 Sara E. Gombash
Sara E. Gombash Priscilla W. Lee
Priscilla W. Lee Elizabeth Sawdai2
Elizabeth Sawdai2 Amy E. Lovett-Racke
Amy E. Lovett-Racke
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Neurol., 16 May 2022
Sec. Multiple Sclerosis and Neuroimmunology
Volume 13 - 2022 | https://doi.org/10.3389/fneur.2022.796933
This article is part of the Research TopicPreventing Multiple SclerosisView all 12 articles
Vitamin D insufficiency during childhood has been linked to the development of multiple sclerosis (MS), typically an adult-onset inflammatory demyelinating disease of the central nervous system (CNS). Since vitamin D was known to have immunoregulatory properties on both innate and adaptive immunity, it was hypothesized that low vitamin D resulted in aberrant immune responses and the development of MS. However, vitamin D receptors are present on many cell types, including neurons, oligodendrocytes, astrocytes and microglia, and vitamin D has profound effects on development and function of the CNS. This leads to the possibility that low vitamin D may alter the CNS in a manner that makes it vulnerable to inflammation and the development of MS. This review analysis the role of vitamin D in the immune and nervous system, and how vitamin D insufficiency in children may contribute to the development of MS.
The importance of vitamin D (VitD) in health was formally recognized in 1922 when it was determined that cod liver oil and sunlight cured rickets (1, 2). Our understanding of the role of VitD has expanded well beyond bone health with the observation that VitD receptors (VDR) are widely expressed on many cell types in many tissues. VitD is a fat-soluble vitamin with limited availability in foods. Thus, the predominant source of VitD is synthesis in the skin after sun exposure. VitD is a steroid hormone that regulates numerous genes important in cell differentiation, proliferation and homeostasis. Unfortunately, VitD deficiency is prevalent worldwide, with infants, pregnant women, the elderly, and dark-skinned individuals being the most affected (3). There is substantial literature describing the importance of VitD as an immune regulator with a growing body of evidence on the importance of VitD on the development and function of the nervous system. While VitD deficiency has been correlated with a variety of human diseases, there is substantial evidence that VitD deficiency, particularly in children, may be a major risk factor for the development of multiple sclerosis (MS), which is typically diagnosed in young adults. Historically, it has been postulated that low VitD may be promoting a dysregulated and/or hyperactivated immune system that leads to CNS inflammation. However, the fact that low VitD in children appears to be more closely associated with MS than other autoimmune diseases suggests that VitD insufficiency may be playing an important role in the central nervous system (CNS), making it more vulnerable to inflammation. Thus, VitD deficiency in children may be contributing to the risk of developing MS as an adult by dysregulation of genes in both the immune system and CNS.
Multiple sclerosis (MS) is an inflammatory, demyelinating disease affecting an estimated 2.8 million people worldwide (4). Clinically, MS is characterized by relapsing and progressive neurological dysfunction. In most patients, the disease begins with episodes of neurological dysfunction followed by complete or partial remission— the relapsing/remitting form of the disease (RRMS). In some RRMS patients, the disease is later transformed into uninterrupted progression of neurological deficits — the secondary progressive phase of the disease (SPMS). Other patients' disease initiates with a slow, progressive accumulation of neurological dysfunction — primary progressive multiple sclerosis (PPMS) (5). Pathologically, MS is characterized by focal plaques of demyelination with activated microglia and abundant peripheral inflammatory cells in the CNS. The cause of the disease is unknown and therapies are limited to disease modifying medications that reduce the number inflammatory lesions and slow disease progression.
Many decades ago it was found that MS has the lowest frequency along the equator, and increases prevalence with increasing latitude (6). The relationship between latitude and MS risks has been observed in several studies. MS frequency among French farmers displayed a north-south gradient and was inversely correlated with exposure to sunlight, though the gene pools and life styles of individuals were broadly comparable (7). UK migrants who live in Tasmania in the south had greater MS frequency than those that migrated to tropical Queensland (8). Although MS risks seem to decrease with migration from high to low latitudes (9), the timing of migration has critical effects on this change. Migration studies have shown that people who are younger than 15 years at the time of migration tend to adopt MS risks of the country to which they migrate, whereas those who are older than 15 years retain similar incidence as their country of origin. A recent study in New Zealand confirmed the latitude gradient, but also found that the prevalence gradient was strongest at birth (10). A comprehensive meta-analysis of 94 studies published through 2018 confirmed the latitude gradient in MS (11). Analysis of sun exposure based on age found that living in an area with high UV-B before MS onset was associated with a 45% lower risk of MS, and a 51% reduction in risk when living in a medium to high UV-B area from 5-15 year of age (12). Overall, the risk of developing MS is largely determined before the age of 15 years (13–17) or at least within the first two decades (18), suggesting a role for the environment in modifying MS risks during childhood and adolescence.
The cause of MS is unknown. It remains unclear what triggers the immune system to attack the myelin sheath. Twin studies reveal that genetic factors have important roles in MS risk. The rate of concordance for MS among monozygotic twins is 25–40%, which is much higher than the 5% concordance rate among dizygotic twins (19). Genome-wide sequencing studies have further identified human leukocyte antigen (HLA) class II exerting the strongest association with MS risks (20, 21). On the other hand, the concordance rate among monozygotic twins is not 100%, which means MS risks are not fully determined by genetics.
Unquestionably, the environment is also influential on disease susceptibility. Although the identity of environmental factors involved in MS is not yet unequivocally known, accumulating evidence lends strong supports to several candidates: Epstein-Barr virus (EBV) infection, cigarette smoking and VitD. The relationship between EBV seropositivity and MS risks is now firmly established (p < 10–23). Virtually all (99.5%) patients with MS are seropositive for antibodies directed against EBV (22). A recent study analyzed multiple environmental factors that may contribute to MS risk, including VitD levels and EBV antibody titers (23). EBV antibody titers were significantly higher in MS patients and there was an inverse relationship between VitD levels and EDSS, yet no correlation between VitD levels and EBV antibody titers. The prevalence of EBV infection is high (94%) in age/gender-matched controls, so the vast majority of infected individuals do not develop MS, which suggests that EBV infection may be a necessary contributing factor to MS risk but not a cause of MS. For cigarette smoking, a positive association between smoking before age of onset and MS risks is found in some case-control studies (24, 25). Individuals with RRMS have an increased risk of developing SPMS if they have ever smoked, compared with non-smokers (26). These factors— genetics, EBV infection and smoking— may work interactively to determine MS susceptibility, but none of them can fully explain the geographic variations in MS frequency and the changes in risk that occur with migration.
Much of our fundamental understanding of MS is based on observations in rodent models of MS such as experimental autoimmune encephalomyelitis (EAE). EAE resembles MS in both clinical and pathological aspects (27, 28). Susceptibility to EAE and clinical course vary among strains of mice. For instance, B10.PL and SJL/J mouse strains are two of the more commonly used susceptible strains, whereas BALB/c is much less susceptible (29). SJL/J mice develop a relapsing-remitting disease that can transition into a progressive disease over time, closely resembling RRMS and the transition to the SPMS form (28). C57B/6 mice have become the most utilized EAE model due to the availability of genetically modified mice on the C57B/6 background that allows for defining the role of specific molecules in CNS autoimmunity. The downside of using the C57B/6 mouse model is that disease course and inflammatory components of the lesion have distinct differences from the human condition. Instead, C57B/6 mice develop a rapid-onset, chronic neurological disease without relapses. Furthermore, antibodies and neutrophils contribute significantly to lesion formation which is not typical of MS (30, 31). Some have speculated that C57B/6 EAE may actually be a better model for neuromyelitis optica (NMO), a rare autoimmune neurodegenerative disease very similar in phenotype to MS in which antibodies to aquaporin 4 and neutrophils are known to contribute to the formation of demyelinating lesions (32). These different EAE models have contributed to our understanding of CNS autoimmunity, yet we should be cognizant of how they may or may not reflect MS.
EAE can be induced by several methods. Active induction of EAE is done by direct immunization with myelin proteins or peptides emulsified in complete Freund's adjuvant. The myelin protein and/or peptide used differs because of variations in MHC between strains of mice. Passive induction of EAE is done by transfer of activated myelin-specific CD4 T cells into a naïve mouse. The myelin-specific CD4 T cells can be generated by immunization with a myelin protein/peptide, followed by removal of the draining lymph nodes, reactivation of the myelin-specific T cells in vitro, and injection of the myelin-specific T cells into naïve mice resulting in EAE. Alternatively, T cell receptor transgenic T cells specific for a myelin peptide can be used. For example, CD4 T cells from a T cell receptor transgenic B10.PL mouse in which all the CD4 T cells recognize myelin basic protein (MBP) Ac1-11 peptide can be activated in vitro and injected into naïve B10.PL mice, resulting in classical EAE (33–35). In EAE, both myelin-specific Th1 and Th17 cells contribute to pathogenesis, and both cell types have been implicated in MS (35, 36). While myelin-specific T cells can be found in both healthy individuals and MS patients, myelin-reactive CD4 T cells from MS patients have an activated and/or memory phenotype, whereas those cells are naïve in healthy individuals (37–41), supporting the idea that myelin-specific CD4 T cells are contributing to disease pathology in MS.
For most people, exposure to sunlight is their major source of VitD. Ultraviolet B (UVB) photolyses 7-dehydrocholesterol to pre-vitamin D3 in the epidermis, which then isomerizes to vitamin D3 (Figure 1). VitD can be also obtained from diet through ingestion of vitamin D2 (ergocalciferol) derived from plants, colecalciferol supplements/fortified foods and oily fish. Vitamin D3 in the body then undergoes a series of hydroxylations, first to 25-hydroxyvitamin D3 (25(OH)D3) in the liver, the main circulating form of the vitD with relatively long half-life, and then to biologically active hormone 1,25-dihydroxyvitamin D3 (1,25(OH)2D3, also known as calcitriol) in the kidney (42). 1,25-dihydroxyvitamin D3 is the ligand for VitD receptor (VDR), a member of the nuclear receptor family of transcription factors which activates or represses the expression of many genes (43), and exerts rapid non-genomic effects via the membrane VDR (44).
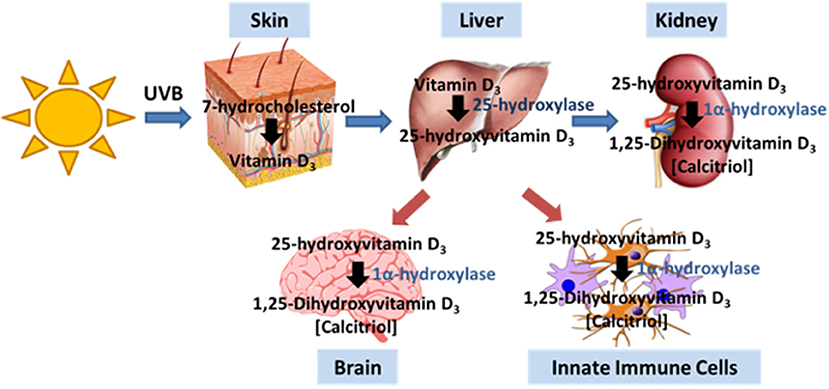
Figure 1. Vitamin D production pathway. Vitamin D3 is produced in the skin by UVB irradiation. Typically, the liver and kidney generate intermediates that ultimately generate calcitriol, the active form of VitD. The brain and immune cells also express the enzyme that allows for the generation of calcitriol.
VitD primarily acts as a hormone that regulates gene transcription. VitD enters cells using carrier proteins or diffusion where it can bind VDR in the cytoplasm (Figure 2). VitD/VDR complexes are translocated to the nucleus where VDR dimerizes with retinoid X receptors (RXR). VDR/RXR complexes bind to VitD response elements which are present in nearly 1,000 genes, thus playing a major transcriptional role in many cell types. There are non-genomic roles for VitD which occur within minutes, far too soon to be mediated by altered gene expression. The most noteworthy non-genomic effect of VitD is calcium regulation. VitD binds to protein disulfide isomerase family A, member 3 (PDIA3), resulting in the upregulation of PKA, pI3K and p38MAPK which contribute to the intracellular flux of calcium (Figure 2). While changes in intracellular calcium may be independent of VDR engagement, calcium homeostasis is affected by VDR signaling as seen in people with type II genetic rickets and VDR-deficient mice which have severe hypocalcemia (45–48).
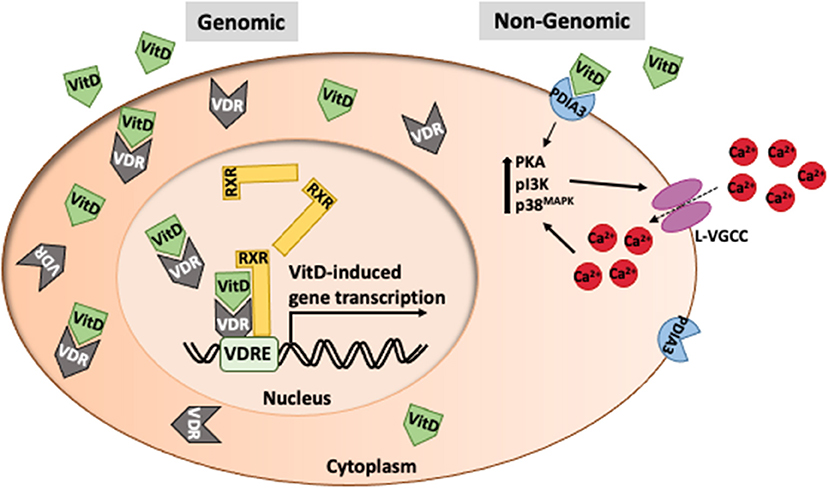
Figure 2. Intracellular function of vitamin D. VitD typically acts as a transcription factor in association with retinoid-X-receptors (RXR) to mediate gene transcription at VitD response elements in promoter regions of genes. However, VitD can have immediate effects on cell function (non-genomic) via interaction with PDIA3 that leads to changes in calcium transport.
