 Qi Yang
Qi Yang Qinle Zhang1,2
Qinle Zhang1,2 Shang Yi
Shang Yi Fei Shen
Fei Shen Jingsi Luo
Jingsi Luo- 1Guangxi Key Laboratory of Birth Defects Research and Prevention, Guangxi Key Laboratory of Reproductive Health and Birth Defects Prevention, Maternal and Child Health Hospital of Guangxi Zhuang Autonomous Region, Nanning, China
- 2Department of Genetic and Metabolic Central Laboratory, Maternal and Child Health Hospital of Guangxi Zhuang Autonomous Region, Nanning, China
The Poirier–Bienvenu neurodevelopmental syndrome is an autosomal dominant disorder characterized by intellectual disability and epilepsy. The disease is caused by mutations in the CSNK2B gene, which encodes the beta subunit of casein kinase II, and it has important roles in neuron development and synaptic transmission. In this study, five Chinese patients were diagnosed with Poirier–Bienvenu neurodevelopmental syndrome caused by CSNK2B mutations by whole exome sequencing. We detected four different de novo variants of the CSNK2B gene in these five unrelated Chinese patients: two novel mutations, namely, c.100delT (p.Phe34fs*16) and c.158_159insA (p.Asp55fs*4), and two recurrent mutations, namely, c.1A>G (p.Met1?) and c.332 G >C (p.R111P). All five patients showed mild-to-profound intellectual disabilities/or learning disabilities and developmental delays, with or without seizures. Although intellectual disability/developmental delay and epilepsy are the most common manifestations of CSNK2B deficiency, the clinical phenotypes of probands are highly variable, and there is no significant correlation between genotype and phenotype. An abnormal stature may be another common manifestation of CSNK2B deficiency. Here, we report the effects of growth hormone (GH) therapy on the patients' linear height. In conclusion, Poirier–Bienvenu neurodevelopmental syndrome is a highly heterogeneous disease caused by mutations in the CSNK2B gene. The phenotype was highly variable, and no significant correlation of genotype and phenotype was found. Patients with short-stature and CSNK2B deficiency may benefit from GH therapy. The identification and characterization of these novel variants will expand the genotypic and phenotypic spectrum of Poirier–Bienvenu neurodevelopmental syndrome.
Introduction
Poirier–Bienvenu neurodevelopmental syndrome (MIM 618732) is a very rare autosomal dominant disorder characterized by early-onset seizures and variably impaired intellectual development, and it is caused by the deficiency of the casein kinase 2β (1, 2), CSNK2B (CSNK2B; MIM 115441, NM_001320.6), which is ~4 kb in length, contains seven coding exons, and is located on chromosome 6p21.33. It encodes the β subunit of the casein kinase II (CK2) protein complex, and participates in biological processes including signal transduction, metabolic processes, replication, transcription, and translation (3–8). CSNK2B is abundantly expressed in the brain, especially in neurons and neuroepithelial cells (9), and it has essential roles in the development of neuronal processes (10, 11). CSNK2B deficiency alters neuron development and synaptic transmission, resulting in severe neurodevelopmental deficiencies (12, 13). Recently, 57 unrelated patients with Poirier–Bienvenu neurodevelopmental syndrome and de novo mutations in the CSNK2B gene have been described (1, 2, 14–20). The phenotypes of these patients were heterogeneous and included treatable or untreatable seizures, mild-to-profound intellectual disabilities/or learning disabilities, language delays, and other symptoms (1, 2, 14–20). The severity of the phenotypes caused by mutations in the CSNK2B gene and the treatment of patients with Poirier–Bienvenu neurodevelopmental syndrome remain to be fully explored.
Here, we present five unrelated Chinese patients diagnosed with Poirier–Bienvenu neurodevelopmental syndrome, and the phenotypes were highly variable. Molecular analyses identified four CSNK2B variants, namely, two novel variants and two recurrent variants. We also evaluated the efficacy of growth hormone (GH) therapy and summarized the genotypes, phenotypes, and clinical features of Poirier–Bienvenu neurodevelopmental syndrome.
Materials and Methods
Editorial Policies and Ethical Considerations
Peripheral blood was collected from 1,230 individuals with early childhood-onset epilepsy and/or intellectual disability and the patients' family medical histories were investigated. Written informed consent for the publication of data was obtained from the patients' family. This study was approved by the Department of Genetic Metabolic Central Laboratory of Maternal and Child Health Hospital of Guangxi Zhuang Autonomous Region.
