based NCL Mutation Database (established in 1998 by prof S.E. Mole; http://ucl.ac.uk/ncl-disease/) lists all published and reported NCL patients as well as mutations.
The impressive amount of data collected over the last two decades has allowed the NCL community to generate a new nomenclature of NCL based on genetic variations (33), followed by comprehensive reviews which have described the clinical features, the cell localization and functions of the mutated gene products, as well as the historical markers which characterize NCL among other progressive neurological diseases with endo-lysosomal storage. For a systematic review of the clinical and diagnostic features of each NCL disease (including the phenotypic heterogeneity), readers are referred to recent publications (1, 63–69). A comprehensive classification of NCL is given in Table 3.
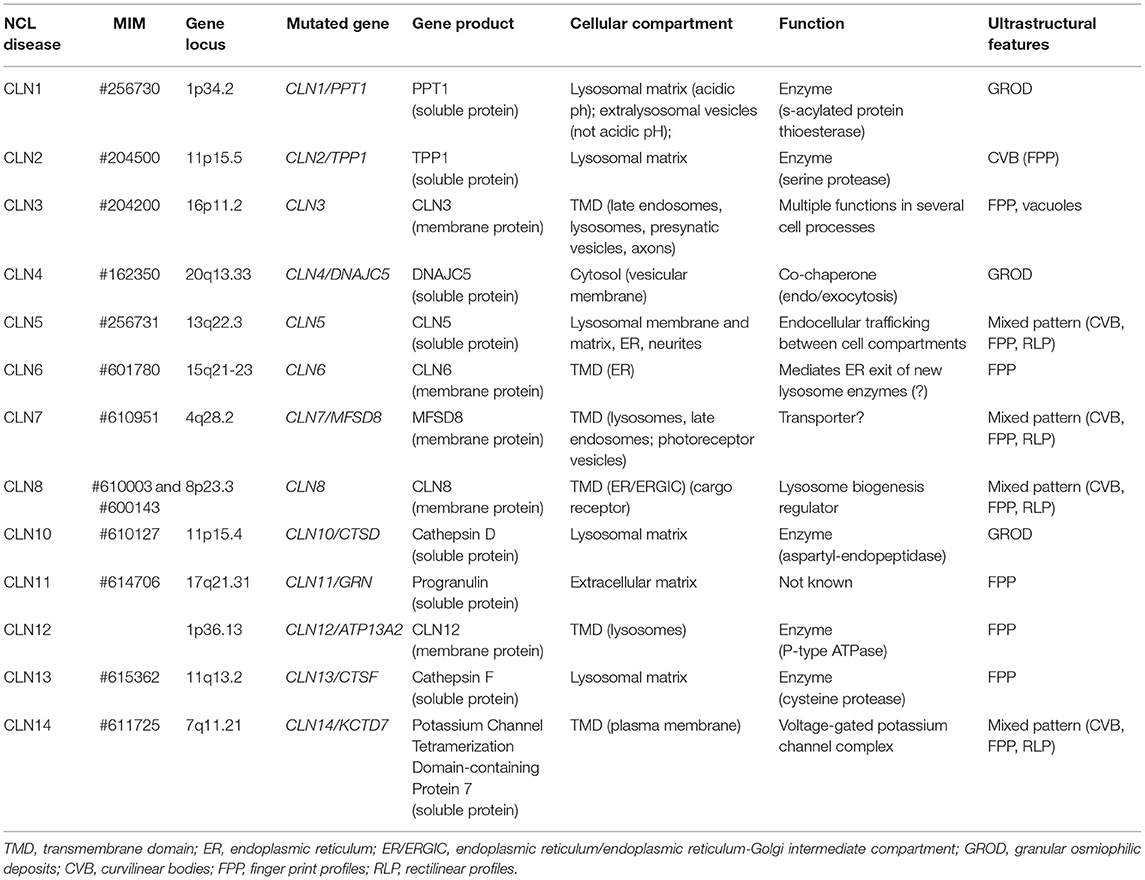
Table 3. NCL: genetic, biochemical and ultrastructural features of each form.
As mentioned in the introduction, the NCLs were grouped as amaurotic idiocies around the end of the 19th century, along with other diseases which were recognized as different disorders later. “Idiocy”, progressive blindness, and seizures, remain the cardinal symptoms of the NCLs. The NCLs are the most common neurodegenerative diseases in childhood, and they are one of the main causes of childhood dementia worldwide. They are progressive with severe physical decline, and an early death. Infantile and late infantile onset NCLs show the most rapid rate of disease progression. Most patients are bedridden by their second decade, and death occurs during the late second-early third decade. A less rapid course is seen in patients affected with CLN3 disease. Staging systems based on severity scores of selected clinical manifestations have been developed to monitor the disease evolution quantitatively. These tools are helpful in describing the natural history of a disease and are of particular relevance when the efficacy of “new” treatments is tested (70–72).
Cognitive decline, ataxia, amaurosis, and early seizures are the clinical manifestations at onset in most cases. They become evident sequentially over a short time frame (6–24 months) in infantile and late-infantile NCLs; spasticity follows leading to loss of motor function, dependence on carers for all activities of daily living within 5–8 years of symptom onset.
In this review we will discuss the clinical symptoms which characterize the NCLs in more detail, and are shared between the forms, focusing on their temporal evolution at onset and during the early stages of the disease course. Seizures, motor deterioration (including ataxia, movement disorders, spasticity), cognitive decline, progressive blindness, and behavioral problems are the major clinical features of infancy and childhood onset NCLs. The identification of the clinical markers and their relationship with patients' age at onset may help to drive diagnostic procedures toward a specific NCL type, which can be confirmed molecularly. Such an approach reflects the clinical experience of the authors in the field of childhood neurodegeneration.
Epilepsy is common to almost all NCL forms. Several seizure types are seen in the NCLs. The severity of epilepsy (in terms of age of onset, seizure semiology, seizure burden and response to anti-seizure medications) is not shared uniformly. Seizures tend to start earlier and semiology is more varied in infantile and late-infantile onset NCLs. With time seizures tend to become less frequent, except for myoclonus (either spontaneous or evoked) which remains the only paroxysmal manifestation in the late stages of the disease. The background EEG progressively deteriorates leading to a nearly isoelectric pattern, as an expression of progressive cortical atrophy and degeneration of the “generators” of cortical electric activity.
Frequent perinatal convulsions (or even status epilepticus) associated with severe cortical and cerebellar atrophy are observed in the ultrarare congenital NCL, which leads to death within the first weeks of life and are usually associated with mutations in CTSD (73).
Generalized seizures, including myoclonus, starting 2 to 6 months after the earliest clinical manifestations, characterize infantile onset NCL (infantile CLN1 and CLN10 diseases). Before the onset of clinical seizures, EEG abnormalities can be detected, which evolve over time into a characteristic pattern, the “vanishing EEG”, observed also in the late infantile variant of CLN1 disease (74–76).
Generalized motor seizures, absences, and myoclonus (including negative myoclonus) are the main seizure types observed in classical CLN2 disease. They are also seen in CLN6, CLN7 and CLN8 late infantile variants. In these forms seizures can be present at disease onset, or appear within 1–2 years. A characteristic EEG feature is the paroxysmal spike-wave response, which is evoked by low-frequency (1–3 Hz) intermittent photic stimulation which is observed in CLN2 and CLN6 (late infantile variant) patients during the early stages of the disease (77, 78).
A prevalent myoclonic epilepsy with progressive features characterizes the late juvenile (teen age) onset CLN6 disease and its allelic adult Kufs A disease. Both forms present with progressive myoclonic epilepsy at onset gradually increasing in frequency and intensity, and less frequent bilateral tonic-clonic seizures. A low-frequency (1–5 Hz) photo-paroxysmal response is present in the vast majority of cases (79). The disease has a slowly progressive course, and patients become wheel-chair bound within few years due to the relentless myoclonus and progressive ataxia (80). Progressive myoclonus epilepsy associated with rapidly progressive dementia is also the clinical hallmark of the dominantly inherited NCL (Parry disease) (42).
Bilateral tonic-clonic seizures (even with focal onset) and absences are the most frequent seizure type in CLN3 disease, rarely occurring as presenting symptoms; myoclonus is less frequent than in other NCLs (81, 82).
Ataxia, weakness, loss of acquired motor abilities and spasticity are the most common motor symptoms in NCL patients. The consequence is the loss of motor autonomy and increasing dependence on caregivers. Signs and symptoms of motor impairment become evident in children who had normal motor development previously.
Ataxia is the most frequent clinical symptom of CLN2, CLN7, CLN8 disease and of the rarer late-infantile variant of CLN1 disease whose age of onset is between 2 and 5 years. It is also evident in CLN6 disease but onset is 1–2 years later. Ataxia in the NCLs is secondary to spino-cerebellar involvement. Purkinje cells and neurons of the deep cerebellar nuclei are severely affected and undergo early death. Cerebellar atrophy is an early neuro-radiological sign, preceding the enlargement of cortical sulci and of the lateral ventricles. Sometimes the term “ataxia” is incorrect, and the transient spells of motor instability which a child shows should be considered as clinical expression of “negative myoclonus”, which is commonly present in these forms.
Motor weakness affecting limbs, eye muscles (strabismus) and deglutitory muscles (dysphagia) is likely to occur because of impaired cortico-descending connectivity due to the involvement of motor cortex and the neurons of motor nuclei. The progressive degeneration of motor pathways is followed by spasticity which represents further disease progression. Changes in gait phenotypes in different forms of Batten disease has been recently considered as a “marker of disease progression” (83).
The progressive involvement of cortical neurons also leads to a progressive loss of motor skills, motor initiative, motor planning and strategies, which represent the “cognitive foundation” of motor function. This same process also leads to dementia. Declining motor function is observed in all NCL forms, leading to loss of independent ambulation, subsequent loss of all voluntary movement and posture control, the rate of decline dependent on the NCL type. Children with the infantile and late infantile NCL types become wheel-chair bound at 6–12 years of age (according to the severity of the form) before becoming “bed-ridden” (early teens in late infantile forms, early-mid second decade in CLN3 patients).
Extrapyramidal symptoms, such as tremor and rigidity, are known to occur in NCL, being particularly relevant in CLN3 disease. They are responsive to available treatments, such as anti-cholinergic drugs, Dopamine. Dystonia and chorea can be recognized at different stages of NCL disease course (84, 85). The reasons why basal ganglia structures and/or neuronal connections underlying these clinical manifestations are relatively spared in NCLs is a matter which requires further investigation.
Cognitive decline is hallmark of the NCLs. In younger children the loss of cognitive abilities related to school-learning occurs rapidly, whereas the relatively slow pace of disease evolution observed in “juvenile” patients allows them often to attend mainstream school whilst continuing to learn new facts and skills into their teenage years. From this perspective two major issues should be considered, which further differentiate early onset NCL from juvenile forms.
Cognitive decline occurs as a two-phase problem. Early in the disease, children's developmental trajectory slows, they acquire new learning more slowly than their peers but often remain within the normal range for some time before professionals become concerned. They go on to plateau and then begin to lose the cognitive competences they have acquired during the earliest months and/or years of their life. This is often the time at which diagnostic investigations are triggered. In CLN2 disease however many children show a delayed acquisition of cognitive skills at a very early stage and their profile is characteristically uneven with delays more evident in the domain of expressive language compared with motor skills. This has been reported in recent years and is of huge importance for driving early diagnosis. Nickel et al. (86) reported that about 40% of a cohort of CLN2 patients presented early with delayed language acquisition. It raises the potential to consider language delay as an early warning sign, leading to careful clinical examination of such children, in order to detect any additional concerning features which would justify a deeper diagnostic workup including genetic screening. The atypical early developmental profiles of affected children may also give some clues about the molecular basis of language development, and the role that cellular pathways involved in the development of this complex function may play. There are no studies available for NCL late infantile variants, but it is tempting to hypothesize similar findings in some NCL types at least, e.g. CLN7 and CLN8 disease. Delayed language development in CLN2 disease children does not seem to be related to any specific CLN2 genotype.
Different clinical issues are related to the decline of cognition in CLN3 disease, which can be considered as the prototype of amaurotic idiocy. The rate of disease progression is commonly slow and children can attend school, with support for the intellectual, visual and sometimes behavioral difficulties. There is no evidence to date of an uneven developmental profile early in life and before the onset of visual impairment. The scores of the neuropsychological function tend to diverge from typically-developing peers, reflecting the failure to achieve the expected achievements, but a marked drop in their cognitive abilities occurs during mid-adolescence, at around the same time as rapid neurological decline (87).