Although the best-known function of VitD is to regulate calcium physiology, it also has important effects on the development and function of CNS. Neurons and microglia express VDR. In addition, they can directly metabolize 25(OH)D3 because they express 1-α hydroxylase (49). 1,25(OH)2D3 has been shown to regulate glial cell line-derived neurotrophic factor (GDNF) (50) and nerve growth factor (NGF) (51) expression. The ability of 1,25(OH)2D3 to regulate certain neurotrophic factors and influence inflammation has led to the hypothesis that 1,25(OH)2D3 is neuroprotective (52). In fact, it has shown a reduction in ROS induced cell death and increased anti-oxidant species in glia cell by 1,25(OH)2D3 (53). VitD insufficiency is associated with several other neurological disorders beside MS, including Parkinson disease, schizophrenia, depression and cognitive decline (54), suggesting its essential role in maintaining normal CNS function.
How much VitD is optimal is somewhat controversial. The most commonly published normal range for blood VitD levels is 20–40 ng/mL (50–100 nmol/L) with levels below 20 ng/mL (50 nmol/L) considered VitD deficient. The Endocrine Society considers VitD levels of 20–29 ng/mL (50–74 nmol/L) to indicate VitD insufficiency, and that VitD levels should be 30-100 ng/mL (75–250 nmol/L) for optimal health benefit (55). Based on these guidelines, it is estimated that 30–50% of Americans may be VitD deficient. A New England Journal of Medicine article suggested that VitD deficiency may be overstated, and perhaps our current metrics for VitD-deficiency are incorrect, because of misinterpretation of the Institute of Medicine's reference values (56). It should be noted that most of the studies evaluating VitD levels in health are based on intestinal uptake of calcium and bone health, so it remains unclear what the optimal dose may be for optimal overall health.
One of the strongest correlates of latitude is the duration and intensity of sunlight, and the synthesis of ViD is subsequently affected by ultraviolet B (UVB) radiation. The incidence gradient according to latitude and the effect of migration within genetically uniform groups can be explained by VitD— as the link between latitude and MS risk. The VitD hypothesis is supported by studies of sunlight exposure history. The seasonal fluctuations in VitD levels resulted in decreased VitD concentrations in utero, which may contribute the month-of birth effect in MS (57). While not all studies are in agreement, a large meta-analysis found that individuals born in the Spring have a significantly higher risks of developing MS compared to individuals born in the fall (58–60). Insufficient maternal 25-hydroxyvitamin D during early pregnancy is associated with a 2-fold increased risk of MS in offspring (61). Similarly, a Danish study used dried blood spot samples collected near the time of birth to measure VitD in individuals who later developed MS and matched controls (62). Neonatal VitD levels were inversely associated risk of developing MS, supporting the notion that maternal VitD levels may be important to prevent MS in children. Higher sun exposure during childhood (age of 6–15 years) was shown associated with reduced MS risks (63). Time spent on outdoor activities during childhood and adolescence (significant for age of 6–20 years) in the summer was inversely related to the risks, whereas there was no such effect in the winter (64). A study of monozygotic twins who were discordant with MS has shown that twins with MS reported significantly lower levels of childhood sun exposure than their healthy sibling (65). However, sunlight may have benefits to prevent CNS autoimmunity beyond VitD. A study in EAE found that UV light suppressed EAE independent of VitD and VDR (66).
Further evidence for the VitD hypothesis comes from the studies of dietary VitD intake. At high latitudes, prevalence of MS was lower than expected in populations with high consumption of VitD-rich oily fish (67). A 40% reduction in MS risks was found among women who used supplemental VitD, compared with women who did not use supplements (68). Lastly, a study directly measured the circulating 25(OH)D3 (the circulating form of VitD) concentrations in individuals who served in the US military, and concluded that serum level of 25(OH)D3 in healthy young white adults is an important predictor of their risk of developing MS (69). These epidemiology studies (latitude, migration, history of sunlight exposure, VitD intake and serum concentration of VitD) give credibility to the hypothesis that VitD, especially in early life, has protective effect against MS development. Nevertheless, due to often confounding variables in epidemiology studies, prospective experimental studies are needed to validate the effect of VitD in determining MS risks. A mendelian randomization study in which single nucleotide polymorphisms associated with 25-hydroxyvitamin D were identified and analyzed in the International Multiple Sclerosis Genetics Consortium found that there was a significant increased susceptibility to MS in individuals with a genetically lowered level of 25-hydroxyvitamin D (70). This genetic study supports the epidemiology data that optimal VitD levels are protective against the development of MS. Interestingly, a study of polymorphisms in the VitD-binding protein found an association with MS risk in whites, but not blacks or Hispanics, indicating that VitD may not be a significant risk factor in all ethnicities (71).
After disease onset, VitD also acts in modulating MS clinical course. Serum concentrations of 25-hydroxyvitamin D3 in MS patients were lower during relapses than during remissions (72), and correlated inversely with disease severity (73) and frequency of relapse (74, 75). Although these results might indicate lower sun exposure in patients with severe MS rather than a beneficial effect of VitD, convincing studies with EAE have demonstrated the immunomodulatory effect of VitD in inflammatory CNS disease. Expression of VDR has been described in immune cells, including dendritic cells, macrophages and activated T and B cells (76). VitD supplementation clearly suppressed EAE preventively (77, 78) and therapeutically (79). Moreover, the therapeutic effects of VitD required VDR function in T cells (80), and were through promoting IL-4, TGF-β (81) and IL-10 (82) production, and inhibiting TH1 cells differentiation (83, 84). With these results established from EAE, experimental basis supports the beneficial role of VitD in modulating disease progression. Yet, there are numerous studies that indicate that VitD supplementation in MS patients has little, if any benefit to reducing symptoms (85). The SOLAR trial found that VitD supplementation (14,000 IU/d) in MS patients on interferon beta-1a was beneficial in reducing new lesions, but no change in progression of disability or annualized relapse rate was observed (86). In the 2-year CHOLINE trial, MS patients on interferon beta-1a were given 100,000 IU of oral cholecalciferol or placebo biweekly. The end point (changed in annualized relapse rate) was not met, yet there were positive benefits observed on imaging and the average EDSS score was significantly lower in the treatment group (87). Analysis of 12 random controlled trials evaluating VitD supplementation concluded that VitD supplementation had no significant benefit on relapse rates, progression of disability or MRI lesions (88). Thus, the benefit of VitD supplementation in MS patients is unclear, but given that most MS patients are VitD deficient, supplementation is prudent and likely benefits that overall health of the patients.
The first evidence that VitD may affect the immune system came from a study in 1983 in which VitD promoted the fusion of macrophages (89). In 1986, it was shown that VitD inhibited IL-2 production and proliferation by T cells (90), providing solid evidence that VitD had the capacity to regulate T cells. There is now substantial evidence that VitD is a major regulator of both innate and adaptive immune cells and influences the outcomes of infections, cancer and autoimmunity.
Activated macrophages and monocytes upregulate expression of CYP27B1, the gene that encodes 1α-hydroxylase, the enzyme that converts 25-hydroxyvitamin D3 to 1,25-dihydroxyvitamin D3 (calcitriol), the active form of VitD (Figure 1), indicating that macrophages/monocytes have the capacity to increase VitD at site of inflammation (91). The local expression of VitD by activated macrophages/monocytes sets up an autocrine pathway since macrophages/monocytes express VDR, resulting in the production of anti-microbial products such as cathelicidin and defensins. Cathelicidin is particularly important against infections by destablilizing microbial membranes and disrupting viral envelopes (92–94). Equally important, VitD regulates the maturation and activation of macrophages and dendritic cells, compromising their ability to be effective antigen presenting cells (Figure 3). Upon TLR engagement, macrophages and dendritic cells upregulate MHCII, CD80/CD8, CD40 and cytokines which are critical to antigen presentation to T cells. VitD suppresses these molecules, promoting macrophages and dendritic cells that are immature and somewhat tolerogenic (95, 96). This suppression of macrophages and dendritic cells may be due to the suppression of toll-like receptors (97–99) or inhibition of IL-12 via NF-κB (100) mediated by VitD.
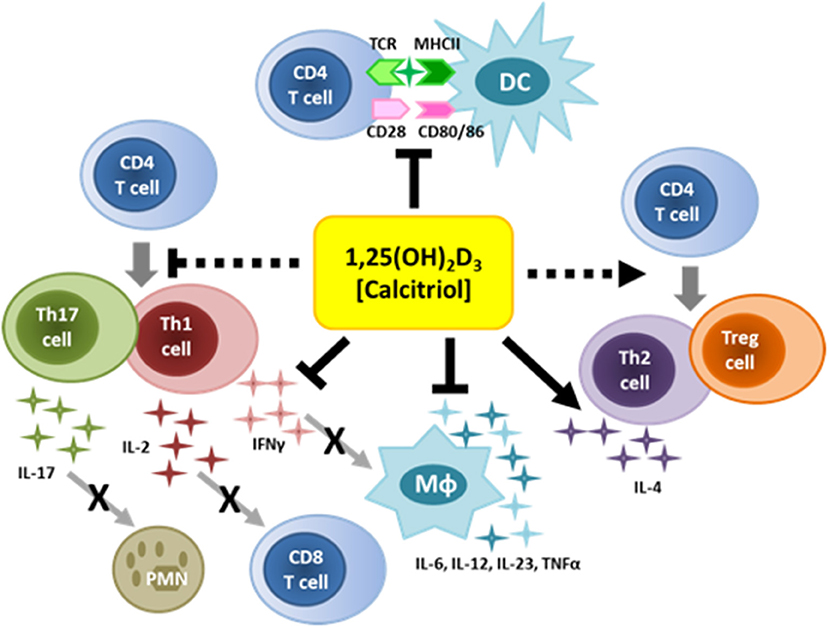
Figure 3. Immunoregulatory functions of vitamin D. VitD can regulate the function of numerous immune cells. VitD suppresses inflammatory cytokines and antigen-presentation by innate immune cells. VitD also suppresses T cell activation, and favors the generation of Th2 cells and Tregs.
VitD affects the function of microglia which are known to secrete inflammatory mediators that contribute to myelin damage during CNS autoimmunity. Mice with EAE treated with calcitriol immediately following EAE induction have reduced microglia activation and oxidative stress, and less blood brain barrier permeability (101). Partial deletion of VDR in young mice attenuated microglia activation and reduced the incidence and severity of EAE (102). In various models of CNS diseases and injury, VitD has been shown to regulate microglia phenotype and oxidative stress (103–106). Thus, vitD appears to effectively skew microglia from a pro-inflammatory M1 phenotype to a reparative M2 phenotype, reducing inflammation and limiting demyelination.
Both T cells and B cells can express VDR and respond to VitD (Figure 3). There is actually very little, if any, VDR expression on resting human T and B cells; however, VDR is rapidly upregulated upon activation (107–109). Similar to innate immune cells, activated T cells express CYP27B1 and can make active VitD. VitD suppresses T cell proliferation via reduced IL-2. In addition, VitD alters the differentiation of CD4 T cells by skewing CD4 T cells toward Th2 and away from Th1 and Th17, the phenotypes associated with MS (110, 111). Also of particular relevance to MS, VitD promotes the differentiation of Tregs (112), which are known to be defective in MS patients (113–118). The mechanism by which VitD promotes Treg development appears to be via altered APCs, since VitD added to human dendritic cells alters glucose metabolism favoring the differentiation of Tregs over Teff cells (119). VitD status in MS patients positively correlates with Treg function, supporting the observation the VitD promotes Treg development (120). VitD in association with CD46 was shown to promote Type I regulatory T cells (Tr1) cells in MS patients (121), indicating that optimal VitD may be an important component of immune regulation.
B cell differentiation and maturation into plasma cells is also regulated by VDR, thus affecting antibody production. In addition, VitD downregulates co-stimulatory molecules on B cells, similar to what is observed for macrophages and dendritic cells (122). Thus, optimal VitD may compromise the ability of B cells to act as antigen presenting cells. B cell –depletion therapies have been very beneficial in the treatment of MS. Given that the beneficial affects appear to be independent of antibody production, there is speculation and evidence that B cells are critical antigen-presenting cells in MS (123–125). An immune profile study on MS patients on B cell depletion therapy indicated that the T cell profile showed a favorable change, reflected by a reduction in memory T cells and an increase in Tregs (124, 125), consistent with the role of B cells as antigen-presenting cells. Thus, low VitD may enhance the ability of B cells to drive the activation and differentiation of T cells, increasing the probability developing MS.
Substantial evidence indicates that VitD acts as a neurosteroid. VDR is expressed throughout the developing and mature brain, including the hippocampus, amygdala, hypothalamus, cortex and cerebellum (49, 126, 127), implicating VitD as an important modulator of gene expression throughout the CNS. Furthermore, 1α-hydroxylase and 25-hydroxylase are both expressed in the brain providing the critical enzymes to generate VitD locally (49, 128). While a major role of VitD is gene regulation, it also has non-genomic functions, particularly regulation of calcium signaling which is critical in normal cellular function.