Whole Exome Sequencing and Sanger Sequencing
Genomic DNA was extracted from 5 ml of peripheral blood samples using the Lab-Aid DNA kit (Zeesan Biotech Co., Ltd., Xiamen, China). Whole exome sequencing was performed using the Agilent SureSelect Human Exon V6 kit (Agilent Technologies, Santa Clara, CA, USA), according to the manufacturer's protocol. The prepared libraries were sequenced by the HiSeq 2500 system (Illumina, San Diego, CA, USA), with a read depth of 120 × and a sequencing depth > 20 × for 95% of the captured regions. The sequenced reads were mapped to the human reference genome assembly build hg19 GRCh37 with the Burrows–Wheeler Aligner (http://bio-bwa.sourceforge.net/). Data analysis was performed with TGex software (LifeMap Sciences, Alameda, VA, USA). Variants with minor allele frequencies of <5% in the gnomAD database (http://gnomad.broadinstitute.org/) were selected. Synonymous variants and intronic variants not located within the splice site regions were removed, and all the protein-altering variants [non-synonymous single nucleotide polymorphisms (SNPs) and indels] were systematically evaluated. The effects of the mutations were examined using PolyPhen-2 (http://genetics.bwh.harvard.edu/pph2/), SIFT (http://provean.jcvi.org/), and CADD (https://cadd.gs.washington.edu/snv), (https://cadd.gs.washington.edu/snv) databases. The pathogenicity of each variant in the patient was scored according to American College of Medical Genetics and Genomics/Association for Molecular Pathology (ACMG/AMP) guidelines (21).
Results
Clinical Features
Each Chinese patient was the first child of healthy parents without significant medical family history and diagnosed by whole exome sequencing. All patients underwent uneventful full-term gestations. Patients with a mutation in the CSNK2B gene are summarized in Table 1.
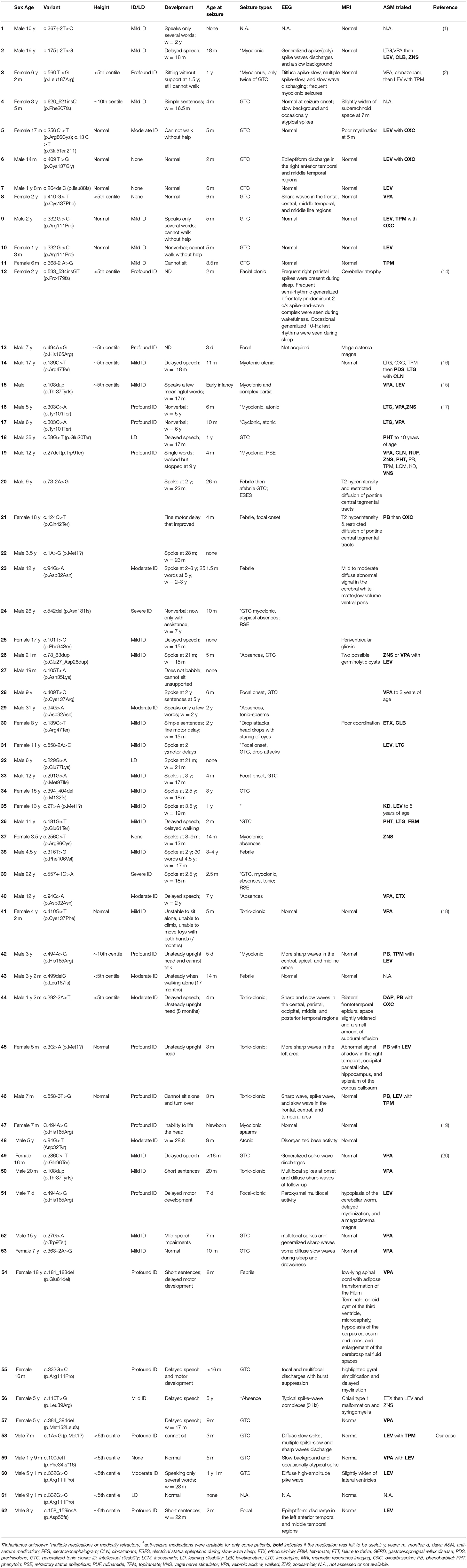
Table 1. Genotype and phenotype details for individuals with CSNK2B variants.
