 John Kim
John Kim Clara Erice
Clara Erice Ursula K. Rohlwink
Ursula K. Rohlwink Elizabeth W. Tucker
Elizabeth W. Tucker
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
MINI REVIEW article
Front. Neurol., 17 February 2022
Sec. Neuroinfectious Diseases
Volume 13 - 2022 | https://doi.org/10.3389/fneur.2022.805786
This article is part of the Research TopicBroadening Our Understanding of the Impact of Infections on the Developing Central Nervous System - from Basic to Clinical SciencesView all 9 articles
Central nervous system (CNS) infections occur more commonly in young children than in adults and pose unique challenges in the developing brain. This review builds on the distinct vulnerabilities in children's peripheral immune system (outlined in part 1 of this review series) and focuses on how the developing brain responds once a CNS infection occurs. Although the protective blood-brain barrier (BBB) matures early, pathogens enter the CNS and initiate a localized innate immune response with release of cytokines and chemokines to recruit peripheral immune cells that contribute to the inflammatory cascade. This immune response is initiated by the resident brain cells, microglia and astrocytes, which are not only integral to fighting the infection but also have important roles during normal brain development. Additionally, cytokines and other immune mediators such as matrix metalloproteinases from neurons, glia, and endothelial cells not only play a role in BBB permeability and peripheral cell recruitment, but also in brain maturation. Consequently, these immune modulators and the activation of microglia and astrocytes during infection adversely impact normal neurodevelopment. Perturbations to normal brain development manifest as neurodevelopmental and neurocognitive impairments common among children who survive CNS infections and are often permanent. In part 2 of the review series, we broadly summarize the unique challenges CNS infections create in a developing brain and explore the interaction of regulators of neurodevelopment and CNS immune response as part of the neuro-immune axis.
The development of the central nervous system (CNS) is a tightly regulated complex and dynamic process. The CNS begins to develop in the third week of gestation and continues to mature postnatally into late adolescence and adulthood (1). The critical periods of perinatal and early postnatal neurodevelopment, together with adolescent maturation of the brain, are particularly sensitive to environmental influences. Therefore, it is not surprising that CNS infections represent a significant source of mortality and morbidity in children worldwide (2–5). In children, these infections lead to devastating neurological sequelae, such as motor deficits or cognitive impairment, that can last a lifetime (6–8). Moreover, immunocompromised children are at higher risk of developing more severe CNS infections with poorer outcomes (9). In this review, we explore the idea that key components of the CNS immune response are also regulators of early neurodevelopment, interacting along the neuro-immune axis. Moreover, we provide an overview of current evidence suggesting that this dual role is a key driver of the unique pediatric (0–18 years) susceptibility to CNS infections and the resultant sequelae. Specifically, we highlight how a dysregulated immune response secondary to two CNS infections cause long-term neurological sequelae.
Innate immunity in the CNS begins at the blood-brain barrier (BBB), a semipermeable barrier consisting of brain microvascular endothelial cells (BMECs), stabilized by astrocytic end-feet and pericytes. BMECs line the cerebral vasculature and form a cellular monolayer via tight junctions (TJs) and adherents junctions. The resulting barrier allows for bidirectional regulation of molecular flow and hinders the hematogenous entry of pathogens and toxins (10). The stage at neurodevelopment where the BBB becomes functional remains unclear. Historically, an immature BBB was considered to contribute to an increased risk of CNS infections in children. This notion can be traced back to early studies of dye-injection experiments and observations of higher cerebrospinal fluid (CSF) protein concentrations in newborn animals (11). Conclusions on BBB maturity drawn from these experiments, however, have since been challenged, and mounting evidence supports the existence of a functional BBB early in embryonic development (11).
Nevertheless, even a “mature” BBB is not impregnable. The predominant cause of meningitis in older infants and children is Streptococcus pneumoniae, which interacts with the endothelial receptors polymeric immunoglobulin receptor (pIgR) and platelet endothelial cell adhesion molecule-1 (PECAM-1 or CD31) to invade the CNS (12). The gram-positive bacterium Listeria monocytogenes, a leading cause of meningitis in neonates, enters the CNS by triggering endocytosis in host epithelial cells (13, 14) and hijacking infiltrating monocytes (15). The protozoan parasite Toxoplasma gondii similarly utilizes this “trojan horse” method of CNS entry via infected dendritic cells and monocytes (16) while also replicating in endothelial cells (17).
Virulence factors which enable host extracellular matrix-pathogen interactions and the resulting neuroinflammatory response can also disrupt the BBB at the molecular level. For example, Group B Streptococcus upregulates Snail1 on BMECs which in turn represses the expression of TJ proteins including occludin, zonula occludens, and claudin-5, leading to a leaky BBB (18). Additionally, the parasite Plasmodium falciparum, the major cause of pediatric cerebral malaria (CM), does not cross the BBB. Rather, P. falciparum binds to endothelial cells, triggering an inflammatory cascade that ultimately leads to BBB damage, vascular leakage, and often lethal cerebral edema (19). Even peripheral infections such as sepsis can lead to neuroinflammation with similar BBB damage, as demonstrated by the absence of occludin on brain autopsies from septic patients (20).
Once pathogens compromise the BBB or/and enter the CNS, the CNS must mount a sufficient immune response to control the infection. Unfortunately, the host immune response may become dysregulated and initiate various injury cascades that compromise normal neurodevelopment.
Recent evidence describing dynamic interactions between the CNS and immune system together with the discovery of the CNS lymphatic system (i.e., glymphatic system and meningeal lymphatics) (21, 22) have rendered it necessary to rethink the long-held notion that the CNS is an immune privileged site (23, 24). Instead, it is now suggested that the CNS is a site of active, highly regulated immune surveillance (25). This framework is essential to contextualizing processes whereby microglia, astrocytes, and secreted immune mediators are intimately involved in neurodevelopment (26) (summarized in Figure 1).