VitD promotes cell differentiation and apoptosis which are critical for embryonic development. When VitD is removed during gestation in model systems, multiple regions of the brain have increased cell proliferation and decreased apoptosis, as well as enhance cell proliferation, leading to CNS anomalies (129–131). The increased proliferation was mediated by increased expression of cyclin genes which were regulated by VDR signaling (132), while the reduction in apoptosis may have been due to increased levels of BAX and Bcl-2 (131). Low VitD also leads to more neural stem cells which may be due to a loss of regulation of cell proliferation or a failure in neural stems to efficiently differentiate into neural cell progenitors (133). Changes in brain morphology have been observed in rodents with VitD deficiency (129, 134). In humans, VitD deficiency is associated with decreased brain volume and enlarged ventricles in older adults (135). Ex vivo studies illustrated that VitD inhibits the proliferation of hippocampal neurons, while promoting neurite outgrowth (130). Analysis of dopaminergic neurons found that VitD-deficiency reduced Nurr1 and P57kip2 gene expression during embryogenesis which are critical to the development and homeostasis of dopaminergic neurons (136). In addition, VitD has been found to regulate the expression of genes essential in the normal function of dopaminergic neurons (137, 138). Interestingly, while it appears that VitD promotes the differentiation of neurons, astrocyte differentiation appears to be impaired by VitD when using adult neural stem cells (139). VitD also promotes the differentiation of neural stem cells into oligodendrocytes (139), the myelinating cells of the CNS. Oligodendrocyte precursor cells fail to differentiate into oligodendrocytes when VDR signaling is blocked (140). These studies implicate VitD as an essential regulator of neuron and oligodendrocyte development. The signaling between neurons and oligodendrocytes is essential to the development of a properly myelinated CNS during early life, as well as remyelination that occurs following CNS damage.
In addition to the development and differentiation of CNS cells, VitD plays a role in their ability to function properly. The release of several neurotransmitters is affected by VitD. In dopaminergic neurons, VitD promotes the release of dopamine (141). VitD appears to regulate neurotransmitter synthesis, for example, VitD regulates the inhibitory neurotransmitter GABA, via upregulation of GAD65 and GAD67 (142–144). Neurotrophic factors, which are essential for CNS homeostasis and communication between cells in the CNS, are also regulated by VitD. VitD appears to induce nerve growth factor (NGF) expression in neurons (129, 135). Neural stem cells upregulate brain-derived growth factor (BDNF), Glia cell line-derived nerve factor (GDNF), and ciliary neurotropic factor (CNTF) in the presence of VitD (139). In astrocytes, VitD appears to regulate the expression of neurotrophin receptors, as well GDNF, NT-3 and NT-4 (145–147).
VitD also plays a role in neuronal plasticity. In cultured cortical neurons, VitD increased the expression of microtubule associated protein-2, growth-associated protein-43, and synapsin 1 which are important in synaptic vesicle transport and axonal growth (148). Low VitD during embryogenesis resulted in altered expression of proteins important in cytoskeletal integrity, organelle transport, and synaptic plasticity (149, 150), suggesting that VitD is critical to the normal development and function of the CNS.
VitD plays a vital role in regulating calcium levels in the CNS which is particularly important given that high levels of calcium are neurotoxic. In neurons, VitD modulates L-type voltage-gated calcium channels by downregulation of A1C subunits (151). Mice lacking VitD have upregulated L-type voltage-gated calcium channels and elevated calcium influx in neurons (152) (Figure 2). In vitro treatment of neurons with VitD downregulated L-type voltage-gated calcium channels and protected neurons from excitotoxicity (153). VitD was shown to very rapidly increase the uptake of extracellular calcium via L-type voltage gated calcium channels (154). Since this occurred within a few minutes, it was clear that the effect was independent of gene transcription. The increase in calcium influx was dependent on the PKA, pI3K, and p38MAPK. The non-genomic effects of VitD have largely been attributed to VitD interaction with PDIA3 (also known as 1,25D3-Marrs) on the plasma membrane (155, 156). In addition, VitD regulates the expression of numerous genes associated with calcium homeostasis vital to the proper regulation of calcium in the CNS and other tissues (150, 154, 157).
Epidemiological data indicate that VitD has neuroprotective properties. Optimal VitD levels during early life appear to be important to minimize the risk of several psychiatric and neurodegenerative diseases. Schizophrenia, depression and autism spectrum disorders have all been associated with low VitD, particularly during embryogenesis and infancy (158–162). There is an inverse correlation between Parkinson's disease risk and VitD levels (163, 164). Given that VitD protects dopaminergic neurons by upregulating genes numerous genes associated with the function of dopaminergic neurons (137, 138), it is logical that VitD may be a critical factor in minimizing the risk of developing Parkinson's disease. Similarly, Alzheimer's disease patients tend to have low serum VitD levels compared to matched healthy controls (165). The risk of dementia and symptoms of neurodegenerative diseases, such as cognitive and memory impairments and impaired motor function, increases with low serum VitD levels (166–169). Serum VitD deficiency is linked to greater infarct volumes, increased overall stroke severity, and worse long-term outcomes in stroke patients (170–172). Impacts on the risks and outcomes in these neurological conditions are clearly multifactorial, but it stands to reason that VitD plays a role in susceptibility and outcome.
The neuroprotective properties of VitD take effect though several mechanisms. Direct neuroprotective action of VitD is associated with the regulation of neurotrophic factors and reduction in oxidative stress. Neurotrophic factors are critical for the differentiation, survival and maintenance of neural and glial cells. VitD stimulates expression of nerve growth factor (NGF), brain-derived neurotrophic factor (BDNF), glial cell line-derived neurotrophic factor (GDNF) and neurotrophin-3 (NT3) (173). Neurotrophic factors downregulate their expression into adulthood, therefore remaining levels have critical cell maintenance functions. Reduction in VitD-assisted neurotrophic factor expression due to deficiency may leave neurons more vulnerable to insult.
Neurons are particularly susceptible to oxidative damage because of increased oxygen consumption, bi-products of neurotransmitter production, excitotoxicity and high overall lipid content (174). Additional neuroinflammation will increase the reactive oxygen species (ROS) load. Adequate VitD levels downregulate intracellular oxidative-stress related activities, while suboptimal levels result in increased oxidative damage and neuronal apoptosis (175). Increased reactive oxygen production has been implicated in the pathogenesis of multiple neurodegenerative conditions, including Parkinson's disease (176), Alzheimer's disease (177), Huntington's disease (178), stroke (179) and Multiple Sclerosis (180), and suboptimal serum VitD levels have been linked to these conditions. VitD is a potent regulator of the nuclear factor erythroid-2-related factor 2 (Nrf2) antioxidant pathway in neurons and glial cells, and intracellular Nrf2 levels are inversely correlated with the accumulation of mitochondrial ROS. Within the CNS, upregulation of Nrf2 target genes superoxide dismutase (SOD), catalase (CAT) and heme oxygenase-1 (HMOX1) to make neurons more resistant to oxidative insults (181). Furthermore, neurotrophic signaling pathways, such as the BDNF-TrkB pathway that is essential for mature neuron survival and normal function, also signal Nrf2 activation (182, 183). VitD then has double the influence on neuronal survival – first in neurotrophic action by increasing levels of BDNF, and second in oxidative defense by direct activation of Nrf2. The neuroprotective properties of VitD center around its antioxidant function, and in conjunction with neurotrophic factor expression, likely enhances neural defense and repair mechanisms.
Oxidative stress and mitochondrial dysfunction are prominent features of MS. T cells can produce ROS and T cell activity and proliferation are influenced by ROS, adding an increased level of complexity to the impact of ROS in MS (184). Activated microglia and macrophages are the major contributors to the elevated ROS observed in the disease. These cells produce oxidating radicals such as superoxide, hydrogen peroxide and nitric oxide with the help of ROS-generating enzymes, such as myeloperoxidase, NADPH oxidase and nitric oxide synthase. ROS have shown to mediate demyelination in both MS and its animal models (184, 185). Studies in postmortem brains of MS patients have identified myeloperoxidase expression in activated macrophages and microglia near lesions. Elevated expression of myeloperoxidase was detected in demyelinated regions of postmortem MS brain homogenates when compared to unaffected regions from the same individual (186, 187). Other markers of oxidative damage, such as 4-hydroxy-2-noneal (4-HNE), produced by lipid peroxidation of cell membranes, and nitrotyrosine, the product of nitric oxide and superoxide reactions, increase and accumulate in macrophages and astrocytes in MS lesions (188–190).
ROS in later stages of MS stems from mitochondrial dysfunction within neurons themselves. Mitochondrial dysfunction and associated ROS have been implicated in non-inflammatory mediated axonal degeneration that occurs with chronic demyelination. It is believed that mitochondria become taxed after sodium channel redistribution in response to demyelination. Sodium channel redistribution causes large influxes of sodium, taxing the ATP dependent sodium-potassium pump (191). Increased ATP needs trigger mitochondria production and proliferation, resulting in increased ROS (192, 193). Notably, increased mitochondrial heat shock protein 70, a marker of mitochondrial stress, has been observed in astrocytes and axons within MS lesions (192). There is some controversy regarding whether increased ROS from mitochondria exists primarily due to mitochondrial proliferation and ATP production after demyelination (chronic injury) or if mitochondria actually acquire oxidative damage during the inflammatory stage of the disease (acute injury) (194).
Antioxidant enzymes are the endogenous ROS defense system in the CNS. ROS exposure activates Nrf2, which then translocates to the nucleus and activates antioxidant response element promoters (ARE) for antioxidant enzyme production. Expression of hundreds Nrf2 responsive antioxidant genes have already been identified (195). Numerous studies have suggested a role for Nrf2 inactivity in the pathogenesis of MS. Nrf2 knock-out EAE mice experience more rapid disease onset, a more severe clinical course, increased glial activation, increased pro-inflammatory cytokine expression and increased axonal degeneration compared to Nrf2 inclusive controls (196–198). Conversely, increasing the activity of Nrf2 in the CNS of EAE mice lessened the clinical severity (199). In postmortem brain tissue of MS patients, NRF2 is strongly upregulated in active MS lesions and expression is most pronounced in degenerating neurons and glial cells, including oligodendrocytes (200, 201).
NRF2 activity is already relevant in MS clinical treatment. Both VitD and dimethyl fumerate (DMF or Tecfidera™) are NRF2 activators. DMF treatment is an approved oral RRMS therapy known to reduce disease activity and progression, and accomplishes these outcomes via immunomodulatory and neuroprotective mechanisms (202–207). VitD and DMF both signal through the NRF2/KEAP1 pathway to generate antioxidant action and specifically increase glutathione signaling for neuroprotection. DMF and VitD can also exert protective effects by reducing proinflammatory cytokine expression and increasing neurotrophic factor expression (208). DMF and VitD derivatives have demonstrated a cooperative effect on increased VDR expression and Nrf2 activity that limit leukemia progression (209). Although mechanisms of action overlap, DMF is a safer therapeutic option for avoiding calcemic toxicity that can occur with long-term use of high levels of VitD. In fact, excess VitD can exacerbate EAE, emphasizing the point that a measured approach to VitD-based therapies is critical (210). It should also be noted that melatonin which is produced during the dark has similar anti-oxidant properties as VitD in EAE (211, 212). There is contradicting data on the relationship between melatonin and VitD, yet both appear to be beneficial in reducing CNS inflammation. Thus, the interplay between appropriate sunlight to optimize VitD levels and appropriate darkness to optimize melatonin to maintain circadium rhythm should be considered in strategies to prevent and/or treat MS. It is unlikely that DMF mimics every function of VitD, but similarities in function between DMF and VitD suggest that VitD has a critical role as an endogenous neuroprotective mediator in MS.
An important consideration is the temporal influence of VitD, NRF2 activity, and the endogenous antioxidant system during the course of MS. Upregulated expression of NRF2 in MS brain lesions suggests that the NRF2 pathway is already highly active in distressed cells (200, 201) and it appears that endogenous antioxidant mechanisms are not enough to halt demyelination and axonal degeneration at end stages of disease. However early in the disease, VitD and NRF2 signaling may have increased potential for neuroprotection because less neuroinflammatory-induced oxidative damage has occurred. Understanding the neuroprotective potential of VitD in early stages of MS is complicated by the striking frequency of VitD deficiency in patients at the time of diagnosis. Therefore, whether preventing VitD deficiency prior to disease onset can enhance neuroprotection and alter disease progression warrants further investigation.
MS is a complex disease in which immune and nervous system components interact to form and sometimes, resolve CNS inflammatory, demyelinating lesions. Genetic studies have largely implicated immune genes as susceptibility factors, supporting the hypothesis that MS is an immune-mediated disease and that the CNS is the unfortunate target of the aberrant immune response. There is significant data to support that VitD has a profound immunoregulatory role on both innate and adaptive immune cells, indicating that low VitD may alter normal immune regulation leading to autoimmunity. Given that the CNS is the sole target of the aberrant immune response in MS, it is important to consider if the CNS is somehow more vulnerable to inflammation in some individuals that may make them at an increased risk of developing MS. While the answer is still unknown, the literature clearly indicates that VitD impacts CNS development and function.