Patient 1 was a Chinese boy of non-consanguineous parents with a birth weight of 3.5 kg and a gestational age of 38+5 weeks. He had his first focal seizure at 3 months of age, and seizures occurred 6 to 15 times a day for the first 10 days. Each seizure lasted for approximately 30 s to 5 min. At the time of writing, he was 7 months old and presented with a severely short stature (height, 63 cm <2.5 SD). He has been hospitalized three times due to repeated seizures without fever. The electroencephalogram (EEG) results were abnormal at 3 months of age, showing a diffuse slow spike, multiple slow spikes, and sharp wave discharges. The results of brain magnetic resonance imaging (MRI), metabolic assays, and chromosomal karyotype analysis were all normal. The Gesell Developmental Assessment Scale for Children was used at 4 months of age, and the patient was diagnosed with severe global developmental delay. The seizures were controlled from 3 months of age until the present time with levetiracetam (LEV) and topiramate (TPM).
Patient 2 was a 1-year-and-9-month-old boy who had epilepsy without intellectual and developmental delays. The patient had his first seizure at 5 months of age, and the type of seizure was generalized tonic-clonic seizure (GTCS) with a frequency of nine times a day, each lasting for approximately 10 s to 1 min. EEG abnormalities (a slow background and occasionally, an atypical spike) were observed. The seizures were controlled with valproate (VPA) and LEV treatment. The patient did not have a developmental delay, and the brain MRI results were normal.
CSNK2B mutations in patients 3 and 4 were identical; however, the patients had different clinical presentations. Patient 3 was a 5-year-and-1-month-old boy who presented with mild proportionate short stature with a Height Standard Deviation Score of −2.1 SD. GH deficiency was ruled out by the arginine clonidine GH stimulation test. The endocrine test results revealed an insulin-like growth factor 1 (IGF-1) level of 1,140 ng/ml (reference range, 49–283 ng/ml) and an insulin-like growth factor-binding protein 3 (IGFBP-3) level of 3.5 μg/ml (reference range, 1–4.7 μg/ml), and the highest GH level (insulin combined with arginine) in the stimulation test was 6.93 ng/ml. Patient 3 had epilepsy and mild DD. He started walking at 28 months and speaking at 3 years of age, and at the time of writing, he still could not construct long sentences. The results of the Wechsler Intelligence Scale for Children-IV test revealed that his full-scale IQ was 70. The patient had his first seizure at 1 year and 1 month of age, with a frequency of five times a day, each lasting for approximately 2 s. The seizures were characterized as myoclonic epilepsy. The patient was not taken to the hospital for treatment until half a year later, because of neglect by the parents. The EEG results were abnormal at 2 years of age with medium-to-high amplitude sharp waves in the frontal pole, as well as frontal and central regions. The brain MRI results revealed a slight widening of the lateral ventricles at 2 years of age, which was relieved at 4 years of age. Patient 4 was a 9-year-and-1-month-old boy without epilepsy and intellectual and developmental delays. At 6 years of age, he was taken to the hospital for genetic counseling of short stature (height, 102 cm, <3 SD) and learning disabilities. The results of the GH provocative test, which employed arginine and L-dopa as biomarkers, revealed that the highest GH level was 4.47 ng/ml (2.21 ng/ml at 0 min, 4.47 ng/ml at 30 min, 3.25 ng/ml at 60 min, 1.10 ng/ml at 90 min, 2.05 ng/ml at 120 min, 1.85 ng/ml at 150 min, and 0.88 ng/ml at 180 min), suggestive of a complete GH deficiency. The other hormonal measurements were an IGF-1 level of 115 ng/ml (reference range, 49–283 ng/ml) and an IGFBP-3 level of 2.8 μg/ml (reference range, 1–4.7 μg/ml). Recombinant human growth hormone (rhGH) therapy (0.2 mg kg−1 week−2, Subcutaneous injection) was initiated when the patient was 6 years and 1 month of age. The growth velocity was 5.31 cm/year before treatment (from 4 years and 11 months to 6 years of age). The growth velocity during the first year of treatment was 7.2 cm/year, and the height increased by 0.5 SD. By the end of the third year of treatment (9 years and 2 months of age), the height was 124.5 cm, and the height increased from −3.46 SD to −2 SD (Figure 1).