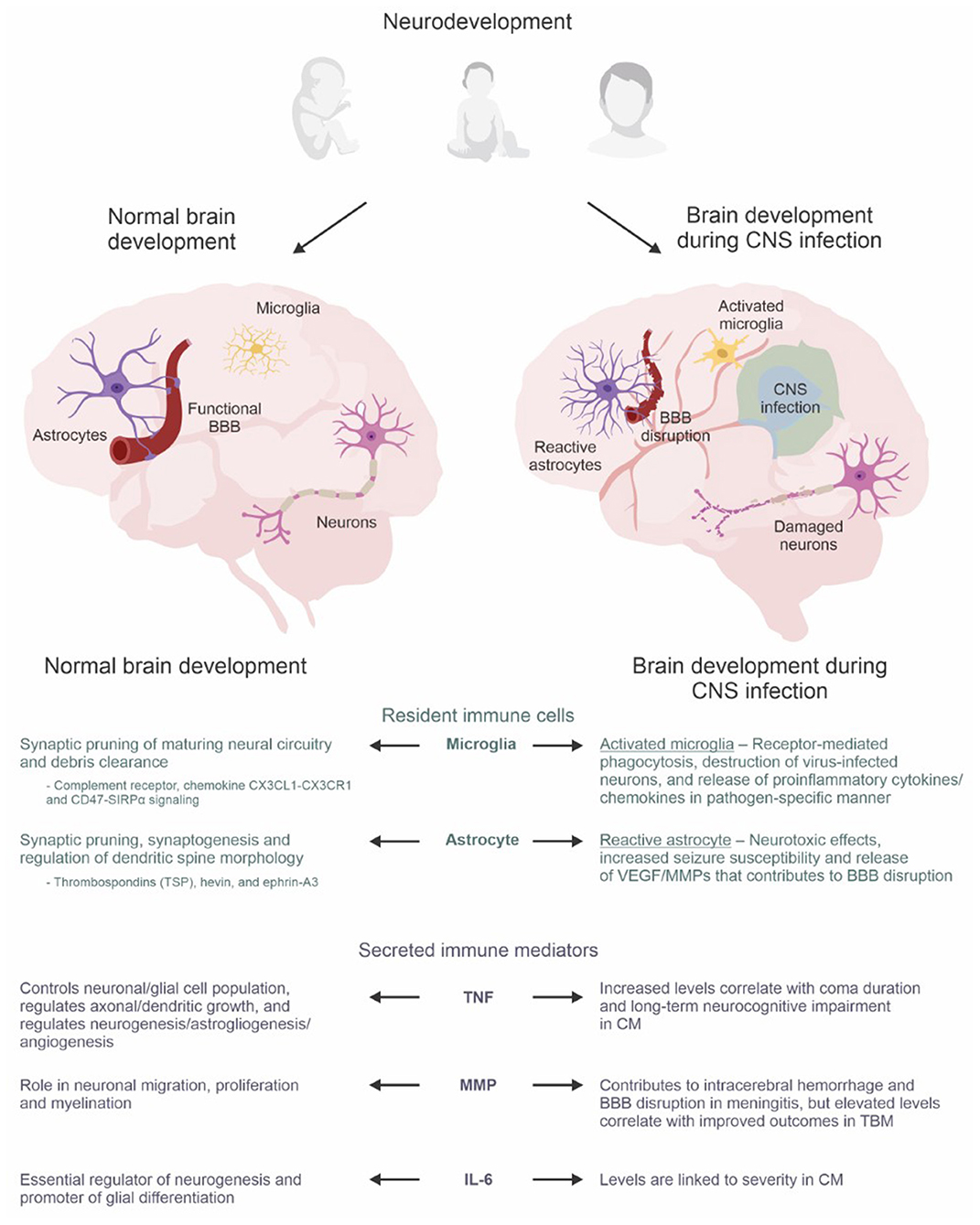
Figure 1. The neuro-immune axis in the developing brain. Astrocytes, microglia, and secreted immune mediators have dual roles during normal brain development and the immune response triggered by CNS infections. During normal brain development, they are integral to synaptic pruning, regulation of dendritic growth/morphology, neurogenesis, astrogliogenesis, angiogenesis, myelination, and glial differentiation. However, amid the immune response during infection, brain development becomes dysregulated which can lead to neurological, neurodevelopmental, and psychiatric sequelae. The lower panel summarizes the roles of resident immune cells (microglia and astrocytes) and secreted immune mediators (TNF super family, MMP, IL-6) during normal brain development compared to dysregulated brain development during CNS infection. Abbreviations: BBB, blood-brain barrier; CM, cerebral malaria; CNS, central nervous system; MMP, matrix metalloproteinases; TBM, tuberculous meningitis; TNF, tumor necrosis factor; VEGF, vascular endothelial growth factors. Created with BioRender.com and CorelDRAW Graphics Suite 2021.
Furthermore, the proliferation and maturation of microglia and astrocytes themselves are shaped by complex local and systemic cues which originate from diverse sources, ranging from the developing CNS itself to the maternal microbiome (27, 28). In response to key signals including but not limited to transforming growth factor-β (TGF-β) and colony-stimulating factor-1 (CSF-1), microglia undergo distinct, sequential stages of differentiation with morphological, transcriptomic, and functional transformations (29, 30). Similarly, recent studies have further characterized the temporo-spatial heterogeneity of transcriptomic profiles in astrocytes during perinatal synaptic development (31), circuitry-specific synaptic association (32), and even throughout normal aging (33, 34). Amidst the complex milieu that is the developing CNS, further appreciation of the roles of immunologically active glia and signaling molecules in neurodevelopment is quintessential to understanding the susceptibility of the pediatric CNS to infections and devastating sequelae.
Microglia are resident immune cells of the CNS and an important defense against invading pathogens and tissue injury. Microglial distribution in the brain is heterogenous, ranging from 0.5 to 16.6% of cells in the parenchyma (35). The colonization of the developing brain by embryonic microglia precedes neurogenesis, astrogliogenesis, and vasculogenesis, alluding to the crucial role of microglia in mediating early developmental and homeostatic processes of the CNS (36).
Microglia are dynamic cells that constantly survey their microenvironment and respond to environmental cues. Under pathological conditions they undergo a phenotypic shift from the ramified (or “quiescent”) form to the ameboid (or “activated”) form (37). Their intrinsic role as active surveyors and phagocytes of the brain parenchyma makes microglia key players in synaptic pruning (38–40). Synaptic pruning constitutes the elimination of excess connectivity and its functional structures (i.e., synaptic terminals, axonal and dendritic branches), and is a critical process during the prenatal and postnatal maturation of neural circuitry. Accordingly, depletion of microglia leads to defective pruning in the developing auditory brainstem (41), the visual cortex (42), and the somatosensory cortex (43). To date, numerous studies have identified microglia-specific signaling molecules that underpin the normal development of neural circuitry including complement receptor 3-C3 (44), the chemokine CX3CL1-CX3CR1 (45), and CD47-SIRPα signaling (46).
In order to respond to environmental cues, microglia express extensive molecular tools critical to sampling and interpreting brain milieu. For example, pattern recognition receptors (PRRs) expressed on microglia cell membranes, such as toll-like receptors (TLRs), enable microglia to mount a rapid response to microbial invasion and endogenous cellular damage. These receptors recognize pathogen-associated molecular patterns (PAMPs) and damaged-associated molecular patterns (DAMPs) (47, 48). Moreover, microglia express a cluster of genes, dubbed the microglial “sensome,” that allow them to sense CNS perturbations and include genes encoding purinergic receptors (e.g., P2RY12, ADORA3, TMEM173), cytokine and chemokine receptors (e.g., CSF1R, TGFBR1, IFNGR1, CX3CR1, CMKLR1), and Fc receptors (e.g., FCGR3, FCER1G) (49, 50).
During CNS infections, microglia clear bacteria via receptor-mediated phagocytosis (51, 52), identify and destroy virus-infected neurons (53), and even pave the way for remyelination post-infection by clearing debris and recruiting oligodendrocytes (54). In addition, microglia mediate a broader immune response by secreting key signaling molecules including proinflammatory cytokines and chemokines in a pathogen-specific manner. For instance, microglial activation by lipopolysaccharide (LPS) results in significantly different expression of tumor necrosis factor (TNF) and interleukin (IL)-1α/β compared to stimulation by neurotropic Semliki Forest virus (55). Furthermore, microglia can also cross-present viral antigens from infected neurons to recruit CD8+ T cells (56) and phagocytose infiltrating neutrophils to counteract ischemic injury (57).
Astrocytes comprise up to 30% of the mammalian CNS. Once thought to be mere scaffolds holding neurons together, astrocytes are now recognized for their dynamic roles in BBB maintenance, neuroinflammation, neurotransmission, and other essential CNS processes (58–60).
Although microglia are important for synaptic pruning, astrocytes are vital to the establishment and fine-tuning of synapses and broader cortical circuitry (61). Therefore, the role of astrocytes during neurodevelopment has primarily been studied in the context of synaptogenesis and synaptic pruning, leading to the identification of various astrocyte-expressed prosynaptogenic molecules (62). For example, astrocytic extracellular glycoproteins called thrombospondins (TSPs) have been characterized to promote excitatory synaptogenesis via interaction with the neuronal α2δ-1 receptor (63, 64). Additionally, the protein SPARCL1 (hevin) is highly expressed by astrocytes during critical periods of early synaptic refinement and is sufficient to selectively induce excitatory synapse formation (65–67). Furthermore, astrocytic ephrin-A3 activates EphA4 receptors on neuronal dendritic spines (DS) and regulates DS morphology and lifetime (68, 69). Notably, many of these molecules are now being examined for their roles during CNS injury and infection. For instance, TSP-1 and TSP-2 are antiangiogenic factors and are upregulated after intracerebral hemorrhage (70). Astrocytic hevin is also pivotal during synaptic remodeling after ischemic injury in an adult stroke model (71). This suggests the sustained importance of astrocytes in synapse integrity beyond perinatal and early postnatal neurodevelopment, though the effect of infection warrants further investigation.
