 Kenya Nishioka
Kenya Nishioka Yuzuru Imai
Yuzuru Imai Hiroyo Yoshino3
Hiroyo Yoshino3 Manabu Funayama
Manabu Funayama Nobutaka Hattori
Nobutaka Hattori- 1Department of Neurology, Juntendo University School of Medicine, Tokyo, Japan
- 2Department of Research for Parkinson's Disease, Juntendo University Graduate School of Medicine, Tokyo, Japan
- 3Research Institute for Diseases of Old Age, Graduate School of Medicine, Juntendo University, Tokyo, Japan
Over the past 20 years, numerous robust analyses have identified over 20 genes related to familial Parkinson's disease (PD), thereby uncovering its molecular underpinnings and giving rise to more sophisticated approaches to investigate its pathogenesis. α-Synuclein is a major component of Lewy bodies (LBs) and behaves in a prion-like manner. The discovery of α-Synuclein enables an in-depth understanding of the pathology behind the generation of LBs and dopaminergic neuronal loss. Understanding the pathophysiological roles of genes identified from PD families is uncovering the molecular mechanisms, such as defects in dopamine biosynthesis and metabolism, excessive oxidative stress, dysfunction of mitochondrial maintenance, and abnormalities in the autophagy–lysosome pathway, involved in PD pathogenesis. This review summarizes the current knowledge on familial PD genes detected by both single-gene analyses obeying the Mendelian inheritance and meta-analyses of genome-wide association studies (GWAS) from genome libraries of PD. Studying the functional role of these genes might potentially elucidate the pathological mechanisms underlying familial PD and sporadic PD and stimulate future investigations to decipher the common pathways between the diseases.
Introduction
The nature of Parkinson's disease (PD) was initially described by James Parkinson in his “Essay on the shaking palsy” in 1817. Since then, efforts have been made to understand the clinical symptoms and pathophysiology of this disease. However, currently, only incomplete symptomatic treatments are available. The common symptoms of PD are tremor, rigidity, akinesia, and unsteadiness. Age is an important prognostic factor that increases the prevalence of PD, with 41 patients in their 40s, 107 patients in their 50s, 428 patients in their 60s, 1,087 patients in their 70s, and 1,903 patients older than 80 years being detected (all per 100,000) (1, 2). PD is pathologically characterized by the degeneration of dopamine neurons in the substantia nigra and the deposition of Lewy bodies (LBs) or Lewy neurites, a pathological hallmark of PD, which are often observed in the affected regions (3). The major component of LBs is α-synuclein, encoded by the SNCA gene located in 4q21-22 (4). α-Synuclein is thought to be the key protein involved in the pathological mechanisms underlying PD and other neurodegenerative disorders.
The development of molecular genetics technologies and family tree analysis for PD have identified genes linked to PD (5–9). Over 20 genes, namely PARK genes from PARK1 to PARK23 from Online Mendelian Inheritance in Man (OMIM) (https://www.omim.org), are associated with the development of PD. However, the PARK genes include heterogeneous genes such as Mendelian genes, candidate loci, or genes not confirmed to mediate the disease pathogenicity (10). The PARK genes also include genes confirmed as genes not associated with typical PD (i.e., ATP13A2, associated with atypical parkinsonism) (11). SNCA and LRRK2 have been identified using positional cloning in families with PD (7, 12–14) and were also later detected as major risk factors for PD using genome-wide association studies (GWAS) (15–18). The autosomal recessive genes inherited in families, PRKN (6) or PINK1 (9), were not identified through the GWAS as common genetic risk variants probably due to their low prevalence. There are several large studies that reported a lack of association between heterozygous PRKN and PINK1 variants with PD (19–21), while PD risk might be increased with heterozygous variants in these genes (22).
This review aimed to describe the clinical differences among patients with various pathogenic genes associated with PD or Parkinsonism to highlight potential underlying mechanisms regulating these genes, with a particular focus on SNCA, LRRK2, VPS13C, glucosylceramidase beta (GBA1), GCH1, and microtubule-associated protein tau (MAPT). These genes have been identified as PD causative or susceptible genes in PD families and were found through meta-analyses of GWAS (15–18). We aimed to identify the common pathological pathways governed by these genes between familial and sporadic PD.
PARK Genes
Genes associated with familial PD were historically categorized as PARK. To date, the genes belonging to the PARK category range from PARK1 to PARK24 (Table 1) (OMIM: https://www.ncbi.nlm.nih.gov/omim), with PARK1 being the same as PARK4. The PARK category includes twelve autosomal dominant inheritances, nine autosomal recessive inheritances, one X-linked, and four unidentified genes. Although the PARK16 locus (1q32) is a prominent risk locus associated with PD, responsible genes have not been determined (15). Other genes excluded from the PARK category, such as GBA1, GTP cyclohydrolase 1 (GCH1), and MAPT, were also significantly linked to PD or parkinsonism through meta-analyses of GWAS (17, 18, 23).
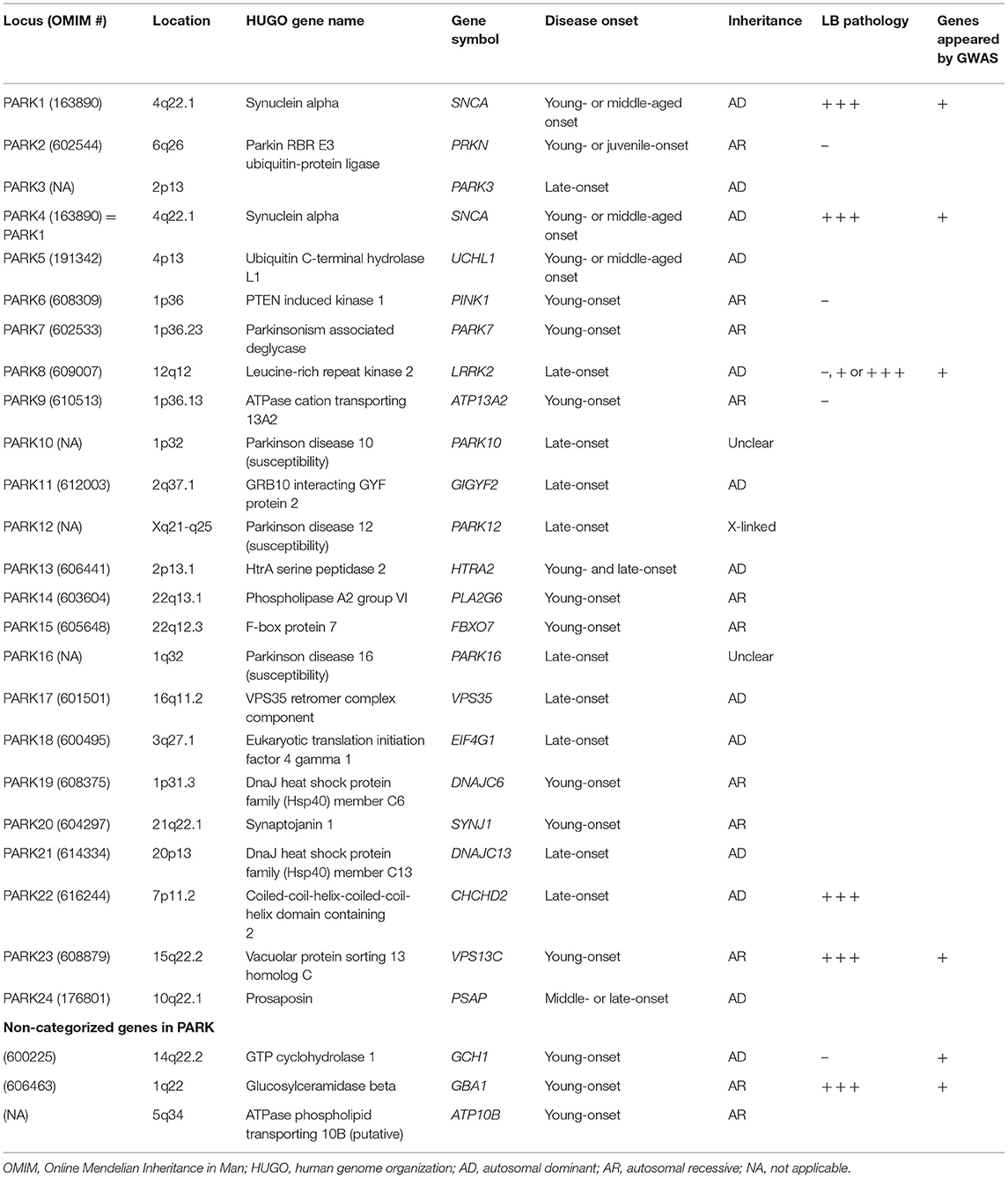
Table 1. PARK categories from the genes related to PD.
The prevalence of familial PD among all patients with PD is ~10–20% (24), whereas the rest of the cases without any family history are considered sporadic PD (80–90%). LRRK2 p.G2019S is the most common mutation in specific populations, such as in 30% cases of the Ashkenazi Jews or Arab Berbers. In other populations, the prevalence of LRRK2 was estimated at 2–5% (25). There are very few other pathogenic genes involved in PD, showing a prevalence of 1–3% among familial PD (26–34). Overall, the prevalence of pathogenic genes is extremely low among both familial and sporadic PD.
Genome-Wide Association Studies
Several meta-analyses of GWAS have been performed to identify the molecular mechanisms regulating PD (15–18, 23). Based on the analyses from over a million patients and controls, common genes associated with the PD cohort were PARK16, GBA1, SNCA, LRRK2, GCH1, and VPS13C, with SNCA and LRRK2 showing a significantly higher association with PD than other genes across populations (15, 18). Moreover, another gene, MAPT, has been identified to be associated with the PD cohort. In the European cohort, SNCA, GBA1, and LRRK2 are significantly associated with PD (17, 23). In the Asian cohort, SC2C, WBSCR17, and BST1 showed a robust association with PD (15, 18). Intriguingly, the fact that familial PD genes have been identified by GWAS means that familial PD genes are involved in the pathogenesis of sporadic PD, strongly suggesting common pathogenic pathways between familial and sporadic PD, or that multiple concurrent variants of familial PD genes may relate to the rapid motor progression of sporadic PD (35).
In the next section, we have described the genetic evidence, clinical and pathological features, and molecular backgrounds in terms of PD-associated genes. The main clinical features are also summarized in Table 2.

Table 2. Major clinical features for each gene.
Synuclein Alpha
Clinical Symptoms of Patients With SNCA Variants
Synuclein alpha (SNCA) variants associated with PD are of two types: one has missense mutations, such as p.A30G, p.A30P, p.E46K, p.H50Q, p.G51D, p.A53T/E/G/V, and p.E83Q, whereas the other has amplifications, including duplication and triplication (5, 8, 12, 36–43). Patients with missense variants are likely to develop parkinsonism in young- or middle-aged adults, along with cognitive decline or psychosis (26, 44–46). Patients with genetic amplifications showed young- or middle-aged onset of parkinsonism, psychosis, and consciousness fluctuation, resembling the symptoms of PD with dementia (PDD), along with LBs (47, 48). The amplified genes contain two- or three-fold tandem repeat replication of an SNCA locus (49). SNCA locus amplification induces an increased expression of α-synuclein in the brain or peripheral blood and accumulations of α-synuclein in the detergent-insoluble fraction (50). The clinical severity of patients with SNCA multiplications obeys the gene-dosage-dependent phenomenon (51). Patients with four copies of the gene show a more severe PD onset at a younger age (the 20–30s) than those with three copies (the 40–50s) (51). More copy numbers of SNCA may induce more severe symptoms, indicating that the increased intracellular concentration of α-synuclein is responsible for PD development.