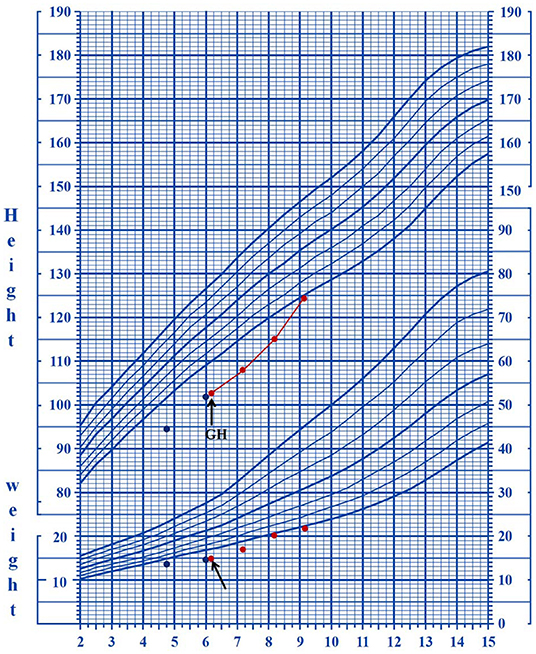
Figure 1. The growth curves of patient four who underwent rhGH treatment. The arrows indicate the starting date of the treatment.
Patient 5 was an 8-year-old boy with epilepsy and moderate intellectual and developmental delays. He started walking at 22 months and talking at 2 years and 6 months of age. The Gesell Developmental Quotient Score was 52 at 5 years and 4 months of age. He continued to exhibit growth retardation, speech delays, comprehension deficits, and learning disabilities. The patient had his first focal seizure at 2 months and presented with tetanic twitching of the limbs with a frequency of 5 to 8 times a day, each lasting for approximately 30 s to 1 min. The results of brain MRI and EEG were normal. The seizures were controlled with LEV.
Molecular Analysis
By whole-exome sequencing, we detected heterozygous mutations in the CSNK2B gene in probands as follows: (RefSeq NM_001320.6): c.1A>G (p.Met1?) in patient 1, c.100delT (p.Phe34fs*16) in patient 2, c.332 G>C (p.R111P) in patients 3 and 4, and c.158_159insA (p.Asp55fs*4) in patient 5 (Figure 2). We confirmed the four mutations by Sanger sequencing and sequenced the parental samples to validate that the four variants were de novel. Specifically, c.100delT (p.Phe34fs*16) and c.158_159insA (p.Asp55fs*4) were novo variants, which were not deposited in the Human Gene Mutation database, 1000 Genomes Database, ClinVar database, and Single Nucleotide Polymorphism database. However, c.1A>G (p.Met1?) and c.332 G>C (p.R111P) were previously reported in affected individuals (2, 17). These de novo variants were predicted as deleterious through multiple functional prediction tools, including SIFT, PolyPhen 2.0, CADD, and MutationTaster (Table 2). All variants were classified as pathogenic according to ACMG/AMP standards and guidelines (Table 2).
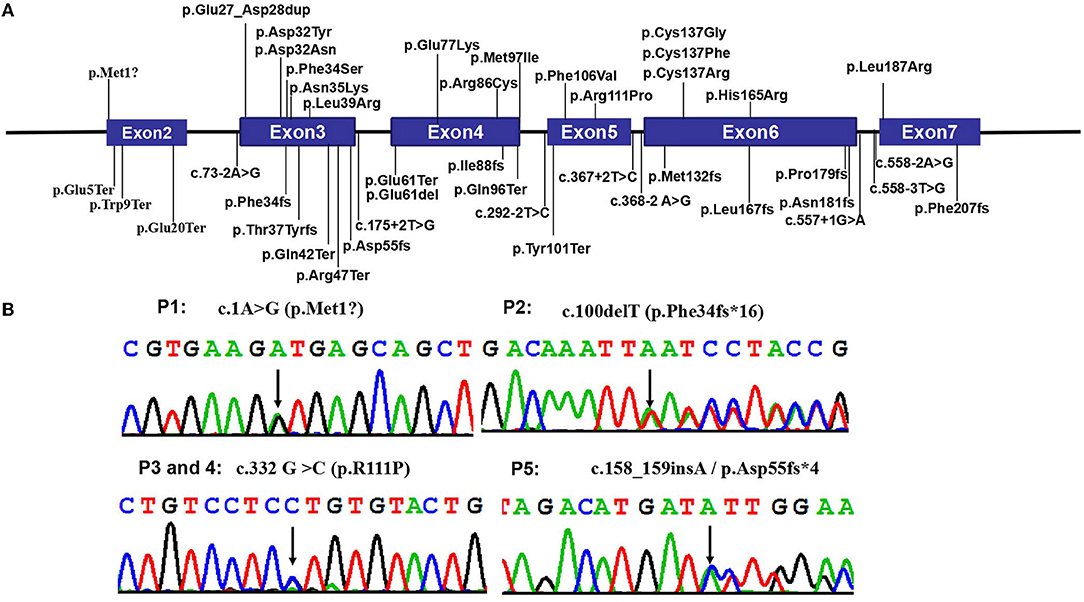
Figure 2. Pathogenic CSNK2B variants. (A) The distribution of all CSNK2B variants detected so far in the 57 reported patients and 5 in this study. Boxes represent six different exons as indicated, and solid lines connecting these boxes represent the introns of CSNK2B gene. The numbers above the boxes indicate the positions of the CSNK2B complementary DNA at the start-stop sites and exon-intron boundaries. Vertical lines represent the locations of missense (above the boxes) or deletion/nonsense/frameshift/splicing (below the boxes) variants. (B) The Sanger chromatograms of the detected variants in patients 1–5. Among them, P1 [NM_001320:c.1A>G (p.Met1?)] had a start loss variant, and P3 and P4 [c.332 G >C (p.R111P)] had missense variant; P2 [c.100delT (p.Phe34fs*16)] and P5 [c.158_159insA(p.Asp55fs*4)] had frameshift variants. Moreover, c.1A>G (p.Met1?) and c.332 G >C (p.R111P) were reported by Li et al. and Michelle et al.