Astrocytes are also indispensable to CNS innate immunity. They constitute an integral component of the BBB, the first line of defense against pathogen invasion (59). Ensheathment of cerebral microvasculature by astrocytic terminal processes (“end-feet”) is critical to BBB integrity (72). While neurotropic pathogens can bypass this barrier, its integrity during infection and inflammation remains a crucial bulwark against pathological leukocyte infiltration and hematogenous entry of additional pathogens. During neuroinflammation and ischemia, astrocytes secrete factors such as vascular endothelial growth factors (VEGFs) and matrix metalloproteinases (MMPs), that contribute to BBB disruption (73–76). Conversely, astrocyte-derived factors including sonic hedgehog can support BBB recovery (77). Therefore, astrocytes play a pivotal role in regulating BBB permeability and can help or harm BBB integrity during brain insults.
Following infection or injury, astrocytes undergo a morphological, transcriptomic, and functional shift through a process referred to as “reactive astrogliosis” (78–80). Reactive astrogliosis is often categorized by the dichotomous A1/A2 system, which many consider too simplistic (80, 81). Typically, A1 astrocytes upregulate proinflammatory genes which mediate neurotoxic outcomes in response to neuroinflammation, and A2 astrocytes are induced upon ischemic injury and favor a neuroprotective transcriptomic profile that promotes tissue repair. The cytokines TNF, IL-1α, and the complement component 1 subcomponent q (C1q), secreted by activated microglia, robustly induce an A1 phenotype (81). Interestingly, the A1 shift is associated with alterations in prosynaptogenic factors (i.e., TSP and hevin) which may prove pathological if it occurs during critical periods of neurodevelopment (81).
Similar to microglia, astrocytes express a variety of PRRs such as TLRs, mannose receptor, and nucleotide-binding and oligomerization domain-like receptors (NLRs) which allow them to respond directly to PAMPs and DAMPs (82). For instance, Staphylococcus aureus infection augments TLR2 mRNA expression in astrocytes, and TLR2 knock out mice exhibit attenuated release of TNF and IL-1β (83). Pathogenic activation of astrocytic PRRs and downstream immune signaling pathways may, however, pathologically alter neurodevelopment. Astrocytic TLR4 recognizes LPS, which leads to the expression of TNF, IL-15, and IL-27 through TLR4-MyD88 signaling (84). Early postnatal activation of astrocytic TLR4-MyD88 promotes hippocampal excitatory synaptogenesis and dendritic branching which may underpin increased seizure susceptibility accompanying many CNS infections (85).
During the CNS immune response to infection, microglia, astrocytes, neurons and BMECs secrete a plethora of cytokines, chemokines and MMPs. The roles of these mediators in innate immunity are well documented, therefore, we will concentrate on their involvement during neurodevelopment. Due to the wide breadth of these secreted factors, we will focus on three classical examples: the TNF superfamily (TNFSF), MMPs, and IL-6.
The TNFSF consists of 19 cytokines which interact with the TNF receptor superfamily (TNFRSF) (86). Today, TNFSF/TNFRSF are known to have variegated roles beyond innate immunity and inflammation. Most members of TNFSF and TNFRSF are constitutively expressed in the mammalian CNS and play non-immunological roles critical to healthy brain development such as neuronal and glial cell population control (87). In addition, they regulate axonal and dendritic growth both in the CNS (88) and peripheral nervous system (89). TNF, the best understood member of this family, regulates neurogenesis, astrogliogenesis, and angiogenesis (90–92). Indeed, TNF−/− mice display reduced numbers of neurons and microglia, and pharmacological TNF inhibition impairs learning and memory in in vivo models (93). However, human studies have associated chronic expression of TNF with the development and severity of autism and schizophrenia (94).
MMPs are a family of endopeptidases which mediate broad physiological processes (e.g., bone remodeling, wound healing, embryonic development) via the proteolytic cleavage of extracellular matrix proteins, cytokines, and chemokines. Research into the role of MMPs in the developing brain and spinal cord has revealed their homeostatic and spatiotemporal specific expression, and their roles in neuronal migration, proliferation, and myelination (95–99). Increased levels of MMP-9, one of the most widely studied MMPs, has been linked to various neurodevelopmental disorders; from autism spectrum disorder (100), Fragile X syndrome (101), to schizophrenia (102). In animal models of meningitis, MMP-9 activity has also been shown to contribute to intracerebral hemorrhage and BBB disruption (103, 104). Similarly, elevated MMP-9 and the ratio of MMP-9 to tissue inhibitor of MMP-1 (TIMP-1) have been observed in the CSF of pediatric patients with bacterial meningitis, including tuberculous meningitis (TBM) (105–107). Interestingly, increased MMP-9 during treatment in pediatric TBM patients was associated with improved outcomes, hypothesized to be secondary to MMP's role in neurodevelopment (107).
IL-6 is a cytokine of the interleukin family with both proinflammatory and anti-inflammatory properties that is constitutively expressed at low levels by microglia, neurons, astrocytes, and BMECs (108). In the developing CNS, IL-6 is an essential regulator of neurogenesis (109, 110) and promotor of glial differentiation (111). Notably, neural stem cells are known to self-regulate progenitor pools via autocrine IL-6 signaling, and transient exposure to increased maternal IL-6 is sufficient to dysregulate neural precursor cell pools in the developing forebrain (112). Under pathological conditions, IL-6 levels dramatically increase in the CNS and may be neuroprotective. For instance, microglial IL-6 prevents neuronal loss of neural progenitor cells during herpes simplex virus type 1 infection (113), and endogenous upregulation of IL-6 in response to cerebral ischemia is neuroprotective against excitotoxicity (114). However, elevated IL-6 levels in the fetal brain after maternal immune activation is a key mediator of transcriptional and behavioral alterations in the adult offspring brain (115, 116).
Both the immune system and neurodevelopment are tightly regulated and versatile processes that must respond to changing microenvironmental cues. Therefore, as outlined above, it is not surprising that they share mechanistic overlap (117) which has left the developing brain vulnerable. It is becoming well accepted that infections during pregnancy and childhood can profoundly affect neurodevelopmental outcome of offspring as well as later in adolescence and adulthood (118). Below we highlight two CNS infections to exemplify how components of these processes are dysregulated and associated with adverse clinical outcomes.
CM is one of the deadliest forms of malaria affecting children under 5 years in low- and middle-income countries (LMICs) disproportionately. Despite aggressive antiparasitic therapy and parasite clearance, over 50% of children surviving CM suffer from neurocognitive deficits, seizures, and neurobehavioral disorders (19, 119, 120). Both activated microglia and reactive astrocytes have been linked to the pathobiology of CM in both in vivo models and post-mortem human studies (121). The classical proinflammatory cytokines TNF and IL-6 have also been associated with CM outcomes in human studies (122). Specifically, higher CSF levels of TNF correlate with coma duration and long-term neurocognitive impairments (123). Additionally, IL-6 has been linked to severity, where children with CM have increased levels of serum IL-6 compared to those with uncomplicated malaria (124).
TBM is the most aggressive form of extrapulmonary TB. Like CM, children from LMICs are those most at risk. Pediatric survivors of TBM also suffer from neurodevelopmental deficits in locomotor, personal-social, and language function despite intensive antimicrobial treatment (125, 126). Although numerous factors drive pathogenesis, preclinical and clinical studies suggest the host inflammatory response as a primary contributor to clinical manifestations and sequelae in TBM (127, 128). A study utilizing a murine model showed uncontrolled tissue pathology without the use of treatment targeting neuroinflammation (129), and a rabbit model of pediatric TBM has demonstrated elevated levels of activated microglia in the brains of infected rabbits compared to those of healthy rabbits (130).
Furthermore, a study of South African children with TBM performed transcriptomic analysis and found compartmentalized immune response with increased cytokine signaling in lumbar CSF and increased neuronal excitotoxicity associated with glutamate release in ventricular CSF (131). This glutamate dysregulation within the ventricular milieu may be a manifestation of injury to astrocytes, who are known to be key regulators of glutamate homeostasis (132). In fact, glial fibrillary acidic protein (GFAP), an astrocyte surface marker, along with neuronal injury biomarkers [e.g., S100B and neuron-specific enolase (NSE)] increased in the CSF of pediatric TBM patients with poor outcomes (133). While serum S100B levels fail to reflect the progressive injury in TBM (133), serial measurement of S100B has been proposed to have prognostic value in pediatric patients after traumatic brain injury (134). Hence, sequential evaluation of plasma/serum S100B may prove more informative in TBM.