The neuroimaging reports regarding familial PD are scarce. Most of the analyses were from the cross-sectional study without considering the duration between the disease onset and examination time. However, these differences may suggest that each variant has a different prognosis or a different spread of α-synucleinopathy. SNCA amplifications may present specific neuroimaging patterns related to dementia with LBs (DLBs) or PDD (26, 48). The brain magnetic resonance imaging (MRI) showed progressive atrophic changes in the hippocampus (26, 48), whereas [123I]N-ω-fluoropropyl-2β-carbomethoxy-3β-(4-iodophenyl) tropane (123I-FP-CIT) single-photon emission computed tomography (SPECT) showed a reduced expression of the dopamine transporter. [123I]metaiodobenzylguanidine (MIBG) myocardial scintigraphy showed a reduced heart-to-mediastinum ratio (52). The brain SPECT or positron emission tomography (PET) revealed hypoperfusion in the bilateral occipital lobes (48). Patients with a missense variant of SCNA, p.A53T, showed atrophic changes in the hippocampus and the temporal lobes in the brain MRI, a decreased heart-to-mediastinum ratio in MIBG myocardial scintigraphy, and hypoperfusion in the parieto-occipital lobe in the brain SPECT (53, 54). The findings infer that SNCA variants cause the widespread propagation of α-synuclein, with patients showing symptoms similar to DLB.
Pathology of Patients With SNCA Variants
Patients with SNCA variants commonly show a severe neuronal loss in the substantia nigra or the hippocampus and widespread appearances of LBs and Lewy neurites (47, 55) with Braak's stage 5 or 6 (46, 54). Braak's staging is advocated to confirm the severity of LB formation (56) localized in the medulla oblongata in stage 1, the pontine tegmentum in stage 2, the midbrain in stage 3, the basal prosencephalon and mesocortex in stage 4, the neocortex in sensory association areas of the neocortex and prefrontal neocortex in stage 5, and the premotor and motor areas of the neocortex in stage 6. The higher stages include the pre-stage areas. The staging is based on the LB pathology that is widespread from the medulla oblongata to neocortices and depends on disease severity. Patients with SNCA variants commonly show the higher Braak's staging with DLB (57). Patients with SNCA triplication showed higher expression levels of α-synuclein in the blood and brain tissue (50). Moreover, disease onset correlates with SNCA gene dosage (51). The findings support the hypothesis that the expression levels of α-synuclein direct the clinical severity of PD in patients with SNCA multiplications.
α-Synuclein and Lysosomal Storage Disorders
The abnormal expression and aggregation of α-synuclein are critical factors for PD, PDD, or DLB. α-synuclein-positive inclusions or LBs have been identified in several other disorders, such as multiple system atrophy (MSA) or pure autonomic failure, Alzheimer's disease, Down's syndrome, Hallervorden–Spatz disease, and Gaucher's disease (58–63). The physiological function and accumulation of α-synuclein are only partially understood. α-Synuclein is predominantly localized in presynaptic termini of neurons and regulates neurotransmitter release promoting sensitive factor attachment protein receptor (SNARE)-complex assembly (64, 65). α-Synuclein is subjected to lysosomal degradation by the autophagy–lysosomal systems (66) and the chaperon-mediated autophagy (67). Lysosomes play a central role in maintaining cellular metabolism, degradation, and recycling of amino acids and lipids, eliminating damaged proteins/organelles or proteins with pathogenic properties (66, 68). The lysosomes collaborate with micro-autophagy and macro-autophagy, chaperone-mediated autophagy, and endosomes to conduct their functions (67). Impaired lysosomal function induces the accumulation of aggregated α-synuclein and the formation of LB. Thus, lysosomal dysfunction induces dysfunctional protein and organelle accumulation, leading to lysosomal storage disorders. Several genes, such as SNCA, LRRK2, GBA1, ATP13A2, and VPS35, among the pathogenic ones related to familial PD, are associated with lysosomal storage disorders (68). Genetic screening for 54 genes related to lysosomal storage disorders has identified PD-related genes, such as GBA1, SMPD1, CTSD, SLC17A5, and ASAH1 (69). Most patients with PD (56%), including 40% with familial and 60% with sporadic PD, have at least one putative damaging variant related to lysosomal storage disorders (69).
Formation of LBs and Propagation of α-Synuclein Pathologies
It has been reported that a patient's brain having DLB shows a high accumulation of insoluble α-synuclein (70, 71). The membrane unbound form of α-synuclein is natively unfolded, whereas the elevated protein levels or pathogenic mutations of α-synuclein promote structural conversion to crossed β-sheets, leading to the accumulation of insoluble α-synuclein fibrils (72). Electron microscopy analysis reveals that the introduction of α-synuclein p.A53T mutation accelerates fibril formation with a twisted appearance (73). Other SNCA variants are also likely to facilitate the structural conversion and subsequent LB formation. The degrees of aggregation and fibril propagation by α-synuclein in the central nervous system probably determine the clinical severity of PD, PDD, or DLB obeying Braak's hypothesis rule (56). PD is now recognized as a systemic disease (74). The accumulation of α-synuclein aggregates is observed in the brain and the cardiac nerves, or Auerbach's or Meissner's plexus (75, 76). Concurrently, patients with PD show both motor symptoms and nonmotor symptoms (77). Motor symptoms include gait disturbance, tremor, and rigidity, whereas the nonmotor symptoms include persistent pain, insomnia, constipation, urinary incontinence, and orthostatic hypotension accompanied by syncope or faintness (77). The propagation and expansion of α-synuclein aggregates may be essential factors in determining the clinical severity and symptoms of PD.
Propagation of α-Synuclein and Prion-Like Hypothesis
Animal models of α-synuclein propagation suggest that PD is a prion-like disease. Inoculation of α-synuclein derived from PD brain tissues with LBs replicates progressive nigral degeneration and triggers the pathological conversion of endogenous α-synuclein in mouse and monkey models (78). The inoculation of insoluble α-synuclein from the DLB brains also causes hyperphosphorylated α-synuclein pathology in mice (79). The inoculation of α-synuclein fibrils in mice expressing pathological human p.A53T mutant α-synuclein causes rapid propagation (80). These previous studies support the “prion-like hypothesis,” indicating how pathological α-synuclein derived from PD, DLB, or MSA, as well as fibrils prepared from recombinant protein, induces the cell-to-cell transmission, the spreading of α-synuclein, and amyloid-like formation.
Genetic Evidence, Clinical and Pathological Features, and Molecular Backgrounds of Other Genes Associated With PD
Glucosylceramidase Beta
The GBA1 gene consists of 11 exons, 7.6 kb in length, and is located on chromosome 1q21 (81). GBA1 pathogenic variants cause Gaucher disease (82, 83), a lysosomal storage disorder characterized by the deficiency of the enzyme glucocerebrosidase (GCase) (84). It is categorized into three types: type 1, non-neuropathic Gaucher disease with various types of symptoms and courses; type 2, acute neuropathic Gaucher disease with an infantile-onset and rapidly progressive neurological symptoms; and type 3, chronic neurological symptoms (84). Patients with type 2 and type 3 Gaucher disease commonly show neurological symptoms (84), such as parkinsonism, hydrocephalus, eye movement disorder, epilepsy, dementia, or ataxia. Pathologically, type 1 Gaucher disease presented numerous α-synuclein-positive inclusions similar to LBs in the hippocampus (60). Moreover, GBA1 variants have a higher odds ratio, with approximately five-fold OD between PD vs. controls (85). Patients with GBA1 pathogenic variants likely induce cognitive decline and short survival times, whose symptoms resemble DLBs with no or low levels of Alzheimer's disease (86–88). GBA1 is involved in the glucolipid metabolism and hydrolyzes glucosylceramide to ceramide and glucose and glucosylsphingosine to sphingosine and glucose (84). It has been proposed that lysosomal impairment directly causes α-synuclein aggregation, leading to the pathogenesis of synucleinopathies (66, 89).
LRRK2 Gene
The pathogenic variants in the LRRK2 gene are the most common genetic cause of familial PD (90). The prevalence of LRRK2 p.G2019S is over 30% in the Ashkenazi Jews or Arab Berber. Other populations essentially showed ~0–4% prevalence among sporadic and familial PD (25). LRRK2 is located on 12q12, consists of 51 exons, and encodes a large protein with 2,527-amino acids that belong to the ROCO protein family and include seven domains: armadillo, ankyrin, leucine-rich repeat (LRR), Ras in complex proteins (Roc), C-terminal of Roc (COR), kinase, and WD40 (14). We originally mapped the region around 12p11.2–q13.1 from the Sagamihara family in Japan (7). Two reports concurrently identified the causative gene and mutations from Spanish, German–Canadian, and American families (13, 14). After numerous screening analyses, to date, seven missense mutations (p.N1437H, p.R1441C/G/H, p.Y1699C, p.G2019S, and p.I2020T) are thought to be pathogenic variants from the pathological observations (91).
Patients with LRRK2 variants show middle- or late-onset parkinsonism with an excellent response to levodopa (25, 90). Their clinical course resembles that of sporadic PD. LRRK2 showed broad types of brain pathologies, including LB pathology, tau pathology, TDP-43 pathology, or isolated nigral degeneration (91, 92). LRRK2 p.G2019S, the most prevalent variant, commonly showed LB pathology with broad severities of Braak's stage from 3 to 6 and rarely involves tau pathology (91). On the other hand, tau pathology is found in almost 100% of the p.G2019S carriers (93). A Japanese PD family with LRRK2 p.I2020T also showed a variety of pathological changes, including LB formation and glial cytoplasmic inclusion (94). Moreover, patients with LRRK2 p.R1441G or p.R1441H showed isolated nigral degeneration in the absence of LB pathology (92, 95, 96). Different domain mutations may induce different pathologies.
Neuroimaging of patients with LRRK2 variants shows heterogeneous results. Three of the six patients with p.G2019S show a reduced heart-to-mediastinum ratio of MIBG myocardial scintigraphy (97), whereas patients with p.R1441G/H show no reduction of heart-to-mediastinum ratio (90, 92). The brain MRI commonly show no atrophic changes even over 10 years from disease onset (90, 92).
Rab GTPase, a branch of the Ras superfamily, is a crucial regulator of membrane trafficking (98). A subset of Rab proteins, including Rab3, Rab8, Rab10, and Rab12, have been reported as physiological substrates of LRRK2 (99–101). Although most pathogenic mutants of LRRK2 appear to have enhanced kinase activity toward substrates, mutations in each domain could determine the clinical phenotype and produce differential effects in terms of neuropathology. p.R1441H/G/C localized in the Rab-like ROC domain, which stimulates the LRRK2 kinase, is thought to function as a molecular switch of LRRK2 (102). The ROC domain mutant, p.R1441G, phosphorylates Rab10 more strongly than the kinase domain mutant, p.G2019S, and appears to be a potent activator of these Rab proteins (103). LRRK2 has been reported to be involved in various organelle functions and membrane dynamics in cells (104). These include mitochondria, endo-lysosomes, trans-Golgi network, microtubules, phagocytosis, endocytosis, and exocytosis of synaptic vesicles (105–112). At present, these reports do not provide a unified understanding of the molecular function of LRRK2, and the critical molecular function involved in the pathogenesis is expected to be analyzed in the future.