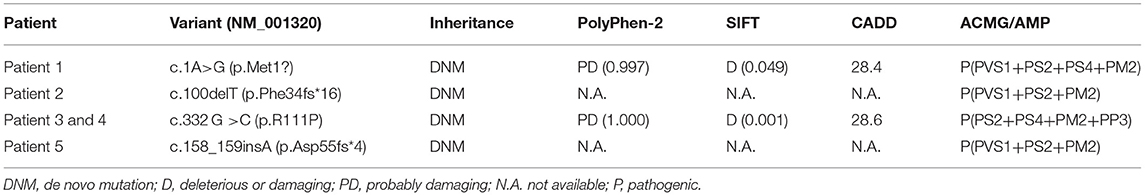
Table 2. Predicted pathogenicity of de novo CSNK2B variants.
Genotype–Phenotype Correlations
To date, a total of 57 patients with pathogenic CSNK2B mutations have been reported in the literature (1, 2, 14–20). Clinical and molecular features of the reported 62 patients and of our patients are summarized in Table 1. By extensive literature analysis, we compared the phenotypes of 24 patients with missense mutations and 38 patients with loss-of-function mutations (LoF; including deletions, duplications, frameshifts, nonsense mutations, start-loss mutations, and splice sites). The phenotypes were classified into five categories, namely, intellectual disabilities/learning disabilities, speech delay or disability, motor delay or disability, seizures, and short stature (Table 3). However, no significant differences in these phenotypes between the missense mutation group and the LoF mutation group were observed.
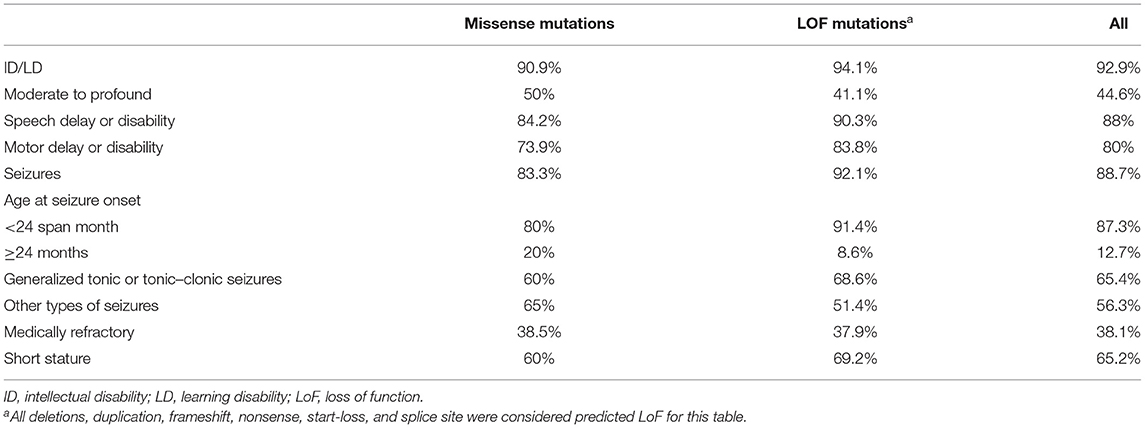
Table 3. Comparison of effects of variant type on clinical manifestations.