In both CM and TBM, appropriate antimicrobial therapy does not prevent devastating neurological injury and highlights the need for adjunctive therapy. Currently, there are no approved adjunctive host-directed therapeutics for CM and to date, corticosteroids are the only standard of care host-directed therapy for TBM. Yet, corticosteroids cause side effects and, although they improve TBM mortality, they do not reduce neurological sequelae in survivors and exhibit poor BBB penetration (126, 128, 135, 136). Modulating chemokine and cytokine responses via monoclonal antibodies is an attractive avenue for treating CNS infections. However, these biologics do not cross the BBB and have not shown conclusive results in clinical trials for sepsis and CM (19).
Understanding the dual role that microglia, astrocytes, and secreted immune mediators play during development and the CNS immune response may additionally aid in identifying novel or repurposed drugs to attenuate neurological sequelae. The window of opportunity may also be greatest in children, as they carry the greatest risk of CNS infection and the greatest need to maintain capacity for proper neurodevelopment. Most importantly, any proposed adjunctive therapeutic should aim to have good BBB penetration, and efforts to overcome this bottleneck via novel drug delivery technologies are ongoing (137).
The developing brain of a child is particularly vulnerable to CNS infections because the cells, molecules and signaling pathways that govern the host response to infection also coordinate key aspects of neurodevelopment. This is evident by the fact that survivors of CNS infections, regardless of etiology, often develop not only neurological disorders but long-term neurological sequelae. Thus, it is imperative to further our understanding of the neuro-immune axis during development to design more effective host-directed adjunctive therapeutics that not only attenuate neuroinflammation but also restore key neurodevelopmental processes.
JK, CE, UR, and ET conceptualized the manuscript. JK and CE wrote the initial draft and all coauthors edited the manuscript. ET and UR provided funding. All authors contributed to the article and approved the submitted version.
ET is supported by the NIH NIAID K08AI139371 and the Hartwell Foundation−2018 Individual Biomedical Research Award. UR is supported by the Crick African Career Accelerated Award.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
1. Stiles J, Jernigan TL. The basics of brain development. Neuropsychol Rev. (2010) 20:327–48. doi: 10.1007/s11065-010-9148-4
2. Soeters HM, Diallo AO, Bicaba BW, Kadadé G, Dembélé AY, Acyl MA, et al. Bacterial meningitis epidemiology in five countries in the meningitis belt of Sub-Saharan Africa, 2015–2017. J Infect Dis. (2019) 220(Supplement_4):S165–74. doi: 10.1093/infdis/jiz358
3. Collaborators GM. Global, regional, and national burden of meningitis, 1990–2016: a systematic analysis for the Global Burden of Disease Study 2016. Lancet Neurol. (2018) 17:1061–82. doi: 10.1016/S1474-4422(18)30387-9
4. de Blauw D, Bruning AHL, Busch CBE, Kolodziej LM, Jansen NJG, van Woensel JBM, et al. Epidemiology and etiology of severe childhood encephalitis in the Netherlands. Pediatr Infect Dis J. (2020) 39:267–72. doi: 10.1097/INF.0000000000002551
5. Mameli C, Genoni T, Madia C, Doneda C, Penagini F, Zuccotti G. Brain abscess in pediatric age: a review. Child's Nervous System. (2019) 35:1117–28. doi: 10.1007/s00381-019-04182-4
6. Rand KM, Austin NC, Inder TE, Bora S, Woodward LJ. Neonatal infection and later neurodevelopmental risk in the very preterm infant. J Pediatr. (2016) 170:97–104. doi: 10.1016/j.jpeds.2015.11.017
7. Lee I, Neil JJ, Huettner PC, Smyser CD, Rogers CE, Shimony JS, et al. The impact of prenatal and neonatal infection on neurodevelopmental outcomes in very preterm infants. J Perinatol. (2014) 34:741–7. doi: 10.1038/jp.2014.79
8. van Vliet EOG, de Kieviet JF, Oosterlaan J, van Elburg RM. Perinatal infections and neurodevelopmental outcome in very preterm and very low-birth-weight infants: a meta-analysis. JAMA Pediatr. (2013) 167:662–8. doi: 10.1001/jamapediatrics.2013.1199
9. Walker M, Zunt JR. Parasitic central nervous system infections in immunocompromised hosts. Clin Infect Dis. (2005) 40:1005–15. doi: 10.1086/428621
10. Kadry H, Noorani B, Cucullo L. A blood–brain barrier overview on structure, function, impairment, and biomarkers of integrity. Fluids Barriers CNS. (2020) 17:69. doi: 10.1186/s12987-020-00230-3
11. Saunders NR, Dreifuss J-J, Dziegielewska KM, Johansson PA, Habgood MD, Møllgård K, et al. The rights and wrongs of blood-brain barrier permeability studies: a walk through 100 years of history. Front Neurosci. (2014) 8:404. doi: 10.3389/fnins.2014.00404
12. Iovino F, Engelen-Lee J-Y, Brouwer M, van de Beek D, van der Ende A, Valls Seron M, et al. pIgR and PECAM-1 bind to pneumococcal adhesins RrgA and PspC mediating bacterial brain invasion. J Exp Med. (2017) 214:1619–30. doi: 10.1084/jem.20161668
13. Dinner S, Kaltschmidt J, Stump-Guthier C, Hetjens S, Ishikawa H, Tenenbaum T, et al. Mitogen-activated protein kinases are required for effective infection of human choroid plexus epithelial cells by Listeria monocytogenes. Microbes Infect. (2017) 19:18–33. doi: 10.1016/j.micinf.2016.09.003
14. Ghosh P, Halvorsen EM, Ammendolia DA, Mor-Vaknin N, O'Riordan MXD, Brumell JH, et al. Invasion of the brain by Listeria monocytogenes is mediated by InlF and host cell vimentin. MBio. (2018) 9:e00160–18. doi: 10.1128/mBio.00160-18
15. Drevets DA, Dillon MJ, Schawang JS, Van Rooijen N, Ehrchen J, Sunderkötter C, et al. The Ly-6Chigh monocyte subpopulation transports Listeria monocytogenes into the brain during systemic infection of mice. J Immunol. (2004) 172:4418–24. doi: 10.4049/jimmunol.172.7.4418
16. Sanecka A, Frickel EM. Use and abuse of dendritic cells by Toxoplasma gondii. Virulence. (2012) 3:678–89. doi: 10.4161/viru.22833
17. Konradt C, Ueno N, Christian DA, Delong JH, Pritchard GH, Herz J, et al. Endothelial cells are a replicative niche for entry of Toxoplasma gondii to the central nervous system. Nat Microbiol. (2016) 1:16001. doi: 10.1038/nmicrobiol.2016.1
18. Kim BJ, Bee OB, McDonagh MA, Stebbins MJ, Palecek SP, Doran KS, et al. Modeling group B streptococcus and blood-brain barrier interaction by using induced pluripotent stem cell-derived brain endothelial cells. mSphere. (2017) 2:6. doi: 10.1128/mSphere.00398-17