VPS13C Gene
The VPS13C gene belongs to the VPS13 family, consisting of VPS13A, VPS13B, VPS13C, and VPS13D (113). The size of each gene is considerably huge, including over 70–80 exons and 200–800 kb of genomic DNA sequence (113). The VPS13 gene is conserved from yeasts and is evolutionarily divided into four types in human. Lesage et al. (114) identified a truncated variant in VPS13C from a large Turkish pedigree of PD via linkage mapping and whole-exome sequencing (114). Patients exhibited early- or middle-age onset of PD and severe cognitive decline, with their brain pathology showing abundant expression of LB pathology. The burden analysis proved the statistical significance of variants in VPS13C among the Chinese early-onset PD cohorts (115). Another meta-analysis report proved the statistical significance of VPS13C among the Han Chinese population (116). Conversely, there is no association between VPS13C variants and late-onset PD (117). These findings strongly suggested that the VPS13C variants possibly relate to the early-onset PD and not late-onset.
The VPS13A variants are associated with chorea-acanthocytosis of hyperkinetic involuntary movements and abnormal morphology of erythrocytes (118). VPS13B variants with Cohen disease of developmental delay, microcephaly, retinal dystrophy, and intermittent neutropenia (119). VPS13D variants induce heterogeneous neurodegenerative disorders such as ataxia, developmental delay, spastic paraplegia, or spinocerebellar ataxia (120, 121).
It has been reported that the loss of VPS13C causes oxidative stress-mediated mitochondrial deterioration and upregulated PINK1/PRKN-dependent mitophagy (114). VPA13A and VPS13C are related to lipid transport between the endoplasmic reticulum and other organelles (122). VPA13A is also involved in the actin dynamics (123) and loss of VPA13A impaired autophagy and phagocytosis (124). Mitochondrial dysfunction is commonly observed in the loss-of-function of VPS13 genes and is a major pathogenic cascade to induce dopaminergic cell loss, which may be associated with the mitochondrial quality control pathway regulated by PRKN and PINK1 (125, 126). Loss-of-function of VPS13B induces dysfunction of Golgi-trafficking (127). Loss-of-function of VPS13D induced peroxisome loss and mitochondrial morphological abnormality (128).
The yeast VPS13 gene is thought to be involved in lipid transport by forming contact sites between organelles. Like yeast VPS13, the human VPS13 paralogue genes are thought to be involved in lipid transport, but the details of their molecular functions are still not clearly understood. VPS13A is associated with the endoplasmic reticulum (ER)-mitochondria contacts (122); VPS13B is mainly localized in the Golgi complex (127, 129); VPS13C is localized at ER-late endosome/lysosome contacts (122); and VPS13D is localized at ER-mitochondria and ER-peroxisome contact sites (130). They may be involved in lipid transport at the different sites, and these differences may be responsible for distinct pathophysiologies.
The neuroimaging reports of patients with VPS13C variants are unavailable.
GCH1 Gene
The GCH1 gene was initially identified in a patient with dopa-responsive dystonia (DRD), distinctively known as Segawa's disease or DYT5a (131). The patients show unique symptoms, such as juvenile or young-age onset, dystonia initially in the feet, and excellent response to a low levodopa dosage (132). It was also reported that other symptoms include diurnal fluctuations, cramps, dystonic tremors, and sleep benefits (133). The characteristic symptoms resemble those of patients with PRKN or PINK1 variants (6, 9). Patients with PRKN or PINK1 also manifested the juvenile- (under 20 years of age at onset) or young-onset parkinsonism (under 40 years) with excellent response to even the low doses of levodopa, which leads to the brain pathology in the absence of LBs (6, 9).
A large population study showed a high frequency of GCH1 variants in patients with PD compared to controls (134). The variants in GCH1 are related to an increased risk of PD. Some GWAS also showed the association between the GCH1 locus and PD (16, 17). In a large population study from China, GCH1 deletions or non-coding region variants were associated with early-onset or familial PD (135). Although the GCH1 variants are rare, they have been a proven risk factor for the onset of DRD and PD. DRD and PD may involve a common pathway causing abnormal dopamine metabolism (136).
Continuous monitoring for 32 years revealed that many patients showed no alteration or mild progression of dystonia (133), with a mild prognosis. The pedigrees primarily show autosomal dominant inheritance and female predominance (132). Some pedigrees harbor the complex appearance of patients with DRD and PD (133, 137). Adult-onset patients with GCH1 variants show upper-limb tremors or non-tremulous parkinsonian syndrome (133). The brain pathology mostly shows the absence of LB pathology, and none to minor changes of morphological abnormalities, but only a few cases were reported (138, 139). In brief, patients with DRD and GCH1 variants show distinctive symptoms compared to PD. The patients with PD and GCH1 variants may involve neuronal loss in the striatum or the substantia nigra due to the reduction of dopamine transporter expression, although there are no brain pathology reports of PD phenotype with GCH1 variants. It has been indicated in reports that “age” may be a factor in distinguishing DRD from PD. Patients with young-age onset likely belong to the DRD phenotype, whereas those with older-age onset likely belong to the PD phenotype (137). Both the disorders would be improved by oral administration of levodopa.
Studies on GCH1 reported that half of the patients with PD show a reduction in heart-to-mediastinum ratio (137). Patients with DRD commonly showed normal values of dopamine transporter uptake in 123I-FP-CIT SPECT (140). However, patients with PD phenotype showed a reduction in dopamine transporter expression (134).
The enzymatic deficiency of dopamine production is the main pathogenesis of DRD (141). GCH1-encoded GTP cyclohydrolase 1 functions upstream of the dopamine synthesis (138) (Figure 1). The deficiency of GTP cyclohydrolase 1 reduces the production of tetrahydrobiopterin, an essential co-factor in dopamine production by tyrosine hydroxylase (142). The reduction in tyrosine hydroxylase levels caused by GCH1 mutations also contributes to the symptoms related to DRD (141). Thus, deleterious variants of GCH1 are likely responsible for the decrease in dopamine production more directly than other genes like SNCA, LRRK2, or MAPT.
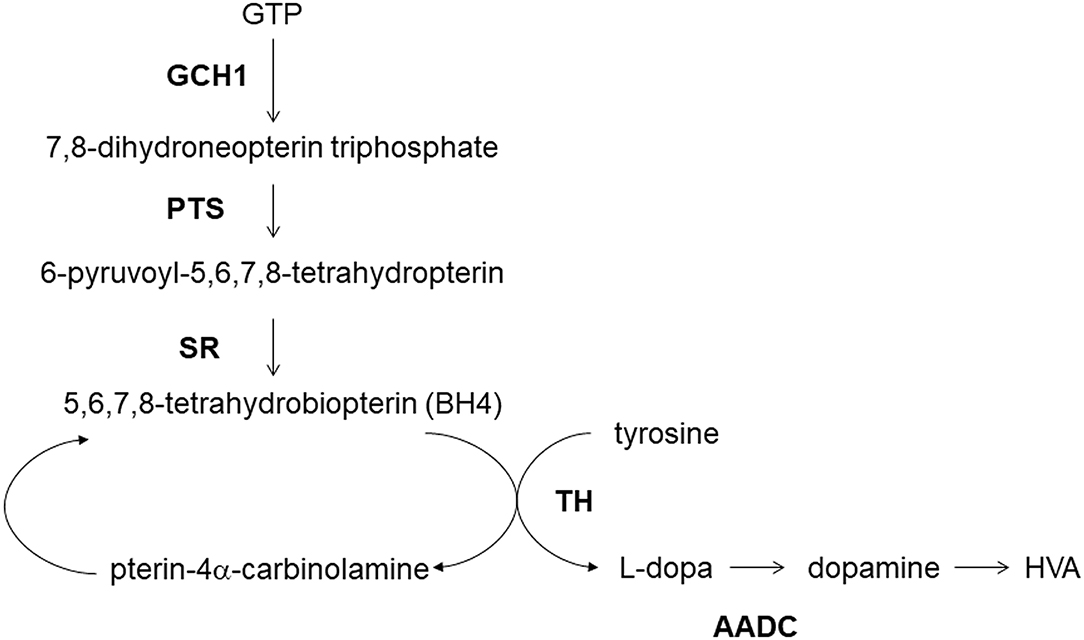
Figure 1. Dopamine metabolism and GCH1. GCH1, GTP cyclohydrolase 1; PTS, 6-pyruvoyltetrahydropterin synthase; SR, sepiapterin reductase; TH, tyrosine hydroxylase; AADC, aromatic L-amino acid decarboxylase; HVA, homovanillic acid.
MAPT Gene
The MAPT gene, which encodes tau protein, is not a PD causative gene and is linked to frontotemporal dementia. However, MAPT is a gene that should not be ignored as a basis for PD pathology. Patients with MAPT, which was detected by GWAS, are sometimes indistinguishable from patients with PD in terms of clinical symptoms. Moreover, tauopathy is frequently observed in LRRK2 pathology, and MAPT variants were reported to correlate with the severity of PD (143, 144). Historically, the region of chromosome 17q21–22 has been identified as a locus related to familial frontotemporal dementia and parkinsonism by the linkage analysis (145–148). In 1998, three missense mutations and three mutations in the 5′-splice site of exon 10 in MAPT were identified in large Dutch kindred with hereditary frontotemporal dementia (149). Tau is fundamentally associated with multiple neurodegenerative disorders, such as Alzheimer's disease, progressive supranuclear palsy, corticobasal degeneration, frontotemporal dementia, and prion disease (150).
Patients with MAPT mutations showed middle-aged onset of progressive parkinsonism and cognitive decline with a high penetrance ratio (151–153). Patients likely involve psychiatric symptoms and rigid–akinesic parkinsonism (154) and show a partial response to levodopa at early-onset PD (153, 155).
Tau maintains the stability of microtubules in neurons and promotes axonal outgrowth (156). The brain pathology of patients with MAPT mutations shows hyperphosphorylated tau inclusions, such as neurofibrillary tangles.
It has been highlighted that patients with MAPT mutations or tauopathy-related disorders show no abnormalities of MIBG myocardial scintigraphy. Patients with MAPT mutations commonly show atrophic changes in the frontotemporal lobes in the brain MRI within a few years from disease onset. 123I-FP-CIT SPECT showed a severe reduction in dopamine transporter from an early stage (153, 157). Thus, patients with MAPT mutations may be diagnosed with PD and treated with levodopa at an early clinical stage. Our research has identified patients with MAPT N279K or p.K298_H299insQ from patients with middle-aged onset of parkinsonism or those clinically diagnosed with familial PD (153, 158). Tau imaging SPECT revealed a high tau accumulation from the brain stem to the basal ganglia (153). The distribution of tau pathology may relate to the onset of parkinsonism and disease severity. In vivo, tau imaging analysis will expand our understanding of tau-related disorders (159).
Genetic Interactions Among Pathogenic Genes
The brain pathology of patients with SNCA mutations, GBA1 variants, LRRK2 p.G2019S, or VPS13C variants shows LB formation. Excessive α-synuclein or α-synuclein aggregation is suggested to impair cellular vesicular transport, by which the transport of newly synthesized lysosomal enzyme GCase, encoded by GBA1, from the ER to the lysosomes may be inhibited (89). On the other hand, the perturbation of transport of GCase, involved in the metabolism of glycosphingolipids, could also lead to a reduction in lysosomal function and inhibit the lysosomal degradation of α-synuclein (89). This vicious cycle of GBA1 variants has been proposed to be a risk factor for theLB formation. The GBA1 pathogenic variants reportedly accumulate glucosylceramide and glucosylsphingosine, probably in lysosomes (160). These lipids could promote the aggregation of α-synuclein (161, 162). Nevertheless, the aforementioned considerations are speculative and await further experimental validation.