Discussion
POBINDS is a very rare autosomal dominant genetic disorder (1). Poirier et al. in 2017 provided the first report of mutations in the CSNK2B gene in two unrelated patients with neurodevelopmental abnormality and epilepsy; since then, a total of 57 patients with pathogenic mutations in this gene have been described (1, 2, 14–20). The most frequent manifestations of these subjects with mutations in the CSNK2B gene are epilepsy, developmental delays, and intellectual disabilities (Tables 1, 3). Patients are often diagnosed within the first 2 years of life, and the severity of the phenotype is highly variable (Table 1). In the present study, five individuals were diagnosed as POBINDS by performing whole-exome sequencing. These patients showed the common phenotypes associated with POBINDS, including varying degrees of ID/LD, developmental delay with or without seizures, and growth abnormalities.
Neurodevelopmental disabilities are a hallmark of CSNK2B gene deficiency. Many subjects (including our patients) suffer from delays that span multiple areas. For instance, most patients have delayed motor development, more than four-fifths of the patients have speech deficits, and one-thirds of patients have cognitive impairments that range from moderate to profound (1, 2, 14–20) (Table 3). Seizures, which start in infancy, are observed in approximately 88.7% of patients (Table 3). Different types of seizures have been reported, with GTCS as the most common type (Table 3). In this study, three out of four patients with epilepsy presented with GTCS. Although many patients have severe epilepsy with recurrent episodes of refractory status epilepticus (17), four patients in this study became seizure-free with LEV or combination therapy. Intellectual disabilities are usually associated with seizure severity. However, the intelligence and development of POBINDS patients with intellectual/developmental disabilities and treatable epilepsy do not always improve after the seizures are controlled with therapy, and some patients with normal intelligence still experience seizures, indicating that intellectual/developmental disabilities and seizures may be comorbidities. Further studies are needed to define the genotypic and phenotypic determinants, as well as the mechanisms of action.
In addition to intellectual/developmental disabilities and epilepsy, short stature may be another common feature of the disease. Short stature has been observed in most patients (15/23, 65.2%), including our patients; however, it was not reported by Ernst et al. (17). Human GH is often used to treat growth disorders in children with GH deficiency. Thus, far, only one patient with CSNK2B deficiency has been tested and ruled out for GH deficiency (16). The patient received GH therapy, and his height increased significantly. Of the five patients in this study, two subjects (patients 3 and 4) were subjected to the GH stimulation test, and the results revealed that patient 3 showed partial GH deficiency and patient 4 showed complete GH deficiency. Patient 4 underwent rhGH treatment for 3 years and showed a good response with no side effect. This study provides further evidence for GH therapy in patients with mutations in the CSNK2B gene.
To date, 46 CSNK2B gene mutations have been identified in 62 Poirier–Bienvenu neurodevelopmental syndrome patients, including 8 splice site mutations, 9 frameshift mutations, 15 missense mutations, 8 nonsense mutations, 1 in-frame duplication, 1 in-frame deletion, and 3 start-loss mutations. CSNK2B deficiency is associated with clinically heterogeneous deficits, including intellectual disabilities, multiple congenital anomalies, development delays, seizures, and short stature (1, 2, 14–20) (Table 1). We tried to correlate the mutations in the CSNK2B gene with the clinical symptoms of Poirier–Bienvenu neurodevelopmental syndrome patients. We analyzed the genetic and clinical information of all reported patients and our cases (Tables 1, 3). Mild or severe phenotypes can be evident in patients with missense or LoF mutations, and although severe phenotypes were associated more often with LoF mutations than with missense mutations, this finding was not statistically significant. Even patients with the same mutation can have different phenotypes. For instance, patients (including our patients) with the L111P mutation have varying degrees of intellectual impairment with delayed cognitive, language, and motor development, and growth abnormalities with or without seizures (Table 1). As such, the clinical features of patients with heterozygous mutations in the CSNK2B gene are highly variable with no obvious genotype–phenotype correlation. It is unclear how these mutations cause neurodevelopmental deficits and epilepsy. Nakashima et al. reported that the disease can be explained by several mechanisms, although LoF mutations are responsible for CSNK2B haploinsufficiency (15). Furthermore, the reduced expression of CSNK2B results in fewer CK2 tetrameric complexes. On the other hand, missense mutations and other types of mutations, such as p.His165Arg and p.Pro179Tyrfs*49, can have a dominant negative effect and impair CK2 enzyme activity. Further functional studies are needed to enhance our understanding of the disease and its mechanisms of action.