The epidemiology studies indicate that optimal VitD is particularly important during embryogenesis and childhood in determining risk of developing MS. During childhood, our immune system is repeatedly being challenged by pathogens, most of which are cleared with minimal clinical consequences. Some childhood viruses, such as Epstein-Barr, varicella zoster, and some strains of influenza, can infect neurons, yet they typically do not cause clinical CNS manifestations. Even in the absence of CNS clinical signs of illness, do these viruses affect the CNS differently in children with low VitD? Our recent study in which partial VDR deletion was induced in young mice specifically in neurons resulted in an enhanced susceptibility to EAE in adult mice, suggesting that low VitD signaling in neurons in early life may increase the vulnerability of the CNS to inflammation (102). There is substantial evidence that viral infections are affected by VitD levels. Of particular interest in MS is EBV which has been long speculated to be a vital risk factor for the onset of disease and this has been confirmed in a new study of >10 million young adults (213). While many theories have been explored, a recent study identified EBV infection as a precipitating factor that may be driving molecular mimicry (214–217). EBV antibody titers are negatively correlated with VitD levels in MS patients (218) suggesting that these two environmental factors may be synergistic. EBV is also associated with activating endogenous retroviruses (ERVs) and ERV levels in MS plaques correlates with disease activity (219). These ERVs may act as novel antigens that drive CNS inflammation. VitD supplementation mitigates EBV reactivation (220), which may in turn limit ERV activation and the associated inflammation. In a humanized mouse model, it was shown that HLA-DR15-restricted T cells fail to control EBV infection, suggesting that there is potential relationship between the strongest genetic factor (HLA-DR15) and EBV with respect to MS risk (221). The relationship between ERVs, VitD and MS has become of increasing interest and some speculate that EBV may be the missing link between ERVs and VitD that trigger the development of MS (222). While many autoimmune diseases have been associated with low VitD to some extent, the epidemiology data in MS is far more convincing suggesting that low VitD is likely increasing the risk of developing MS due to the negative impact on immune regulation and CNS homeostasis.
A recent study of >1,900 subjects demonstrated that sun exposure negatively correlated with development of MS, and high VitD levels (>30.31 ng/mL) in MS patients reduced the risk of relapses and accumulation of disability (223). The evidence that VitD level is important in risk of developing MS and disease severity appears well established, yet questions still remain as to whether VitD supplementation is beneficial to patients with MS. Since most MS patients have low VitD levels, supplementation is now common practice. Perhaps the more important question is how do we prevent low VitD? Although VitD is currently a common food supplement in many countries, it is unclear whether we are doing enough to ensure that children are getting sufficient VitD. Rickets is rare in countries in which food is supplemented with VitD, indicating that the levels of VitD provided via food supplementation is sufficient for bone health. However, it is unclear whether these levels of VitD are adequate for optimal neuroprotection, given that countries like the United States still have a high incidence of MS. Additional VitD supplementation may be essential for children in higher latitudes where sunlight is very limited for several months each year, and perhaps in children with a family history of MS in which genetic risks are highest. We should also balance pros and cons of sunscreen which reduces UVB induced VitD synthesis by 95% and may negatively impact health of our immune and nervous systems. Sunlight remains the best source of VitD so ensuring that children play outside daily may be the best solution to the epidemic of low VitD.
AL-R, SG, PL, and ES each reviewed the literature and wrote sections of the manuscripts. All authors contributed to the article and approved the submitted version.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
1. Chick H, Dalyell EJH, Hume EM, Mackay HMM, Henderson-Smith H. The aetiology of rickets in infants: prophylactic and curative observations at the Vienna University Kinderklinik. Lancet. (1922) ii:7–11. doi: 10.1016/S0140-6736(01)00835-2
2. McCollum EV, Simmonds N, Becker JE, Shipley PG. Studies on experimental rickets. XXI An experimental demonstration of the existence of a vitamin which promotes calcium deposition. J Biol Chem. (1922) 53:293–312. doi: 10.1016/S0021-9258(18)85783-0
3. Lips P. Worldwide status of vitamin D nutrition. J Steroid Biochem Mol Biol. (2010) 124:197–300. doi: 10.1016/j.jsbmb.2010.02.021
4. Walton C, King R, Rechtman L, Kaye W, Leray E, Marrie RA, et al. Rising prevalence of multiple sclerosis worldwide: Insights from the atlas of MS, third edition. Mult Scler. (2020) 26:1816–21. doi: 10.1177/1352458520970841
5. Lublin FD, Reingold SC. Defining the clinical course of multiple sclerosis: results of an international survey. National Multiple Sclerosis Society (USA) Advisory Committee on clinical trials of new agents in multiple sclerosis. Neurology. (1996) 46:907–11. doi: 10.1212/WNL.46.4.907
6. Archeson ED. Bachrach CA. The distribution of multiple sclerosis in US veterans by birthplace. Am J Hyg. (1960) 72:88–99. doi: 10.1093/oxfordjournals.aje.a120137
7. Vukusic S, Van Bockstael V, Gosselin S, Confavreux C. Regional variations in the prevalence of multiple sclerosis in French farmers. J Neurol Neurosurg Psychiatry. (2007) 78:707–9. doi: 10.1136/jnnp.2006.101196
8. Hammond SR, de Wytt C, Maxwell IC, Landy PJ, English E, McLeod G, et al. The epidemiology of multiple sclerosis in Queensland, Australia. J Neurol Sci. (1987) 80:185–204. doi: 10.1016/0022-510X(87)90154-7
9. Gale CR, Martyn CN. Migrant studies in multiple sclerosis. Prog Neurobiol. (1995) 47:425–48. doi: 10.1016/0301-0082(95)80008-V
10. Sabel CE, Pearson JF, Mason DF, Willoughby E, Abernathy DA, Taylor BV. The latitude gradient for multiple sclerosis prevalence is established in the early life course. Brain. (2021) 144:2038–46. doi: 10.1093/brain/awab104
11. Simpson S, Wang W, Otahal P, Blizzard L, van der Mei IAF, Taylor BV. Latitude continues to be significantly associated with the prevalence of multiple sclerosis: an updated meta-analysis. J Neurol Neurosurg Psychiatry. (2019) 90:1193–200. doi: 10.1136/jnnp-2018-320189
12. Tremlett H, Zhu F, Ascherio A, Munger KL. Sun exposure over the life course and associations with multiple sclerosis. Neurol. (2018) 90:e1191–9. doi: 10.1212/WNL.0000000000005257
13. Alter M, Leibowtz U, Speer J. Risk of multiple sclerosis related to age at immigration to Israel. Arch Neurol. (1966) 15:234–7. doi: 10.1001/archneur.1966.00470150012002
14. Dean G, Elian M. Age at immigration to England of Asian and Caribbean immigrants and the risk of developing multiple sclerosis. J Neurol Neurosurg Psychiatry. (1997) 63:565–8. doi: 10.1136/jnnp.63.5.565
15. Hammond SR, English DR, McLeod JG. The age-range of risk of developing multiple sclerosis: evidence from a migrant population in Australia. Brain. (2000) 123:968–74. doi: 10.1093/brain/123.5.968
16. McLeod JG, Hammond SR, Kurtzke JF. Migration and multiple sclerosis in immigrants to Australia from United Kingdom and Ireland: a reassessment. I Risk of MS by age at immigration. J Neurol. (2011) 258:1140–9. doi: 10.1007/s00415-010-5898-4
17. McLeod JG, Hammond SR, Kurtzke JF. Migration and multiple sclerosis in United Kingdom and Ireland immigrants to Australia: a reassessment. II. Characteristics of early (pre-1947) compared to later migrants. J Neurol. (2012) 259:684–93. doi: 10.1007/s00415-011-6244-1
18. Kurtzke JF, Beebe GW, Norman JE. Epidemiology of multiple sclerosis in US veterans III. Migration and the risk of MIS. Neurology. (1985) 35:672. doi: 10.1212/WNL.35.5.672
19. Willer CJ, Dyment DA, Rsch NJ, Sadovnick AD, Ebers GC. Canadian Collaborative Study Group. Proc Natl Acad Sci USA. (2003) 100:12877–82. doi: 10.1073/pnas.1932604100
20. International Multiple Sclerosis Genetics Consortium, Hafler DA, Compston A, Sawcer S, Lander ES, Daly MJ, et al. Risk alleles for multiple sclerosis identified by a genomewide study. N Engl J Med. (2007) 357:851–62. doi: 10.1056/NEJMoa073493
21. Australia and New Zealand Multiple Sclerosis Genetics Consortium, Bahlo M, Booth DR, Broadley SA, Brown MA, Foote SJ, et al. Genome-wide association study identifies new multiple sclerosis susceptibility loci on chromosomes 12 and 20. Nat Genet. (2009) 41:824–8. doi: 10.1038/ng.396
22. Goodin DS. The Causal Cascade to Multiple Sclerosis: a model for MS pathogenesis. PLoS ONE. (2009) 4:e4565. doi: 10.1371/journal.pone.0004565
23. Dominguez-Mozo N, Perez-Perez S, Villarrubia N, Costa-Frossard L, Fernandez-Velasco JI, Ortega-Madueno I, et al. Herpesvirus antibodies, vitamin d and short-chain fatty acids: their correlation with cell subsets in multiple sclerosis patients and healthy controls. Cells. (2021) 10:119. doi: 10.3390/cells10010119
24. Riise T, Nortvedt MW, Ascherio A. Smoking is a risk factor for multiple sclerosis. Neurology. (2003) 61:1122–4. doi: 10.1212/01.WNL.0000081305.66687.D2
25. Hedstrom AK, Baarnhielm M, Olsson T, Alfredsson L. Tobacco smoking, but not Swedish snuff use, increases the risk of multiple sclerosis. Neurology. (2009) 73:696–701. doi: 10.1212/WNL.0b013e3181b59c40
26. Hernán MA, Jick SS, Logroscino G, Olek MJ, Ascherio A, Jick H. Cigarette smoking and the progression of multiple sclerosis. Brain. (2005) 128:1461–5. doi: 10.1093/brain/awh471
27. Bjelobaba I, Begovic-Kupresanin V, Pekovic S, Lavrnja I. Animal models of multiple sclerosis: focus on experimental autoimmune encephalomyelitis. Neurosci Res. (2018) 96:1021–42. doi: 10.1002/jnr.24224
28. Kipp M, Nyamoya S, Hochstrasser T, Amor S. Multiple sclerosis models: a clinical and histopathological perspective. Brain Pathol. (2016) 27:123–37. doi: 10.1111/bpa.12454
29. Lindsey JW. Characteristics of initial and reinduced experimental autoimmune encephalomyelitis. Immunogenetics. (1996) 44:292–7. doi: 10.1007/BF02602559
30. Wu F, Cao W, Yang Y, Liu A. Extensive infiltration of neutrophils in the acute phase of experimental autoimmune encephalomyelitis in C57BL/6 mice. Histochem Cell Biol. (2010) 133:313–22. doi: 10.1007/s00418-009-0673-2
31. Cross AH, Trotter JL, Lyons JA. B cells and antibodies in CNS demyelinating disease. J Neuroimmunol. (2001) 112:1–14. doi: 10.1016/S0165-5728(00)00409-4
32. Carnero Contentti E, Correale J. Neuromyelitis optica spectrum disorders: from pathophysiology to therapeutic strategies. J Neuroinflammation. (2021) 18:208. doi: 10.1186/s12974-021-02249-1
33. Yang Y, Weiner J, Liu Y, Smith AJ, Huss DJ, Winger R, et al. T-bet is essential for encephalitogenicity of both Th1 and Th17 cells. J Exp Med. (2009) 206:1549–64. doi: 10.1084/jem.20082584
34. Huss DJ, Winger RC, Peng H, Yang Y, Racke MK, Lovett-Racke AE. TGF-beta enhances effector Th1 cell activation but promotes self-regulation via Il-10. J Immunol. (2010) 184:5628–36. doi: 10.4049/jimmunol.1000288
35. Huss DJ, Winger RC, Mavrikis Cox G., Guerau-de-Arellano, Yang Y, Racke MK, et al. TGF-β signaling via Smad4 drives IL-10 production in effector Th1 cells and reduces T-cell trafficking in EAE. Eur J Immunol. (2011) 41:2987–96. doi: 10.1002/eji.201141666
36. Lovett-Racke AE, Yang Y, Racke MK. Th1 versus Th17: are T cell cytokines relevant in multiple sclerosis? Biochim Biophys Acta. (2011) 1812:246–51. doi: 10.1016/j.bbadis.2010.05.012
37. Allegretta M, Nicklas JA, Sriram S, Albertini RJ. T cell responsive to myelin basic protein in patients with multiple sclerosis. Science. (1990) 247:718–21. doi: 10.1126/science.1689076
38. Lovett-Racke AE, Trotter JL, Lauber J, Perrin PJ, June CH, Racke MK. Decreased dependence of myelin basic protein-reactive T cells on CD28-mediated costimulation in multiple sclerosis patients. A marker of activated/memory T cells. J Clin Invest. (1998) 101:725–30. doi: 10.1016/S0165-5728(98)91630-7
39. Perrin PJ, Lovett-Racke A, Phillip SM, Racke MK. Differential requirements of naïve and memory T cells for CD28 costimulation in autoimmune pathogenesis. Histol Histopathol. (1999) 14:1269–76. doi: 10.14670/HH-14.1269
40. Burns J, Bartholomew B, Lobo S. Isolation of myelin basic protein-specific T cells predominantly from the memory T-cell compartment in multiple sclerosis. Ann Neurol. (1999) 45:33–9.