Behavioral problems at onset characterize CLN3 and CLN5 diseases, and to a less extent atypical CLN2 disease, three NCLs where first symptoms occur in school age children. There are well described and differentiated disease natural history, and which are caused by mutations in three different nuclear genes whose products have different functions and are located in different cell compartments.
CLN3 disease is the most prevalent NCL in Northern Europe and USA. It has a juvenile (school age) onset with visual impairment and behavioral problems, followed by cognitive decline. Motor impairment and epilepsy occur later. Behavioral problems include anxiety, depressed mood, bursts of aggressive behavior and psychotic manifestations. These symptoms tend to remain stable or even worsen during the early years after disease onset, and then become less significant as the disease advances and functional and cognitive abilities are lost. Behavioral problems represent a major challenge to the quality of life for those patients (and their families and carers) where a slow disease course and longer survival is expected (87, 88), and seems also to be unrelated to the genotype (89). The disease evolution is much slower than in other forms, and death may occur in the fourth or even sixth decade, unless cardiac involvement results in premature death (see below). There is a relevant phenotypic homogeneity (possibly related to a common mutation which is observed in the vast majority of cases). Some gender differences have been reported, the female patients presenting a more severe clinical course (90).
Behavioral manifestations and impaired language are the early clinical manifestations of a late infantile “variant”, of pre-school age onset, CLN5 disease. Seizures and visual impairment occur 3–4 years after disease onset, followed by progressive motor impairment with spasticity with loss of ambulation by 11–12 years of age. The predicted survival is about 15–20 years after disease onset. In this disease there is some evidence that phenotypic variation (as far as the rapidity of neurological decline and survival) is related to the severity of mutations (91, 92).
Behavioral disorders (along with seizures and, language abnormalities) at onset were also reported in a large South American cohort of children with atypical CLN2 disease (93).
Progressive visual impairment is one of the classical symptoms of NCL of childhood onset. It affects all forms, and in the large majority of cases it occurs as one of the early clinical signs. Retinal structures, visual pathways and visual cortices are affected. Both ganglionic neurons and receptor cells (cones and rods) are involved. Impaired retinal response is visualized by ERG. Optical Computerized Tomography allows monitoring of the progressive retinal degeneration. Most published evidence relates to CLN3 and CLN2 diseases (94, 95), and to less extent to CLN1 disease (96). A spectrum of retinal disturbances was described in CLN7 disease and related to the severity of mutations (97). Whether maculopathy precedes cognitive impairment in CLN3 disease, or if both visual impairment and mild cognitive failure occur at around the same time (98) is still the subject of discussion. Biallelic variants of the MFSD8 gene may be associated with isolated juvenile maculopathy, evolving into a slowly progressive encephalopathy with protracted course (99). Conversely, late visual impairment, or even long lasting preservation of visual function can occur in CLN5 and CLN6 diseases (92, 100). Notably, in the allelic Kufs A disease the retina is not affected and vision is normal. Recently, patients affected with isolated retinal degeneration (including retinitis pigmentosa) have been identified harboring biallelic CLN3 and CLN10 pathogenic variants (101, 102).
Therapeutic trials are in progress aiming to prevent retinal degeneration in animal models using intravitreal gene or enzyme therapy (103, 104). These approaches represent powerful therapeutic systems suitable for translation into NCL patients. A major question arises regarding the contribution of central nervous system pathology to functional vision, which is unlikely to be corrected by a retinal only approach.
Sleep disturbances are common in all NCL types. There are few studies on this issue (105–107) but the importance of sleep management has been outlined in two recent consensus papers (108, 109). Poor sleep quality and sleep pattern disturbance will impact adversely on the quality of life of affected children and their families directly and indirectly through worsening of seizure control and behavior. Supportive measures (such as respite care) and behavioral strategies (such as sleep hygiene) are necessary given the lack of consistent benefit from commonly used medical interventions, such as Melatonin (110). Anti-seizure medications, often prescribed, may help falling asleep, but side effects (e.g. morning drowsiness) impair the restorative function of sleep.
It has been known for several years that phenotypic heterogeneity is a feature of genetic diseases (including NCL), and some patients were considered as atypical cases. The origin of clinical variation is commonly ascribed to the genetic background of patients as well to the severity of the mutations. Recently polymorphisms or mutations of unrelated genes have been considered as modifiers of gene expression, and the interactions between mutated genes and modifiers may lead to clinical variations and to the observed phenotypic heterogeneity. In addition non-genetic confounding factors may also affect clinical phenotype.
An “atypical case” has been described in a child affected with a congenital form of CLN5 disease (which usually presents with a late infantile onset, with relatively slow disease progression after onset): he was a compound heterozygote in CLN5 and was also carrying an incompletely penetrant variant in POLG1, a nuclear gene coding for mitochondrial polymerase (111). No evidence was provided of how the co-inherited POLG1 variant may have enhanced the putative pathogenetic effect of the CLN5 mutations. However, the interactions between lysosomal and mitochondrial compartments may be implicated, further affecting the physiology and viability of cells hampered by the presence of pathogenic CLN5 mutations. Another atypical case is related to CTSD, commonly associated with congenital or late infantile NCL (CLN10). In a patient with juvenile ataxia associated pigmentary retinopathy and cognitive decline, the evidence of granular osmiophilic deposits (an ultrastructural marker of CLN1, CLN4 and CLN10 disease) in angular atrophic fibers following a muscle biopsy led to the final diagnosis (29). Unusual phenotypes associated with mutations in CLN2/TPP1 with residual leukocyte enzymatic activity raise the issue of the phenotypic heterogeneity, regardless the severity of mutations. A prevailing spino-cerebellar involvement was detected in an adult lady who was suffering from ataxia since adolescence. She was carrying a missense and a splice site variant in CLN2/TPP1 (112). A juvenile onset, progressive and protracted form of cognitive decline with myoclonus, dystonia, bradykinesia and ataxia was reported in three siblings and associated with compound heterozygosity in CLN2/TPP1, leading to stop codon formation and an aminoacid substitution (113).
It is evident from these “atypical cases”, that the detection of a mutated gene represents the starting point to explore the complex mechanisms which ultimately lead to the phenotypic expression, taking into account the unique genetic background which each of us has inherited. Improving our knowledge in this field of “personalized medicine” will help to prevent unexpected and potentially serious adverse events, which might arise when using conventional and novel replacement therapies.
NCL are considered as LSD. However, they do not show the characteristic multi-organ involvement typical of many LSDs (114), and they predominantly affect the central nervous system. Progressive cardiac involvement is observed in some patients affected with CLN3 disease with repolarization disturbances, ventricular hypertrophy, and sinus node dysfunction (115). Cardiac pace-maker implantation has been used in a small number of patients to improve symptoms of syncopy and extreme bradycardia but is not in widespread use. Twenty four hour ECG recordings have been recommended for annual surveillance in young people age 16–18 years and older. Degenerative changes of the myocardial wall along the deposition of lipopigment and cytosomes can be detected histologically (116).
Heart involvement has also been reported in CLN2 disease [progressive conduction defect; (117)] and hypertrophic cardiomyopathy was described in patients with CLN10 disease (29, 76).
Reports on life expectancy and mortality in NCL are scarce (118). This issue is assuming particular relevance in natural history studies when assessing the efficacy of “orphan drugs”.
The ultra-rare congenital form of NCL has the most dramatic course, the very few reported cases having died within two weeks of birth (119). Ongoing studies confirm that infantile-onset NCL (due to mutated CLN1 and CLN10 genes) and classical CLN2 disease have the next most rapidly severe course and shortest survival. Variability within each form does not allow accurate predictions of life expectancy in individual patients According to the Italian NCL database CLNet, death in the majority of CLN2 patients has been around nine years of age, but prolonged survival is reported in a number of patients affected with the classical form, up to the early third decade, regardless the predicted severity of the mutations they were carrying. Intra-familial variability is also reported. Little is known for the remaining NCLs with infantile and late infantile onset. As mentioned before, the longest survival (up to 4th-6th decade) is observed in CLN3 patients.
Over the last two decades overall longer survival has been observed in patients of many NCL forms, regardless the severity of mutations. Such findings may be ascribed in large part to the improved availability of supportive tools for home care which guarantee appropriate feeding (e.g. percutaneous gastrostomy), respiratory support, as well as the improved quality of skin and general care (e.g. prevention of bed sores), the availability of new anti-epileptic drugs, and the overall improved awareness of caregivers to provide care in order to enhance the length and quality of life of those affected (120, 121).
The NCLs are progressive neurodegenerative diseases associated with endolysosomal storage. No cure is available, and major efforts aim to provide the best care to patients. Recently, disease modifying agents have become available or are under investigation for some NCLs (122), by replacing a missing enzyme, the mutated gene, or by reducing the substrate involved in abnormal storage formation. Such approaches reflect the ongoing lack of understanding of the basic disease mechanisms specific to each form, as well as of the shared pathways which characterize the whole group of NCL diseases, despite extensive research efforts.
Experimental paradigms and innovative methodologies have provided major breakthroughs over the last two decades on the knowledge of pathological mechanisms underlying the NCLs (123). In this paragraph we summarize the most relevant data resulting from the current research under different experimental settings, which may contribute to better understanding the rationale of the research toward appropriate strategies to generate new, safe and effective treatments.
Recent studies of outcome on a relatively large population of CLN2 patients are revealing a meaningful slowing down of the rate of disease progression, following the availability of the first “new treatment” (recombinant pTPP1 delivered intrathecally into the ventricular system). Great expectations have risen from the recent announcements of forthcoming treatments whose aim is to both increase the survival and ameliorate the quality of life of the affected children.
Several naturally occurring animal models of NCLs have been discovered and investigated, including non-human primates, ovines, felines, dogs and rodents. The use of relatively large mammals, with convoluted gyral brains represents a powerful tool to examine disease evolution over time (from the early stages until death) and monitor brain pathology using in vivo techniques (head imaging, neurophysiology etc) which are currently in clinical use (124–126). In addition, multiple techniques can be utilized to investigate post-mortem brains (and eye) from different perspectives. Along with classical descriptive neuropathology, methodologies are now available which can help to disentangle the complex network of interacting events accompanying the disruptive effects of the intra-neuronal storage in complex mammalian brains. They include the use of immuno-histochemical probes, which allow the detection of selected neuronal and glial populations, to evaluate the distribution and topography of the storage, to check the expression of markers related to pathological processes (such as autophagy), and more recently omics techniques (see below) which allow exploration of expression at the metabolic, protein or even nucleic acid levels of selected biochemical pathways and/or genetic markers (127).
Moreover, these animals are essential in the evaluation of “new” treatments, testing safety, efficacy and putative effects, before progressing to human clinical trials [(39, 128); see Section Pathology and Pathogenetic Mechanisms]. In Supplementary Table 1 some examples of selected animal models contributing to our understanding of disease mechanisms (and treatment) related to specific human NCL forms are listed.
Most of the present knowledge of the patho-mechanisms of NCL comes from experimental studies using mouse models which were modified in syntenic NCL genes. A number of labs in Finland, UK, USA, and Germany have generated mouse models of NCL diseases (CLN1, CLN2, CLN3, CLN5, CLN6, CLN7, CLN8, CLN10). Gene targeting technology has allowed selected mutations within a gene to be engineered, offering an experimental background more alike to that in human disease. Generally large numbers of experimental animals can be created, and survival is long enough for a high number of experiments to be carried out. This is the major advantage as compared to spontaneously occurring large mammals, where the number of animals carrying homozygous mutations for a selected gene is relatively low and experiments need to gather as much information as possible from each animal. Advances in technology allow engineered mice to be investigated according to the same methodologies described for large mammalian brains (for example imaging and neurophysiological studies).
The availability of miniaturized technology has allowed the use of high throughput systems including invertebrate such as Zebrafish, Drosophila, the social amoeba Dictyostelium, yeast, in vitro cell systems, such as SH-SY5Y Neuroblastoma cells, and from human patient fibroblasts (obtained from skin biopsies). The use of these simple cellular models make them suitable to address selected questions and obtain results from targeted experiments which are easily repeatable and provide high statistical power (129).