The LRRK2 was reported to inhibit the GCase activity via Rab10 phosphorylation in dopaminergic neurons differentiated from iPS cells harboring LRRK2 pathogenic mutations (162). Although the details of the inhibitory mechanism of GCase by Rab10 remain unknown, the reduction of the GCase activity by LRRK2 may be indirectly involved in α-synuclein accumulation and aggregation. As mentioned above, the relationship between LRRK2 and α-synuclein aggregation is complex because LRRK2 causes various pathologies, such as LB pathology, tau pathology, and TDP-43 pathology. According to a recent systematic pathological analysis, α-synuclein pathology is observed in 63.6% of LRRK2 mutation carriers (144). On the other hand, tau pathology is found in ~100% of carriers. Most LRRK2 mutation carriers show comorbid AD pathology with amyloid-β. These observations suggest that the pathology caused by LRRK2 mutations is fundamental to neurodegenerative diseases. An interesting observation is the high frequency of AD-type phosphorylated tau accumulation (144). LRRK2 surrounds microtubules and inhibits neuronal axonal transport (110, 112). Microtubule modification by LRRK2 may affect the binding of tau to microtubules or tau phosphorylation after dissociation (163–165).
The molecular relationship between VPS13C and α-synuclein has not been elucidated so far. Because VPS13C is also localized to the lysosomes, its variant may impair lysosomal function, leading to the consequent accumulation of α-synuclein (166). Alternatively, altered lipid transport and metabolism caused by mutations in VPS13C may lead to the aggregation of α-synuclein. These possibilities should be explored in the future. Since GCH1 is involved in dopamine synthesis, it is different from the pathologies caused by the genes mentioned above. However, a report shows decreased BH4 contents in the cerebrospinal fluids of patients with LRRK2 p.N1437H and p.G2019S, and patients with sporadic PD (136). This may result from dopaminergic neurodegeneration, but it may also be possible that pathogenic LRRK2 impairs the function of GCH1.
Perspectives
The GWAS has bridged the gap between molecular-based studies of familial PD and sporadic PD. The multiple genes discovered from the familial PD studies induce dopaminergic neuronal loss and the formation of LB pathology or nigral degeneration (Figure 2). The pathogenic genes yield symptoms related to parkinsonism. Moreover, “aging” is the most critical factor for the deterioration of mitochondrial maintenance or disturbance of intracellular transports during neuronal activity. However, there have been numerous unsolved questions regarding the molecular mechanism of PD pathogenesis, such as how multiple genes interact with each other to induce the dopaminergic neuronal loss, how they yield a single phenotype, what is the precise molecular model of sporadic PD, or how the genes cause LB pathology.
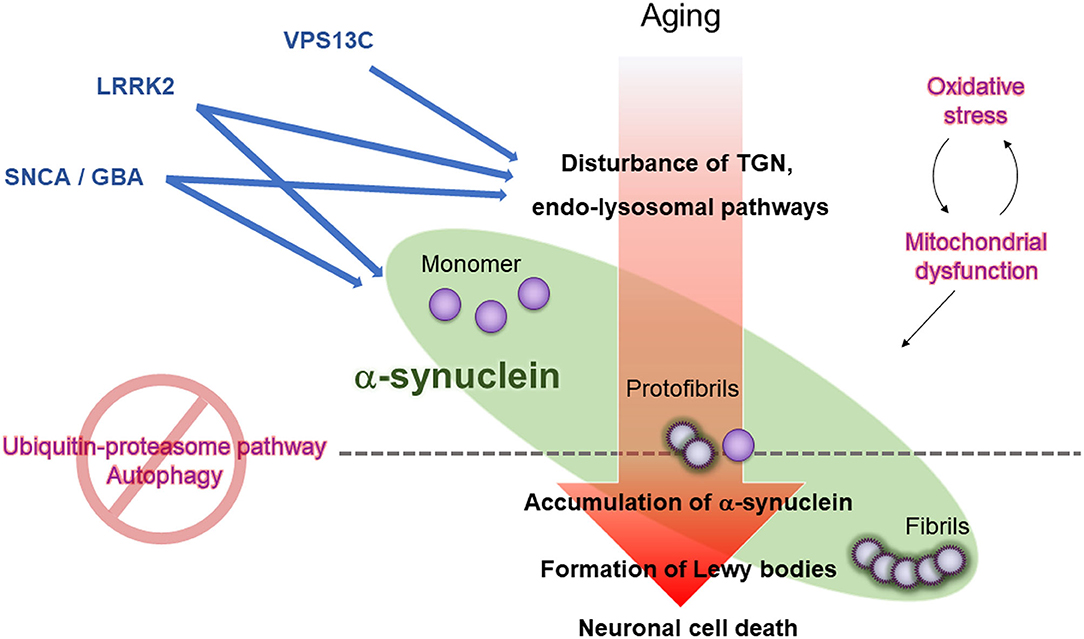
Figure 2. Working hypothesis for LB formation and neuronal cell death in PD. Aging, oxidative stress, and mitochondrial dysfunction lead to dysregulation of the trans-Golgi network (TGN) and endo-lysosomes in neurons, ultimately impairing the removal of the precursor of pathogenic α-synuclein (protofibrils) through ubiquitin-proteasome pathway and autophagy, promoting LB formation, and propagating pathogenic α-synuclein to neighboring neurons. Mutations in PD causative genes and risk-related genes (VPS13C, LRRK2, SNCA, and GBA1) accelerate oxidative stress, mitochondrial dysfunction, and dysregulation of the TGN and endo-lysosomes.
The next generation of GWAS research will lead to analyzing the interaction among multiple PD risk genes. As a leading example, a GWAS for the LRRK2 modifier genes has found that the WD40 protein CORO1C or DNM3 may modulate the penetrance or age-of-onset of LRRK2 mutations (167, 168). New advances in GWASs may come from other fields of research. The loss-of-function of a preferred promoter has been reported to release its partner enhancer, which loops to a neighboring alternative promoter and activates it (169). This target switching process has been termed “enhancer release and retargeting” (169). This study shows that SNPs on the promoter of PARK16 alter the balance of expression intensity of the genes, NUCKS1 and RAB7L1, in PARK16 (169). This phenomenon may explain the unresolved questions about PARK16-mediated disease susceptibility. Thus, new concepts in genomic research can lead to novel interpretations of the data from GWAS for PD that remain mainly unexplored. On the other hand, it is challenging to identify recessively inherited PD genes such as PRKN and PINK1, which GWAS did not detect, and it is desirable to develop new methods.
A more thorough identification of risk-associated genes that cause PD will provide a clearer picture of the molecular pathogenesis of PD, yielding better and more sophisticated molecular-targeted therapies. These would include oligonucleotide therapeutics (170), antibody therapies against α-synuclein and tau (171, 172), or replacement therapies of induced pluripotent stem cells (173). Hence, a growing body of literature hints at increasing expectations for future GWAS research to help overcome PD.
Author Contributions
KN and YI: designed the study, wrote the first draft of the manuscript, and revised the manuscript. HY, YL, MF, and NH: revised the manuscript. All authors contributed to the article and approved the submitted version.
Funding
KN was supported by Japan Society for the Promotion of Science (JSPS KAKENHI) Grant Number 20K07893. The study was partly supported by a research grant from Biogen Japan Ltd (KN). The funders were not involved in the study design, collection, analysis, interpretation of data, the writing of this article or the decision to submit it for publication.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher's Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. de Lau LM, Breteler MM. Epidemiology of Parkinson's disease. Lancet Neurol. (2006) 5:525–35. doi: 10.1016/S1474-4422(06)70471-9
2. Pringsheim T, Jette N, Frolkis A, Steeves TD. The prevalence of Parkinson's disease: a systematic review and meta-analysis. Mov Disord. (2014) 29:1583–90. doi: 10.1002/mds.25945
3. Braak H, Del Tredici K. Invited Article: Nervous system pathology in sporadic Parkinson disease. Neurology. (2008) 70:1916–25. doi: 10.1212/01.wnl.0000312279.49272.9f
4. Spillantini MG, Schmidt ML, Lee VM, Trojanowski JQ, Jakes R, Goedert M. Alpha-synuclein in Lewy bodies. Nature. (1997) 388:839–40. doi: 10.1038/42166
5. Polymeropoulos MH, Higgins JJ, Golbe LI, Johnson WG, Ide SE, Di Iorio G, et al. Mapping of a gene for Parkinson's disease to chromosome 4q21-q23. Science. (1996) 274:1197–9. doi: 10.1126/science.274.5290.1197
6. Kitada T, Asakawa S, Hattori N, Matsumine H, Yamamura Y, Minoshima S, et al. Mutations in the parkin gene cause autosomal recessive juvenile parkinsonism. Nature. (1998) 392:605–8. doi: 10.1038/33416
7. Funayama M, Hasegawa K, Kowa H, Saito M, Tsuji S, Obata F. A new locus for Parkinson's disease (PARK8) maps to chromosome 12p11.2-q13.1. Ann Neurol. (2002) 51:296–301. doi: 10.1002/ana.10113
8. Singleton AB, Farrer M, Johnson J, Singleton A, Hague S, Kachergus J, et al. alpha-Synuclein locus triplication causes Parkinson's disease. Science. (2003) 302:841. doi: 10.1126/science.1090278
9. Valente EM, Abou-Sleiman PM, Caputo V, Muqit MM, Harvey K, Gispert S, et al. Hereditary early-onset Parkinson's disease caused by mutations in PINK1. Science. (2004) 304:1158–60. doi: 10.1126/science.1096284
10. Deng H, Wang P, Jankovic J. The genetics of Parkinson disease. Ageing Res Rev. (2018) 42:72–85. doi: 10.1016/j.arr.2017.12.007
11. Wittke C, Petkovic S, Dobricic V, Schaake S, Group MD-ePS, Respondek G, et al. Genotype-phenotype relations for the atypical Parkinsonism genes: MDSGene systematic review. Mov Disord. (2021) 36:1499–510. doi: 10.1002/mds.28517
12. Polymeropoulos MH, Lavedan C, Leroy E, Ide SE, Dehejia A, Dutra A, et al. Mutation in the alpha-synuclein gene identified in families with Parkinson's disease. Science. (1997) 276:2045–7. doi: 10.1126/science.276.5321.2045
13. Paisan-Ruiz C, Jain S, Evans EW, Gilks WP, Simon J, van der Brug M, et al. Cloning of the gene containing mutations that cause PARK8-linked Parkinson's disease. Neuron. (2004) 44:595–600. doi: 10.1016/j.neuron.2004.10.023
14. Zimprich A, Biskup S, Leitner P, Lichtner P, Farrer M, Lincoln S, et al. Mutations in LRRK2 cause autosomal-dominant parkinsonism with pleomorphic pathology. Neuron. (2004) 44:601–7. doi: 10.1016/j.neuron.2004.11.005
15. Satake W, Nakabayashi Y, Mizuta I, Hirota Y, Ito C, Kubo M, et al. Genome-wide association study identifies common variants at four loci as genetic risk factors for Parkinson's disease. Nat Genet. (2009) 41:1303–7. doi: 10.1038/ng.485
16. Nalls MA, Pankratz N, Lill CM, Do CB, Hernandez DG, Saad M, et al. Large-scale meta-analysis of genome-wide association data identifies six new risk loci for Parkinson's disease. Nat Genet. (2014) 46:989–93.