41. Racke MK, Ratts RB, Arredondo L, Perrin PJ, Lovett-Racke A. The role of costimulation in autoimmune demyelination. J Neuroimmunol. (2000) 107:205–15. doi: 10.1016/S0165-5728(00)00230-7
42. Holick MF. The vitamin D epidemic and its health consequences. J Nutr. (2005) 135:2739S−48S. doi: 10.1093/jn/135.11.2739S
43. Wang TT. Large-scale in silico and microarray-based identification of direct 1,25-dihydroxyvitamin D3 target genes. Mol Endocrinol. (2005) 19:2685–95. doi: 10.1210/me.2005-0106
44. Norman AW. Minireview: vitamin D receptor: new assignments for an already busy receptor. Endocrinology. (2006) 147:5542–8. doi: 10.1210/en.2006-0946
45. Li YC, Pirro AE, Amling M, Delling G, Baron R, Bronson R, et al. Targeted ablation of the vitamin D receptor: an animal model of vitamin D-dependent rickets type II with alopecia. Proc Natl Acad Sci USA. (1997) 94:9831–5. doi: 10.1073/pnas.94.18.9831
46. Yoshizawa T, Handa Y, Uematsu Y, Takeda S, Sekine K, Yoshihara Y, et al. Mice lacking the vitamin D receptor exhibit impaired bone formation, uterine hypoplasia and growth retardation after weaning. Nat Genet. (1997) 16:391–6. doi: 10.1038/ng0897-391
47. Van Cromphaut SJ, Dewerchin M, Hoenderop JG, Stockmans I, Van Herch E, Kato S, et al. Duodenal calcium absorption in vitamin D receptor-knockout mice: functional and molecular aspects. Proc Natl Acad Sci USA. (2001) 98:13324–9. doi: 10.1073/pnas.231474698
48. Tiosano D, Hada S, Chen Z, Nemirovsky A, Gepstin V, Militianu D, et al. Calcium absorption, kinetics, bone density, and bone structure in patients with hereditary vitamin D-resistant rickets. J Clin Endocrinol Metab. (2011) 96:3701–9. doi: 10.1210/jc.2011-1432
49. Eyles DW, Smith S, Kinobe R, Hewison M, McGrath JJ. Distribution of the Vitamin D receptor and 1α-hydroxylase in human brain. J Chem Neuroanat. (2005) 29:21–30. doi: 10.1016/j.jchemneu.2004.08.006
50. Sanchez B, Relova JL, Gallego R, Ben-Batalla I, Perez-Fernandez R. 1,25-Dihydroxyvitamin D 3administration to 6-hydroxydopamine-lesioned rats increases glial cell line-derived neurotrophic factor and partially restores tyrosine hydroxylase expression in substantia nigra and striatum. J Neurosci Res. (2009) 87:723–32. doi: 10.1002/jnr.21878
51. Neveu I, Naveilhan P, Jehan FR, Baudet C, Wion D, De Luca HF, et al. 1,25-Dihydroxyvitamin D3 regulates the synthesis of nerve growth factor in primary cultures of glial cells. Mol Brain Res. (1994) 24:70–6. doi: 10.1016/0169-328X(94)90119-8
52. Garcion E, Wion-Barbot N, Montero-Menei CN, Berger F, Wion D. New clues about vitamin D functions in the nervous system. Trends Endocrinol and Metab. (2002) 13:100–5. doi: 10.1016/S1043-2760(01)00547-1
53. Garcion E, Sindji L, Montero-Menei C, Andre C, Brachet P, Darcy F. Expression of inducible nitric oxide synthase during rat brain inflammation: regulation by 1,25-dihydroxyvitamin D3. Glia. (1998) 22:282–94.
54. Kesby JP, Eyles DW, Burne THJ, McGrath JJ. The effects of vitamin D on brain development and adult brain function. Mol Cell Endocrinol. (2011) 347:121–7. doi: 10.1016/j.mce.2011.05.014
55. Holick MF, Binkley NC, Bischoll-Ferrari HA, Gordon CM, Hanley DA, Heaney RP, et al. Evaluation, treatment, and prevention of vitamin D deficiency: an Endocrine Society clinical practice guideline. J Clin Endocrinol Metab. (2011) 96:1911–30. doi: 10.1210/jc.2011-0385
56. Manson JE, Brannon PM, Rosen CJ, Taylor CL. Vitamin D deficiency – is there really a pandemic? N Engl J Med. (2016) 375:1817–20. doi: 10.1056/NEJMp1608005
57. Willer CJ, Dyment DA, Sadovnick AD, Rothwell PM, Murray TJ, Ebers GC. Timing of birth and risk of multiple sclerosis: population based study. BMJ. (2005) 330:120–120. doi: 10.1136/bmj.38301.686030.63
58. Dobson R, Giovannoni G, Ramagopalan S. The month of birth effect in multiple sclerosis: systematic review, meta-analysis and effect of latitude. J Neurol Neurosurg Psychiatry. (2013) 84:427–32. doi: 10.1136/jnnp-2012-303934
59. Fiddes B, Wason J, Sawcer S. Confounding in association studies: month of birth and multiple sclerosis. Neurol. (2014) 261:1851–6. doi: 10.1007/s00415-014-7241-y
60. Walleczek NK, Frommlet F, Bsteh G, Eggers C, Rauschka H, Koppi S, et al. Month-of-birth-effect in multiple sclerosis in Austria. Mult Scler. (2019) 25:1870–7. doi: 10.1177/1352458518810924
61. Munger KL, Aivo H, Hongell K, Soilu-Hanninen M, Surcel HM, Ascherio A. Vitamin D status during pregnancy and risk of multiple sclerosis in offspring of women in the Finnish Maternity Cohort. JAMA Neurol. (2016) 73:515–9. doi: 10.1001/jamaneurol.2015.4800
62. Nielsen NM, Munger KL, Koch-Henriksen N, Hougaard DM, Magyari M, Jorgensen KT, et al. Neonatal vitamin D status and risk of multiple sclerosis: a population-based case-control study. Neurol. (2017) 88:44–51. doi: 10.1212/WNL.0000000000003454
63. Van der Mei IA, Ponsonby AL, Dwyer T, Blizzard L, Simmons R, Taylor BV, et al. Past exposure to sun, skin phenotype, and risk of multiple sclerosis: case-control study. BMJ. (2003) 327:316. doi: 10.1136/bmj.327.7410.316
64. Kampman MT, Wilsgaard T, Mellgren SI. Outdoor activities and diet in childhood and adolescence relate to MS risk above the arctic circle. J Neurol. (2007) 254:471–7. doi: 10.1007/s00415-006-0395-5
65. Islam T, Gauderman WJ, Cozen W, Mack TM. Childhood sun exposure influences risk of multiple sclerosis in monozygotic twins. Neurology. (2007) 69:381–8. doi: 10.1212/01.wnl.0000268266.50850.48
66. Irving AA, Marling SJ, Seeman J, Plum LA, DeLuca HF. UV light suppression of EAE (a mouse model of multiple sclerosis) is independent of vitamin D and its receptor. Proc Natl Acad Sci U S A. (2019) 116:22552–5. doi: 10.1073/pnas.1913294116
67. Swank RL, Lerstad O, Strøm A, Backer J. Multiple Sclerosis in Rural Norway – its geographic and occupational incidence in relation to nutrition. N Engl J Med. (1952) 246:721–8. doi: 10.1056/NEJM195205082461901
68. Munger KL, Zhang SM, O'Reilly E, Hernán MA, Olek MJ, Willett WC, et al. Vitamin D intake and incidence of multiple sclerosis. Neurology. (2004) 62:60–5. doi: 10.1212/01.WNL.0000101723.79681.38
69. Munger KL, Levin LI, Hollis BW, Howard NS, Ascherio A. Serum 25-hydroxyvitamin D levels and risk of multiple sclerosis. JAMA Neurol. (2006) 296:2832–8. doi: 10.1001/jama.296.23.2832
70. Mokry LE, Ross S, Ahmad OS, Forgetta V, Smith GD, Goltzman D, et al. Vitamin D and risk of multiple sclerosis: a Mendelian randomization study. PLoS Med. (2015) 12:e1001866. doi: 10.1371/journal.pmed.1001866
71. Langer-Gould A, Lucas RM, Xiang AH, Wu J, Chen LH, Gonzales E, et al. Vitamin D-binding protein polymorphisms, 25-hydroxyvitamin D, sunshine and multiple sclerosis. Nutrients. (2018) 10:184. doi: 10.3390/nu10020184
72. Correale J, Ysrraelit MC, Gaitan MI. Immunomodulatory effects of Vitamin D in multiple sclerosis. Brain. (2009) 132:1146–60. doi: 10.1093/brain/awp033
73. van der Mei IAF, Ponsonby AL, Dwyer T, Blizzard L, Taylor BV, Kilpatrick T, et al. Vitamin D levels in people with multiple sclerosis and community controls in Tasmania, Australia. J Neurol. (2007) 254:581–90. doi: 10.1007/s00415-006-0315-8
74. Simpson S, Taylor B, Blizzard L, Ponsonby AL, Pittas F, Tremlett H, et al. Higher 25-hydroxyvitamin D is associated with lower relapse risk in MS. Ann Neurol. (2010) 68:193–203. doi: 10.1002/ana.22043
75. Runia TF, Hop WCJ, de Rijke YB, Buljevac D, Hintzen RQ. Lower serum vitamin D levels are associated with a higher relapse risk in multiple sclerosis. Neurology. (2012) 79:261–6. doi: 10.1212/WNL.0b013e31825fdec7
76. Provvedini D, Tsoukas C, Deftos L, Manolagas S. 1,25-dihydroxyvitamin D3 receptors in human leukocytes. Science. (1983) 221:1181–3. doi: 10.1126/science.6310748
77. Lemire JM, Archer DC. 1,25-dihydroxyvitamin D3 prevents the in vivo induction of murine experimental autoimmune encephalomyelitis. J Clin Invest. (1991) 87:1103–7. doi: 10.1172/JCI115072
78. Cantorna MT, Hayes CE, DeLuca HF. 1,25-Dihydroxyvitamin D3 reversibly blocks the progression of relapsing encephalomyelitis, a model of multiple sclerosis. Proc Natl Acad Sci USA. (1996) 93:7861–4. doi: 10.1073/pnas.93.15.7861
79. Nashold FE, Miller DJ, Hayes CE. 1,25-dihydroxyvitamin D3 treatment decreases macrophage accumulation in the CNS of mice with experimental autoimmune encephalomyelitis. J Neuroimmunol. (2000) 103:171–9. doi: 10.1016/S0165-5728(99)00247-7
80. Mayne CG, Spanier JA, Relland LM, Williams CB, Hayes CE. 1,25-Dihydroxyvitamin D3 acts directly on the T lymphocyte vitamin D receptor to inhibit experimental autoimmune encephalomyelitis. Eur J Immunol. (2011) 41:822–32. doi: 10.1002/eji.201040632
81. Cantorna MT, Woodward WD, Hayes CE, DeLuca HF. 1,25-dihydroxyvitamin D3 is a positive regulator for the two anti-encephalitogenic cytokines TGF-beta 1 and IL-4. J Immunol. (1998) 160:5314–9.
82. Spach KM, Nashold FE, Dittel BN, Hayes CE. IL-10 Signaling Is Essential for 1,25-Dihydroxyvitamin D3-Mediated Inhibition of Experimental Autoimmune Encephalomyelitis. J Immunol. (2006) 177:6030–7. doi: 10.4049/jimmunol.177.9.6030
83. Mattner F, Smiroldo S, Galbiati F. Inhibition of Th1 development and treatment of chronic-relapsing experimental allergic encephalomyelitis by a non-hypercalcemic analogue of 1,25-dihydroxyvitamin D3. Eur J Immunol. (2000) 30:498–508. doi: 10.1002/1521-4141(200002)30:2<498::AID-IMMU498>3.0.CO;2-Q
84. Muthian G, Raikwar HP, Rajasingh J, Bright JJ. 1,25 dihydroxyvitamin-D3 modulates JAK–STAT pathway in IL-12/IFNγ axis leading to Th1 response in experimental allergic encephalomyelitis. J Neurosci Res. (2006) 83:1299–309. doi: 10.1002/jnr.20826
85. Miclea A, Bagnoud M, Chan A, Hoepner R. A brief review of the effects of vitamin d on multiple sclerosis. Front Immunol. (2020) 11:781. doi: 10.3389/fimmu.2020.00781
86. Hupperts R, Smolders J, Vieth R, Holmoy T, Marhardt K, Schluep M, et al. Randomized trial of daily high-dose vitamin D3 in patients with RRMS receiving subcutaneous interferon beta-1a. Neurol. (2019) 93:e1906–16. doi: 10.1212/WNL.0000000000008445
87. Camu W, Lehert P, Pierrot-Deseilligny C, Hautecoeur P, Besserve A, Deleglise ASJ, et al. Cholecalciferol in relapsing-remitting MS: a randomized clinical trial (CHOLINE). Neurol Neuroimmunol Neuroinflamm. (2019) 6:e597. doi: 10.1212/NXI.0000000000000597
88. Jagannath VA, Filippini G, Di Pietrantonj C, Asokan GV, Robak EW, Whamond L, et al. Vitamin D for the management of multiple sclerosis. Cochrane Database Syst Rev. (2018) 9:CD008422. doi: 10.1002/14651858.CD008422.pub3
89. Abe E., Miyaura C, Tanaka H, Shiina Y, Kuribayashi T, Suda S, et al. 1 alpha,25-diyhdroxyvitamin D3 promotes fusin of mouse alveolar macrophages both by a direct mechanism and by a spleen-mediated indirect mechanism. Proc Natl Acad Sci USA. (1983) 80:5583–7. doi: 10.1073/pnas.80.18.5583
90. Haq AU. 1,25-dihydroxyvitamin D3 (calcitriol) suppresses IL-2 induced murine thymocyte proliferation. Thymus. (1986) 8:295–306.