A major advantage of using human fibroblasts is the ability to transform them into neuronal cells through a complex process in vitro of diversion from the primary cell lineage, regression of the cells into induced pluripotent stem cells (iPSCs) and transformation into neurons using ad-hoc enriched media. Such studies are already in progress: neuronal cells, derived from fibroblasts of CLN5 patients, showed features consistent with a NCL phenotype (130), and transformed neurons, derived from fibroblasts of CLN2 and CLN3 patients, showed impaired activities of the lysosomal-mediated pathways (131).
A major limitation of this technology, however, is that fibroblasts carry the genetic background of the affected patient, so that direct comparisons between the phenotypic variations of a specific mutation among cell lines from different patients and inferences about the functional implications of specific mutations are challenging. This difficulty can be addressed by combining the CRISPR/Cas9 genome editing technology to generate targeted mutations in mammals as well as in healthy human iPSCs or in human embryonic stem cells (132, 133), without the bias of the potential effect of the mutated gene on the native genetic background.
A further development in the in vitro technology, using human cells to investigate the functional effects of individual mutations, is the generation from iPSCs of cerebral organoids (134). Such an approach allows investigation of the effects of a mutated gene not only on the mature cells, but also to evaluate its putative involvement in general neurodevelopmental mechanisms. The available technology makes it possible to test hypotheses multi-Omically, and therefore obtain information about the main pathways and functions modified by the mutated gene during the mini-brain development (135).
Omics approaches represent recently developed technologies which provide high-throughput data related to the genome (DNA) the transcriptome (RNA) the proteome (protein), and the metabolome (metabolic products). The integrated study of data derived from Omics investigations represents the foundation of system biology, and it has been applied to investigate disease mechanisms, particularly the identification of affected biological pathways, which may become therapeutic targets as well as potential biomarkers (127). The new knowledge acquired from these methodologies is predicted to contribute to significant advances in the field of NCL research. The use of Omics technologies to NCLs is relatively recent; several issues have been addressed in both human and experimental animal models of several NCL forms. Major studies performed on tissues from patients affected with different NCL forms are summarized in (Supplementary Table 2).
Neuronal death is the disease hallmark shared among all NCL forms: it affects CNS neurons, ganglionic neuronal cells of the retina and even ganglionic neurons of intramural ganglia of the bowel wall. All gray regions of the brain are affected by neuronal death, showing however differential patterns in the topography of neuronal loss, the rate of progression and the secondary involvement of the white matter. Selective neurodegeneration, targeting specific regions and particular cell populations, can be observed during the early stages of the disease, and the patterns of disease evolution can be monitored by neuroimaging studies (136–140). Whether these features reflect the genetic heterogeneity of the NCL is a matter to be investigated further. As clearly indicated by the clinical symptoms at onset and by the patterns of disease progression, cerebral and cerebellar cortices are the most affected brain regions in humans. Selective hippocampal pathology has also been described in different NCL (141). In some forms, particularly in CLN2 disease, the rate of atrophy of the cerebellar cortex is faster than observed in the telencephalic cortex. Less marked is the involvement of the basal ganglia in the early stages of the disease, but eventually generalized atrophy of all gray structures is observed in all childhood NCLs. The neuronal loss leads also to progressive atrophy of the centra semi-ovale, due to the lack of axonal projections from and to the cortex, which is accompanied by progressive enlargement of the ventricular system. Traditional human neuropathological studies have shown that neurons of the spinal cord are also affected in many NCL forms (1, 27, 142). These findings emphasize further the generalized susceptibility of all neuronal cells to this condition.
Most information concerning the selective involvement of gray structures and the neuronal loss in the NCL brain come from elegant studies carried out using mouse models, which allowed descriptions of the temporal evolution of neuronal loss, established the role of both astroglia and microglia in the brain pathology (143), dissected out the most vulnerable cell compartments involved in neuronal degeneration and proposed hypotheses regarding the cellular mechanisms and pathways underlying the progressive neuronal loss seen in the NCLs.
A gradient of neuronal degeneration is commonly observed, affecting the cortex, the cerebellum and the thalami. Thalamic gliosis seems to precede the onset of cortical involvement (neuronal loss) in both CLN1 and CLN10 mouse models (144) and early thalamic involvement is considered as a radiological marker of LSD (145). A reverse pattern of degeneration is observed in CLN5-/- mice (146). Recently, neuronal loss has also been described in in a mouse model of the disease in the spinal cord, preceding the neuronal loss of the remaining brain regions (147).
Such gradients of evolution are less evident in human post-mortem studies, in which all the brain structures are severely affected in a similar manner: neuropathological examinations are performed several years after the disease onset.
Neuronal death represents the end point of a complex process to which several processes contribute. However, some groups of neuronal cells seem to be more vulnerable to the pathological condition, as shown by some elegant experimental studies.
Axons are affected: early axonal breakdown was shown in the CLN1 KO mouse model (144); impaired elongation and branching, giving origin to a stunted growth was observed in vitro, in a neuronal-like cell system, overexpressing CLN1 gene (148).
Several aspects of synaptic pathology were also reported. Loss of synaptic proteins was reported in both mouse (CLN1) and ovine (CLN5) models of diseases (144, 149), as well as impaired synaptic vesicles recycling (150). Impaired NMDA-R development was described by Koster et al. (151). Recently, a reduction of functional voltage-gated Calcium channel, in differentiated SH-SY5Y cells, overexpressing CLN1/PPT1 gene was reported (152).
Results from different experimental techniques provide evidence that neuronal connectivity is affected in several NCL models. Impaired cellular function due to the distorted topology of the neuronal cells because of the intra-lysosomal storage is amplified by the impaired neuron-to-neuron communication, strongly contributing to neuronal dysfunction and subsequently cell death.
The molecular mechanisms leading to endo-lysosomal storage formation have not so far been fully elucidated. It should be noted that a primary defect of lysosomal proteolytic activity is present in only four NCL forms, whereas in the remaining NCLs, storage accumulation is associated with impaired cellular degradation of large molecules by means of a complex pathway, in which lysosomal hydrolytic enzymes are a major, but not exclusive, component. Likewise, which mechanisms link the formation of the endo-lysosomal storage and death of the neuronal cells remains unknown (38).
Apoptosis was considered to be the main mechanism leading to cell death following the detection of targeted markers in canine, ovine and human brains and photoreceptors (153, 154). Subsequently, the role of autophagy was investigated in the NCLs, possibly because of the evidence of a temporal link between autophagy and apoptosis (155). Activation of unfolded protein response and apoptosis was shown in the brain of an early mouse model of CLN1 disease (156). The presence of apoptotic cells and expression of apoptosis markers along with lysosomal dysfunction and autophagic stress were also detected in the CNS of a mouse model of Cathepsin D deficiency (157).
The evidence of lysosomal accumulation of subunit c of the mitochondrial ATPase F0 complex suggested this occurred secondarily to impaired degradation pathways. Cao et al. (49) showed that autophagy was severely affected in a mouse model of CLN3 disease. Moreover, the detection of cell death following inhibition of autophagy suggested that activated autophagy represents a pro-survival response of the cell to the disease process.
Recently impaired autophagy was demonstrated to occur in different experimental models of a number of NCL forms and supported the increasing evidence that dysregulated autophagy is commonly detected in the LSDs (158).
Fibroblasts of patients mutated in KCTD7 (associated with NCL14) had impaired autophagy (159). Impaired autophagy was also detected in a KO mouse model for CLN1 disease, which is associated to mutated palmitoyl-protein thioesterase-1 (PPT1), a lysosomal enzyme that catalyzes the deacylation of S-palmitoylated proteins. Such a defect was associated with impaired palmitoylation of a protein (Rab7), crucial for autophagosome-lysosome fusion, and therefore leading to impaired degradative function along the autophagic pathway (160).
A role for CLN5 in dysregulating autophagy was recently suggested by studies in patient fibroblasts and CLN5-deficient HeLa cells which showed increased autophagy flux (161). Likewise aberrant development was reported in Dictyostelium deleted in cln5, homologous to human CLN5 gene in which autophagy seems to play a regulatory role in terminal differentiation of the amoeba (162).
Interestingly, none of the recently described four experimental conditions, related to mutations in genes associated with different NCL, and associated with impaired autophagy, are associated with genes belonging to the autophagy degradation pathway. That implies that inhibited and/or dysregulated autophagy is the endpoint of a complex network of molecular interactions occurring in NCL neurons, whose outcome is the death of neuronal cells.
Along with investigations directed to the basic mechanism of cell death (such as apoptosis and autophagy), a large amount of data has been obtained over the last two decades from studies aimed at understanding more about the cell physiology in NCL, and other mechanisms which might hamper cell viability and therefore contribute to the cell death process.
Oxidative stress was investigated early, as it was also suggested by the clinical anecdotal observations of transient, worsening of the general wellbeing of affected children under energy-requiring circumstances (e.g. fever, general anesthesia).
Impaired activities of OxPhos enzymes were reported in CLN1 fibroblasts and its putative role on triggering apoptotic cascade was well established (163, 164). Moreover, structural abnormalities of the mitochondrial reticulum as well as abnormal ROS production were described in human fibroblasts (165, 166). More recently, a quantitative proteomic study showed impaired mitochondrial function in different human cells (knocked out in CLN5) models and in Cln5–/– mouse cerebral cortex. Impaired autophagy machinery coupled with mitophagy activation processes were observed linking the CLN5 protein to the process of neuronal death (37).
The involvement of the mitochondrial machinery is not surprising in neurodegeneration (as either primary or secondary event) because of the high dependance of neurons on energy supplied by the oxidative metabolism. These findings, however, reinforce the recent evidence of cross-talk between different cell compartments (including lysosomes and mitochondria, as well as endoplasmic reticulum) which seem to be affected differentially in the NCLs. Greater understanding of the molecular relationships of such intracellular “dialogues” might provide clues toward targeted treatments for specific NCL forms (35).
As in several neurodegenerative disorders (including Alzheimer's disease), inflammatory changes of the neuropil are significant in PPT-1 deficient mice, including the increased production of pro-cytokines, recruitment of inflammatory cells, microglia activation (167–169). The inflammatory response has therefore been considered a powerful amplifier of the NCL disease pathology. Such findings led to several trials using selected immunomodulatory drugs, which alleviated neurological symptoms in the affected animals, but were more effective if applied before the onset of symptomatology (170). The same rationale lead to a clinical trial for juvenile NCL patients (CLN3 disease), using mychophenolate, an immunosuppressant used off label for autoimmune neurological conditions. The drug was tolerated, but there is no evidence so far for a positive effect on clinical disease progression (171).
The medical management of children and young adults affected by one of the NCLs continues to be mainly symptomatic, delivered through a multidisciplinary and multiagency approach, working closely with family members and carers. Medical care should follow internationally agreed standards and guidelines for individual symptoms and organ systems (for example epilepsy, respiratory, orthopedic and gut) and be delivered in line with the holistic values of palliative care. These approaches with particular reference to the NCLs have been described in a number of publications in recent years (108, 109, 120). Experts in rare diseases should be cognisant of advances in these areas or at least be able to signpost families and carers to appropriate expertise. Some advances have been mentioned in previous sections of this review. In particular, advances in management of chest symptoms in children with complex neurodisability and dependence, including for example use of cough assist devices and non-invasive ventilation, together with cardiac pacing suggest sick sinus syndrome. Newer tone management and anti-seizure treatment modalities (including drugs, dietary therapies and stimulation techniques) should be considered and discussed openly when considering the goals of care and potential medical interventions with families.
Perhaps the most troubling clinical symptoms are seizures throughout the disease course and tone/movement disorder management in the later stages.
These are widely reported by parents to be the most worrying and by professionals as the most challenging. Many NCL forms of early childhood onset are characterized by a progressive myoclonic epilepsy syndrome whereas in CLN3 disease with juvenile onset generalized motor and absence seizures predominate. Clinical experience suggests that anti-seizure medications considered best for generalized genetic epilepsies are most effective in these disorders whereas those which are known to exacerbate myoclonus are best avoided. The most commonly used anti-seizure medications are valproate, levetiracetam and the benzodiazepines in varying combinations. Carbamazepine is avoided. Lamotrigine is reported to be helpful in combination with valproate in the later stages of CLN2 disease and is very effective at high doses in older children and young adults with CLN3 disease. Experience of international clinical experts varies but there is consensus that medications for epilepsy are used to alleviate seizure burden rather than with the goal of complete seizure freedom and that drug combinations are usually necessary. In some patients the seizure burden is less at the late stages of the disease and medication can be reduced. Ketogenic diet is not contraindicated, is often very well tolerated and may also be helpful.