17. Chang D, Nalls MA, Hallgrímsdóttir IB, Hunkapiller J, van der Brug M, Cai F, et al. A meta-analysis of genome-wide association studies identifies 17 new Parkinson's disease risk loci. Nat Genet. (2017) 49:1511–6. doi: 10.1038/ng.3955
18. Foo JN, Chew EGY, Chung SJ, Peng R, Blauwendraat C, Nalls MA, et al. Identification of Risk Loci for Parkinson Disease in Asians and Comparison of Risk Between Asians and Europeans: A Genome-Wide Association Study. JAMA Neurol. (2020) 77:746–54. doi: 10.1001/jamaneurol.2020.0428
19. Krohn L, Grenn FP, Makarious MB, Kim JJ, Bandres-Ciga S, Roosen DA, et al. Comprehensive assessment of PINK1 variants in Parkinson's disease. Neurobiol Aging. (2020) 91:168 e161–8 e165. doi: 10.1016/j.neurobiolaging.2020.03.003
20. Lubbe SJ, Bustos BI, Hu J, Krainc D, Joseph T, Hehir J, et al. Assessing the relationship between monoallelic PRKN mutations and Parkinson's risk. Hum Mol Genet. (2021) 30:78–86. doi: 10.1093/hmg/ddaa273
21. Yu E, Rudakou U, Krohn L, Mufti K, Ruskey JA, Asayesh F, et al. Analysis of Heterozygous PRKN Variants and Copy-Number Variations in Parkinson's Disease. Mov Disord. (2021) 36:178–87. doi: 10.1002/mds.28299
22. Klein C, Lohmann-Hedrich K, Rogaeva E, Schlossmacher MG, Lang AE. Deciphering the role of heterozygous mutations in genes associated with parkinsonism. Lancet Neurol. (2007) 6:652–62. doi: 10.1016/S1474-4422(07)70174-6
23. Blauwendraat C, Heilbron K, Vallerga CL, Bandres-Ciga S, von Coelln R, Pihlstrøm L, et al. Parkinson's disease age at onset genome-wide association study: Defining heritability, genetic loci, and α-synuclein mechanisms. Mov Disord. (2019) 34:866–75. doi: 10.1002/mds.27659
24. Tysnes OB, Storstein A. Epidemiology of Parkinson's disease. J Neural Transm (Vienna). (2017) 124:901–5. doi: 10.1007/s00702-017-1686-y
25. Healy DG, Falchi M, O'Sullivan SS, Bonifati V, Durr A, Bressman S, et al. Phenotype, genotype, and worldwide genetic penetrance of LRRK2-associated Parkinson's disease: a case-control study. Lancet Neurol. (2008) 7:583–90. doi: 10.1016/S1474-4422(08)70117-0
26. Nishioka K, Hayashi S, Farrer MJ, Singleton AB, Yoshino H, Imai H, et al. Clinical heterogeneity of alpha-synuclein gene duplication in Parkinson's disease. Ann Neurol. (2006) 59:298–309. doi: 10.1002/ana.20753
27. Funayama M, Tomiyama H, Wu RM, Ogaki K, Yoshino H, Mizuno Y, et al. Rapid screening of ATP13A2 variant with high-resolution melting analysis. Mov Disord. (2010) 25:2434–7. doi: 10.1002/mds.23106
28. Nishioka K, Funayama M, Vilarino-Guell C, Ogaki K, Li Y, Sasaki R, et al. EIF4G1 gene mutations are not a common cause of Parkinson's disease in the Japanese population. Parkinsonism Relat Disord. (2014) 20:659–61. doi: 10.1016/j.parkreldis.2014.03.004
29. Funayama M, Ohe K, Amo T, Furuya N, Yamaguchi J, Saiki S, et al. CHCHD2 mutations in autosomal dominant late-onset Parkinson's disease: a genome-wide linkage and sequencing study. Lancet Neurol. (2015) 14:274–82. doi: 10.1016/S1474-4422(14)70266-2
30. Conedera S, Apaydin H, Li Y, Yoshino H, Ikeda A, Matsushima T, et al. (2016). FBXO7 mutations in Parkinson's disease and multiple system atrophy. Neurobiol Aging.40, 192 e191–2 e195. doi: 10.1016/j.neurobiolaging.2016.01.003
31. Conedera SA, Li Y, Funayama M, Yoshino H, Nishioka K, Hattori N. Genetic analysis of TMEM230 in Japanese patients with familial Parkinson's disease. Parkinsonism Relat Disord. (2018) 48:107–8. doi: 10.1016/j.parkreldis.2017.12.020
32. Daida K, Nishioka K, Li Y, Yoshino H, Shimada T, Dougu N, et al. (2021). PLA2G6 variants associated with the number of affected alleles in Parkinson's disease in Japan. Neurobiol Aging. 97:147 e141–7 e149. doi: 10.1016/j.neurobiolaging.2020.07.004
33. Hayashida A, Li Y, Yoshino H, Daida K, Ikeda A, Ogaki K, et al. (2021). The identified clinical features of Parkinson's disease in homo-, heterozygous and digenic variants of PINK1. Neurobiol Aging. 97: 146 e141–6 e113. doi: 10.1016/j.neurobiolaging.2020.06.017
34. Ishiguro M, Li Y, Yoshino H, Daida K, Ishiguro Y, Oyama G, et al. Clinical manifestations of Parkinson's disease harboring VPS35 retromer complex component p.D620N with long-term follow-up. Parkinsonism Relat Disord. (2021) 84:139–43. doi: 10.1016/j.parkreldis.2021.02.014
35. Cao LX, Jiang Y, Piao YS, Huang Y. Rapid motor progression of Parkinson's disease associates with clinical and genetic variants. Front Biosci (Landmark Ed). (2021) 26:1503–12. doi: 10.52586/5044
36. Kruger R, Kuhn W, Muller T, Woitalla D, Graeber M, Kosel S, et al. Ala30Pro mutation in the gene encoding alpha-synuclein in Parkinson's disease. Nat Genet. (1998) 18:106–8. doi: 10.1038/ng0298-106
37. Zarranz JJ, Alegre J, Gomez-Esteban JC, Lezcano E, Ros R, Ampuero I, et al. The new mutation, E46K, of alpha-synuclein causes Parkinson and Lewy body dementia. Ann Neurol. (2004) 55:164–73. doi: 10.1002/ana.10795
38. Lesage S, Anheim M, Letournel F, Bousset L, Honore A, Rozas N, et al. G51D alpha-synuclein mutation causes a novel parkinsonian-pyramidal syndrome. Ann Neurol. (2013) 73:459–71. doi: 10.1002/ana.23894
39. Proukakis C, Dudzik CG, Brier T, MacKay DS, Cooper JM, Millhauser GL, et al. A novel alpha-synuclein missense mutation in Parkinson disease. Neurology. (2013) 80:1062–4. doi: 10.1212/WNL.0b013e31828727ba
40. Kiely AP, Ling H, Asi YT, Kara E, Proukakis C, Schapira AH, et al. Distinct clinical and neuropathological features of G51D SNCA mutation cases compared with SNCA duplication and H50Q mutation. Mol Neurodegener. (2015) 10:41. doi: 10.1186/s13024-015-0038-3
41. Martikainen MH, Paivarinta M, Hietala M, Kaasinen V. Clinical and imaging findings in Parkinson disease associated with the A53E SNCA mutation. Neurol Genet. (2015) 1:e27. doi: 10.1212/NXG.0000000000000027
42. Kapasi A, Brosch JR, Nudelman KN, Agrawal S, Foroud TM, Schneider JA. A novel SNCA E83Q mutation in a case of dementia with Lewy bodies and atypical frontotemporal lobar degeneration. Neuropathology. (2020) 40:620–6. doi: 10.1111/neup.12687
43. Liu H, Koros C, Strohäker T, Schulte C, Bozi M, Varvaresos S, et al. A Novel SNCA A30G Mutation Causes Familial Parkinson's Disease. Mov Disord. (2021) 36:1624–33. doi: 10.1002/mds.28534
44. Chartier-Harlin MC, Kachergus J, Roumier C, Mouroux V, Douay X, Lincoln S, et al. Alpha-synuclein locus duplication as a cause of familial Parkinson's disease. Lancet. (2004) 364:1167–9. doi: 10.1016/S0140-6736(04)17103-1
45. Ibanez P, Bonnet AM, Debarges B, Lohmann E, Tison F, Pollak P, et al. Causal relation between alpha-synuclein gene duplication and familial Parkinson's disease. Lancet. (2004) 364:1169–71. doi: 10.1016/S0140-6736(04)17104-3
46. Tambasco N, Nigro P, Romoli M, Prontera P, Simoni S, Calabresi P. A53T in a parkinsonian family: a clinical update of the SNCA phenotypes. J Neural Transm (Vienna). (2016) 123:1301–7. doi: 10.1007/s00702-016-1578-6
47. Obi T, Nishioka K, Ross OA, Terada T, Yamazaki K, Sugiura A, et al. Clinicopathologic study of a SNCA gene duplication patient with Parkinson disease and dementia. Neurology. (2008) 70:238–41. doi: 10.1212/01.wnl.0000299387.59159.db
48. Nishioka K, Ross OA, Ishii K, Kachergus JM, Ishiwata K, Kitagawa M, et al. Expanding the clinical phenotype of SNCA duplication carriers. Mov Disord. (2009) 24:1811–9. doi: 10.1002/mds.22682
49. Nishioka K, Ross OA, Hattori N. SNCA Gene Multiplication: A Model Mechanism of Parkinson Disease. In: Gene Duplication. InTech. (2011). doi: 10.5772/24726
50. Miller DW, Hague SM, Clarimon J, Baptista M, Gwinn-Hardy K, Cookson MR, et al. Alpha-synuclein in blood and brain from familial Parkinson disease with SNCA locus triplication. Neurology. (2004) 62:1835–8. doi: 10.1212/01.WNL.0000127517.33208.F4
51. Book A, Guella I, Candido T, Brice A, Hattori N, Jeon B, et al. A Meta-Analysis of α-Synuclein Multiplication in Familial Parkinsonism. Front Neurol. (2018) 9:1021. doi: 10.3389/fneur.2018.01021
52. Itokawa K, Sekine T, Funayama M, Tomiyama H, Fukui M, Yamamoto T, et al. A case of α-synuclein gene duplication presenting with head-shaking movements. Mov Disord. (2013) 28:384–7. doi: 10.1002/mds.25243
53. Yoshino H, Hirano M, Stoessl AJ, Imamichi Y, Ikeda A, Li Y, et al. Homozygous alpha-synuclein p.A53V in familial Parkinson's disease. Neurobiol Aging. (2017) 57:248.e247–8.e212. doi: 10.1016/j.neurobiolaging.2017.05.022
54. Nishioka K, Hashizume Y, Takanashi M, Daida K, Li Y, Yoshino H, et al. Pathological findings in a patient with alpha-synuclein p.A53T and familial Parkinson's disease. Parkinsonism Relat Disord. (2020) 81, 183-187. doi: 10.1016/j.parkreldis.2020.11.001
55. Farrer M, Kachergus J, Forno L, Lincoln S, Wang DS, Hulihan M, et al. Comparison of kindreds with parkinsonism and alpha-synuclein genomic multiplications. Ann Neurol. (2004) 55:174–9. doi: 10.1002/ana.10846
56. Braak H, Del Tredici K, Rüb U, de Vos RA, Jansen Steur EN, Braak E. Staging of brain pathology related to sporadic Parkinson's disease. Neurobiol Aging. (2003) 24:197–211. doi: 10.1016/S0197-4580(02)00065-9
57. McKeith IG, Dickson DW, Lowe J, Emre M, O'Brien JT, Feldman H, et al. Diagnosis and management of dementia with Lewy bodies: third report of the DLB Consortium. Neurology. (2005) 65:1863–72. doi: 10.1212/WNL.65.12.1992-a
58. Lippa CF, Schmidt ML, Lee VM, Trojanowski JQ. Antibodies to alpha-synuclein detect Lewy bodies in many Down's syndrome brains with Alzheimer's disease. Ann Neurol. (1999) 45:353–7. doi: 10.1002/1531-8249(199903)45:3<:353::AID-ANA11>3.0.CO;2-4
59. Galvin JE, Giasson B, Hurtig HI, Lee VM, Trojanowski JQ. Neurodegeneration with brain iron accumulation, type 1 is characterized by alpha-, beta-, and gamma-synuclein neuropathology. Am J Pathol. (2000) 157:361–8. doi: 10.1016/S0002-9440(10)64548-8