91. Adams JS, Ren S, Liu PT, Chun RF, Lagishetty V, Gombart AF, et al. Vitamin D-directed rheostatic regulation of monocyte antibacterial responses. J Immunol. (2009) 182:4289–95. doi: 10.4049/jimmunol.0803736
92. Barlow PG, Svoboda P, Mackellar A, Nash AA, York IA, Pohl J, et al. Antiviral activity and increased host defense against influenza infection elicited by the human cathelicidin LL-37. PLoS ONE. (2011) 6:e25333. doi: 10.1371/journal.pone.0025333
93. Shahmiri M, Enciso M, Adda CG, Smith BJ, Perugini MA, Mechler A. Membrane core-specific antimicrogial action of cathelicidin LL-37 peptide switches between pore and nanofiber formation. Sci Rep. (2016) 6:38184. doi: 10.1038/srep38184
94. Sousa FH, Casanova V, Findlay F, Stevens C, Svoboda P, Pohl J, et al. Cthelicidins display conserved direct antiviral activity towards rhinovirus. Peptides. (2017) 95:76–83. doi: 10.1016/j.peptides.2017.07.013
95. Piemonti L, Monti P, Sironi M, Fraticelli P, Leone BE, Dal Cin E, et al. Vitamin D3 affects differentiation, maturation, and function of human monocyte-derived dendritic cells. J Immunol. (2000) 164:4443–51. doi: 10.4049/jimmunol.164.9.4443
96. Szeles L, Keresztes G, Torocsik D, Balajthy Z, Krenacs L, Poliska S, et al. 1,25-dihydroxyvitamin D3 is an autonomous regulator of the transcriptional changes leading to a tolerogenic dendritic cell phenotype. J Immunol. (2009) 182:2074–83. doi: 10.4049/jimmunol.0803345
97. Liu PT, Stenger S, Li H, Wenzel L, Tan BH, Krutzik SR, et al. Toll-like receptor triggering of a vitamin D-mediated human antimicrobial response. Science. (2006) 311:1770–3. doi: 10.1126/science.1123933
98. Li P, Xu X, Cao E, Yu B, Li W, Fan M, et al. Vitamin D deficiency causes defective resistant to Aspergillus fumigatus in mice via aggravated and sustained inflammation. PLoS ONE. (2014) 9:e99805. doi: 10.1371/journal.pone.0099805
99. Martinez-Moreno J, Hernandez JC, Urcuqui-Inchima S. Effect of high doses of vitamin D supplementation on dengue virus replication, toll-like receptor expression, and cytokine profiles on dendritic cells. Mol Cell Biochem. (2020) 464:169–80. doi: 10.1007/s11010-019-03658-w
100. D'Ambrosio D, Cippitelli M, Cocciolo MG, Mazzeo D, Di Lucia P, Lang R, et al. Inhibition of IL-12 production by 1,25-dihydroxyvitamin D3. Involvement of NF-kappaB downregulation in transcriptional repression of the p40 gene. J Clin Invest. (1998) 101:252–62. doi: 10.1172/JCI1050
101. de Oliveira RC, Mimura LAN, Fraga-Silva TFC, Ishikawa LLW, Fernandes AAH, Zorzella-Pezavento SFG, et al. Calcitriol prevents neuroinflammation and reduces blood-brain barrier disruption and local macrophage/microglia activation. Front Pharmacol. (2020) 11:161. doi: 10.3389/fphar.2020.00161
102. Lee PW, Selhorst A, Lampe SG, Liu Y, Yang Y, Lovett-Racke AE. Neuron-specific vitamin D signaling attenuates microglia activation and CNS autoimmunity. Front Neurol. (2020) 11:19. doi: 10.3389/fneur.2020.00019
103. Evans MA, Kim HA, Ling YH, Uong S, Vinh A, De Silva TM, et al. Vitamin D(3) supplementation reduces subsequent brain injury and inflammation with ischemic stroke. Neuromolecular Med. (2018) 20:147–59. doi: 10.1007/s12017-018-8484-z
104. Calvello R, Cianciulli A, Nicolardi G, De Nuccio F, Giannotti L, Salvatore R, et al. Vitamin D treatment attenuates nueroinflammation and dopaminergic neurodegeneration in an animal model of Parkinson's disease, shifting M1 to M2 microglia. J Neuroimmune Pharmacol. (2017) 12:327–39. doi: 10.1007/s11481-016-9720-7
105. Cui C, Xu P, Li G, Qiao Y, Han W, Geng C, et al. Vitamin D receptor activation regulates microglia polarization and oxidative stress in spontaneously hypertensive rats and angiotensin II-exposed microglial cells: role of renen-angiotensis system. Redox Biol. (2019) 26:101295. doi: 10.1016/j.redox.2019.101295
106. He MC, Shi Z, Sha NN, Chen N, Peng SY, Liao DF, et al. Paricalcitol alleviates lipopolysaccharide-induced depressive-like behavior by suppressing hypothalamic microglia activation and neuroinflammation. Biochem Pharmacol. (2019) 163:1–8. doi: 10.1016/j.bcp.2019.01.021
107. Bhalla AD, Amento EP, Clemens TL, Holick MF, Krane SM. Specific high-affinity receptors for 1,25-dihydroxyvitamin D3 in human peripheral blood mononuclear cells: presence in monocytes and induction in T lymphocytes following activation. J Clin Endocrinol Metab. (1983) 57:1308–10. doi: 10.1210/jcem-57-6-1308
108. Amento EP, Bhalla AK, Kurnick JT, Kradin RL, Clemens TL, Holick SA, et al. 1 alpha,25-dihydroxyvitamin D3 induces maturation of the human monocyte cell line U937, and, in association with a factor from human T lymphocytes, augments production of the monokine, mononuclear cell factor. J Clin Invest. (1984) 73:731–9. doi: 10.1172/JCI111266
109. Chen S, Sims GP, Chen XX, Gu YY, Chen S, Lipsky PE. Modulatory effects of 1,25-dihydroxyvitamin D3 on human B cell differentiation. J Immunol. (2007) 179:1634–47. doi: 10.4049/jimmunol.179.3.1634
110. Boonstra A, Barrat FJ, Crain C, Heath VL, Savelkoul HF, O'Garra A. 1alpha,25-dihydroxyvitamin D3 has a direct effect on naïve CD4(+) T cells to enhance the development of Th2 cells. J Immunol. (2001) 167:4974–80. doi: 10.4049/jimmunol.167.9.4974
111. Tang J, Zhou R, Luger D, Zhu W, Silver PB, Grajewski RS, et al. Calcitriol suppresses antiretinal autoimmunity through inhibitory effects on the Th17 effector response. J Immunol. (2009) 182:4624–32. doi: 10.4049/jimmunol.0801543
112. Gregori S, Giarratana N, Smiroldo S, Uskokovic M, Adorini L. A 1alpha,25-diydroxyvitamin D(3) analog enhances regulatory T cells and arrests autoimmune diabetes in NOD mice. Diabetes. (2002) 51:1367–74. doi: 10.2337/diabetes.51.5.1367
113. Viglietta V, Baecher-Allan C, Weiner HL. Hafler DA Loss of functional suppression by CD4+CD25+ regulatory T cells in patients with multiple sclerosis. J Exp Med. (2004) 199:971–9. doi: 10.1084/jem.20031579
114. Haas J, Hug A, Viehöver A, Fritzsching B, Falk CS, Filser A, et al. Reduced suppressive effect of CD4+CD25high regulatory T cells on the T cell immune response against myelin oligodendrocyte glycoprotein in patients with multiple sclerosis. Eur J Immunol. (2005) 35:3343–52. doi: 10.1002/eji.200526065
115. Kumar M, Putzki N, Limmroth V, Remus R, Lindemann M, Knop D et al. CD4+CD25+FoxP3+ T lymphocytes fail to suppress myelin basic protein-induced proliferation in patients with multiple sclerosis. J Neuroimmunol. (2006) 180:178–84. doi: 10.1016/j.jneuroim.2006.08.003
116. Huan J, Culbertson N, Spencer L, Bartholomew R, Burrows GG, Chou YK et al. Decreased FOXP3 levels in multiple sclerosis patients. J Neurosci Res. (2005) 81:45–52. doi: 10.1002/jnr.20522
117. Haas J, Fritzsching B, Trübswetter P, Korporal M, Milkova L, Fritz B et al. Prevalence of newly generated naive regulatory T cells (Treg) is critical for Treg suppressive function and determines Treg dysfunction in multiple sclerosis. J Immunol. (2007) 179:1322–30. doi: 10.4049/jimmunol.179.2.1322
118. Venken K, Hellings N, Thewissen M, Somers V, Hensen K, Rummens JL et al. Compromised CD4+ CD25(high) regulatory T-cell function in patients with relapsing-remitting multiple sclerosis is correlated with a reduced frequency of FOXP3-positive cells and reduced FOXP3 expression at the single-cell level. Immunology. (2008) 123:79–89. doi: 10.1111/j.1365-2567.2007.02690.x
119. Vanherwegen AS, Eelen G, Ferreira GB, Ghesquiere B, Cook DP, Nikolic T, et al. Vitamin D controls the capacity of human dendritic cells to induce functional regulatory T cells by regulation of glucose metabolism. J Steroid Biochem Mol Biol. (2019) 187:134–45. doi: 10.1016/j.jsbmb.2018.11.011
120. Smolders J, Thewissen M, Peelen E, Menheere P, Tervaert JW, Kamoiseaux J, et al. Vitamin D status is positively correlated with regulatory T cell function in patients with multiple sclerosis. PLoS ONE. (2009) 4:e6635. doi: 10.1371/journal.pone.0006635
121. Killick J, Hay J, Morandi E, Vermeren S, Kari S, Angles T, et al. Vitamin D/CD46 crosstalk in human T cells in multiple sclerosis. Front Immunol. (2020) 11:598727. doi: 10.3389/fimmu.2020.598727
122. Drozdenko G, Scheel T, Heine G, Baumgrass R, Worm M. Impaired T cell activation and cytokine production by calcitriol-primed human B cells. Clin Exp Immunol. (2014) 178:364–72. doi: 10.1111/cei.12406
123. Cross AH, Stark JL, Lauber J, Ramsbottom MJ, Lyons JA. Rituximab reduces B cells and T cells in cerebrospinal fluid of multiple sclerosis patients. J Neuroimmunol. (2006) 180:63–70. doi: 10.1016/j.jneuroim.2006.06.029
124. Lovett-Racke AE, Gormley M, Liu Y, Yang Y, Graham C, Wray S, et al. B cell depletion with ublituximab reshapes the T cell profile in multiple sclerosis patients. J Neuroimmunol. (2019) 332:187–97. doi: 10.1016/j.jneuroim.2019.04.017
125. Lovett-Racke AE, Yang Y, Liu Y, Gormley M, Kraus E, Graham C, et al. B cell depletion changes the immune cell profile in multiple sclerosis patients: one-year report. J Neuroimmunol. (2021) 359:577676. doi: 10.1016/j.jneuroim.2021.577676
126. Stumpf WE, Sar M, Clark SA, DeLuca HF. Brain target sites for 1,25-kihydroxyvitamin D3. Science. (1982) 215:1403–5. doi: 10.1126/science.6977846
127. McGrath JJ, Feron FP, Burne TH, Mackay-Sim A, Eyles DW. Vitamin D3-implications for brain development. J Steroid Biochem Mol Biol. (2004) 89–90:557–60. doi: 10.1016/j.jsbmb.2004.03.070
128. Zehnder D, Bland R, Williams MC, McNinch RW, Howie AJ, Stewart PM, et al. Extrarenal expression of 25-hydroxyvitamin d(3)-1 alpha-hydroxylase. J Clin Endocrinol Metab. (2001) 86:888–94. doi: 10.1210/jc.86.2.888
129. Eyles D, Brown J, Mackay-Sim A, McGrath JJ, Feron F. Vitamin D3 and brain development. Neuroscience. (2003) 118:641–53. doi: 10.1016/S0306-4522(03)00040-X
130. Brown J, Bianco JI, McGrath JJ, Eyles DW. 1,25-dihydroxyvitamin D3 induces nerve growth factor, promotes neurite outgrowth and inhibits mitosis in embryonic rat hippocampal neurons. Neurosci Lett. (2003) 343:139–43. doi: 10.1016/S0304-3940(03)00303-3
131. Ko P, Burkert R, McGrath J, Eyles D. Maternal vitamin D3 deprivation and the regulation of apoptosis and cell cycle during rat brain development. Brain Res Dev Brain Res. (2004) 153:61–8. doi: 10.1016/j.devbrainres.2004.07.013
132. Sinkkonen L, Malinen M, Saavalainen K, Vaisanen S, Carlberg C. Regulation of the human cyclin C gene via multiple vitamin D3-repsonsive regions in its promoter. Nucleic Acids Res. (2005) 33:2440–51. doi: 10.1093/nar/gki502
133. Cui X, McGrath JJ, Burne TH, Mackay-Sim A, Eyles DW. Maternal vitamin D depletion alters neurogenesis in the developing rat brain. Int J Dev Neurosci. (2007) 25:227–32. doi: 10.1016/j.ijdevneu.2007.03.006