Movement disorders are increasingly recognized in the NCLs. In several NCL forms with onset in the preschool or early school years (CLN2, CLN5, CLN6, CLN7, CLN8) a choreo-athetosis or mixed movement disorder with dystonia becomes evident a few years after symptom onset. Determining which involuntary movements are ictal and which are movement disorder can be challenging and EEG video telemetry with event capture can be very helpful. The movement disorder progresses to spasticity in the late stages of all NCL forms, complicated by spinal scoliosis and joint contractures. Baclofen, trihexyphenidyl, clonidine and botulinum toxin injections are used extensively and in combination.
Gabapentin has proven very useful for irritability and distress in the rapidly deteriorating phase of CLN2 disease.
We are beginning to recognize new clinical phenotypes for the few NCL types now amenable to disease modifying therapies such as enzyme replacement and gene therapy. These treatment approaches are new and the full range of implications on symptoms and quality of life as well as survival and longevity is yet to be established. It may well be that there should be a shift from a mainly palliative approach for symptom control to more aggressive intervention with the expectation of complete symptom control (for example seizure freedom) in treated individuals. Close working with the patients themselves, families, caregivers and family representative organizations will be crucial as we go forward to define what standard care should look like.
In LSD and more recently in NCL therapeutic strategies have emerged with the aim of preventing abnormal storage formation and/or to deplete the abnormal endo-lysosomal accumulation, with the ultimate goal to reduce and/or stop the disease progression (122, 172). Several challenges must be overcome, for example the route of drug delivery to the CNS, safety, outcomes and how best to measure efficacy, and above all the still incomplete knowledge of the patho-mechanisms underlying each NCL form. This is still the age of a “gross” therapeutic approach, using replacement therapies (the mutated gene or the missing enzyme), whereas more classical pharmacological treatments targeting crucial steps of the important metabolic pathways still lag behind. It is likely that a combined approach will be necessary to achieve the good therapeutic outcomes patients, families and professionals would like to see.
Complementation therapies (either as gene replacement therapy or enzyme replacement therapy, ERT) are becoming available for some NCL forms (Table 4). ERT became available as part of a clinical trial almost a decade ago for CLN2 disease. Following the decision of the regulatory agencies (FDA and EMA), the recombinant lysosomal enzyme, cerliponase alpha, replacing the ineffective gene product has been commercially available since 2017 (Brineura©). The enzyme is administered directly to the CNS via an intra cerebro-ventricular catheter every 2 weeks. The safety of this approach is well documented by the most expert group with this procedure (173). As for its efficacy there is good evidence that disease progression is slowed down over a 4 year period, as compared with historical untreated controls (174, 175). Likewise, slowed disease progression was observed in a small cohort of CLN2 children (assessed for a shorter period of time), who received intracerebral injection of adenovirus expressing CLN2 (176). Even more interesting results were reported in a very small group of pre-school children who underwent treatment before or a few months after clinical onset of the disease, with delayed onset of disease symptoms as well as maintenance of early cognitive skills (177, 178).
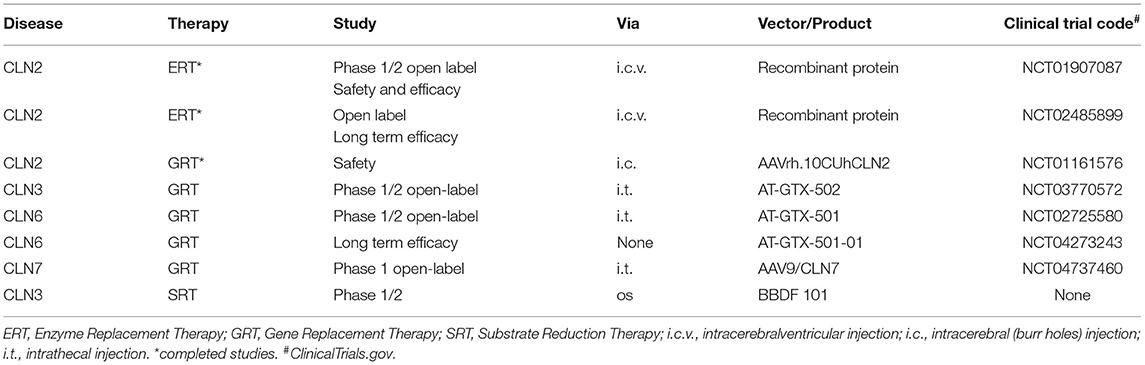
Table 4. Selected therapeutic trials (completed and on-going).
There are ongoing phase 1/2 open label studies to evaluate the safety and efficacy of gene therapy administered intrathecally for CLN3 disease, CLN6 disease and CLN7 disease. These studies have not yet been completed, and the final results are not available (Table 4).
Some preliminary data indicate lysosomal glycosphingolipid accumulation in CLN3 and CLN5 disease. Such findings have suggested an approach using prevention of abnormal lysosomal storage, by interfering with glucosylceramide and ganglioside production using Miglustat a glucosylceramide synthase inhibitor, which is currently used in Niemann-Pick type C disease. This drug is authorized in Europe (but not in USA) for the disease, and beneficial effects of this treatment on juvenile- or adult-onset N-PC were reported on a large cohort of patients (179). Recently it was also shown that trehalose (a disaccharide) promotes lysosomal clearance in storage disease by activation of TFEB, a transcription factor involved in lysosome biogenesis and recruitment (180). By combining the potential benefits of Miglustat and the physiological role of Trehalose a phase 1/2 study is planned using a small number of CLN3 patients using a new recently FDA approved drug (BBDF 101) which contains both components.
There was only one study so far, aiming to deplete the endo-lysosomal storage in CLN1 disease by the combined action of phosphocystemaine (which cleaves thioester linkage in palmitoylated protein and N-acetylcysteine a strong anti-oxidant which also cleaves thiosterer linkage). The long term outcome showed only minor subjective benefits for patients, along with some improvements related to EEG pattern and storage dissolution (181).
NCL are rare, genetically determined, progressive diseases affecting several mammalian species. In humans they affect pre-school and school aged children (or more rarely they start in adulthood) and are defined by clinical criteria supported by pathological features. Amaurosis, seizures, ataxia, behavioral problems, cognitive decline are the major symptoms leading within a relatively short time span to dementia, loss of motor autonomy, blindness and dependence on caregivers. Autofluorescence and selected ultrastructural features of endo-lysosomal storage are the pathological markers detectable both in central and autonomic neurons as well as in several peripheral cells following skin biopsy. Modern diagnosis relies mainly on biochemical (for the NCLs caused by lysosomal enzyme deficiencies) and genetic studies.
Over last two decades dramatic advances in the knowledge of the molecular basis of NCL has been achieved in both humans and several experimental models, but the precise patho-mechanisms leading to cell pathology and neuronal death have not yet been fully elucidated. Nevertheless, a few targeted treatments for the NCLs have become available for human study recently, including enzyme replacement and gene therapies, although only one product is commercially available at present. New pharmacological approaches are foreseen in the near future. The safety and efficacy of such novel treatments are still under scrutiny; there is a conscious effort to ascertain whether new clinical phenotypes may arise as a consequence of these innovative treatments, as recently described in SMA type 1 (182). International collaborations are necessary, recruiting larger cohorts of patients, to generate robust natural history studies to support the regulatory approval process for new drugs (92, 183) and to inform health service delivery.
There is an urgent need to find safe and effective treatments for rare neurodegenerative diseases, such as the NCLs, and mutual agreements between patients, families and advocacy groups, the Health Systems and the pharmaceutical companies are mandatory. With such endeavors, challenges have become evident and will need to be overcome. Examples include ownership and sharing of personal data (natural history data and pre-marketing clinical data), the costs of treatment, the health benefit/cost ratio. These issues are particularly relevant in Europe, where publically funded health systems have to protect and guarantee the privacy of clinical data and, at the same time, have to balance the costs of novel treatments for rare diseases against wider health targets and to guarantee equitable access without discrimination.
AS developed the original concept and design of this study. Both authors have made substantial contributions to further modifications of the work, were actively involved in the revision of this manuscript, approved the submitted version and agreed to be personally accountable for the author's own contributions and to ensure any part of the work are appropriately investigated, documented in accordance with the literature, and read and approved the final manuscript.
This study was supported by the Italian Ministry of University and Research and by a grant from Fondazione Mariani-Child Neurology to AS. AS is member of the European Reference Network MetabERN.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fneur.2022.811686/full#supplementary-material
1. Mole SE, Williams RE, Goebel HH. The Neuronal Ceroid Lipofuscinoses (Batten Disease). New york: Oxford University Press. (2011). doi: 10.1093/med/9780199590018.001.0001
2. Stengel OC. Account of a singular illness among four siblings in the vicinity of Roraas. In: Ceroid Lipofuscinosis (Batten Disease), eds Armstrong D, Koppang N, Rider JA. (Amsterdam: Elsevier/North Holland Biomedical Press). (1826/1982). p. 17–19.
3. Sachs B. A family form of idiocy, generally fatal and associated with early blindness (amaurotic familial idiocy). NY Med J. (1896) 63:697–703.
4. Tay W. Symmetrical changes in the region of the yellow spot in each eye of an infant. Trans Ophtalm Soc. (1881) 1:55–7.
5. Batten FE. Family cerebral degeneration with macular change (so-called juvenile form of family amaurotic idiocy). Q J Med. (1914) 7:444–54.
6. Bielschowski M. Zur Histopathologie und Pathogenese der amaurotischen Idiotie mit besonderer Berucksichtigung der zerebellaren Veranderungen. J Psychol Neurol. (1920) 25:123–99.
7. Jansky J. Dosud nepopsany pripad familiarni amauroticke idiotie komplikovane s hypoplasii mozeckovou. Sborn Lek. (1908) 13:165–96. doi: 10.1111/j.1471-0528.1908.tb14524.x
8. Spielmeyer W. Klinische und amaurotische Untersuchungen uber eine besondere Form von familiarer amaurotischer Idiotie. Histol und Histopathol. (1908) 2:193–251.
9. Vogt H. Familiare amaurotiche Idiotie. Histologische und histopatologischie. Studien Archiv Kinderheilkd. (1909) 51:1–35.
10. Kufs H. Uber eine Spatform der amaurotischen Idiotie und ihre heredofamiliaren Grundlagen. Z Ges Neurol Psychiatr. (1925) 195:165–88. doi: 10.1007/BF02900989
11. Schaffer K. Beitrage zur Nosographie und Histopathologie der amaurotisch-familiaren Idiotieformen. Arch Psychiatr Nervenkr. (1906) 42:127–60. doi: 10.1007/BF02046807
12. Batten FE, Mayou MS. Family cerebral degeneration with macular changes. Proc R Soc Med. (1915) 8:70–90. doi: 10.1177/003591571500801624
13. Sjogren T. Die juvenile amaurotische Idiotie. Klinische und erblichkeitsmedizinische Untersuchungen. Hereditas. (1931) 14:197–426. doi: 10.1111/j.1601-5223.1931.tb02535.x
14. Klenk E. Beitrage zur Chemie der Lipidosen, Niemann-Pickschen Krankheit und amaurotischen Idiotie. Hoppe-Seyler Z Physiol Chem. (1939) 262:128–43. doi: 10.1515/bchm2.1939.262.3-5.128
15. Zeman W, Donohue S. Fine structure of the lipid bodies in juvenile amaurotic idiocy. Acta Neuropathol. (1963) 3:144–9. doi: 10.1007/BF00687063
16. Zeman W. The neuronal ceroid lipofuscinoses. In: Progress in Neuropathology. ed. Zimmermann HM. (New York: Grune Stratton). (1976). p. 207–223.