60. Wong K, Sidransky E, Verma A, Mixon T, Sandberg GD, Wakefield LK, et al. Neuropathology provides clues to the pathophysiology of Gaucher disease. Mol Genet Metab. (2004) 82:192–207. doi: 10.1016/j.ymgme.2004.04.011
61. Mikolaenko I, Pletnikova O, Kawas CH, O'Brien R, Resnick SM, Crain B, et al. Alpha-synuclein lesions in normal aging, Parkinson disease, and Alzheimer disease: evidence from the Baltimore Longitudinal Study of Aging (BLSA). J Neuropathol Exp Neurol. (2005) 64:156–62. doi: 10.1093/jnen/64.2.156
62. Trojanowski JQ, Revesz T. Proposed neuropathological criteria for the post mortem diagnosis of multiple system atrophy. Neuropathol Appl Neurobiol. (2007) 33:615–20. doi: 10.1111/j.1365-2990.2007.00907.x
63. Donadio V, Incensi A, Cortelli P, Giannoccaro MP, Jaber MA, Baruzzi A, et al. Skin sympathetic fiber α-synuclein deposits: a potential biomarker for pure autonomic failure. Neurology. (2013) 80:725–32. doi: 10.1212/WNL.0b013e3182825127
64. Burre J, Sharma M, Tsetsenis T, Buchman V, Etherton MR, Sudhof TC. Alpha-synuclein promotes SNARE-complex assembly in vivo and in vitro. Science. (2010) 329:1663–7. doi: 10.1126/science.1195227
65. Burre J, Sharma M, Sudhof TC. alpha-Synuclein assembles into higher-order multimers upon membrane binding to promote SNARE complex formation. Proc Natl Acad Sci USA. (2014) 111:E4274–4283. doi: 10.1073/pnas.1416598111
66. Moors T, Paciotti S, Chiasserini D, Calabresi P, Parnetti L, Beccari T, et al. Lysosomal Dysfunction and α-Synuclein Aggregation in Parkinson's Disease: Diagnostic Links. Mov Disord. (2016) 31:791–801. doi: 10.1002/mds.26562
67. Cuervo AM, Stefanis L, Fredenburg R, Lansbury PT, Sulzer D. Impaired degradation of mutant alpha-synuclein by chaperone-mediated autophagy. Science. (2004) 305:1292–5. doi: 10.1126/science.1101738
68. Gan-Or Z, Dion PA, Rouleau GA. Genetic perspective on the role of the autophagy-lysosome pathway in Parkinson disease. Autophagy. (2015) 11:1443–57. doi: 10.1080/15548627.2015.1067364
69. Robak LA, Jansen IE, van Rooij J, Uitterlinden AG, Kraaij R, Jankovic J, et al. Excessive burden of lysosomal storage disorder gene variants in Parkinson's disease. Brain. (2017) 140:3191–203.
70. Campbell BC, Li QX, Culvenor JG, Jäkälä P, Cappai R, Beyreuther K, et al. Accumulation of insoluble alpha-synuclein in dementia with Lewy bodies. Neurobiol Dis. (2000) 7:192–200. doi: 10.1006/nbdi.2000.0286
71. Klucken J, Ingelsson M, Shin Y, Irizarry MC, Hedley-Whyte ET, Frosch M, et al. Clinical and biochemical correlates of insoluble alpha-synuclein in dementia with Lewy bodies. Acta Neuropathol. (2006) 111:101–8. doi: 10.1007/s00401-005-0027-7
72. Mori A, Imai Y, Hattori N. Lipids: Key Players That Modulate alpha-Synuclein Toxicity and Neurodegeneration in Parkinson's Disease. Int J Mol Sci. (2020) 21:3301. doi: 10.3390/ijms21093301
73. Conway KA, Harper JD, Lansbury PT. Accelerated in vitro fibril formation by a mutant alpha-synuclein linked to early-onset Parkinson disease. Nat Med. (1998) 4:1318–20. doi: 10.1038/3311
74. Coon EA, Cutsforth-Gregory JK, Benarroch EE. Neuropathology of autonomic dysfunction in synucleinopathies. Mov Disord. (2018) 33:349–58. doi: 10.1002/mds.27186
75. Wakabayashi K, Takahashi H, Takeda S, Ohama E, Ikuta F. Parkinson's disease: the presence of Lewy bodies in Auerbach's and Meissner's plexuses. Acta Neuropathol. (1988) 76:217–21. doi: 10.1007/BF00687767
76. Mitsui J, Saito Y, Momose T, Shimizu J, Arai N, Shibahara J, et al. Pathology of the sympathetic nervous system corresponding to the decreased cardiac uptake in 123I-metaiodobenzylguanidine (MIBG) scintigraphy in a patient with Parkinson disease. J Neurol Sci. (2006) 243:101–4. doi: 10.1016/j.jns.2005.11.034
77. Armstrong MJ, Okun MS. Diagnosis and treatment of Parkinson disease: a review. JAMA. (2020) 323:548–60. doi: 10.1001/jama.2019.22360
78. Recasens A, Dehay B, Bové J, Carballo-Carbajal I, Dovero S, Pérez-Villalba A, et al. Lewy body extracts from Parkinson disease brains trigger α-synuclein pathology and neurodegeneration in mice and monkeys. Ann Neurol. (2014) 75:351–62. doi: 10.1002/ana.24066
79. Masuda-Suzukake M, Nonaka T, Hosokawa M, Oikawa T, Arai T, Akiyama H, et al. Prion-like spreading of pathological alpha-synuclein in brain. Brain. (2013) 136:1128–38. doi: 10.1093/brain/awt037
80. Luk KC, Kehm VM, Zhang B, O'Brien P, Trojanowski JQ, Lee VM. Intracerebral inoculation of pathological α-synuclein initiates a rapidly progressive neurodegenerative α-synucleinopathy in mice. J Exp Med. (2012) 209:975–86. doi: 10.1084/jem.20112457
81. Horowitz M, Wilder S, Horowitz Z, Reiner O, Gelbart T, Beutler E. The human glucocerebrosidase gene and pseudogene: structure and evolution. Genomics. (1989) 4:87–96. doi: 10.1016/0888-7543(89)90319-4
82. Tsuji S, Choudary PV, Martin BM, Stubblefield BK, Mayor JA, Barranger JA, et al. A mutation in the human glucocerebrosidase gene in neuronopathic Gaucher's disease. N Engl J Med. (1987) 316:570–5. doi: 10.1056/NEJM198703053161002
83. Shachar T, Lo Bianco C, Recchia A, Wiessner C, Raas-Rothschild A, Futerman AH. Lysosomal storage disorders and Parkinson's disease: Gaucher disease and beyond. Mov Disord. (2011) 26:1593–604. doi: 10.1002/mds.23774
84. Sidransky E. Gaucher disease: complexity in a “simple” disorder. Mol Genet Metab. (2004) 83:6–15. doi: 10.1016/j.ymgme.2004.08.015
85. Sidransky E, Nalls MA, Aasly JO, Aharon-Peretz J, Annesi G, Barbosa ER, et al. Multicenter analysis of glucocerebrosidase mutations in Parkinson's disease. N Engl J Med. (2009) 361:1651–61. doi: 10.1056/NEJMoa0901281
86. Tsuang D, Leverenz JB, Lopez OL, Hamilton RL, Bennett DA, Schneider JA, et al. GBA mutations increase risk for Lewy body disease with and without Alzheimer disease pathology. Neurology. (2012) 79:1944–50. doi: 10.1212/WNL.0b013e3182735e9a
87. Li Y, Sekine T, Funayama M, Li L, Yoshino H, Nishioka K, et al. Clinicogenetic study of GBA mutations in patients with familial Parkinson's disease. Neurobiol Aging. (2014) 35:935.e933–938. doi: 10.1016/j.neurobiolaging.2013.09.019
88. Cilia R, Tunesi S, Marotta G, Cereda E, Siri C, Tesei S, et al. Survival and dementia in GBA-associated Parkinson's disease: the mutation matters. Ann Neurol. (2016) 80:662–73. doi: 10.1002/ana.24777
89. Mazzulli JR, Xu YH, Sun Y, Knight AL, McLean PJ, Caldwell GA, et al. Gaucher disease glucocerebrosidase and alpha-synuclein form a bidirectional pathogenic loop in synucleinopathies. Cell. (2011) 146:37–52. doi: 10.1016/j.cell.2011.06.001
90. Li Y, Ikeda A, Yoshino H, Oyama G, Kitani M, Daida K, et al. Clinical characterization of patients with leucine-rich repeat kinase 2 genetic variants in Japan. J Hum Genet. (2020) 65:771–81. doi: 10.1038/s10038-020-0772-4
91. Schneider SA, Alcalay RN. Neuropathology of genetic synucleinopathies with parkinsonism: review of the literature. Mov Disord. (2017) 32:1504–23. doi: 10.1002/mds.27193
92. Takanashi M, Funayama M, Matsuura E, Yoshino H, Li Y, Tsuyama S, et al. Isolated nigral degeneration without pathological protein aggregation in autopsied brains with LRRK2 p.R1441H homozygous and heterozygous mutations. Acta Neuropathol Commun. (2018) 6:105. doi: 10.1186/s40478-018-0617-y
93. Ysselstein D, Nguyen M, Young TJ, Severino A, Schwake M, Merchant K, et al. LRRK2 kinase activity regulates lysosomal glucocerebrosidase in neurons derived from Parkinson's disease patients. Nat Commun. (2019) 10:5570. doi: 10.1038/s41467-019-13413-w
94. Hasegawa K, Stoessl AJ, Yokoyama T, Kowa H, Wszolek ZK, Yagishita S. Familial parkinsonism: study of original Sagamihara PARK8 (I2020T) kindred with variable clinicopathologic outcomes. Parkinsonism Relat Disord. (2009) 15:300–6. doi: 10.1016/j.parkreldis.2008.07.010
95. Marti-Masso JF, Ruiz-Martinez J, Bolano MJ, Ruiz I, Gorostidi A, Moreno F, et al. Neuropathology of Parkinson's disease with the R1441G mutation in LRRK2. Mov Disord. (2009) 24:1998–2001. doi: 10.1002/mds.22677
96. Kalia LV, Lang AE, Hazrati LN, Fujioka S, Wszolek ZK, Dickson DW, et al. Clinical correlations with Lewy body pathology in LRRK2-related Parkinson disease. JAMA Neurol. (2015) 72:100–5. doi: 10.1001/jamaneurol.2014.2704
97. Quattrone A, Bagnato A, Annesi G, Novellino F, Morgante L, Savettieri G, et al. Myocardial 123metaiodobenzylguanidine uptake in genetic Parkinson's disease. Mov Disord. (2008) 23:21–7. doi: 10.1002/mds.21701
98. Kiral FR, Kohrs FE, Jin EJ, Hiesinger PR. Rab GTPases and Membrane Trafficking in Neurodegeneration. Curr Biol. (2018) 28:R471–r486. doi: 10.1016/j.cub.2018.02.010
99. Steger M, Tonelli F, Ito G, Davies P, Trost M, Vetter M, et al. Phosphoproteomics reveals that Parkinson's disease kinase LRRK2 regulates a subset of Rab GTPases. Elife. (2016) 5. doi: 10.7554/eLife.12813.023
100. Steger M, Diez F, Dhekne HS, Lis P, Nirujogi RS, Karayel O, et al. Systematic proteomic analysis of LRRK2-mediated Rab GTPase phosphorylation establishes a connection to ciliogenesis. Elife. (2017) 6. doi: 10.7554/eLife.31012.018
101. Kelly K, Chang A, Hastings L, Abdelmotilib H, West AB. Genetic background influences LRRK2-mediated Rab phosphorylation in the rat brain. Brain Res. (2021) 1759:147372. doi: 10.1016/j.brainres.2021.147372
102. Nguyen AP, Moore DJ. Understanding the GTPase Activity of LRRK2: regulation, function, and neurotoxicity. Adv Neurobiol. (2017) 14:71–88. doi: 10.1007/978-3-319-49969-7_4
103. Fan Y, Nirujogi RS, Garrido A, Ruiz-Martinez J, Bergareche-Yarza A, Mondragon-Rezola E, et al. R1441G but not G2019S mutation enhances LRRK2 mediated Rab10 phosphorylation in human peripheral blood neutrophils. Acta Neuropathol. (2021) 142:475–94. doi: 10.1007/s00401-021-02325-z