134. Al-Amin MM, Sullivan RKP, Kurniawan ND, Burne THJ. Adult vitamin D deficiency disrupts hippocampal-dependent learning and structural brain connectivity in BALB/c mice. Brain Struct Funct. (2019) 224:1315–29. doi: 10.1007/s00429-019-01840-w
135. Annweiler C, Bartha R, Karras SN, Gautier J, Roche F, Beauchet O. Vitamin D and white matter abnormalities in older adults: a quantitative volumetric analysis of brain MRI. Exp Gerontol. (2015) 63:41–7. doi: 10.1016/j.exger.2015.01.049
136. Cui X, Pelekanos M, Burne TH, McGrath JJ, Eyles DW. Maternal vitamin D deficiency alters the expression of genes involved in dopamine specification in the developing rat mesencephalon. Neurosci Lett. (2010) 486:220–3. doi: 10.1016/j.neulet.2010.09.057
137. Orme RP, Bhangal MS, Fricker RA. Calcitriol imparts neuroprotection in vitro to midbrain dopaminergic neurons by upregulating GDNF expression. PLoS ONE. (2013) 8:e62040. doi: 10.1371/journal.pone.0062040
138. Pertile RAN, Cui X, Eyles DW. Vitamin D signaling and the differentiation of developing dopamine systems. Neuroscience. (2016) 333:193–203. doi: 10.1016/j.neuroscience.2016.07.020
139. Shirazi HA, Rasouli J, Ciric B, Rostami A, Zhang GX. 1,25-dihydroxyvitamin D3 enhances neural stem cell proliferation and oligodendrocyte differentiation. Exp Mol Pathol. (2015) 98:240–5. doi: 10.1016/j.yexmp.2015.02.004
140. De la Fuenta AG, Errea O, van Wijngaarden P, Gonzalez GA, Kerninon C, Jarjour AA, et al. Vitamin D receptor-retinoid X receptor heterodimer signaling regulates oligodendrocyte progenitor cell differentiation. J Cell Biol. (2015) 211:975–85. doi: 10.1083/jcb.201505119
141. Cass WA, Peters LE, Fletcher AM, Yurek DM. Evoked dopamine overflow is augmented in the striatum of calcitriol treated rats. Neurochem Int. (2012) 60:186–91. doi: 10.1016/j.neuint.2011.11.010
142. Groves NJ, Kesby JP, Eyles DW, McGrath JJ, Mackay-Sin A, Burne TH. Adult vitamin D deficiency leads to beavioural and brain neurochemical alterations in C57BL/6J and BALB/c mice. Behav Brain Res. (2013) 241:120–31. doi: 10.1016/j.bbr.2012.12.001
143. Jiang P, Zhang LH, Cai HL, Li HD, Liu YP, Tang MM, et al. Neurochemical effects of chronic administration of calcitriol in rats. Nutrients. (2014) 6:6048–59. doi: 10.3390/nu6126048
144. Kesby JP, Turner KM, Alexander S, Eyles DW, McGrath JJ, Burne THJ. Developmental vitamin D deficiency alters multiple neurotransmitter systems in the neonatal rat brain. Int J Dev Neurosci. (2017) 62:1–7. doi: 10.1016/j.ijdevneu.2017.07.002
145. Neveu I, Naveilhan P, Baudet C, Brachet P, Metsis M. 1,25-dihydroxyvitamin D3 regulates NT-3, NT-4 but not BDNF mRNA in astrocytes. Neuroreport. (1994) 6:124–6. doi: 10.1097/00001756-199412300-00032
146. Naveilhan P, Neveu I, Wion D, Brachet P. 1,25-kihydroxyvitamin D3, an inducer of glial cell line-derived neurotrophic factor. Neuroreport. (1996) 7:171–5. doi: 10.1097/00001756-199609020-00023
147. Naveilhan P, Neveu I, Baudet C, Funakoshi H, Wion D, Brachet P, et al. 1,25-kihydroxyvitamin D3 regulates the expression of the low-affinity neurotrophin receptor. Brain Res Mol Brain Res. (1996) 41:259–68. doi: 10.1016/0169-328X(96)00103-9
148. Taniura H, Ito M, Sanada N, Kuramoto N, Ohno Y, Nakamichi N, et al. chronic vitamin D3 treatment protects against neurotoxicity by glutamate in association with upregulation of vitamin D receptor mRNA expression in cultured rat corical neurons. J Neurosci Res. (2006) 83:1179–89. doi: 10.1002/jnr.20824
149. Almeras L, Eyles D, Benech P, Laffite D, Villard C, Patatian A, et al. Developmental vitamin D deficiency alters brain protein expression in the adult rat: implications for neuropsychiatric disorders. Proteomics. (2007) 7:769–80. doi: 10.1002/pmic.200600392
150. Eyles D, Almeras L, Benech P, Patatian A, Mackay-Sim A, McGrath J, Feron F. Developmental vitamin D deficiency alters the expression of genes encoding mitochondrial, cytosketetal and synaptic proteins in the adult rat brain. J Steroid Biochem Mol Biol. (2007) 103:538–45. doi: 10.1016/j.jsbmb.2006.12.096
151. Gezen-Ak D, Dursun E, Yilmazer S. The effects of vitamin D receptor silencing on the expression of LVSCC-A1C and LVSCC-A1D and the release of NGF in cortical neurons. PLoS ONE. (2011) 6:e17553. doi: 10.1371/journal.pone.0017553
152. Zhu Y, Zhou R, Yang R, Zhang Z, Bai Y, Chang F, et al. Abnormal neurogenesis in the dentate gyrus of adult mice lacking 1,25-dihydroxy vitamin D3 (1,25-(OH)2 D3). Hippocampus. (2012) 22:421–33. doi: 10.1002/hipo.20908
153. Brewer LD, Thibault V, Chen KC, Langub MC, Landfield PW, Porter NM. Vitamin D hormone confers neuroprotection in parallel with downregulation of L-type calcium channel expression in hippocampal neurons. J Neurosci. (2001) 21:98–108. doi: 10.1523/JNEUROSCI.21-01-00098.2001
154. Zanatta L, Goulart PB, Goncalves R, Pierozan P, Winkelmann-Duarte EC, Woehl VM, et al. 1alpha,25-kihydroxyvitamin D(3) mechanism of action: modulation of L-type calcium channels leading to calcium uptake and intermediate filament phosphorylation in cerebral cortex of young rats. Biochim biophys Acta. (2012) 1823:1708–19. doi: 10.1016/j.bbamcr.2012.06.023
155. Boyan BD, Sylvia VL, Dean DD, Schwartz Z. Membrane mediated signaling mechanisms are used differentially by metabolites of vitamin D(3) in musculoskeletal cells. Steroids. (2002) 67:421–7. doi: 10.1016/S0039-128X(01)00178-7
156. Chen J, Doroudi M, Cheung H, Grozier AL, Schwartz Z, Boyan BD. Plasma membrane Pdia3 and VDR interact to elicit rapid responses to 1alpha,25(OH)(2)D(3). Cell Signal. (2013) 25:2362–73. doi: 10.1016/j.cellsig.2013.07.020
157. Nemere I, Garbi N, Hammerling G, Hintze KJ. Role of the 1,25D3-MARRS receptor in the 125(OH)2D3-stimulated uptake of calcium and phosphate in intestinal cells. Steroids. (2012) 77:897–902. doi: 10.1016/j.steroids.2012.04.002
158. Jozefowicz O, Rabe-Jablonska J, Wozniacka A, Strzelecki D. Analysis of vitamin D status in major depression. J Psychiatr Pract. (2014) 20:329–37. doi: 10.1097/01.pra.0000454777.21810.15
159. Kerr DC, Zava DT, Piper WT, Saturn SR, Frei B, Gombart AF. Associations between vitamin D levels and depressive symptoms in healthy young adult women. Psychiatry Res. (2015) 227:46–51. doi: 10.1016/j.psychres.2015.02.016
160. Coskun S, Simsek S, Camkurt MA, Cim A, Celik SB. Association of polymorphisms in the vitamin D receptor gene and serum 25-hydroxyvitamin D levels in children with autism spectrum disorder. Gene. (2016) 588:109–14. doi: 10.1016/j.gene.2016.05.004
161. Sourander A, Upadhyaya S, Surcel HM, Hinkka-Yli-Salomaki S, Cheslack-Postava K, Silwal S, et al. Maternal vitamin D levels during pregnancy and offspring autism spectrum disorder. Biol Psychiatry. (2021) 90:790–7. doi: 10.1016/j.biopsych.2021.07.012
162. Cui X, McGrath JJ, Burne THJ, Eyles DW. Vitamin D and schizophrenia: 20 years on. Mol Psychiatry. (2021) 26:2708–20. doi: 10.1038/s41380-021-01025-0
163. Evatt ML, Delong MR, Khazai N, Rosen A, Triche S, Tangpricha V. Prevalence of vitamin d insufficiency in patients with Parkinson disease and Alzheimer disease. Arch Neurol. (2008) 65:1348–52. doi: 10.1001/archneur.65.10.1348
164. Lv L, Tan X, Peng X, Bai R, Xiao Q, Tan J, et al. The relationships of vitamin D, vitamin D receptor gene polymorphisms, and vitamin D supplementation with Parkinson's disease. Transl Neurodegener. (2020) 9:34. doi: 10.1186/s40035-020-00213-2
165. Annweiler C, Llewellyn DF, Beauchet O. Low serum vitamin D concentrations in Alzheimer's disease: a systemic review and meta-analysis. J Alzheimers Dis. (2013) 33:659–74. doi: 10.3233/JAD-2012-121432
166. Balion C, Griffith LE, Strifler L, Henderson M, Patterson C, Heckman G, et al. Vitamin D, cognition, and dementia: a systematic review and meta-analysis. Neurology. (2012) 79:1397–405. doi: 10.1212/WNL.0b013e31826c197f
167. Peterson AL, Mancini M, Horak FB. The relationship between balance control and vitamin D in Parkinson's disease-a pilot study. Mov Disord. (2013) 28:1133–7. doi: 10.1002/mds.25405
168. Peterson AL, Murchison C, Zabetian C, Leverenz JB, Watson GS, Montine T, et al. Memory, mood, and vitamin D in persons with Parkinson's disease. J Park Dis. (2013) 3:547–55. doi: 10.3233/JPD-130206
169. Littlejohns TJ, Henley WE, Lang IA, Annweiler C, Beauchet O, Chaves PHM, et al. Vitamin D and the risk of dementia and Alzheimer disease. Neurology. (2014) 83:920–8. doi: 10.1212/WNL.0000000000000755
170. Turetsky A, Goddeau RP, Henninger N. Low serum vitamin D is independently associated with larger lesion volumes after ischemic stroke. J Stroke Cerebrovasc Dis. (2015) 24:1555–63. doi: 10.1016/j.jstrokecerebrovasdis.2015.03.051
171. Huang H, Zheng T, Wang S, Wei L, Wang Q, Sun Z. Serum 25-hydroxyvitamin D predicts early recurrent stroke in ischemic stroke patients. Nutr Metab Cardiovasc Dis. (2016) 26:908–14. doi: 10.1016/j.numecd.2016.06.009
172. Nie Z, Ji XC, Wang J, Zhang HX. Serum levels of 25-hydroxyvitamin D predicts infarct volume and mortality in ischemic stroke patients. J Neuroimmunol. (2017) 313:41–5. doi: 10.1016/j.jneuroim.2017.10.002
173. Di Somma C, Scarano E, Barrea L, Zhukouskaya VV, Savastano S, Mele C, et al. Vitamin D and neurological diseases: an endocrine view. Int J Mol Sci. (2017) 18:11. doi: 10.3390/ijms18112482
174. Sivandzade F, Prasad S, Bhalerao A, Cucullo L. NRF2 and NF-κB interplay in cerebrovascular and neurodegenerative disorders: molecular mechanisms and possible therapeutic approaches. Redox Biol. (2019) 21:101059. doi: 10.1016/j.redox.2018.11.017
175. Wimalawansa SJ. Vitamin D deficiency: effects on oxidative stress, epigenetics, gene regulation, and aging. Biology. (2019) 8:30. doi: 10.3390/biology8020030
176. Trist BG, Hare DJ, Double KL. Oxidative stress in the aging substantia nigra and the etiology of Parkinson's disease. Aging Cell. (2019) 18:e13031. doi: 10.1111/acel.13031
177. Niedzielska E, Smaga I, Gawlik M, Moniczewski A, Stankowicz P, Pera J, et al. Oxidative stress in neurodegenerative diseases. Mol Neurobiol. (2016) 53:4094–125. doi: 10.1007/s12035-015-9337-5
178. Browne SE, Beal MF. Oxidative damage in Huntington's disease pathogenesis. Antioxid Redox Signal. (2006) 8:2061–73. doi: 10.1089/ars.2006.8.2061
179. Rodrigo R, Fernandez-Gajardo R, Guierrez R, Matamala JM, Carrasco R, Miranda-Merchak A, et al. Oxidative stress and pathophysiology of ischemic stroke: novel therapeutic opportunities. CNS Neurol Disord Drug Targets. (2013) 12:698–714. doi: 10.2174/1871527311312050015
180. Di Filippo M, Chiasserini D, Tozzi A, Picconi B, Calabresi P. Mitochondria and the link between neuroinflammation and neurodegeneration. J Alzheimers Dis. (2010) 20:S369–79. doi: 10.3233/JAD-2010-100543