17. Berkovic SF, Carpenter S, Andermann F, Andermann E, Wolfe SE. Kufs' disease: a critical reappraisal. Brain. (1988) 111:27–62. doi: 10.1093/brain/111.1.27
18. Zeman W, Dyken P. Neuronal ceroid-lipofuscinosis (Batten's disease): relationship to amaurotic family idiocy? Pediatrics. (1969) 44:570–83. doi: 10.1542/peds.44.4.570
19. Svennerholm L. The chemical structure of normal brain and Tay-Sachs gangliosides. Biochem Biophys Res Commun. (1962) 9:436–41. doi: 10.1016/0006-291X(62)90030-X
20. Seehafer SS, Pearce DA. You say lipofuscin, we say ceroid: Defining autofluorescent storage material. Neurobiol Aging. (2006) 27:576–88. doi: 10.1016/j.neurobiolaging.2005.12.006
21. Palmer DN, Fearnley IM, Walker JE, Hall NA, Lake BD, Wolfe LS, et al. Mitochondrial ATP synthase subunit c storage in the ceroid-lipofuscinoses (Batten disease). Am J Med Genet. (1992) 42:561–7. doi: 10.1002/ajmg.1320420428
22. Tyynela J, Palmer DN, Baumann M, Haltia M. Storage of saposins A and D in infantile neuronal ceroid lipofuscinosis. FEBS Lett. (1993) 330:8–12. doi: 10.1016/0014-5793(93)80908-D
23. Anderson G, Goebel HH, Simonati A. Human pathology of NCL. Bioch Biophys Acta. (2013) 1832:1807–26. doi: 10.1016/j.bbadis.2012.11.014
24. Brett EM, Lake BD. Reassessment of rectal approach to neuropathology in childhood. Arch Dis Child. (1975) 50:753–762. doi: 10.1136/adc.50.10.753
25. Simonati A, Rizzuto N. Neuronal Ceroidolipofuscinoses: Pathological features of bioptic specimens in 28 patients. Neurol Sci. (2000) 21:S63–70. doi: 10.1007/s100720070042
26. Wang SH, Williams EM, Takahash K, Nelvagal HR, et al. Enteric Nervous System defects underlie bowel dysfunction in Cln1, Cln2 and Cln3 disease mice: a new therapeutic target? In: NCL2021 October 6-10th abstract book, (abstract 013) (2021). p 27.
27. Haltia M. The neuronal ceroid lipofuscinoses. J Neuropathol Exp Neurol. (2003) 62:1–13. doi: 10.1093/jnen/62.1.1
28. Haltia M, Goebel HH. The neuronal ceroid-lipofuscinoses: a historical introduction. Biochim Biophys Acta. (2013) 1832:1795–800. doi: 10.1016/j.bbadis.2012.08.012
29. Hersheson J, Burke D, Clayton R, Anderson G, Jacques TS, Mills MP, et al. Cathepsin D deficiency causes juvenile onset ataxia and distinctive muscle pathology. Neurology. (2014) 83:1873–5. doi: 10.1212/WNL.0000000000000981
30. Kohlschutter A, Williams RE, Goebel HH, Mole SE, Boustany RM, van Diggelen OP, et al. NCL Diagnosis and Algorithms. In: The Neuronal Ceroid Lipofuscinoses (Batten Disease)., eds. Mole SE, Williams RE, Goebel HH. (New York: Oxford University Press). (2011). p. 24–34. doi: 10.1093/med/9780199590018.003.0003
31. Vesa J, Hellsten E, Verkruyse LA, Camp LA, Rapola J, Santavuori P, et al. Mutations in the palmitoyl protein thioesterase gene causing infantile neuronal ceroid lipofuscinosis. Nature. (1995) 376:584–7. doi: 10.1038/376584a0
32. The International Batten Disease Consortium. Isolation of a novel gene underlying Batten disease, CLN3. Cell. (1995) 82:949–57. doi: 10.1016/0092-8674(95)90274-0
33. Williams RE, Mole SE. New nomenclature and classification scheme for the neuronal ceroid lipofuscinoses. Neurology. (2012) 79:183–91. doi: 10.1212/WNL.0b013e31825f0547
34. Gardner E, Mole SE. The genetic basis of phenotypic heterogeneity in the neuronal ceroid lipofuscinoses. Front Neurol. (2021) 12:754045. doi: 10.3389/fneur.2021.754045
35. Bajaj L, Sharma J, di Ronza A, Zhang P, Eblimit A, Pal R, et al. A CLN6-CLN8 complex recruits lysosomal enzymes at the ER for Golgi transfer. J Clin Invest. (2020) 130:4118–32. doi: 10.1172/JCI130955
36. Di Ronza A, Bajaj L, Sharma J, Sanagasetti D, Lotfi P, Adamski CJ, et al. CLN8 is an endoplasmic reticulum cargo receptor that regulates lysosome biogenesis. Nat Cell Biol. (2018) 20:1370–7. doi: 10.1038/s41556-018-0228-7
37. Doccini S, Morani F, Nesti C, Pezzini F, Calza G, Soliymani R, et al. Proteomic and functional analyses in disease models reveal CLN5 protein involvement in mitochondrial dysfunction. Cell Death Discovery. (2020) 6:18. doi: 10.1038/s41420-020-0250-y
38. Mukherjee AB, Appu AP, Sadhukhan T, Casey S, Mondal A, Zhang Z, et al. Molecular Neurodegeneration. Emerging new roles of the lysosome and neuronal ceroid lipofuscinoses. Mol Neurodegen. (2019) 14:4. doi: 10.1186/s13024-018-0300-6
39. Katz ML, Coates JR, Sibigtroth CM, Taylor JD, Carpentier M, Young WM, et al. Enzyme replacement therapy attenuates disease progression in a canine model of late-infantile neuronal ceroid lipofuscinosis (CLN2 disease). J Neurosci Res. (2014) 92:1591–8. doi: 10.1002/jnr.23423
40. Magrinelli F, Pezzini F, Moro F, Santorelli FM, Simonati A. Diagnostic methods and emerging treatments for adult neuronal ceroid lipofuscinosis (Kufs disease). Expert Opinion in Orphan Drugs. (2017) 5:487–501. doi: 10.1080/21678707.2017.1325359
41. Arsov T, Smith KR, Damiano J, Franceschetti S, Canafoglia L, Bromhead CJ, et al. Kufs disease, the major adult form of neuronal ceroid lipofuscinosis, caused by mutations in CLN6. Am J Hum Genet. (2011) 88:1–8. doi: 10.1016/j.ajhg.2011.04.004
42. Nosková L, Stránecký V, Hartmannová H, Pristoupilová A, Barešová V, Ivánek R, et al. Mutations in DNAJC5, encoding cysteine-string protein alpha, cause autosomal-dominant adult-onset neuronal ceroid lipofuscinosis. Am J Hum Genet. (2011) 89:241–252. doi: 10.1016/j.ajhg.2011.07.003
43. Velinov M, Dolzhanskaya N, Gonzalez M, Powell E, Konidari I, Hulme W, et al. Mutations in the gene DNAJC5 cause autosomal dominant Kufs disease in a proportion of cases: study of the Parry family and 8 other families. PLoS ONE. (2012) 7:e29729. doi: 10.1371/journal.pone.0029729
44. Smith KR, Damiano J, Franceschetti S, Carpenter S, Canafoglia L, Morbin M, et al. Strikingly different clinicopathological phenotypes determined by progranulin-mutation dosage. Am J Hum Genet. (2012) 90:1102–7. doi: 10.1016/j.ajhg.2012.04.021
45. Smith KR, Dahl HH, Canafoglia L, Andermann E, Damiano J, Morbin M, et al. Cathepsin F mutations cause type B Kufs disease, an adult-onset neuronal ceroid lipofuscinosis. Hum Mol Genet. (2013) 22:1417–23. doi: 10.1093/hmg/dds558
46. Qureshi YH, Patel VM, Berman DE, Kothiya MJ, Neufeld JL, Vardarajan B, et al. An Alzheimer's disease-linked loss-of-function cln5 variant impairs cathepsin d maturation, consistent with a retromer trafficking defect. Mol Cell Biol. (2018) 38:e00011–18. doi: 10.1128/MCB.00011-18
47. Geier EG, Bourdenx M, Storm NJ, Cochran JN, Sirkis DW, Hwang JH, et al. Rare variants in the neuronal ceroid lipofuscinosis gene MFSD8 are candidate risk factors for frontotemporal dementia. Acta Neuropathol. (2019) 137:71–88. doi: 10.1007/s00401-018-1925-9
48. Bras J, Verloes A, Schneider SA, Mole SE, Guerreiro RJ. Mutation of the parkinsonism gene ATP13A2 causes neuronal ceroid-lipofuscinosis. Hum Mol Genet. (2012) 21:2646–2650. doi: 10.1093/hmg/dds089
49. Cao Y, Espinola JA, Fossale E, Massey AC, Cuervo AM, MacDonald ME, et al. Autophagy is disrupted in a knock-out mouse model of juvenile neuronal ceroid lipofuscinosis. J Biol Chem. (2006) 281:20483–93. doi: 10.1074/jbc.M602180200
50. Thelen M, Damme M, Schweizer M, Hagel C, Wong AMS, Cooper JD, et al. Disruption of the autophagy-lysosome pathway is involved in neuropathology of the nclf mouse model of neuronal ceroid lipofuscinosis. PLoS ONE. (2012) 7:e35493. doi: 10.1371/journal.pone.0035493
51. Giorgi C, Bouhamida E, Danese A, Previati M, Pinton P, Patergnani S. Relevance of autophagy and mitophagy dynamics and markers in neurodegenerative diseases. Biomedicines. (2021) 9:149. doi: 10.3390/biomedicines9020149
52. Root J, Merino P, Nuckols A, Johnson M, Kukar T. Lysosome dysfunction as a cause of neurodegenerative diseases: Lessons from frontotemporal dementia and amyotrophic lateral sclerosis. Neurobiol Dis. (2021) 154:105360. doi: 10.1016/j.nbd.2021.105360
53. Sleat DE, Donnelly RJ, Lackland H, Liu CG, Sohar I, Pullarkat RK, et al. Association of mutations in a lysosomal protein with classical late-infantile neuronal ceroid lipofuscinosis. Science. (1997) 277:1802–5. doi: 10.1126/science.277.5333.1802
54. van Diggelen OP, Keulemans JL, Winchester B, Hofman IL, Vanhanen SL, Santavuori P, et al. A rapid fluorogenic palmitoyl-protein thioesterase assay: pre- and postnatal diagnosis of INCL. Mol Genet Metab. (1999) 66:240–4. doi: 10.1006/mgme.1999.2809
55. Maeser S, Brindusa-Alina P, Ion L, Rawer S, Kohlschutter A, Santorelli FM, et al. Enzymatic diagnosis of neuronal ceroid lipofuscinoses in dried blood specimens using substrates for concomitant tandem mass spectrometry and fluorimetry. J Mass Spectrom. (2021) 56:4675. doi: 10.1002/jms.4675
56. Savukoski M, Klockars T, Holmberg V, Santavuori P, Lander ES, Peltonen L. CLN5, a novel gene encoding a putative transmembrane protein mutated in Finnish variant late infantile neuronal ceroid lipofuscinosis. Nat Genet. (1998) 19:286–8. doi: 10.1038/975
57. Ranta S, Zhang Y, Ross B, Lonka L, Takkunen E, Messer A, et al. The neuronal ceroid lipofuscinoses in human EPMR and mnd mutant mice are associated with mutations in CLN8. Nat Genet. (1999) 23:233–6. doi: 10.1038/13868
58. Teixeira CA, Espinola J, Huo L, Kohlschütter J, Persaud Sawin DAP, Minassian B, et al. Novel mutations in the CLN6 gene causing a variant late infantile neuronal ceroid lipofuscinosis. Hum Mutat. (2003) 21:502–8. doi: 10.1002/humu.10207
59. Williams RE. Appendix 1: NCL incidence and prevalence data. In: The Neuronal Ceroid Lipofuscinoses (Batten Disease)., eds Mole SE, Williams RE, and Goebel HH. (New York: Oxford University Press) (2011) p. 361–365. doi: 10.1093/med/9780199590018.003.0023
60. Santorelli FM, Garavaglia B, Cardona F, Nardocci N, Dalla Bernardina B, Sartori S, et al. Molecular epidemiology of childhood neuronal ceroid-lipofuscinosis in Italy. Orphanet J Rare Dis. (2013) 8:19. doi: 10.1186/1750-1172-8-19