Conclusion
In summary, we presented the clinical phenotypes of five unrelated Chinese patients with de novo heterozygous mutations in the CSNK2B gene. This study expands the mutation spectrum of Poirier–Bienvenu neurodevelopmental syndrome. This is the first report of c.158_159insA/p.Asp55fs*4 and c.100delT (p.Phe34fs*16) mutations in the CSNK2B gene. Our study also summarizes the phenotypes of patients with CSNK2B deficiency and reveals no significant correlation between genotype and phenotype, thereby providing a foundation for clinical diagnosis in the future. Long-term recombinant GH treatment appears safe and effective; however, additional cases should be examined to fully evaluate the benefits of GH therapy.
Data Availability Statement
The datasets presented in this study can be found in online repositories. The names of the repository/repositories and accession number(s) can be found at: https://www.ncbi.nlm.nih.gov/, PRJNA778391.
Ethics Statement
The studies involving human participants were reviewed and approved by Department of Genetic Metabolic Central Laboratory of Guangxi Zhuang Autonomous Region, Women and Children Care Hospital. Written informed consent to participate in this study was provided by the participants' legal guardian/next of kin. Written informed consent was obtained from the individual(s), and minor(s)' legal guardian/next of kin, for the publication of any potentially identifiable images or data included in this article.
Author Contributions
QY and SH designed the study. QY and JL gathered clinical information from the family members and drafted the manuscript. ZQ, SY, QZ, FS, and SO performed the sequencing, as well as analyzed, and interpreted the data. All authors coordinated the study coordination and revised the manuscript, read, and approved the final version of the manuscript.
Funding
This research was supported by the Health Department of Guangxi Province (Grant Nos. ZJC202001, Z20210440, and Z2015238), Guangxi Medical High-level Backbone Talents 139 Plan (G202003023), and Guangxi Key Research and Development Program (GuikeAB17195004).
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher's Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Acknowledgments
We are grateful to the all the patients and their families who participated in this study.
References
1. Poirier K, Hubert L, Viot G, Rio M, Billuart P, Besmond C, et al. CSNK2B splice site mutations in patients cause intellectual disability with or without myoclonic epilepsy. Hum Mutat. (2017) 38:932–41. doi: 10.1002/humu.23270
2. Li J, Gao K, Cai S, Liu Y, Wang Y, Huang S, et al. Germline de novo variants in CSNK2B in Chinese patients with epilepsy. Sci Rep. (2019) 9:17909. doi: 10.1038/s41598-019-53484-9
3. Kusk M, Ahmed R, Thomsen B, Bendixen C, Issinger OG, Boldyreff B. Interactions of protein kinase CK2beta subunit within the holoenzyme and with other proteins. Mol Cell Biochem. (1999) 191:51–8. doi: 10.1023/A:1006840613986
4. Raaf J, Issinger OG, Niefind K. First inactive conformation of CK2 alpha, the catalytic subunit of protein kinase CK2. J Mol Biol. (2009) 386:1212–21. doi: 10.1016/j.jmb.2009.01.033
5. Bodenbach L, Fauss J, Robitzki A, Krehan A, Lorenz P, Lozeman FJ, et al. Recombinant human casein kinase II. A study with the complete set of subunits (alpha, alpha' and beta), ite-directed autophosphorylation mutants and a bicistronically expressed holoenzyme. Eur JBiochem. (1994) 220:263–73. doi: 10.1111/j.1432-1033.1994.tb18622.x
6. Gao Y, Wang HY. Casein kinase 2 is activated and essential for Wnt/beta-catenin signaling. J Biol Chem. (2006) 281:18394–400. doi: 10.1074/jbc.M601112200
7. Ponce DP, Maturana JL, Cabello P, Yefi R, Niechi I, Silva E, et al. Phosphorylation of AKT/PKB by CK2 is necessary for the AKT-dependent up-regulation of β-catenin transcriptional activity. J Cell Physiol. (2011) 226:1953–9. doi: 10.1002/jcp.22527