104. Usmani A, Shavarebi F, Hiniker A. The Cell Biology of LRRK2 in Parkinson's Disease. Mol Cell Biol. (2021) 41 e00660–20. doi: 10.1128/MCB.00660-20
105. Matta S, Van Kolen K, da Cunha R, van den Bogaart G, Mandemakers W, Miskiewicz K, et al. LRRK2 controls an EndoA phosphorylation cycle in synaptic endocytosis. Neuron. (2012) 75:1008–21. doi: 10.1016/j.neuron.2012.08.022
106. Hsieh CH, Shaltouki A, Gonzalez AE, Bettencourt da. Cruz A, Burbulla L.F, St Lawrence E, et al. Functional impairment in miro degradation and mitophagy is a shared feature in familial and sporadic Parkinson's disease. Cell Stem Cell. (2016) 19:709–24. doi: 10.1016/j.stem.2016.08.002
107. Eguchi T, Kuwahara T, Sakurai M, Komori T, Fujimoto T, Ito G, et al. LRRK2 and its substrate Rab GTPases are sequentially targeted onto stressed lysosomes and maintain their homeostasis. Proc Natl Acad Sci USA. (2018) 115:E9115–24. doi: 10.1073/pnas.1812196115
108. Beilina A, Bonet-Ponce L, Kumaran R, Kordich JJ, Ishida M, Mamais A, et al. The Parkinson's Disease Protein LRRK2 Interacts with the GARP Complex to Promote Retrograde Transport to the trans-Golgi Network. Cell Rep. (2020) 31:107614. doi: 10.1016/j.celrep.2020.107614
109. Bonet-Ponce L, Beilina A, Williamson C.D, Lindberg E, Kluss J.H, Saez-Atienzar S, et al. (2020). LRRK2 mediates tubulation and vesicle sorting from lysosomes. Sci Adv 6(46). doi: 10.1126/sciadv.abb2454
110. Deniston CK, Salogiannis J, Mathea S, Snead DM, Lahiri I, Matyszewski M, et al. Structure of LRRK2 in Parkinson's disease and model for microtubule interaction. Nature. (2020) 588:344–9. doi: 10.1038/s41586-020-2673-2
111. Liu Z, Xu E, Zhao HT, Cole T, West AB. LRRK2 and Rab10 coordinate macropinocytosis to mediate immunological responses in phagocytes. EMBO J. (2020) 39:e104862. doi: 10.15252/embj.2020104862
112. Watanabe R, Buschauer R, Bohning J, Audagnotto M, Lasker K, Lu T.W, et al. (2020). The In Situ Structure of Parkinson's Disease-Linked LRRK2. Cell 182:1508-1518 e1516. doi: 10.1016/j.cell.2020.08.004
113. Velayos-Baeza A, Vettori A, Copley RR, Dobson-Stone C, Monaco AP. Analysis of the human VPS13 gene family. Genomics. (2004) 84:536–49. doi: 10.1016/j.ygeno.2004.04.012
114. Lesage S, Drouet V, Majounie E, Deramecourt V, Jacoupy M, Nicolas A, et al. Loss of VPS13C Function in autosomal-recessive parkinsonism causes mitochondrial dysfunction and increases PINK1/Parkin-dependent mitophagy. Am J Hum Genet. (2016) 98:500–13. doi: 10.1016/j.ajhg.2016.01.014
115. Gu X, Li C, Chen Y, Ou R, Cao B, Wei Q, et al. Mutation screening and burden analysis of VPS13C in Chinese patients with early-onset Parkinson's disease. Neurobiol Aging. (2020) 94:311.e311–311.e314. doi: 10.1016/j.neurobiolaging.2020.05.005
116. Zou M, Li R, Wang JY, Wang K, Wang YN, Li Y, et al. Association analyses of variants of SIPA1L2, MIR4697, GCH1, VPS13C, and DDRGK1 with Parkinson's disease in East Asians. Neurobiol Aging. (2018) 68:159.e157–9.e114. doi: 10.1016/j.neurobiolaging.2018.03.005
117. Rudakou U, Ruskey JA, Krohn L, Laurent SB, Spiegelman D, Greenbaum L, et al. Analysis of common and rare VPS13C variants in late-onset Parkinson disease. Neurol Genet. (2020) 6:385. doi: 10.1212/NXG.0000000000000385
118. Rampoldi L, Dobson-Stone C, Rubio JP, Danek A, Chalmers RM, Wood NW, et al. A conserved sorting-associated protein is mutant in chorea-acanthocytosis. Nat Genet. (2001) 28:119–20. doi: 10.1038/88821
119. Kolehmainen J, Black GC, Saarinen A, Chandler K, Clayton-Smith J, Traskelin AL, et al. Cohen syndrome is caused by mutations in a novel gene, COH1, encoding a transmembrane protein with a presumed role in vesicle-mediated sorting and intracellular protein transport. Am J Hum Genet. (2003) 72:1359–69. doi: 10.1086/375454
120. Gauthier J, Meijer IA, Lessel D, Mencacci NE, Krainc D, Hempel M, et al. Recessive mutations in >VPS13D cause childhood onset movement disorders. Ann Neurol. (2018) 83:1089–95. doi: 10.1002/ana.25204
121. Seong E, Insolera R, Dulovic M, Kamsteeg EJ, Trinh J, Bruggemann N, et al. Mutations in VPS13D lead to a new recessive ataxia with spasticity and mitochondrial defects. Ann Neurol. (2018) 83:1075–88. doi: 10.1002/ana.25220
122. Kumar N, Leonzino M, Hancock-Cerutti W, Horenkamp FA, Li P, Lees JA, et al. VPS13A and VPS13C are lipid transport proteins differentially localized at ER contact sites. J Cell Biol. (2018) 217:3625–39. doi: 10.1083/jcb.201807019
123. Shiokawa N, Nakamura M, Sameshima M, Deguchi A, Hayashi T, Sasaki N, et al. Chorein, the protein responsible for chorea-acanthocytosis, interacts with β-adducin and β-actin. Biochem Biophys Res Commun. (2013) 441:96–101. doi: 10.1016/j.bbrc.2013.10.011
124. Samaranayake HS, Cowan AE, Klobutcher LA. Vacuolar protein sorting protein 13A, TtVPS13A, localizes to the tetrahymena thermophila phagosome membrane and is required for efficient phagocytosis. Eukaryot Cell. (2011) 10:1207–18. doi: 10.1128/EC.05089-11
125. Pickrell AM, Youle RJ. The roles of PINK1, parkin, and mitochondrial fidelity in Parkinson's disease. Neuron. (2015) 85:257–73. doi: 10.1016/j.neuron.2014.12.007
126. Imai Y. PINK1-Parkin signaling in Parkinson's disease: Lessons from Drosophila. Neurosci Res. (2020) 159:40–6. doi: 10.1016/j.neures.2020.01.016
127. Seifert W, Kühnisch J, Maritzen T, Horn D, Haucke V, Hennies HC. Cohen syndrome-associated protein, COH1, is a novel, giant Golgi matrix protein required for Golgi integrity. J Biol Chem. (2011) 286:37665–75. doi: 10.1074/jbc.M111.267971
128. Baldwin HA, Wang C, Kanfer G, Shah HV, Velayos-Baeza A, Dulovic-Mahlow M, et al. VPS13D promotes peroxisome biogenesis. J Cell Biol. (2021) 220:e202001188. doi: 10.1083/jcb.202001188
129. Seifert W, Kuhnisch J, Maritzen T, Lommatzsch S, Hennies HC, Bachmann S, et al. Cohen syndrome-associated protein COH1 physically and functionally interacts with the small GTPase RAB6 at the Golgi complex and directs neurite outgrowth. J Biol Chem. (2015) 290:3349–58. doi: 10.1074/jbc.M114.608174
130. Guillen-Samander A, Leonzino M, Hanna MG, Tang N, Shen H, De Camilli P. VPS13D bridges the ER to mitochondria and peroxisomes via Miro. J Cell Biol. (2021) 220:e202010004. doi: 10.1083/jcb.202010004
131. Ichinose H, Ohye T, Takahashi E, Seki N, Hori T, Segawa M, et al. Hereditary progressive dystonia with marked diurnal fluctuation caused by mutations in the GTP cyclohydrolase I gene. Nat Genet. (1994) 8:236–42. doi: 10.1038/ng1194-236
132. Segawa M, Nomura Y, Nishiyama N. Autosomal dominant guanosine triphosphate cyclohydrolase I deficiency (Segawa disease). Ann Neurol. (2003) 54:S32–45. doi: 10.1002/ana.10630
133. Trender-Gerhard I, Sweeney MG, Schwingenschuh P, Mir P, Edwards MJ, Gerhard A, et al. Autosomal-dominant GTPCH1-deficient DRD: clinical characteristics and long-term outcome of 34 patients. J Neurol Neurosurg Psychiatry. (2009) 80:839–45. doi: 10.1136/jnnp.2008.155861
134. Mencacci NE, Isaias IU, Reich MM, Ganos C, Plagnol V, Polke JM, et al. Parkinson's disease in GTP cyclohydrolase 1 mutation carriers. Brain. (2014) 137:2480–92. doi: 10.1093/brain/awu179
135. Pan HX, Zhao YW, Mei JP, Fang ZH, Wang Y, Zhou X, et al. GCH1 variants contribute to the risk and earlier age-at-onset of Parkinson's disease: a two-cohort case-control study. Transl Neurodegener. (2020) 9:31. doi: 10.1186/s40035-020-00212-3
136. Ichinose H, Inoue KI, Arakawa S, Watanabe Y, Kurosaki H, Koshiba S, et al. Alterations in the reduced pteridine contents in the cerebrospinal fluids of LRRK2 mutation carriers and patients with Parkinson's disease. J Neural Transm (Vienna). (2018) 125:45–52. doi: 10.1007/s00702-017-1784-x
137. Yoshino H, Nishioka K, Li Y, Oji Y, Oyama G, Hatano T, et al. GCH1 mutations in dopa-responsive dystonia and Parkinson's disease. J Neurol. (2018) 265:1860–70. doi: 10.1007/s00415-018-8930-8
138. Rajput AH, Gibb WR, Zhong XH, Shannak KS, Kish S, Chang LG, et al. Dopa-responsive dystonia: pathological and biochemical observations in a case. Ann Neurol. (1994) 35:396–402. doi: 10.1002/ana.410350405
139. Furukawa Y, Nygaard TG, Gutlich M, Rajput AH, Pifl C, DiStefano L, et al. Striatal biopterin and tyrosine hydroxylase protein reduction in dopa-responsive dystonia. Neurology. (1999) 53:1032–41. doi: 10.1212/WNL.53.5.1032
140. Jeon BS, Jeong JM, Park SS, Kim JM, Chang YS, Song HC, et al. Dopamine transporter density measured by [123I]beta-CIT single-photon emission computed tomography is normal in dopa-responsive dystonia. Ann Neurol. (1998) 43:792–800. doi: 10.1002/ana.410430614
141. Wijemanne S, Jankovic J. Dopa-responsive dystonia–clinical and genetic heterogeneity. Nat Rev Neurol. (2015) 11:414–24. doi: 10.1038/nrneurol.2015.86
142. Sato K, Sumi-Ichinose C, Kaji R, Ikemoto K, Nomura T, Nagatsu I, et al. Differential involvement of striosome and matrix dopamine systems in a transgenic model of dopa-responsive dystonia. Proc Natl Acad Sci USA. (2008) 105:12551–6. doi: 10.1073/pnas.0806065105
143. Wang G, Huang Y, Chen W, Chen S, Wang Y, Xiao Q, et al. Variants in the SNCA gene associate with motor progression while variants in the MAPT gene associate with the severity of Parkinson's disease. Parkinsonism Relat Disord. (2016) 24:89–94. doi: 10.1016/j.parkreldis.2015.12.018
144. Henderson MX, Sengupta M, Trojanowski JQ, Lee VMY. Alzheimer's disease tau is a prominent pathology in LRRK2 Parkinson's disease. Acta Neuropathol Commun. (2019) 7:183. doi: 10.1186/s40478-019-0836-x
145. Wilhelmsen KC, Lynch T, Pavlou E, Higgins M, Nygaard TG. Localization of disinhibition-dementia-parkinsonism-amyotrophy complex to 17q21-22. Am J Hum Genet. (1994) 55:1159–65.