61. Simpson NA, Wheeler ED, Pearce DA. Screening, diagnosis and epidemiology of Batten disease. Expert Opinion in Orphan Drug. (2014) 2:903–10. doi: 10.1517/21678707.2014.935762
62. Sleat DE, Gedvilaite E, Zhang Y, Lobel P, Xing J. Analysis of large-scale whole exome sequencing data to determine the prevalence of genetically-distinct forms of neuronal ceroid lipofuscinosis. Gene. (2016) 593:284–91. doi: 10.1016/j.gene.2016.08.031
63. Mole SE, Williams RE, Goebel HH. Correlations between genotype, ultrastructural morphology and clinical phenotype in the neuronal ceroid lipofuscinoses. Neurogenetics. (2005) 6:107–26. doi: 10.1007/s10048-005-0218-3
64. Cotman SL, Karaa A, Staropoli JF, Sims KB. Neuronal Ceroid Lipofuscinosis: Impact of recent genetic advances and expansion of the clinicopathologic spectrum. Curr Neurol Neurosci Rep. (2013) 13:366. doi: 10.1007/s11910-013-0366-z
65. Mink JW, Augustine EF, Adams HR, Marshall FJ, Kwon JM. Classification and natural history of the neuronal ceroid lipofuscinoses. J Child Neurol. (2013) 28:1101–5. doi: 10.1177/0883073813494268
66. Schulz A, Kohlschütter A, Mink J, Simonati A, Williams RE. NCL diseases - clinical perspectives. Bioch Biophys Acta. (2013) 1832:1801–6. doi: 10.1016/j.bbadis.2013.04.008
67. Simonati A, Pezzini F, Moro F, Santorelli FM. Neuronal Ceroid Lipofuscinosis: the increasing spectrum of an old disease. Curr Mol Med. (2014) 8:1043–51. doi: 10.2174/1566524014666141010154913
68. Nita AD, Mole SE, Minassian BA. Neuronal ceroid lipofuscinoses. Epileptic Disord. (2016) 18:S73–88. doi: 10.1684/epd.2016.0844
69. Specchio N, Ferretti A, Trivisano M, Pietrafusa N, Pepi C, Calabrese C, et al. Neuronal Ceroid Lipofuscinosis: Potential for Targeted Therapy. Drugs. (2021) 81:101–23. doi: 10.1007/s40265-020-01440-7
70. Steinfeld R, Heim P, von Gregory H, Meyer K, Ullrich K, Goebel HH, et al. Late infantile neuronal ceroid lipofuscinosis: quantitative description of the clinical course in patients with CLN2 mutations. Am J Med Genet. (2002) 112:347–54. doi: 10.1002/ajmg.10660
71. Marshall FJ, de Blieck EA, Mink JW, Dure L, Adams HR, Messing S, et al. A clinical rating scale for Batten disease: reliable and relevant for clinical trials. Neurology. (2005) 65:275–9. doi: 10.1212/01.wnl.0000169019.41332.8a
72. Masten MC, Williams JD, Vermilion J, Adams HR, Vierhile A, Collins A, et al. The CLN3 Disease Staging System: A new tool for clinical research in Batten disease. Neurology. (2020) 94:e2436–40. doi: 10.1212/WNL.0000000000009454
73. Varvagiannis K, Hanquinet S, Billieux MH, De Luca R, Rimensberger P, Lidgren M, et al. Congenital neuronal ceroid lipofuscinosis with a novel CTSD gene mutation: a rare cause of neonatal-onset neurodegenerative disorder. Neuropediatrics. (2018) 49:150–3. doi: 10.1055/s-0037-1613681
74. Vanhanen S-L, Sainio K, Lappi M, Santavuori P. EEG and evoked potentials in infantile ceroid-lipofuscinosis. Dev Med Child Neurol. (1997) 39:456–63. doi: 10.1111/j.1469-8749.1997.tb07465.x
75. Simonati A, Tessa A, Dalla Bernardina B, Biancheri R, Veneselli E, Tozzi G, et al. Variant late infantile neuronal ceroidlipofuscinosis because of CLN1 mutations. Pediatr Neurol. (2009) 40:271–6. doi: 10.1016/j.pediatrneurol.2008.10.018
76. Doccini S, Sartori S, Maeser S, Pezzini F, Rossato S, Moro F, et al. Early infantile Neuronal Ceroid Lipofuscinosis (CLN10 Disease) associated with a novel mutation in CTSD. J Neurol. (2016) 263:1029–1032. doi: 10.1007/s00415-016-8111-6
77. Pampiglione G, Harden A. Neurophysiological identification of a late infantile form of 'neuronal lipidosis'. J Neurol Neurosurg Psychiatry. (1973) 36:68–74. doi: 10.1136/jnnp.36.1.68
78. Specchio N, Bellusci M, Pietrafusa N, Trivisano M, de Palma L, Vigevano F. Photosensitivity is an early marker of neuronal ceroid lipofuscinosis type 2 disease. Epilepsia. (2017) 58:1380–8. doi: 10.1111/epi.13820
79. Peña JA, Cardozo JJ, Montiel CM, Molina OM, Boustany R-M. Serial MRI findings in the Costa Rican variant of neuronal ceroid-lipofuscinosis. Pediatr Neurol. (2001) 25:78–80. doi: 10.1016/S0887-8994(01)00284-3
80. Berkovic SF, Oliver KL, Canafoglia L, Krieger P, Damiano JA, Hildebrand MS, et al. Kufs disease due to mutation of CLN6: clinical, pathological and molecular features. Brain. (2019) 142:59–69. doi: 10.1093/brain/awy297
81. Augustine EF, Adams HR, Beck CA, Vierhile A, Kwon JM, Rothberg PG, et al. Standardized assessment of seizures in patients with juvenile neuronal ceroid lipofuscinosis. Dev Med Child Neurol. (2015) 57:366–71. doi: 10.1111/dmcn.12634
82. Arntsen V, Strandheim J, Helland IB, Sand T, Brodtkorb E. Epileptological aspects of juvenile neuronal ceroid lipofuscinosis (CLN3 disease) through the lifespan. Epilepsy Behav. (2019) 94:59–64. doi: 10.1016/j.yebeh.2019.02.020
83. Ostergaard JR. Gait phenotype in Batten disease: a marker of disease progression. Eur J Pediatr Neurol. (2021) 35:1–7. doi: 10.1016/j.ejpn.2021.09.004
84. Simonati A, Santorum E, Tessa A, Polo A, Simonetti F, Dalla Bernardina B, et al. A CLN2 nonsense mutation is associated with severe caudate atrophy and dystonia. Neuropediatrics. (2000) 31:199–201. doi: 10.1055/s-2000-7453
85. Saini AG, Sankhyan N, Singhi P. Chorea in late infantile neuronal ceroid lipofuscinosis: an atypical presentation. Pediatr Neurol. (2016) 60:75–8. doi: 10.1016/j.pediatrneurol.2016.02.015
86. Nickel M, Simonati A, Jacoby D, Lezius S, Kilian D, Van de Graaf B, et al. Disease characteristics and progression in patients with late infantile neuronal ceroid lipofuscinosis type 2 (CLN2) disease: an observational cohort study. Lancet Child Adolesc Health. (2018) 2:582–90. doi: 10.1016/S2352-4642(18)30179-2
87. Adams HR, Mink JW. Neurobehavioral features and natural history of juvenile neuronalceroid lipofuscinosis (Batten disease). J Child Neurol. (2013) 28:1128–36. doi: 10.1177/0883073813494813
88. Ostergaard JR. Juvenile neuronal ceroid lipofuscinosis (Batten disease): current insights. Degener Neurol Neuromuscul Dis. (2016) 6:73–83. doi: 10.2147/DNND.S111967
89. Adams HR, Beck CA, Levy E, Jordan R, Kwon JM, Marshall FJ, et al. Genotype does not predict severity of behavioural phenotype in juvenile neuronal ceroid lipofuscinosis (Batten disease). Dev Med Child Neurol. (2010) 52:637–43. doi: 10.1111/j.1469-8749.2010.03628.x
90. Cialone J, Adams HR, Augustine EF, Marshall FJ, Kwon JM, Newhouse N, et al. Females experience a more severe disease course in Batten disease. J Inherit Metab Dis. (2012) 35:549–55. doi: 10.1007/s10545-011-9421-6
91. Xin W, Mullen TE, Kiely R, Min J, Feng X, Cao Y, et al. CLN5 mutations are frequent in juvenile and late-onset non-Finnish patients with NCL. Neurology. (2010) 74:565–71. doi: 10.1212/WNL.0b013e3181cff70d
92. Simonati A, Williams RE, Nardocci N, Laine M, Battini R, Schulz A, et al. Disease natural history and phenotypic profiles in a cohort of patients affected with variant late infantile ceroid-lipofuscinosis 5 (CLN5). Dev Med Child Neurol. (2017) 58:815–21. doi: 10.1111/dmcn.13473
93. Lourenço CM, Pessoa A, Mendes CC, Rivera-Nieto C, Vergara D, Troncoso M, et al. Revealing the clinical phenotype of atypical neuronal ceroid lipofuscinosis type 2 disease: Insights from the largest cohort in the world. J Paediatr Child Health. (2021) 57:519–25. doi: 10.1111/jpc.15250
94. Ouseph MM, Kleinman ME, Wang QJ. Vision loss in juvenile neuronal ceroid lipofuscinosis (Batten disease). Ann N Y Acad Sci. (2016) 1371:55–67. doi: 10.1111/nyas.12990
95. Kovacs KD, Patel S, Orlin A, Kim K, Van Everen S, Conner T, et al. Symmetric age association of retinal degeneration in patients with CLN2-associated batten disease. Ophthalmol Retina. (2020). 4:728–36. doi: 10.1016/j.oret.2020.01.011
96. Weleber RG, Gupta N, Trzupek KM, Wepner MS, Kurz DE, Milam AH. Electroretinographic and clinicopathologic correlations of retinal dysfunction in infantile neuronal ceroid lipofuscinosis (infantile Batten disease). Mol Genet Metab. (2004) 83:128–37. doi: 10.1016/j.ymgme.2004.06.019
97. Khan KN, El-Asrag ME, Ku CA, Holder GE, McKibbin M, Arno G, et al. Specific alleles of CLN7/MFSD8, a protein that localizes to photoreceptor synaptic terminals, cause a spectrum of nonsyndromic retinal dystrophy. Invest Ophthalmol Vis Sci. (2017) 58:2906–14. doi: 10.1167/iovs.16-20608
98. Kuper WFE, van Alfen C, Rigterink RH, Fuchs SA, van Genderen MM, van Hasselt PM. Timing of cognitive decline in CLN3 disease. J Inherit Metab Dis. (2018) 41:257–61. doi: 10.1007/s10545-018-0143-x
99. Bauwens M, Storch S, Weisschuh N, Ceuterick-de Groote C, De Rycke R, Guillemyn B, et al. Functional characterization of novel MFSD8 pathogenic variants anticipates neurological involvement in juvenile isolated maculopathy. Clin Genet. (2020) 97:426–436. doi: 10.1111/cge.13673
100. Chin JJ, Behnam B, Davids M, Sharma P, Zein WM, Wang C, et al. Novel mutations in CLN6 cause late-infantile neuronal ceroid lipofuscinosis without visual impairment in two unrelated patients. Mol Genet Metab. (2019) 126:188–95. doi: 10.1016/j.ymgme.2018.12.001
101. Lin T-Y, Chang Y-C, Hsiao Y-R, Chien Y, Cheng Y-C, Wu J-R, et al. Identification of novel genomic-variant patterns of OR56A5, OR52L1, and CTSD in retinitis pigmentosa patients by whole-exome sequencing. Int J Mol Sci. (2021) 22:5594. doi: 10.3390/ijms22115594
102. Wang F, Wang H, Tuan H-F, Nguyen DH, Sun V, Keser V, et al. Next generation sequencing-based molecular diagnosis of retinitis pigmentosa: Identification of a novel genotypephenotype correlation and clinical refinements. Hum Genet. (2014) 133:331–345. doi: 10.1007/s00439-013-1381-5
103. Murray SJ, Russell KN, Melzer TR, Gray SJ, Heap SJ, Palmer DN, et al. Intravitreal gene therapy protects against retinal dysfunction and degeneration in sheep with CLN5 Batten disease. Exp Eye Res. (2021) 207:108600. doi: 10.1016/j.exer.2021.108600
104. Whiting RE, Kick GR, Ota-Kuroki J, Lim S, Castaner LJ, Jensen CA, et al. Intravitreal enzyme replacement inhibits progression of retinal degeneration in canine CLN2 neuronal ceroid lipofuscinosis. Exp Eye Res. (2020) 198:108135. doi: 10.1016/j.exer.2020.108135