19. Erice C, Kain KC. New insights into microvascular injury to inform enhanced diagnostics and therapeutics for severe malaria. Virulence. (2019) 10:1034–46. doi: 10.1080/21505594.2019.1696621
20. Erikson K, Tuominen H, Vakkala M, Liisanantti JH, Karttunen T, Syrjälä H, et al. Brain tight junction protein expression in sepsis in an autopsy series. Critical Care. (2020) 24:385. doi: 10.1186/s13054-020-03101-3
21. Louveau A, Smirnov I, Keyes TJ, Eccles JD, Rouhani SJ, Peske JD, et al. Structural and functional features of central nervous system lymphatic vessels. Nature. (2015) 523:337–41. doi: 10.1038/nature14432
22. Iliff JJ, Wang M, Liao Y, Plogg BA, Peng W, Gundersen GA, et al. A paravascular pathway facilitates CSF flow through the brain parenchyma and the clearance of interstitial solutes, including amyloid β. Sci Transl Med. (2012) 4:147ra11. doi: 10.1126/scitranslmed.3003748
23. Carson MJ, Doose JM, Melchior B, Schmid CD, Ploix CC. CNS immune privilege: hiding in plain sight. Immunol Rev. (2006) 213:48–65. doi: 10.1111/j.1600-065X.2006.00441.x
24. Louveau A, Harris TH, Kipnis J. Revisiting the mechanisms of CNS immune privilege. Trends Immunol. (2015) 36:569–77. doi: 10.1016/j.it.2015.08.006
25. Negi N, Das BK, CNS. Not an immunoprivilaged site anymore but a virtual secondary lymphoid organ. Int Rev Immunol. (2018) 37:57–68. doi: 10.1080/08830185.2017.1357719
26. Garay PA, McAllister AK. Novel roles for immune molecules in neural development: implications for neurodevelopmental disorders. Front Synaptic Neurosci. (2010) 2:136. doi: 10.3389/fnsyn.2010.00136
27. Molofsky AV, Krencik R, Ullian EM, Tsai H-h, Deneen B, Richardson WD, et al. Astrocytes and disease: a neurodevelopmental perspective. Genes Dev. (2012) 26:891–907. doi: 10.1101/gad.188326.112
28. Thion MS, Low D, Silvin A, Chen J, Grisel P, Schulte-Schrepping J, et al. Microbiome influences prenatal and adult microglia in a sex-specific manner. Cell. (2018) 172:500–16.e16. doi: 10.1016/j.cell.2017.11.042
29. Thion MS, Ginhoux F, Garel S. Microglia and early brain development: an intimate journey. Science. (2018) 362:185–9. doi: 10.1126/science.aat0474
30. Wurm J, Konttinen H, Andressen C, Malm T, Spittau B. Microglia development and maturation and its implications for induction of microglia-like cells from human iPSCs. Int J Mol Sci. (2021) 22:6. doi: 10.3390/ijms22063088
31. Farhy-Tselnicker I, Boisvert MM, Liu H, Dowling C, Erikson GA, Blanco-Suarez E, et al. Activity-dependent modulation of synapse-regulating genes in astrocytes. Elife. (2021) 10:70514. doi: 10.7554/eLife.70514
32. Chai H, Diaz-Castro B, Shigetomi E, Monte E, Octeau JC, Yu X, et al. Neural circuit-specialized astrocytes: transcriptomic, proteomic, morphological, and functional evidence. Neuron. (2017) 95:531–49 e9. doi: 10.1016/j.neuron.2017.06.029
33. Clarke LE, Liddelow SA, Chakraborty C, Munch AE, Heiman M, Barres BA. Normal aging induces A1-like astrocyte reactivity. Proc Natl Acad Sci USA. (2018) 115:E1896–E905. doi: 10.1073/pnas.1800165115
34. Boisvert MM, Erikson GA, Shokhirev MN, Allen NJ. The aging astrocyte transcriptome from multiple regions of the mouse brain. Cell Rep. (2018) 22:269–85. doi: 10.1016/j.celrep.2017.12.039
35. Mittelbronn M, Dietz K, Schluesener HJ, Meyermann R. Local distribution of microglia in the normal adult human central nervous system differs by up to one order of magnitude. Acta Neuropathol. (2001) 101:249–55. doi: 10.1007/s004010000284
36. Menassa DA, Gomez-Nicola D. Microglial dynamics during human brain development. Front Immunol. (2018) 9:1014. doi: 10.3389/fimmu.2018.01014
37. Fernández-Arjona MdM, Grondona JM, Fernández-Llebrez P, López-Ávalos MD. Microglial morphometric parameters correlate with the expression level of IL-1β, and allow identifying different activated morphotypes. Front Cell Neurosci. (2019) 13:472. doi: 10.3389/fncel.2019.00472
38. Tremblay M, Lowery RL, Majewska AK. Microglial interactions with synapses are modulated by visual experience. PLoS Biol. (2010) 8:e1000527. doi: 10.1371/journal.pbio.1000527
39. Wake H, Moorhouse AJ, Jinno S, Kohsaka S, Nabekura J. Resting microglia directly monitor the functional state of synapses in vivo and determine the fate of ischemic terminals. J Neurosci. (2009) 29:3974–80. doi: 10.1523/JNEUROSCI.4363-08.2009
40. Sipe GO, Lowery RL, Tremblay M, Kelly EA, Lamantia CE, Majewska AK. Microglial P2Y12 is necessary for synaptic plasticity in mouse visual cortex. Nat Commun. (2016) 7:10905. doi: 10.1038/ncomms10905
41. Milinkeviciute G, Henningfield CM, Muniak MA, Chokr SM, Green KN, Cramer KS. Microglia regulate pruning of specialized synapses in the auditory brainstem. Front Neural Circuits. (2019) 13:55. doi: 10.3389/fncir.2019.00055
42. Ma X, Chen K, Cui Y, Huang G, Nehme A, Zhang L, et al. Depletion of microglia in developing cortical circuits reveals its critical role in glutamatergic synapse development, functional connectivity, and critical period plasticity. J Neurosci Res. (2020) 98:1968–86. doi: 10.1002/jnr.24641
43. Favuzzi E, Huang S, Saldi GA, Binan L, Ibrahim LA, Fernández-Otero M, et al. GABA-receptive microglia selectively sculpt developing inhibitory circuits. Cell. (2021) 184:4048–63.e32. doi: 10.1016/j.cell.2021.06.018
44. Schafer Dorothy P, Lehrman Emily K, Kautzman Amanda G, Koyama R, Mardinly Alan R, Yamasaki R, et al. Microglia sculpt postnatal neural circuits in an activity and complement-dependent manner. Neuron. (2012) 74:691–705. doi: 10.1016/j.neuron.2012.03.026
45. Paolicelli RC, Bolasco G, Pagani F, Maggi L, Scianni M, Panzanelli P, et al. Synaptic pruning by microglia is necessary for normal brain development. Science. (2011) 333:1456–8. doi: 10.1126/science.1202529
46. Lehrman EK, Wilton DK, Litvina EY, Welsh CA, Chang ST, Frouin A, et al. CD47 protects synapses from excess microglia-mediated pruning during development. Neuron. (2018) 100(1):120–34.e6. doi: 10.1016/j.neuron.2018.09.017
47. Esen N, Kielian T. Central role for MyD88 in the responses of microglia to pathogen-associated molecular patterns. J Immunol. (2006) 176:6802–11. doi: 10.4049/jimmunol.176.11.6802
48. Hanisch U-K, Gertig U. Microglial diversity by responses and responders. Front Cell Neurosci. (2014) 8:101. doi: 10.3389/fncel.2014.00101
49. Hickman SE, Kingery ND, Ohsumi TK, Borowsky ML. Wang L-c, Means TK, et al. The microglial sensome revealed by direct RNA sequencing. Nat Neurosci. (2013) 16:1896–905. doi: 10.1038/nn.3554