8. Shehata M, Schnabl S, Demirtas D, Hilgarth M, Hubmann R, Ponath E, et al. Reconstitution of PTEN activity by CK2 inhibitors and interference with the PI3-K/ Akt cascade counteract the antiapoptotic effect of human stromal cells in chronic lymphocytic leukemia. Blood. (2010) 116:2513–21. doi: 10.1182/blood-2009-10-248054
9. Mestres P, Boldyreff B, Ebensperger C, Hameister H, Issinger OG. Expression of casein kinase 2 during mouse embryogenesis. Acta Anat. (1994) 149:13–20. doi: 10.1159/000147550
10. Eiber N, Rehman M, Kravic B, Rudolf R, Sandri M, Hashemolhosseini S. Loss of protein kinase Csnk2b/CK2β at neuromuscular junctions affects morphology and dynamics of aggregated nicotinic acetylcholine receptors, neuromuscular transmission, and synaptic gene expression. Cells. (2019) 8:940. doi: 10.3390/cells8080940
11. Ulloa L, Díaz-Nido J, Avila J. Depletion of casein kinase II by antisense oligonucleotide prevents neuritogenesis in neuroblastoma cells. EMBO J. (1993) 12:1633–40. doi: 10.1002/j.1460-2075.1993.tb05808.x
12. Huillard E, Ziercher L, Blond O, Wong M, Deloulme JC, Souchelnytskyi S, et al. Disruption of CK2beta in embryonic neural stem cells compromises proliferation and oligodendrogenesis in the mouse telencephalon. Mol Cell Biol. (2010) 30:2737–49. doi: 10.1128/MCB.01566-09
13. Yang CP, Li X, Wu Y, Shen Q, Zeng Y, Xiong Q, et al. Comprehensive integrative analyses identify GLT8D1 and CSNK2B as schizophrenia risk genes. Nat Commun. (2018) 9:838. doi: 10.1038/s41467-018-03247-3
14. Nakashima M, Tohyama J, Nakagawa E, Watanabe Y, Siew CG, Kwong CS, et al. Identification of de novo CSNK2A1 and CSNK2B variants in cases of global developmental delay with seizures. J Hum Genet. (2019) 64:313–22. doi: 10.1038/s10038-018-0559-z
15. Sakaguchi Y, Uehara T, Suzuki H, Kosaki K, Takenouchi T. Truncating mutation in CSNK2B and myoclonic epilepsy. Hum Mutat. (2017) 38:1611–2. doi: 10.1002/humu.23307
16. Selvam P, Jain A, Cheema A, Atwal H, Forghani I, Atwal PS. Poirier-Bienvenu neurodevelopmental syndrome: a report of a patient with a pathogenic variant in CSNK2B with abnormal linear growth. Am J Med Genet A. (2021) 185:539–43. doi: 10.1002/ajmg.a.61960
17. Ernst ME, Baugh EH, Thomas A, Bier L, Lippa N, Stong N, et al. CSNK2B: a broad spectrum of neurodevelopmental disability and epilepsy severity. Epilepsia. (2021) 62:e103–9. doi: 10.1111/epi.16931
18. Yang S, Wu L, Liao H, Lu X, Zhang X, Kuang X, et al. Clinical and genetic analysis of six Chinese children with Poirier-Bienvenu neurodevelopmental syndrome caused by CSNK2B mutation. Neurogenetics. (2021) 22:323–32. doi: 10.1007/s10048-021-00649-2
19. Wilke MVMB, Oliveira BM, Pereira A, Doriqui MJR, Kok F, Souza CFM. Two different presentations of de novo variants of CSNK2B: two case reports. J Med Case Rep. (2022) 16:4. doi: 10.1186/s13256-021-03184-8
20. Alessandro O, Andrea S, Francesca B, Alice B, Diego P, Roberta B, et al. Expanding phenotype of poirier–bienvenu syndrome: new evidence from an italian multicentrical cohort of patients. Genes. (2022) 13:276. doi: 10.3390/genes13020276
21. Richards S, Aziz N, Bale S, Bick D, Das S, Gastier-Foster J, et al. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet Med. (2015) 17:405–24. doi: 10.1038/gim.2015.30
Keywords: CSNK2B, seizure, intellectual disability, short stature, growth hormone (GH) therapy
Citation: Yang Q, Zhang Q, Yi S, Qin Z, Shen F, Ou S, Luo J and He S (2022) De Novo CSNK2B Mutations in Five Cases of Poirier–Bienvenu Neurodevelopmental Syndrome. Front. Neurol. 13:811092. doi: 10.3389/fneur.2022.811092
Received: 08 November 2021; Accepted: 16 February 2022;
Published: 16 March 2022.
Edited by:
Gijs Van Haaften, University Medical Center Utrecht, NetherlandsReviewed by:
Bobby Koeleman, University Medical Center Utrecht, NetherlandsAlessandro Orsini, Pisana University Hospital, Italy
Copyright © 2022 Yang, Zhang, Yi, Qin, Shen, Ou, Luo and He. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Sheng He, aHN0Z3lzMTgzQDEyNi5jb20=; Jingsi Luo, NDkwODgwMjU5QHFxLmNvbQ==