105. Kirveskari E, Partinen M, Salmi T, Sainio K, Telakivi T, Hämäläinen M, et al. Sleep alterations in juvenile neuronal ceroid-lipofuscinosis. Pediatr Neurol. (2000) 22:347–54. doi: 10.1016/S0887-8994(00)00138-7
106. Kirveskari E, Partinen M, Santavuori P. Sleep and its disturbance in a variant form of late infantile neuronal ceroid lipofuscinosis (CLN5). J Child Neurol. (2001) 16:707–13. doi: 10.1177/088307380101601001
107. Lehwald LM, Pappa R, Steward S, de Los Reyes E. Neuronal ceroid lipofuscinosis and associated sleep abnormalities. Pediatr Neurol. (2016) 59:30–5. doi: 10.1016/j.pediatrneurol.2016.02.009
108. Augustine EF, Adams HR, de Los Reyes E, Drago K, Frazier M, Guelbert N, et al. Management of CLN1 disease: international clinical consensus. Paediatr Neurol. (2021) 120:38–51. doi: 10.1016/j.pediatrneurol.2021.04.002
109. Mole SE, Schulz A, Badoe E, Berkovic SF, de Los Reyes EC, Dulz S, et al. Guidelines on the diagnosis, clinical assessments, treatment and management for CLN2 disease patients. Orphanet J Rare Dis. (2021) 16:185. doi: 10.1186/s13023-021-01813-5
110. Hätönen T, Kirveskari E, Heiskala H, Sainio K, Laakso ML, Santavuori P. Melatonin ineffective in neuronal ceroid lipofuscinosis patients with fragmented or normal motor activity rhythms recorded by wrist actigraphy. Mol Genet Metab. (1999) 66:401–6. doi: 10.1006/mgme.1999.2815
111. Staropoli JF, Xin W, Barone R, Cotman SL, Sims KB. An atypical case of neuronal ceroid lipofuscinosis with co-inheritance of a variably penetrant POLG1 mutation. BMC Med Genet. (2012) 13:50. doi: 10.1186/1471-2350-13-50
112. Sun Y, Almomani R, Breedveld GJ, Santen GW, Aten E, Lefeber DJ, et al. Autosomal recessive spinocerebellar ataxia 7 (SCAR7) is caused by variants in TPP1, the gene involved in classic late-infantile neuronal ceroid lipofuscinosis 2 disease (CLN2 Disease). Hum Mutat. (2013) 34:706–13. doi: 10.1002/humu.22292
113. Di Giacopo R, Cianetti L, Caputo V, La Torraca I, Piemonte F, Ciolfi A, et al. Protracted late infantile ceroid lipofuscinosis due to TPP1 mutations: Clinical, molecular and biochemical characterization in three sibs. J Neurol Sci. (2015) 356:65–71. doi: 10.1016/j.jns.2015.05.021
114. Platt FM, d'Azzo A, Davidson BL, Neufeld EF, Tifft CJ. Lysosomal storage diseases. Nat Rev Dis Primers. (2018) 4:27. doi: 10.1038/s41572-018-0025-4
115. Østergaard JR, Rasmussen TB, Mølgaard H. Cardiac involvement in juvenile neuronal ceroid lipofuscinosis (Batten disease). Neurology. (2011) 76:1245–51. doi: 10.1212/WNL.0b013e31821435bd
116. Ofman IL, Van Der Wal AC, Dingemans KP, Becker AE. Cardiac pathology in neuronal ceroid lipofuscinoses-a clinicopathologic correlation in three patients. Eur J Paediatr Neurol. (2001) 5:213–7. doi: 10.1053/ejpn.2000.0465
117. Fukumura S, Saito Y, Saito T, Komaki H, Nakagawa E, Sugai K, et al. Progressive conduction defects and cardiac death in late infantile neuronal ceroid lipofuscinosis. Dev. Med. Child Neurol. (2021) 54:663–666. doi: 10.1111/j.1469-8749.2011.04170.x
118. Augestad LB, Flanders WD. Occurrence of and mortality from childhood neuronal ceroid lipofuscinosis in Norway. J Child Neurol. (2006) 21:917–22. doi: 10.1177/08830738060210110801
119. Siintola E, Partanen S, Strömme P, Haapanen A, Haltia M, Maehlen J, et al. Cathepsin D deficiency underlies congenital human neuronal ceroid-lipofuscinosis. Brain. (2006) 129:1438–45. doi: 10.1093/brain/awl107
120. Williams RE, Adams HR, Blohm M, Cohen-Pfeffer JL, de Los Reyes E, Denecke J, et al. Management Strategies for CLN2 Disease. Pediatr Neurol. (2017) 69:102–12. doi: 10.1016/j.pediatrneurol.2017.01.034
121. Gissen P, Specchio N, Olaye A, Jain M, Butt T, Ghosh W, et al. Investigating health-related quality of life in rare diseases: a case study in utility value determination for patients with CLN2 disease (neuronal ceroid lipofuscinosis type 2). Orphanet J Rare Dis. (2021) 16:217. doi: 10.1186/s13023-021-01829-x
122. Mole SE, Anderson G, Band HA, Berkovic SF, Cooper JD, Kleine Holthaus SM, et al. Clinical challenges and future therapeutic approaches for neuronal ceroid lipofuscinosis. Lancet Neurol. (2019) 18:107–16. doi: 10.1016/S1474-4422(18)30368-5
123. Nelvagal HR, Lange J, Takahashi K, Tarczyluk-Wells MA, Cooper JD. Pathomechanisms in the neuronal ceroid lipofuscinoses. Biochim Biophys Acta Mol Basis Dis. (2020) 1866:165570. doi: 10.1016/j.bbadis.2019.165570
124. Sawiak SJ, Perumal SR, Rudiger SR, Matthews L, Mitchell NL, McLaughlan CJ, et al. Rapid and progressive regional brain atrophy in CLN6 Batten disease affected sheep measured with longitudinal magnetic resonance imaging. PLoS ONE. (2015) 10:e0132331. doi: 10.1371/journal.pone.0132331
125. Perentos N, Martins AQ, Watson TC, Bartsch U, Mitchell NL, Palmer DN, et al. Translational neurophysiology in sheep: measuring sleep and neurological dysfunction in CLN5 Batten disease affected sheep. Brain. (2015) 138:862–74. doi: 10.1093/brain/awv026
126. Russell KN, Mitchell NL, Anderson NG, Bunt CR. Computed tomography provides enhanced techniques for longitudinal monitoring of progressive intracranial volume loss associated with regional neurodegeneration in ovine neuronal ceroid lipofuscinoses. Brain Behav. (2018) 8:e01096. doi: 10.1002/brb3.1096
127. Kline RA, Wishart TM, Mills K, Heywood WE. Applying modern Omic technologies to the Neuronal Ceroid Lipofuscinoses. Biochim Biophys Acta Mol Basis Dis. (2020) 1866:165498. doi: 10.1016/j.bbadis.2019.06.012
128. Mitchell NL, Russell KN, Wellby MP, Wicky HE, Schoderboeck L, Barrell GK, et al. Longitudinal in vivo monitoring of the CNS demonstrates the efficacy of gene therapy in a sheep model of CLN5 batten disease. Mol Ther. (2018) 26:2366-78. doi: 10.1016/j.ymthe.2018.07.015
129. Minnis CJ, Thornton CD, FitzPatrick LM, Tristan R, McKay TR. Cellular models of Batten disease. Biochim Biophys Acta Mol Basis Dis. (2020) 1866:165559. doi: 10.1016/j.bbadis.2019.165559
130. Uusi-Rauva K, Blom T, Schantz-Fant V, Blom T, Jalanko A, Kyttälä A. Induced pluripotent stem cells derived from a CLN5 patient manifest phenotypic characteristics of neuronal ceroid lipofuscinoses. Int J Mol Sci. (2017) 18:955. doi: 10.3390/ijms18050955
131. Lojewski X, Staropoli JF, Biswas-Legrand S, Simas AM, Haliw L, Selig MK, et al. Human iPSC models of neuronal ceroid lipofuscinosis capture distinct effects of TPP1 and CLN3 mutations on the endocytic pathway. Hum Mol Genet. (2014) 23:2005–22. doi: 10.1093/hmg/ddt596
132. Eaton SL, Proudfoot C, Lillico SG, Skehel P, Kline RA, Hamer K, et al. CRISPR/Cas9 mediated generation of an ovine model for infantile neuronal ceroid lipofuscinosis (CLN1 disease). Sci Rep. (2019) 9:9891. doi: 10.1038/s41598-019-45859-9
133. Ma L, Prada AM, Schmidt M, Morrow EM. Generation of pathogenic TPP1 mutations in human stem cells as a model for neuronal ceroidlipofuscinosis type 2 disease. Stem Cell Res. (2021) 53:102323. doi: 10.1016/j.scr.2021.102323
134. Chiaradia I, Lancaster MA. Brain organoids for the study of human neurobiology at the interface of in vitro and in vivo. Nat Neurosci. (2020) 23:1496–508. doi: 10.1038/s41593-020-00730-3
135. Gomez-Giro G, Arias-Fuenzalida J, Jarazo J, Zeuschner D, Ali M, Possemis N, et al. Synapse alterations precede neuronal damage and storage pathology in a human cerebral organoid model of CLN3-juvenile neuronal ceroid lipofuscinosis. Acta Neuropathol Commun. (2019) 7:222. doi: 10.1186/s40478-019-0871-7
136. Autti TH, Hämäläinen J, Mannerkoski M, Van Ven Leemput K, Aberg LE. JNCL patients show marked brain volume alterations on longitudinal MRI in adolescence. J Neurol. (2008) 255:1226–30. doi: 10.1007/s00415-008-0891-x
137. Baker EH, Levin SW, Zhang Z, Mukherjee AB. MRI brain volume measurements in infantile neuronal ceroid lipofuscinosis. AJNR Am J Neuroradiol. (2017) 38:376–82. doi: 10.3174/ajnr.A4978
138. Dyke JP, Sondhi D, Voss HU, Yohay K, Hollmann C, Mancenido D, et al. Brain region-specific degeneration with disease progression in late infantile neuronal ceroid lipofuscinosis (CLN2 Disease). AJNR Am J Neuroradiol. (2016) 37:1160–9. doi: 10.3174/ajnr.A4669
139. Löbel U, Sedlacik J, Nickel M, Lezius S, Fiehler J, Nestrasil I, et al. Volumetric description of brain atrophy in Neuronal Ceroid Lipofuscinosis 2: supratentorial gray matter shows uniform disease progression. AJNR Am J Neuroradiol. (2016) 37:1938–43. doi: 10.3174/ajnr.A4816
140. Roine T, Roine U, Tokola A, Balk MH, Mannerkoski M, Åberg L, et al. Topological alterations of the structural brain connectivity network in children with juvenile neuronal ceroid lipofuscinosis. AJNR Am J Neuroradiol. (2019) 40:2146–53. doi: 10.3174/ajnr.A6306
141. Tyynelä J, Cooper JD, Khan MN, Shemilt SJ, Haltia M. Hippocampal pathology in the human neuronal ceroid-lipofuscinoses: distinct patterns of storage deposition, neurodegeneration and glial activation. Brain Pathol. (2004) 14:349–57. doi: 10.1111/j.1750-3639.2004.tb00077.x
142. Radke J, Stenzel W, Goebel HH. Human NCL neuropathology. Biochim Biophys Acta. (2015) 1852:2262–6. doi: 10.1016/j.bbadis.2015.05.007
143. Lange J, Haslett LJ, Lloyd-Evans E, Pocock JM, Sands MS, Williams BP, et al. Compromised astrocyte function and survival negatively impact neurons in infantile neuronal ceroid lipofuscinosis. Acta Neuropathol Commun. (2018) 6:74. doi: 10.1186/s40478-018-0575-4
144. Kielar C, Wishart TM, Palmer A, Dihanich S, Wong AM, Macauley SL, et al. Molecular correlates of axonal and synaptic pathology in mouse models of Batten disease. Hum Mol Genet. (2009) 18:4066–80. doi: 10.1093/hmg/ddp355
145. Autti T, Joensuu R, Aberg J. Decreased T2 signal. in the thalami. may be a sign of lysosomal storage disease. Neuroradiology. (2007) 49:571–8. doi: 10.1007/s00234-007-0220-6