181. Brandes MS, Gray NE. NRF2 as a Therapeutic Target in Neurodegenerative Diseases. ASN Neuro. (2020) 12:1759091419899782. doi: 10.1177/1759091419899782
182. Khairy EY, Attia MM. Protective effects of vitamin D on neurophysiologic alterations in brain aging: role of brain-derived neurotrophic factor (BDNF). Nutr Neurosci. (2021) 24:650–9. doi: 10.1080/1028415X.2019.1665854
183. Hannan A, Dash R, Al Mamun Sohag A, Haque N, Soo Moon I. Neuroprotection Against Oxidative Stress: Phytochemicals Targeting TrkB Signalaing and the Nrf2-ARE Antioxidant System. Fronter Mol Neurosci. (2020). doi: 10.3389/fnmol.2020.00116
184. Ohl K, Tenbrock K, Kipp M. Oxidative stress in multiple sclerosis: Central and peripheral mode of action. Exp Neurol. (2016) 277:58–67. doi: 10.1016/j.expneurol.2015.11.010
185. Genestra M. Oxyl radicals, redox-sensitive signaling cascades and antioxidants. Cell Signal. (2007) 19:1807–19. doi: 10.1016/j.cellsig.2007.04.009
186. Gray E, Thomas TL, Betmouni S, Scolding N, Love S. Elevated activity and microglial expression of myeloperoxidase in demyelinated cerebral cortex in multiple sclerosis. Brain Path. (2008) 18:86–95. doi: 10.1111/j.1750-3639.2007.00110.x
187. Gray E, Thomas TL, Betmouni S, Scolding N, Love S. Elevated myeloperoxidase activity in white matter in multiple sclerosis. Neurosci Lett. (2008) 444:195–8. doi: 10.1016/j.neulet.2008.08.035
188. Cross AH, Manning PT, Keeling RM, Schmidt RE, Misko TP. Peroxynitrite formation within the central nervous system in active multiple sclerosis. J Neuroimmunol. (1998) 88:45–56. doi: 10.1016/S0165-5728(98)00078-2
189. Liu JS, Zhao ML, Brosnan CF, Lee SC. Expression of inducible nitric oxide synthase and nitrotyrosine in multiple sclerosis lesions. Amer J Pathol. (2001) 158:2057–66. doi: 10.1016/S0002-9440(10)64677-9
190. van Horssen J, Schreibelt G, Drexhage J, Hazes T, Dijkstra CD, van der Valk P, et al. Severe oxidative damage in multiple sclerosis lesions coincides with enhanced antioxidant enzyme expression. Free Radical Biol Med. (2008) 45:1729–37. doi: 10.1016/j.freeradbiomed.2008.09.023
191. Craner MJ, Newcombe J, Black JA, Hartle C, Cuzner ML, Waxman SG. Molecular changes in neurons in multiple sclerosis: altered axonal expression of Nav1.2 and Nav1.6 sodium channels and Na+/Ca2+ exchanger. Proc Natl Acad Sci USA. (2004) 101:8168–73. doi: 10.1073/pnas.0402765101
192. Witte ME, Bø L, Rodenburg RJ, Belien JA, Musters R, Hazes T. Enhanced number and activity of mitochondria in multiple sclerosis lesions. J Pathol. (2009) 219:193–204. doi: 10.1002/path.2582
193. Campbell G, Mahad DJ. Mitochondrial dysfunction and axon degeneration in progressive multiple sclerosis. FEBS Lett. (2018) 592:1113–21. doi: 10.1002/1873-3468.13013
194. Waxman SG. Ions, energy and axonal injury: towards a molecular neurology of multiple sclerosis. Trends Mol Med. (2006) 12:192–5. doi: 10.1016/j.molmed.2006.03.001
195. Hybertson BM, Gao B, Bose SK, McCord JM. Oxidative stress in health and disease: the therapeutic potential of Nrf2 activation. Mol Aspects Med. (2011) 32:234–46. doi: 10.1016/j.mam.2011.10.006
196. Johnson DA, Amirahmadi S, Ward C, Fabry Z, Johnson JA. The absence of the pro-antioxidant transcription factor Nrf2 exacerbates experimental autoimmune encephalomyelitis. Toxicological Sci. (2010) 114:237–46. doi: 10.1093/toxsci/kfp274
197. Larabee CM, Desai S, Agasing A, Georgescu C, Wren JD, Axtell RC, et al. Loss of Nrf2 exacerbates the visual deficits and optic neuritis elicited by experimental autoimmune encephalomyelitis. Molecular Vision. (2016) 22:1503–13.
198. Morales Pantoja IE, Hu CL, Perrone-Bizzozero NI, Zheng J, Bizzozero OA. Nrf2-dysregulation correlates with reduced synthesis and low glutathione levels in experimental autoimmune encephalomyelitis. J Neurochem. (2016) 139:640–50. doi: 10.1111/jnc.13837
199. Linker RA, Lee DH, RyanS, van Dam AM, Conrad R, Bista P, et al. Fumaric acid esters exert neuroprotective effects in neuroinflammation via activation of the Nrf2 antioxidant pathway. Brain. (2011) 134:678–92. doi: 10.1093/brain/awq386
200. van Horssen J, Drexhage JA, Flor T, Gerritsen W, van der Valk P, de Vries HE. Nrf2 and DJ1 are consistently upregulated in inflammatory multiple sclerosis lesions. Free Radical Biol Med. (2010) 49:1283–9. doi: 10.1016/j.freeradbiomed.2010.07.013
201. Licht-Mayer S, Wimmer I, Traffehn S, Metz I, Brück W, Bauer J, et al. Cell type-specific Nrf2 expression in multiple sclerosis lesions. Acta neuropathological. (2015) 130:263–77. doi: 10.1007/s00401-015-1452-x
202. Ghoreschi K, Bruck J, Kellerer C, Deng C, Peng H, Rothfuss O, et al. Fumarates improve psoriasis and multiple sclerosis by inducing type II dendritic cells. J Exp Med. (2011) 208:2291–303. doi: 10.1084/jem.20100977
203. Kappos L, Gold R, Miller DH, Macmanus DG, Havrdova E, Limmroth V, et al. Efficacy and safety of oral fumarate in patients with relapsing-remitting multiple sclerosis: a multicentre, randomised, double-blind, placebo-controlled phase IIb study. Lancet. (2008) 372:1463–72. doi: 10.1016/S0140-6736(08)61619-0
204. Albrecht P, Bouchachia I, Goebels N, Henke N, Hofstetter HH, Issberner A, et al. Effects of dimethyl fumarate on neuroprotection and immunomodulation. J neuroinflammation. (2012) 9:163. doi: 10.1186/1742-2094-9-163
205. Fox RJ, Miller DH, Phillips JT, Hutchinson M, Havrdova E, Kita M, et al. Placebo-controlled phase 3 study of oral BG-12 or glatiramer in multiple sclerosis. N Engl J Med. (2012) 367:1087–97. doi: 10.1056/NEJMoa1206328
206. Gold R, Kappos L, Arnold DL, Bar-Or A, Giovannoni G, Selmaj K, et al. Placebo-controlled phase 3 study of oral BG-12 for relapsing multiple sclerosis. N Engl J Med. (2012) 367:1098–107. doi: 10.1056/NEJMoa1114287
207. Peng H., Guerau-de-Arellano M, Mehta VB, Yang Y, Huss DJ, Papenfuss TL, et al. Dimethyl fumarate inhibits dendritic cell maturation via nuclear factor kB (NF-kB)and extracellular signal-regulated kinase 1 and 2 (ERK1/2) and mitogen stress-activated kinase 1 (MSK1) signaling. J Biol Chem. (2012) 287:28017–26. doi: 10.1074/jbc.M112.383380
208. Salehi MS, Borhani-Haghighi A, Pandamooz S, Safari A, Dargahi L, Dianatpour M, et al. Dimethyl fumarate up-regulates expression of major neurotrophic factors in the epidermal neural crest stem cells. Tissue cell. (2019) 56:114–20. doi: 10.1016/j.tice.2019.01.004
209. Nachliely M, Trachtenberg A, Khalfin B, Nalbandyan K, Cohen-Lahav M, Yasuda K, et al. Dimethyl fumarate and vitamin D derivatives cooperatively enhance VDR and Nrf2 signaling in differentiating AML cells in vitro and inhibit leukemia progression in a xenograft mouse model. J Steroid Biochem Mol Biol. (2019) 188:8–16. doi: 10.1016/j.jsbmb.2018.11.017
210. Hausler D, Torke S, Peelen E, Bertsch T, Djukic M, Nau R, et al. High dose vitamin D exacerbates central nervous system autoimmunity by raising T-cell excitatory calcium. Brain. (2019) 142:2737–55. doi: 10.1093/brain/awz190
211. Long T, Yang Y, Peng L, Li Z. Neuroprotective effects of melatonin on experimental allergic encephalomyelitis mice via anti-oxidative stress activity. J Mol Neurosci. (2018) 64:233–41. doi: 10.1007/s12031-017-1022-x
212. Ghareghani M, Reiter RJ, Zibara K, Farhadi N. Latitude, vitamin D, melatonin, and gut microbiota act in concert to initiate multiple sclerosis: a new mechanistic pathway. Front Immunol. (2018) 9:2484. doi: 10.3389/fimmu.2018.02484
213. Bjornevik K, Cortese M, Healy BC, Kuhle J, Mina MJ, Leng Y, et al. Longitudinal analysis reveals high prevalence of Epstein-Barr virus associated with multiple sclerosis. Science. (2022) 375:296–301. doi: 10.1126/science.abj8222
214. Lang HLE, Jacobsen H, Ikemizu S, Andersson C, Harlos K, Madsen L, et al. A functional and structural basis for TCR cross-reactivity in multiple sclerosis. Nat Immunol. (2002) 3:940–3. doi: 10.1038/ni835
215. Lindsey JW. Antibodies to the Epstein-Barr virus proteins BFRF3 and BRRF2 cross-react with human proteins. J Neuroimmunol. (2017) 10:131–4. doi: 10.1016/j.jneuroim.2017.07.013
216. Tengvall K, Huang J, Hellstrom C, Kammer P, Bistrom M, Ayoglu B, et al. Molecular mimicry between anoctamin 2 and Epstein-Barr virus nuclear antigen 1 associates with multiple sclerosis risk. Proc Natl Acad Sci U S A. (2019) 116:16955–60. doi: 10.1073/pnas.1902623116
217. Lanz TV, Brewer RC, Ho PP, Moon JS, Jude KM, Fernandez D, et al. Clonally expanded B cells in multiple sclerosis bind EBV EBNA1 and GlialCAM. Nature. (2022) doi: 10.21203/rs.3.rs-1239863/v1
218. Perez-Perez S, Dominguez-Mozo MI, Garcia-Martinez MA, Aladro Y, Martinez-Gines M, Garcia-Dominguez JM, et al. Study of the possible link of 25-hydroxyvitamin D with Epstain-Barr virus and human herpesvirus 6 in patients with multiple sclerosis. Eur J Neurol. (2018) 25:1446–53. doi: 10.1111/ene.13749
219. Mameli G, Madeddu G, Mei A, Uleri E, Poddighe L, Delogu LG, et al. Activation of MSRV-type endogenous retroviruses during infectious mononucleosis and Epstein-Barr virus latency: the missing link with multiple sclerosis. PLoS ONE. (2013) 8:e78474. doi: 10.1371/journal.pone.0078474
220. Zwart SR, Mehta S, Ploutz-Snyder R, Bourbeau Y, Locke JP, Pierson DL, et al. Response to vitamin D supplementation during Antarctic winter is related to BMI, and supplementation can mitigate Epstein-Barr virus reactivation. J Nutr. (2011) 141:692–7. doi: 10.3945/jn.110.134742
221. Zdimerova H, Murer A, Engelmann C, Raykova A, Deng Y, Gujer C, et al. Attenuated immune control of Epstein-Barr virus in humanized mice is associated with the multiple sclerosis risk factor HLA-DR15. Eur J Immunol. (2021) 51:64–75. doi: 10.1002/eji.202048655
222. Brutting C, Stangl GI, Staege MS. Vitamin D, Epstein-Barr virus, and endogenous retroviruses in multiple sclerosis – facts and hypothesis. J Integr Neurosci. (2021) 20:233–8. doi: 10.31083/j.jin.2021.01.392
Keywords: multiple sclerosis, vitamin D, neuroprotection, immune regulation, oxidative stress
Citation: Gombash SE, Lee PW, Sawdai E and Lovett-Racke AE (2022) Vitamin D as a Risk Factor for Multiple Sclerosis: Immunoregulatory or Neuroprotective? Front. Neurol. 13:796933. doi: 10.3389/fneur.2022.796933
Received: 18 October 2021; Accepted: 13 April 2022;
Published: 16 May 2022.
Edited by:
Bruno Gran, Nottingham University Hospitals NHS Trust, United KingdomReviewed by:
Radu Tanasescu, University of Nottingham, United KingdomCopyright © 2022 Gombash, Lee, Sawdai and Lovett-Racke. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Amy E. Lovett-Racke, YW15LmxvdmV0dC1yYWNrZUBvc3VtYy5lZHU=
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.