50. Hickman SE, El Khoury J. Analysis of the microglial sensome. Methods Mol Biol. (2019) 2034:305–23. doi: 10.1007/978-1-4939-9658-2_23
51. Weinstein JR, Quan Y, Hanson JF, Colonna L, Iorga M, Honda S-i, et al. IgM-dependent phagocytosis in microglia is mediated by complement receptor 3, not Fcα/μ receptor. J Immunol. (2015) 195:5309–17. doi: 10.4049/jimmunol.1401195
52. Cockram TOJ, Puigdellívol M, Brown GC. Calreticulin and galectin-3 opsonise bacteria for phagocytosis by microglia. Front Immunol. (2019) 10:2647. doi: 10.3389/fimmu.2019.02647
53. Fekete R, Cserép C, Lénárt N, Tóth K, Orsolits B, Martinecz B, et al. Microglia control the spread of neurotropic virus infection via P2Y12 signalling and recruit monocytes through P2Y12-independent mechanisms. Acta Neuropathol. (2018) 136:461–82. doi: 10.1007/s00401-018-1885-0
54. Sariol A, Mackin S, Allred M-G, Ma C, Zhou Y, Zhang Q, et al. Microglia depletion exacerbates demyelination and impairs remyelination in a neurotropic coronavirus infection. Proc Nat Acad Sci. (2020) 117:24464–74. doi: 10.1073/pnas.2007814117
55. McKimmie CS, Roy D, Forster T, Fazakerley JK. Innate immune response gene expression profiles of N9 microglia are pathogen-type specific. J Neuroimmunol. (2006) 175:128–41. doi: 10.1016/j.jneuroim.2006.03.012
56. Moseman EA, Blanchard AC, Nayak D, McGavern DB. T cell engagement of cross-presenting microglia protects the brain from a nasal virus infection. Sci Immunol. (2020) 5:eabb1817. doi: 10.1126/sciimmunol.abb1817
57. Neumann J, Sauerzweig S, Rönicke R, Gunzer F, Dinkel K, Ullrich O, et al. Microglia cells protect neurons by direct engulfment of invading neutrophil granulocytes: a new mechanism of CNS immune privilege. J Neurosci. (2008) 28:5965–75. doi: 10.1523/JNEUROSCI.0060-08.2008
58. Dallérac G, Chever O, Rouach N. How do astrocytes shape synaptic transmission? Insights from electrophysiology. Front Cell Neurosci. (2013) 7:159. doi: 10.3389/fncel.2013.00159
59. Heithoff BP, George KK, Phares AN, Zuidhoek IA, Munoz-Ballester C, Robel S. Astrocytes are necessary for blood–brain barrier maintenance in the adult mouse brain. Glia. (2021) 69:436–72. doi: 10.1002/glia.23908
60. Hasel P, Rose IVL, Sadick JS, Kim RD, Liddelow SA. Neuroinflammatory astrocyte subtypes in the mouse brain. Nat Neurosci. (2021) 24:1475–87. doi: 10.1038/s41593-021-00905-6
61. Farhy-Tselnicker I, Allen NJ. Astrocytes, neurons, synapses: a tripartite view on cortical circuit development. Neural Dev. (2018) 13:7. doi: 10.1186/s13064-018-0104-y
62. Chung WS, Allen NJ, Eroglu C. Astrocytes control synapse formation, function, and elimination. Cold Spring Harb Perspect Biol. (2015) 7:a020370. doi: 10.1101/cshperspect.a020370
63. Christopherson KS, Ullian EM, Stokes CC, Mullowney CE, Hell JW, Agah A, et al. Thrombospondins are astrocyte-secreted proteins that promote CNS synaptogenesis. Cell. (2005) 120:421–33. doi: 10.1016/j.cell.2004.12.020
64. Eroglu C, Allen NJ, Susman MW, O'Rourke NA, Park CY, Ozkan E, et al. Gabapentin receptor alpha2delta-1 is a neuronal thrombospondin receptor responsible for excitatory CNS synaptogenesis. Cell. (2009) 139:380–92. doi: 10.1016/j.cell.2009.09.025
65. Eroglu C. The role of astrocyte-secreted matricellular proteins in central nervous system development and function. J Cell Commun Signal. (2009) 3:167–76. doi: 10.1007/s12079-009-0078-y
66. Kucukdereli H, Allen NJ, Lee AT, Feng A, Ozlu MI, Conatser LM, et al. Control of excitatory CNS synaptogenesis by astrocyte-secreted proteins Hevin and SPARC. Proc Natl Acad Sci USA. (2011) 108:E440–9. doi: 10.1073/pnas.1104977108
67. Gan KJ, Südhof TC. SPARCL1 promotes excitatory but not inhibitory synapse formation and function independent of neurexins and neuroligins. J Neurosci. (2020) 40:8088–102. doi: 10.1523/JNEUROSCI.0454-20.2020
68. Murai KK, Nguyen LN, Irie F, Yamaguchi Y, Pasquale EB. Control of hippocampal dendritic spine morphology through ephrin-A3/EphA4 signaling. Nat Neurosci. (2003) 6:153–60. doi: 10.1038/nn994
69. Nishida H, Okabe S. Direct astrocytic contacts regulate local maturation of dendritic spines. J Neurosci. (2007) 27:331–40. doi: 10.1523/JNEUROSCI.4466-06.2007
70. Zhou H-J, Zhang H-N, Tang T, Zhong J-H, Qi Y, Luo J-K, et al. Alteration of thrombospondin-1 and−2 in rat brains following experimental intracerebral hemorrhage: Laboratory investigation. J Neurosurg JNS. (2010) 113:820–5. doi: 10.3171/2010.1.JNS09637
71. Kim J-H, Jung H-G, Kim A, Shim HS, Hyeon SJ, Lee Y-S, et al. Hevin–calcyon interaction promotes synaptic reorganization after brain injury. Cell Death Different. (2021) 28:2571–88. doi: 10.1038/s41418-021-00772-5
72. Segarra M, Aburto MR, Cop F, Llaó-Cid C, Härtl R, Damm M, et al. Endothelial Dab1 signaling orchestrates neuro-glia-vessel communication in the central nervous system. Science. (2018) 361:eaao2861. doi: 10.1126/science.aao2861
73. Argaw AT, Asp L, Zhang J, Navrazhina K, Pham T, Mariani JN, et al. Astrocyte-derived VEGF-A drives blood-brain barrier disruption in CNS inflammatory disease. J Clin Invest. (2012) 122:2454–68. doi: 10.1172/JCI60842
74. Min H, Hong J, Cho I-H, Jang YH, Lee H, Kim D, et al. TLR2-induced astrocyte MMP9 activation compromises the blood brain barrier and exacerbates intracerebral hemorrhage in animal models. Mol Brain. (2015) 8:23. doi: 10.1186/s13041-015-0116-z
75. Chelluboina B, Klopfenstein JD, Pinson DM, Wang DZ, Vemuganti R, Veeravalli KK. Matrix metalloproteinase-12 induces blood–brain barrier damage after focal cerebral ischemia. Stroke. (2015) 46:3523–31. doi: 10.1161/STROKEAHA.115.011031
76. Paul R, Lorenzl S, Koedel U, Sporer B, Vogel U, Frosch M, et al. Matrix metalloproteinases contribute to the blood—brain barrier disruption during bacterial meningitis. Ann Neurol. (1998) 44:592–600. doi: 10.1002/ana.410440404
77. Xing G, Zhao T, Zhang X, Li H, Li X, Cui P, et al. Astrocytic sonic hedgehog alleviates intracerebral hemorrhagic brain injury via modulation of blood brain barrier integrity. Front Cell Neurosci. (2020) 14:364. doi: 10.3389/fncel.2020.575690
78. Sterpka A, Yang J, Strobel M, Zhou Y, Pauplis C, Chen X. Diverged morphology changes of astrocytic and neuronal primary cilia under reactive insults. Mol Brain. (2020) 13:28. doi: 10.1186/s13041-020-00571-y
79. Zamanian JL, Xu L, Foo LC, Nouri N, Zhou L, Giffard RG, et al. Genomic analysis of reactive astrogliosis. J Neurosci. (2012) 32:6391–410. doi: 10.1523/JNEUROSCI.6221-11.2012