146. von Schantz C, Kielar C, Hansen SN, Pontikis CC, Alexander NA, Kopra O, et al. Progressive thalamocortical neuron loss in Cln5 deficient mice: distinct effects in Finnish variant late infantile NCL. Neurobiol Dis. (2009) 34:308–19. doi: 10.1016/j.nbd.2009.02.001
147. Shyng C, Nelvagal HR, Dearborn JT, Tyynelä J, Schmidt RE, Sands MS, et al. Synergistic effects of treating the spinal cord and brain in CLN1 disease. Proc Natl Acad Sci U S A. (2017) 114:E5920–E5929. doi: 10.1073/pnas.1701832114
148. Pezzini F, Bettinetti L, Di Leva F, Bianchi M, Zoratti E, Carrozzo R, et al. Transcriptomic profiling discloses molecular and cellular events related to neuronal differentiation in SH-SY5Y neuroblastoma cells. Cell Mol Neurobiol. (2017) 37:665–82. doi: 10.1007/s10571-016-0403-y
149. Amorim IS, Mitchell NL, Palmer DN, Sawiak SJ, Mason R, Wishart TM, et al. Molecular neuropathology of the synapse in sheep with CLN5 Batten disease. Brain Behav. (2015) 5:e00401. doi: 10.1002/brb3.401
150. Kim SJ, Zhang Z, Sarkar C, Tsai PC, Lee YC, Dye L, et al. Palmitoyl protein thioesterase-1 deficiency impairs synaptic vesicle recycling at nerve terminals, contributing to neuropathology in humans and mice. J Clin Invest. (2008) 118:3075–86. doi: 10.1172/JCI33482
151. Koster KP, Francesconi W, Berton F, Alahmadi S, Srinivas R, Yoshii A. Developmental NMDA receptor dysregulation in the infantile neuronal ceroid lipofuscinosis mouse model. Elife. (2019) 8:e40316. doi: 10.7554/eLife.40316
152. Demontis GC, Pezzini F, Margari E, Bianchi M, Longoni B, Doccini S, et al. Electrophysiologic profile remodeling via selective suppression of voltage-gated currents by CLN1/PPT1 over expression in human neuronal-like cells. Front Cell Neurosci. (2020) 14:569598 doi: 10.3389/fncel.2020.569598
153. Lane SC, Jolly RD, Schmechel DE, Alroy J, Boustany RM. Apoptosis as the mechanism of neurodegeneration in Batten's disease. J Neurochem. (1996) 67:677–83. doi: 10.1046/j.1471-4159.1996.67020677.x
154. Hachiya Y, Hayashi M, Kumada S, Uchiyama A, Tsuchiya K, Kurata K. Mechanisms of neurodegeneration in neuronal ceroid-lipofuscinoses. Acta Neuropathol. (2006) 111:168–77. doi: 10.1007/s00401-005-0024-x
155. Boya P, González-Polo RA, Casares N, Perfettini JL, Dessen P, Larochette N, et al. Inhibition of macroautophagy triggers apoptosis. Mol Cell Biol. (2005) 25:1025–40. doi: 10.1128/MCB.25.3.1025-1040.2005
156. Zhang Z, Lee YC, Kim SJ, Choi MS, Tsai PC, Xu Y, et al. Palmitoyl-protein thioesterase-1 deficiency mediates the activation of the unfolded protein response and neuronal apoptosis in INCL. Hum Mol Genet. (2006) 15:337–46. doi: 10.1093/hmg/ddi451
157. Shacka JJ, Klocke BJ, Young C, Shibata M, Olney JW, Uchiyama Y, et al. Cathepsin D deficiency induces persistent neurodegeneration in the absence of Bax-dependent apoptosis. J Neurosci. (2007) 27:2081–90. doi: 10.1523/JNEUROSCI.5577-06.2007
158. Seranova E, Connolly KJ, Zatyka M, Rosenstock TR, Barrett T, Tuxworth RI, et al. Dysregulation of autophagy as a common mechanism in lysosomal storage diseases. Essays Biochem. (2017) 61:733–49. doi: 10.1042/EBC20170055
159. Metz KA, Teng X, Coppens I, Lamb HM, Wagner BE, Rosenfeld JA, et al. KCTD7 deficiency defines a distinct neurodegenerative disorder with a conserved autophagy-lysosome defect. Ann Neurol. (2018) 84:766–80. doi: 10.1002/ana.25351
160. Sarkar C, Sadhukhan T, Bagh MB, Appu AP, Chandra G, Mondal A, et al. Cln1-mutations suppress Rab7-RILP interaction and impair autophagy contributing to neuropathology in a mouse model of infantile neuronal ceroid lipofuscinosis. J Inherit Metab. (2020) 43:1082–101. doi: 10.1002/jimd.12242
161. Adams J, Feuerborn M, Molina JA, Wilden AR, Adhikari B, Budden T, et al. Autophagy-lysosome pathway alterations and alpha-synuclein up-regulation in the subtype of neuronal ceroid lipofuscinosis, CLN5 disease. Sci Rep. (2019) 9:151. doi: 10.1038/s41598-018-36379-z
162. McLaren MD, Mathavarajah S, Kim WD, Yap SQ, Huber RJ. Aberrant autophagy impacts growth and multicellular development in a dictyostelium knockout model of CLN5 disease. Front Cell Dev Biol. (2021) 9:657406. doi: 10.3389/fcell.2021.657406
163. Jolly RD, Brown S, Das AM, Walkley SU. Mitochondrial dysfunction in the neuronal ceroidlipofuscinoses (Batten disease). Neurochem Internat. (2002) 40:565–71. doi: 10.1016/S0197-0186(01)00128-0
164. Wei H, Kim SJ, Zhang Z, Tsai PC, Wisniewski KE, Mukherjee AB. ER and oxidative stresses are common mediators of apoptosis in both neurodegenerative and non-neurodegenerative lysosomal storage disorders and are alleviated by chemical chaperones. Hum Mol Genet. (2008) 17:469–77. doi: 10.1093/hmg/ddm324
165. Pezzini F, Gismondi F, Tessa A, Tonin P, Carrozzo R, Mole SE, et al. Involvement of the mitochondrial compartment in human NCL fibroblasts. Biochem Biophys Res Commun. (2011) 416:159–64. doi: 10.1016/j.bbrc.2011.11.016
166. Vidal-Donet JM, Cárcel-Trullols J, Casanova B, Aguado C, Knecht E. Alterations in ROS activity and lysosomal pH account for distinct patterns of macroautophagy in LINCL and JNCL fibroblasts. PLoS ONE. (2013) 8:e55526. doi: 10.1371/journal.pone.0055526
167. Macauley SL, Wong AM, Shyng C, Augner DP, Dearborn JT, Pearse Y, et al. An anti-neuroinflammatory that targets dysregulated glia enhances the efficacy of CNS-directed gene therapy in murine infantile neuronal ceroid lipofuscinosis. J Neurosci. (2014) 34:13077–82. doi: 10.1523/JNEUROSCI.2518-14.2014
168. Groh J, Kühl TG, Ip CW, Nelvagal HR, Sri S, Duckett S, et al. Immune cells perturb axons and impair neuronal survival in a mouse model of infantile neuronal ceroid lipofuscinosis. Brain. (2013) 136:1083–101. doi: 10.1093/brain/awt020
169. Sadhukhan T, Bagh MB, Appu AP, Mondal A, Zhang W, Liu A, et al. In a mouse model of INCL reduced S-palmitoylation of cytosolic thioesterase APT1 contributes to microglia proliferation and neuroinflammation. J Inherit Metab Dis. (2021) 44:1051–69. doi: 10.1002/jimd.12379
170. Groh J, Berve K, Martini R. Immune modulation attenuates infantile neuronal ceroid lipofuscinosis in mice before and after disease onset. Brain Commun. (2021) 3:fcab047. doi: 10.1093/braincomms/fcab047
171. Augustine EF, Beck CA, Adams HR, Defendorf S, Vierhile A, Timm D, et al. Short-term administration of mycophenolate is well-tolerated in CLN3 disease (juvenile neuronal ceroid lipofuscinosis). JIMD Rep. (2019) 43:117–24. doi: 10.1007/8904_2018_113
172. Geraets RD, yon Koh S, Hastings ML, Kielian T, Pearce DA, Weimer JM. Moving towards effective therapeutic strategies for Neuronal Ceroid Lipofuscinosis. Orphanet J Rare Dis. (2016) 11:40. doi: 10.1186/s13023-016-0414-2
173. Schwering C, Kammler G, Wibbeler E, Christner M, Knobloch JK, Nickel M, et al. Development of the “Hamburg best practice guidelines for ICV-enzyme replacement therapy (ERT) in CLN2 disease” based on 6 years treatment experience in 48 patients. J Child Neurol. (2021) 36:635–41. doi: 10.1177/0883073821989154
174. Schulz A, Ajayi T, Specchio N, de Los Reyes E, Gissen P, Ballon D, et al. Study of intraventricular cerliponase alfa for CLN2 Disease. N Engl J Med. (2018) 378:1898–907. doi: 10.1056/NEJMoa1712649
175. Kohlschutter A, Schulz A, Bartsch U, Storch S. Current and emerging treatment strategies for neuronal ceroid lipofuscinoses. CNS Drugs. (2019) 33:315–25. doi: 10.1007/s40263-019-00620-8
176. Sondhi D, Kaminsky SM, Hackett NR, Pagovich OE, Rosenberg JB, De BP, et al. Slowing late infantile Batten disease by direct brain parenchymal administration of a rh.10 adeno-associated virus expressing CLN2. Sci Transl Med. (2020) 12:eabb5413. doi: 10.1126/scitranslmed.abb5413
177. Schaefers J, van der Giessen LJ, Klees C, Jacobs EH, Sieverdink S, Dremmen MHG, et al. Presymptomatic treatment of classic late-infantile neuronal ceroid lipofuscinosis with cerliponase alfa. Orphanet J Rare Dis. (2021) 16:221. doi: 10.1186/s13023-021-01858-6
178. Estublier B, Cano A, Hoebeke C, Pichard S, Scavarda D, Desguerre I, et al. Cerliponase alfa changes the natural history of children with neuronal ceroid lipofuscinosis type 2: the first French cohort. Eur J Paediatr Neurol. (2021) 30:17–21. doi: 10.1016/j.ejpn.2020.12.002
179. Patterson MC, Mengel E, Vanier MT, Schwierin B, Muller A, Cornelisse P, et al. Stable or improved neurological manifestations during miglustat therapy in patients from the international disease registry for Niemann-Pick disease type C: an observational cohort study. Orphanet J Rare Dis. (2015) 10:65. doi: 10.1186/s13023-015-0284-z
180. Bajaj L, Lotfi P, Pal R, Ronza AD, Sharma J, Sardiello M. Lysosome biogenesis in health and disease. J Neurochem. (2019) 148:573–89. doi: 10.1111/jnc.14564
181. Levin SW, Baker EH, Zein WM, Zhongjian Z, Quezado ZMNC, Ning M, et al. Oral cysteamine bitartrate and N-acetylcysteine for patients with infantile neuronal ceroid lipofuscinosis: a pilot study. Lancet Neurol. (2014) 13:777–87. doi: 10.1016/S1474-4422(14)70142-5
182. Masson R, Brusa C, Scoto M, Baranello G. Brain, cognition and language development in spinal muscular atrophy type 1: a scoping review. Dev. Med. Child Neurol. (2021) 63:527–36. doi: 10.1111/dmcn.14798
Keywords: neuronal ceroid lipofuscinosis, NCL clinical features, NCL pathogenetic mechanisms, NCL treatments, NCL review
Citation: Simonati A and Williams RE (2022) Neuronal Ceroid Lipofuscinosis: The Multifaceted Approach to the Clinical Issues, an Overview. Front. Neurol. 13:811686. doi: 10.3389/fneur.2022.811686
Received: 09 November 2021; Accepted: 11 January 2022;
Published: 11 March 2022.
Edited by:
Nicola Specchio, Bambino Gesù Children's Hospital (IRCCS), ItalyReviewed by:
Sara Mole, University College London, United KingdomCopyright © 2022 Simonati and Williams. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Alessandro Simonati, YWxlc3NhbmRyby5zaW1vbmF0aUB1bml2ci5pdA==; Ruth E. Williams, cnV0aC53aWxsaWFtc0Bnc3R0Lm5ocy51aw==
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.