146. Foster NL, Wilhelmsen K, Sima AA, Jones MZ, D'Amato CJ, Gilman S. Frontotemporal dementia and parkinsonism linked to chromosome 17: a consensus conference. Conf Part Ann Neurol. (1997) 41:706–15. doi: 10.1002/ana.410410606
147. Poorkaj P, Bird TD, Wijsman E, Nemens E, Garruto RM, Anderson L, et al. Tau is a candidate gene for chromosome 17 frontotemporal dementia. Ann Neurol. (1998) 43:815–25. doi: 10.1002/ana.410430617
148. Spillantini MG, Murrell JR, Goedert M, Farlow MR, Klug A, Ghetti B. Mutation in the tau gene in familial multiple system tauopathy with presenile dementia. Proc Natl Acad Sci USA. (1998) 95:7737–41. doi: 10.1073/pnas.95.13.7737
149. Hutton M, Lendon CL, Rizzu P, Baker M, Froelich S, Houlden H, et al. Association of missense and 5'-splice-site mutations in tau with the inherited dementia FTDP-17. Nature. (1998) 393:702–5. doi: 10.1038/31508
150. Lee VM, Goedert M, Trojanowski JQ. Neurodegenerative tauopathies. Annu Rev Neurosci. (2001) 24:1121–59. doi: 10.1146/annurev.neuro.24.1.1121
151. van Swieten JC, Stevens M, Rosso SM, Rizzu P, Joosse M, de Koning I, et al. Phenotypic variation in hereditary frontotemporal dementia with tau mutations. Ann Neurol. (1999) 46:617–26. doi: 10.1002/1531-8249(199910)46:4<617::AID-ANA10>3.0.CO;2-I
152. Janssen JC, Warrington EK, Morris HR, Lantos P, Brown J, Revesz T, et al. Clinical features of frontotemporal dementia due to the intronic tau 10(+16) mutation. Neurology. (2002) 58:1161–8. doi: 10.1212/WNL.58.8.1161
153. Ikeda A, Shimada H, Nishioka K, Takanashi M, Hayashida A, Li Y, et al. Clinical heterogeneity of frontotemporal dementia and Parkinsonism linked to chromosome 17 caused by MAPT N279K mutation in relation to tau positron emission tomography features. Mov Disord. (2019) 34:568–74. doi: 10.1002/mds.27623
154. Kasuga K, Kikuchi M, Tokutake T, Nakaya A, Tezuka T, Tsukie T, et al. Systematic review and meta-analysis of Japanese familial Alzheimer's disease and FTDP-17. J Hum Genet. (2015) 60:281–3. doi: 10.1038/jhg.2015.15
155. Arima K, Kowalska A, Hasegawa M, Mukoyama M, Watanabe R, Kawai M, et al. Two brothers with frontotemporal dementia and parkinsonism with an N279K mutation of the tau gene. Neurology. (2000) 54:1787–95. doi: 10.1212/WNL.54.9.1787
156. Wang Y, Mandelkow E. Tau in physiology and pathology. Nat Rev Neurosci. (2016) 17:5–21. doi: 10.1038/nrn.2015.1
157. Takeshige H, Nakayama S, Nishioka K, Li Y, Motoi Y, Hattori N. Marked Reduction in the Striatal Dopamine Transporter Uptake During the Early Stage of Motor Symptoms in Patients with the MAPT N279K Mutation. Intern Med. (2018) 57:3015–9. doi: 10.2169/internalmedicine.0454-17
158. Nakayama S, Shimonaka S, Elahi M, Nishioka K, Oji Y, Matsumoto S.E, et al. (2019). Tau aggregation and seeding analyses of two novel MAPT variants found in patients with motor neuron disease and progressive parkinsonism. Neurobiol Aging 84, 240.e213-240.e222. doi: 10.1016/j.neurobiolaging.2019.02.016
159. Tagai K, Ono M, Kubota M, Kitamura S, Takahata K, Seki C, et al. (2021). High-Contrast In Vivo Imaging of Tau Pathologies in Alzheimer's and Non-Alzheimer's Disease Tauopathies. Neuron 109:42-58.e48. doi: 10.1016/j.neuron.2020.09.042
160. Polinski NK, Martinez TN, Gorodinsky A, Gareus R, Sasner M, Herberth M, et al. Decreased glucocerebrosidase activity and substrate accumulation of glycosphingolipids in a novel GBA1 D409V knock-in mouse model. PLoS ONE. (2021) 16:e0252325. doi: 10.1371/journal.pone.0252325
161. Taguchi YV, Liu J, Ruan J, Pacheco J, Zhang X, Abbasi J, et al. Glucosylsphingosine promotes alpha-synuclein pathology in mutant GBA-associated Parkinson's disease. J Neurosci. (2017) 37:9617–31. doi: 10.1523/JNEUROSCI.1525-17.2017
162. Zunke F, Moise AC, Belur NR, Gelyana E, Stojkovska I, Dzaferbegovic H, et al. Reversible conformational conversion of alpha-synuclein into toxic assemblies by glucosylceramide. Neuron. (2018) 97:92–107 e110. doi: 10.1016/j.neuron.2017.12.012
163. Kett LR, Boassa D, Ho CC, Rideout HJ, Hu J, Terada M, et al. LRRK2 Parkinson disease mutations enhance its microtubule association. Hum Mol Genet. (2012) 21:890–9. doi: 10.1093/hmg/ddr526
164. Law BM, Spain VA, Leinster VH, Chia R, Beilina A, Cho HJ, et al. A direct interaction between leucine-rich repeat kinase 2 and specific beta-tubulin isoforms regulates tubulin acetylation. J Biol Chem. (2014) 289:895–908. doi: 10.1074/jbc.M113.507913
165. Shanley MR, Hawley D, Leung S, Zaidi NF, Dave R, Schlosser KA, et al. LRRK2 Facilitates tau Phosphorylation through Strong Interaction with tau and cdk5. Biochemistry. (2015) 54:5198–208. doi: 10.1021/acs.biochem.5b00326
166. Yang RY, Xue H, Yu L, Velayos-Baeza A, Monaco AP, Liu FT. Identification of VPS13C as a Galectin-12-binding protein that regulates galectin-12 protein stability and adipogenesis. PLoS ONE. (2016) 11:e0153534. doi: 10.1371/journal.pone.0153534
167. Trinh J, Gustavsson EK, Vilarino-Guell C, Bortnick S, Latourelle J, McKenzie MB, et al. DNM3 and genetic modifiers of age of onset in LRRK2 Gly2019Ser parkinsonism: a genome-wide linkage and association study. Lancet Neurol. (2016) 15:1248–56. doi: 10.1016/S1474-4422(16)30203-4
168. Lai D, Alipanahi B, Fontanillas P, Schwantes-An TH, Aasly J, Alcalay RN, et al. Genomewide association studies of LRRK2 modifiers of Parkinson's disease. Ann Neurol. (2021) 90:76–88. doi: 10.1002/ana.26094
169. Oh S, Shao J, Mitra J, Xiong F, D'Antonio M, Wang R, et al. Enhancer release and retargeting activates disease-susceptibility genes. Nature. (2021) 595:735–40. doi: 10.1038/s41586-021-03577-1
170. Mercuri E, Darras BT, Chiriboga CA, Day JW, Campbell C, Connolly AM, et al. Nusinersen versus sham control in later-onset spinal muscular atrophy. N Engl J Med. (2018) 378:625–35. doi: 10.1056/NEJMoa1710504
171. Congdon EE, Sigurdsson EM. Tau-targeting therapies for Alzheimer disease. Nat Rev Neurol. (2018) 14:399–415. doi: 10.1038/s41582-018-0013-z
172. Vaikath NN, Hmila I, Gupta V, Erskine D, Ingelsson M, El-Agnaf OMA. Antibodies against alpha-synuclein: tools and therapies. J Neurochem. (2019) 150:612–25. doi: 10.1111/jnc.14713
Keywords: familial Parkinson's disease, genetics, GWAS, dopamine, alpha-synuclein, LRRK2
Citation: Nishioka K, Imai Y, Yoshino H, Li Y, Funayama M and Hattori N (2022) Clinical Manifestations and Molecular Backgrounds of Parkinson's Disease Regarding Genes Identified From Familial and Population Studies. Front. Neurol. 13:764917. doi: 10.3389/fneur.2022.764917
Received: 26 August 2021; Accepted: 29 April 2022;
Published: 02 June 2022.
Edited by:
Yue Huang, Capital Medical University, ChinaReviewed by:
Gonzalo Arboleda, National University of Colombia, ColombiaMasato Hasegawa, Tokyo Metropolitan Institute of Medical Science, Japan
Copyright © 2022 Nishioka, Imai, Yoshino, Li, Funayama and Hattori. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Kenya Nishioka, bmlzaGlva2FAanVudGVuZG8uYWMuanA=; Yuzuru Imai, eXppbWFpQGp1bnRlbmRvLmFjLmpw