80. Escartin C, Galea E, Lakatos A, O'Callaghan JP, Petzold GC, Serrano-Pozo A, et al. Reactive astrocyte nomenclature, definitions, and future directions. Nat Neurosci. (2021) 24:312–25. doi: 10.1038/s41593-020-00783-4
81. Liddelow SA, Guttenplan KA, Clarke LE, Bennett FC, Bohlen CJ, Schirmer L, et al. Neurotoxic reactive astrocytes are induced by activated microglia. Nature. (2017) 541:481–7. doi: 10.1038/nature21029
82. Farina C, Aloisi F, Meinl E. Astrocytes are active players in cerebral innate immunity. Trends Immunol. (2007) 28:138–45. doi: 10.1016/j.it.2007.01.005
83. Esen N, Tanga FY, DeLeo JA, Kielian T. Toll-like receptor 2 (TLR2) mediates astrocyte activation in response to the Gram-positive bacterium Staphylococcus aureus. J Neurochem. (2004) 88:746–58. doi: 10.1046/j.1471-4159.2003.02202.x
84. Gorina R, Font-Nieves M, Márquez-Kisinousky L, Santalucia T, Planas AM. Astrocyte TLR4 activation induces a proinflammatory environment through the interplay between MyD88-dependent NFκB signaling, MAPK, and Jak1/Stat1 pathways. Glia. (2011) 59:242–55. doi: 10.1002/glia.21094
85. Shen Y, Qin H, Chen J, Mou L, He Y, Yan Y, et al. Postnatal activation of TLR4 in astrocytes promotes excitatory synaptogenesis in hippocampal neurons. J Cell Biol. (2016) 215:719–34. doi: 10.1083/jcb.201605046
86. Yi F, Frazzette N, Cruz AC, Klebanoff CA, Siegel RM. Beyond cell death: new functions for TNF family cytokines in autoimmunity and tumor immunotherapy. Trends Mol Med. (2018) 24:642–53. doi: 10.1016/j.molmed.2018.05.004
87. Twohig JP, Cuff SM, Yong AA, Wang EC. The role of tumor necrosis factor receptor superfamily members in mammalian brain development, function and homeostasis. Rev Neurosci. (2011) 22:509–33. doi: 10.1515/RNS.2011.041
88. Carriba P, Davies AM. How CD40L reverse signaling regulates axon and dendrite growth. Cell Mol Life Sci. (2021) 78:1065–83. doi: 10.1007/s00018-020-03563-2
89. Erice C, Calhan OY, Kisiswa L, Wyatt S, Davies AM. Regional differences in the contributions of TNF reverse and forward signaling to the establishment of sympathetic innervation. Dev Neurobiol. (2019) 79:317–34. doi: 10.1002/dneu.22680
90. Bernardino L, Agasse F, Silva B, Ferreira R, Grade S, Malva JO. Tumor necrosis factor-alpha modulates survival, proliferation, and neuronal differentiation in neonatal subventricular zone cell cultures. Stem Cells. (2008) 26:2361–71. doi: 10.1634/stemcells.2007-0914
91. Lan X, Chen Q, Wang Y, Jia B, Sun L, Zheng J, et al. TNF-α Affects Human Cortical Neural Progenitor Cell Differentiation through the Autocrine Secretion of Leukemia Inhibitory Factor. PLoS ONE. (2012) 7:e50783. doi: 10.1371/journal.pone.0050783
92. Wang L, Chopp M, Teng H, Bolz M, Francisco MA, Aluigi DM, et al. Tumor necrosis factor α primes cerebral endothelial cells for erythropoietin-induced angiogenesis. J Cereb Blood Flow Metab. (2011) 31:640–7. doi: 10.1038/jcbfm.2010.138
93. Yli-Karjanmaa M, Larsen KS, Fenger CD, Kristensen LK, Martin NA, Jensen PT, et al. TNF deficiency causes alterations in the spatial organization of neurogenic zones and alters the number of microglia and neurons in the cerebral cortex. Brain Behav Immun. (2019) 82:279–97. doi: 10.1016/j.bbi.2019.08.195
94. Ratnayake U, Quinn T, Walker DW, Dickinson H. Cytokines and the neurodevelopmental basis of mental illness. Front Neurosci. (2013) 7:180. doi: 10.3389/fnins.2013.00180
95. Cañete Soler R, Gui Y-H, Linask KK, Muschel RJ. MMP-9 (gelatinase B) mRNA is expressed during mouse neurogenesis and may be associated with vascularization. Dev Brain Res. (1995) 88:37–52. doi: 10.1016/0165-3806(95)00079-S
96. Ayoub AE, Cai TQ, Kaplan RA, Luo J. Developmental expression of matrix metalloproteinases 2 and 9 and their potential role in the histogenesis of the cerebellar cortex. J Comp Neurol. (2005) 481:403–15. doi: 10.1002/cne.20375
97. Ulrich R, Gerhauser I, Seeliger F, Baumgärtner W, Alldinger S. Matrix metalloproteinases and their inhibitors in the developing mouse brain and spinal cord: a reverse transcription quantitative polymerase chain reaction study. Dev Neurosci. (2005) 27:408–18. doi: 10.1159/000088455
98. Larsen PH, DaSilva AG, Conant K, Yong VW. Myelin formation during development of the CNS is delayed in matrix metalloproteinase-9 and−12 null mice. J Neurosci. (2006) 26:2207–14. doi: 10.1523/JNEUROSCI.1880-05.2006
99. Bednarek N, Clément Y, Lelièvre V, Olivier P, Loron G, Garnotel R, et al. Ontogeny of MMPs and TIMPs in the murine neocortex. Pediatr Res. (2009) 65:296–300. doi: 10.1203/PDR.0b013e3181973aee
100. Abdallah MW, Pearce BD, Larsen N, Greaves-Lord K, Nørgaard-Pedersen B, Hougaard DM, et al. Amniotic fluid MMP-9 and neurotrophins in autism spectrum disorders: an exploratory study. Autism Research. (2012) 5:428–33. doi: 10.1002/aur.1254
101. Dziembowska M, Pretto DI, Janusz A, Kaczmarek L, Leigh MJ, Gabriel N, et al. High MMP-9 activity levels in fragile X syndrome are lowered by minocycline. Am J Med Genet Part A. (2013) 161a:1897–903. doi: 10.1002/ajmg.a.36023
102. Dwir D, Giangreco B, Xin L, Tenenbaum L, Cabungcal J-H, Steullet P, et al. MMP9/RAGE pathway overactivation mediates redox dysregulation and neuroinflammation, leading to inhibitory/excitatory imbalance: a reverse translation study in schizophrenia patients. Mol Psychiatry. (2020) 25:2889–904. doi: 10.1038/s41380-019-0393-5
103. Leib SL, Leppert D, Clements J, Täuber MG. Matrix metalloproteinases contribute to brain damage in experimental pneumococcal meningitis. Infect Immun. (2000) 68:615–20. doi: 10.1128/IAI.68.2.615-620.2000
104. Ricci S, Grandgirard D, Wenzel M, Braccini T, Salvatore P, Oggioni MR, et al. Inhibition of matrix metalloproteinases attenuates brain damage in experimental meningococcal meningitis. BMC Infect Dis. (2014) 14:726. doi: 10.1186/s12879-014-0726-6
105. Roine I, Pelkonen T, Lauhio A, Lappalainen M, Cruzeiro ML, Bernardino L, et al. Changes in MMP-9 and TIMP-1 concentrations in cerebrospinal fluid after 1 week of treatment of childhood bacterial meningitis. J Clin Microbiol. (2015) 53:2340–2. doi: 10.1128/JCM.00714-15
106. Leppert D, Leib SL, Grygar C, Miller KM, Schaad UB, Holländer GA. Matrix metalloproteinase (MMP)-8 and MMP-9 in cerebrospinal fluid during bacterial meningitis: association with blood-brain barrier damage and neurological sequelae. Clin Infect Dis. (2000) 31:80–4. doi: 10.1086/313922
107. Li YJ, Wilkinson KA, Wilkinson RJ, Figaji AA, Rohlwink UK. Elevated matrix metalloproteinase concentrations offer novel insight into their role in pediatric tuberculous meningitis. J Pediatric Infect Dis Soc. (2019) 9:82–6.
108. Erta M, Quintana A, Hidalgo J. Interleukin-6, a major cytokine in the central nervous system. Int J Biol Sci. (2012) 8:1254–66. doi: 10.7150/ijbs.4679
109. Islam O, Gong X, Rose-John S, Heese K. Interleukin-6 and neural stem cells: more than gliogenesis. Mol Biol Cell. (2009) 20:188–99. doi: 10.1091/mbc.e08-05-0463
110. Mouihate A, Kalakh S. Maternal interleukin-6 hampers hippocampal neurogenesis in adult rat offspring in a sex-dependent manner. Dev Neurosci. (2021) 43:106–15. doi: 10.1159/000516370
111. Nakanishi M, Niidome T, Matsuda S, Akaike A, Kihara T, Sugimoto H. Microglia-derived interleukin-6 and leukaemia inhibitory factor promote astrocytic differentiation of neural stem/progenitor cells. Eur J Neurosci. (2007) 25:649–58. doi: 10.1111/j.1460-9568.2007.05309.x
112. Gallagher D, Norman AA, Woodard CL, Yang G, Gauthier-Fisher A, Fujitani M, et al. Transient maternal IL-6 mediates long-lasting changes in neural stem cell pools by deregulating an endogenous self-renewal pathway. Cell Stem Cell. (2013) 13:564–76. doi: 10.1016/j.stem.2013.10.002
113. Chucair-Elliott AJ, Conrady C, Zheng M, Kroll CM, Lane TE, Carr DJJ. Microglia-induced IL-6 protects against neuronal loss following HSV-1 infection of neural progenitor cells. Glia. (2014) 62:1418–34. doi: 10.1002/glia.22689
114. Ali C, Nicole O, Docagne F, Lesne S, MacKenzie ET, Nouvelot A, et al. Ischemia-induced interleukin-6 as a potential endogenous neuroprotective cytokine against NMDA receptor-mediated excitoxicity in the brain. J Cereb Blood Flow Metab. (2000) 20:956–66. doi: 10.1097/00004647-200006000-00008
115. Smith SE Li J, Garbett K, Mirnics K, Patterson PH. Maternal immune activation alters fetal brain development through interleukin-6. J Neurosci. (2007) 27:10695–702. doi: 10.1523/JNEUROSCI.2178-07.2007
116. Connor CM, Dincer A, Straubhaar J, Galler JR, Houston IB, Akbarian S. Maternal immune activation alters behavior in adult offspring, with subtle changes in the cortical transcriptome and epigenome. Schizophr Res. (2012) 140:175–84. doi: 10.1016/j.schres.2012.06.037
117. Zengeler KE, Lukens JR. Innate immunity at the crossroads of healthy brain maturation and neurodevelopmental disorders. Nat Rev Immunol. (2021) 21:454–68. doi: 10.1038/s41577-020-00487-7
118. Kohler-Forsberg O, Petersen L, Gasse C, Mortensen PB, Dalsgaard S, Yolken RH, et al. A nationwide study in denmark of the association between treated infections and the subsequent risk of treated mental disorders in children and adolescents. JAMA Psychiat. (2019) 76:271–9. doi: 10.1001/jamapsychiatry.2018.3428
119. Ssenkusu JM, Hodges JS, Opoka RO. Long-term behavioral problems in children with severe malaria. Pediatrics. (2017) 138:e20161965. doi: 10.1542/peds.2017-2709
120. Idro R, Kakooza-Mwesige A, Asea B, Ssebyala K, Bangirana P, Opoka RO, et al. Cerebral malaria is associated with long-term mental health disorders: a cross sectional survey of a long-term cohort. Malaria J. (2016) 15:1–11. doi: 10.1186/s12936-016-1233-6
121. Andoh NE, Gyan BA. The potential roles of glial cells in the neuropathogenesis of cerebral malaria. Front Cell Infect Microbiol. (2021) 11:957. doi: 10.3389/fcimb.2021.741370
122. Dunst J, Kamena F, Matuschewski K. Cytokines and chemokines in cerebral malaria pathogenesis. Front Cell Infect Microbiol. (2017) 7:324. doi: 10.3389/fcimb.2017.00324
123. Shabani E, Ouma BJ, Idro R, Bangirana P, Opoka RO, Park GS, et al. Elevated cerebrospinal fluid tumour necrosis factor is associated with acute and long-term neurocognitive impairment in cerebral malaria. Parasite Immunol. (2017) 39:12438. doi: 10.1111/pim.12438
124. Lyke KE, Burges R, Cissoko Y, Sangare L, Dao M, Diarra I, et al. Serum levels of the proinflammatory cytokines interleukin-1 beta (IL-1 beta), IL-6, IL-8, IL-10, tumor necrosis factor alpha, and IL-12(p70) in Malian children with severe Plasmodium falciparum malaria and matched uncomplicated malaria or healthy controls. Infect Immun. (2004) 72:5630–7. doi: 10.1128/IAI.72.10.5630-5637.2004
125. Rohlwink UK, Donald K, Gavine B, Padayachy L, Wilmshurst JM, Fieggen GA, et al. Clinical characteristics and neurodevelopmental outcomes of children with tuberculous meningitis and hydrocephalus. Dev Med Child Neurol. (2016) 58:461–8. doi: 10.1111/dmcn.13054
126. Van Well GT, Paes BF, Terwee CB, Springer P, Roord JJ, Donald PR, et al. Twenty years of pediatric tuberculous meningitis: a retrospective cohort study in the western cape of South Africa. Pediatrics. (2009) 123:e1–8. doi: 10.1542/peds.2008-1353
127. Be NA, Kim KS, Bishai WR, Jain SK. Pathogenesis of central nervous system tuberculosis. Curr Mol Med. (2009) 9:94–9. doi: 10.2174/156652409787581655
128. Jain SK, Tobin DM, Tucker EW, Venketaraman V, Ordonez AA, Jayashankar L, et al. Tuberculous meningitis: a roadmap for advancing basic and translational research. Nat Immunol. (2018) 19:521–5. doi: 10.1038/s41590-018-0119-x
129. Majeed S, Radotra BD, Sharma S. Adjunctive role of MMP-9 inhibition along with conventional anti-tubercular drugs against experimental tuberculous meningitis. Int J Exp Pathol. (2016) 97:230–7. doi: 10.1111/iep.12191
130. Tucker EW, Pokkali S, Zhang Z, DeMarco VP, Klunk M, Smith ES, et al. Microglia activation in a pediatric rabbit model of tuberculous meningitis. Dis Model Mech. (2016) 9:1497–506. doi: 10.1242/dmm.027326
131. Rohlwink UK, Figaji A, Wilkinson KA, Horswell S, Sesay AK, Deffur A, et al. Tuberculous meningitis in children is characterized by compartmentalized immune responses and neural excitotoxicity. Nat Commun. (2019) 10:3767. doi: 10.1038/s41467-019-11783-9
132. Mahmoud S, Gharagozloo M, Simard C, Gris D. Astrocytes maintain glutamate homeostasis in the CNS by controlling the balance between glutamate uptake and release. Cells. (2019) 8:184. doi: 10.3390/cells8020184
133. Rohlwink UK, Mauff K, Wilkinson KA, Enslin N, Wegoye E, Wilkinson RJ, et al. Biomarkers of cerebral injury and inflammation in pediatric tuberculous meningitis. Clin Infect Dis. (2017) 65:1298–307. doi: 10.1093/cid/cix540
134. Park S-H, Hwang S-K. Prognostic value of serum levels of S100 calcium-binding protein B, neuron-specific enolase, and interleukin-6 in pediatric patients with traumatic brain injury. World Neurosurg. (2018) 118:e534–e42. doi: 10.1016/j.wneu.2018.06.234
135. Torok ME, Nguyen DB, Tran TH, Nguyen TB, Thwaites GE, Hoang TQ, et al. Dexamethasone and long-term outcome of tuberculous meningitis in Vietnamese adults and adolescents. PLoS ONE. (2011) 6:e27821. doi: 10.1371/journal.pone.0027821
136. Prasad K, Singh MB, Ryan H. Corticosteroids for managing tuberculous meningitis. Cochrane Database Syst Rev. (2016) 4:Cd002244. doi: 10.1002/14651858.CD002244.pub4
Keywords: microglia, astrocyte, development, central nervous system (CNS) infection, neurological sequelae, pediatrics
Citation: Kim J, Erice C, Rohlwink UK and Tucker EW (2022) Infections in the Developing Brain: The Role of the Neuro-Immune Axis. Front. Neurol. 13:805786. doi: 10.3389/fneur.2022.805786
Received: 31 October 2021; Accepted: 24 January 2022;
Published: 17 February 2022.
Edited by:
Peter R. Williamson, National Institutes of Health (NIH), United StatesReviewed by:
Todd Peterson, University of North Carolina at Wilmington, United StatesCopyright © 2022 Kim, Erice, Rohlwink and Tucker. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Elizabeth W. Tucker, ZXR1Y2tlcjlAamguZWR1
†These authors have contributed equally to this work and share first authorship
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.